

Abstract
Post‐translational modifications in intestinal epithelial cells (IECs) allow for precise control in intestinal homeostasis, the breakdown of which may precipitate the pathological damage and inflammation in inflammatory bowel disease. The O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) modification on intracellular proteins controls diverse biological processes; however, its roles in intestinal homeostasis are still largely unexplored. Here, we found that levels of protein O‐GlcNAcylation and the expression of O‐GlcNAc transferase (OGT), the enzyme adding the O‐GlcNAc moiety, were reduced in IECs in human IBD patients. Deletion of OGT specifically in IECs resulted in disrupted epithelial barrier, microbial dysbiosis, Paneth cell dysfunction, and intestinal inflammation in mice. Using fecal microbiota transplantation in mice, we demonstrated that microbial dysbiosis although was insufficient to induce spontaneous inflammation but exacerbated chemical‐induced colitis. Paneth cell‐specific deletion of OGT led to Paneth cell dysfunction, which might predispose mice to chemical‐induced colitis. On the other hand, the augmentation of O‐GlcNAc signaling by inhibiting O‐GlcNAcase, the enzyme removing O‐GlcNAcylation, alleviated chemical‐induced colitis. Our data reveal that protein O‐GlcNAcylation in IECs controls key regulatory mechanisms to maintain mucosal homeostasis.
Keywords: epithelial barrier function, gut microbiota, inflammatory bowel disease, Paneth cells, STAT signaling
Subject Categories: Digestive System; Immunology; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), is a group of conditions characterized by chronic or recurring inflammation of the gastrointestinal tract. The induction and perpetuation of intestinal inflammation require the convergence of several abnormalities that affect overlapping layers of homeostatic modules including genetic predisposition, barrier dysfunction, microbial dysbiosis, and immune over‐activation (Khor et al, 2011; Maloy & Powrie, 2011; Kayama & Takeda, 2012; Kamada et al, 2013; Knights et al, 2013; Sonnenberg & Artis, 2015). Intestinal epithelial cells (IECs) establish a barrier between luminal environment and the internal milieu, placing IECs at the center of interactions between the mucosal immune system and luminal antigens and metabolites. A healthy and robust layer of IECs maintains multiple layers of intestinal homeostasis. Dysfunction in IEC biology, such as epithelial barrier malfunction, uncontrolled cell death, and defective autophagy in Paneth cells, drives intestinal inflammation (Khor et al, 2011; Gilbert et al, 2012; Peterson & Artis, 2014).
The epithelial barrier is primarily mediated by the formation of junction complexes between IECs, which connect adjacent IECs to form a continuous physical barrier that restricts luminal pathogens from invading the intestine. The turnover of IECs, such as apoptosis, provides an additional challenge to the maintenance of epithelial continuity. In IBD, dysregulation of junction complexes and cell death both contribute to the “leaky gut” and intestinal inflammation (Turner, 2009).
The Paneth cell is a type of secretory IECs found at the base of the small intestine crypt. It contains large granules high in anti‐microbial peptides (AMPs), which can alter the composition of gut microbiota and counteract enteric pathogens (Bevins & Salzman, 2011). A recent study has demonstrated Paneth cells as a site of origin for intestinal inflammation (Adolph et al, 2013). Several genetic susceptibility alleles for human IBD, such as ATG16L1, NOD2, and XBP1, all lead to Paneth cell dysfunction (Bevins & Salzman, 2011; Khor et al, 2011). Taken together, IECs control multi‐layers of intestinal homeostasis, the disruption of which make a major contribution to the IBD pathogenesis.
O‐GlcNAcylation is the post‐translational modification of serine and threonine residues with β‐N‐acetylglucosamine (O‐GlcNAc) on intracellular proteins (Torres & Hart, 1984; Hart et al, 2007). This dynamic modification is attached by O‐GlcNAc transferase (OGT) and removed by O‐GlcNAcase (OGA). Protein O‐GlcNAcylation acts as a hormone and nutrient sensor to control many biological processes including cell signaling, metabolism, development, and aging (Hanover et al, 2012; Ruan et al, 2012, 2013b, 2014; Yang & Qian, 2017). Nevertheless, the role of intestinal epithelial O‐GlcNAcylation in barrier function and inflammation is still largely unexplored. Herein, we found that levels of OGT and protein O‐GlcNAcylation were downregulated in IECs of IBD patients. IEC‐specific knockout of OGT in mice resulted in permeable epithelial barrier, Paneth cell dysfunction, microbial dysbiosis, and ultimately intestinal inflammation. Elevating intestinal O‐GlcNAcylation levels increased barrier function and protected mice from chemical‐induced inflammation. Our data demonstrate that protein O‐GlcNAcylation in IECs is important for the intestinal homeostasis.
Results
Defective O‐GlcNAc modification in IECs in IBD patients
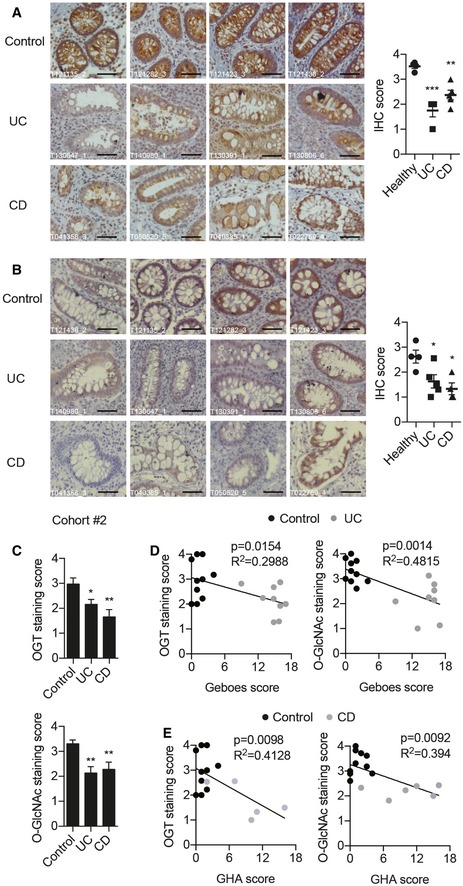
To explore the potential involvement of O‐GlcNAc modification in intestinal inflammation, we performed the immunohistochemistry staining of OGT and protein O‐GlcNAcylation on colon tissues of IBD patients (Appendix Table S1). In both UC and CD, levels of OGT and O‐GlcNAc modification were robustly downregulated in IECs, compared to those in controls (Fig 1A and B). We also observed a similar reduction in levels of OGT and O‐GlcNAcylation in a separate cohort of Chinese UC patients (Fig EV1A and B). Although not statistically significant, the expression of OGT gene in UC patients tended to decrease when compared to healthy subjects (Fig EV1C). To further evaluate whether there was any correlation between disease severity and levels of OGT and O‐GlcNAcylation, we performed the immunohistochemistry staining on another set of intestine biopsies (Appendix Table S2) and confirmed the reduction in OGT and O‐GlcNAcylation levels in IECs of both UC and CD (Fig 1C). Interestingly, intensities of both OGT and O‐GlcNAcylation were negatively correlated with histological disease activities in UC and CD that were determined by Geboes and global histological activity (GHA) scores, respectively (Fig 1D and E, and Dataset EV1; van Loosdregt & Coffer, 2014). These data demonstrate that O‐GlcNAc dysfunction in IECs is associated with IBD.
Figure 1. Defective O‐GlcNAc signaling in intestinal epithelial cells in IBD patients.
-
A, BRepresentative images of OGT (A) and O‐GlcNAc (B) immunohistochemistry on paraffin‐embedded colon sections from control, UC, and CD patients (n = 4). Intensities of staining were scored on the right. Scale bars = 50 μm. (One‐way ANOVA, OGT: Healthy versus UC P = 0.0002, Healthy versus CD P = 0.003; O‐GlcNAc: Healthy versus UC P = 0.0365, Healthy versus CD P = 0.0125).
-
CLevels of intestinal epithelial OGT and O‐GlcNAcylation in a second cohort of control, UC, and CD patients. (One‐way ANOVA, OGT: Control n = 10, UC n = 9, CD n = 6; Control versus UC P = 0.035, Control versus CD P = 0.003; O‐GlcNAc: Control n = 10, UC n = 8, CD n = 8; Control versus UC P = 0.0027, Control versus CD P = 0.008).
-
D, ERegression plots of average immune‐staining scores of OGT and O‐GlcNAcylation against histological scores in UC (D) and CD (E) (D: OGT n = 19, O‐GlcNAc n = 18; E: OGT n = 15, O‐GlcNAc n = 16).
Figure EV1. Defective O‐GlcNAc signaling in epithelial cells of Chinese UC patients.
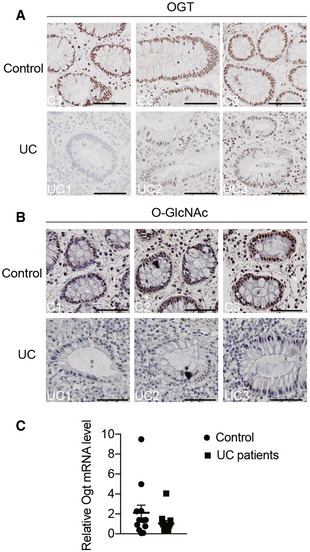
-
A, BRepresentative images of OGT (A) and O‐GlcNAc (B) immunohistochemistry in colon tissues from Chinese normal controls and UC subjects. Scale bars = 50 μm.
-
CmRNA levels of OGT in the colon from Chinese healthy and UC subjects (n = 12).
Epithelial deficiency in OGT causes intestinal damages in mice
To directly examine the role of protein O‐GlcNAcylation in the intestinal epithelia, we generated IEC‐specific Ogt gene knockout mice (Vil‐Ogt KO) by crossing the Villin‐Cre and Ogt‐floxed mouse lines. Immunohistochemistry demonstrated that OGT and O‐GlcNAcylation were specifically and efficiently depleted in both ileum and colon in Vil‐Ogt KO mice (Fig EV2A and B). Male Vil‐Ogt KO mice were viable, but substantially lighter in weight (Fig 2A) and gradually developed rectal prolapse, rectal bleeding, and diarrhea (Fig 2B and C). In females, heterozygous KO mice appeared normal and healthy, while homozygous KO females showed similar phenotypes as KO males including the body weight loss and the progressive rectal prolapse (Fig 2D and E). Later, we used male mice for most experiments. Histological analyses showed that intestinal architecture was disrupted in Vil‐Ogt KO mice, such as the irregularity of the size and shape of crypts and increased crypt branching (Fig 2F).
Figure EV2. Knockout specificity and efficiency in Vil‐OGT KO mice.
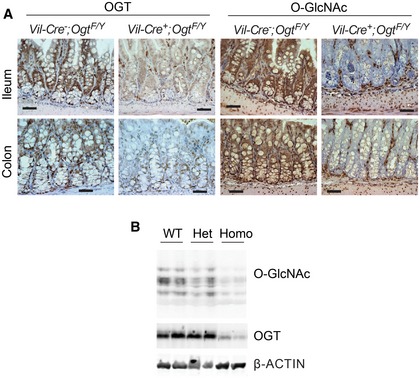
- Immunostaining of OGT and O‐GlcNAc in the ileum and colon sections from male wild‐type and Vil‐Ogt KO mice. Scale bars = 50 μm.
- Immunoblotting of O‐GlcNAc and OGT in the colon of female wild‐type and Vil‐Ogt KO mice.
Source data are available online for this figure.
Figure 2. Loss of OGT in IECs causes intestinal damages in mice.

- Body weight of male wild‐type and Vil‐Ogt KO mice at 6 weeks of age (n = 6, P = 0.000015).
- Incidence of rectal prolapse in male wild‐type and Vil‐Ogt KO mice (WT n = 17, KO n = 9, P < 0.0001).
- Representative colonoscopy images of male wild‐type and Vil‐Ogt KO mice.
- Body weight of female wild‐type and Vil‐Ogt KO mice at 9 weeks of age (WT n = 11, heterozygous KO n = 4, homozygous KO n = 5, P < 0.0001).
- Incidence of rectal prolapse in female wild‐type and Vil‐Ogt KO mice (WT n = 9, heterozygous KO n = 8, homozygous KO n = 6, P = 0.0439).
- H&E staining of duodenum, jejunum, ileum, colon, and rectum of 10‐week‐old male wild‐type and Vil‐Ogt KO mice. Scale bars = 100 μm.
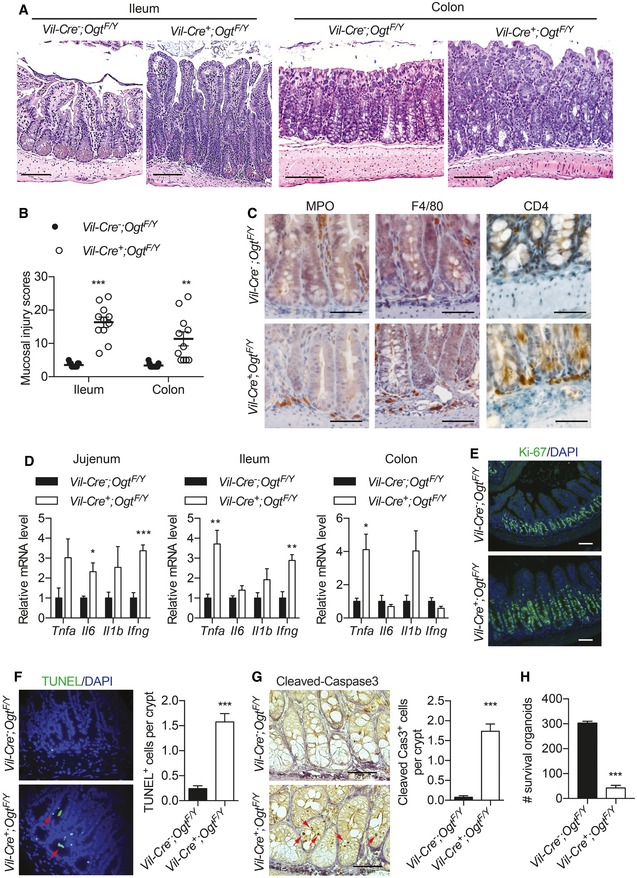
We then utilized semi‐quantitative pathology to qualify the pathological alterations in ileal and colonic mucosa (Erben et al, 2014; Gilbert et al, 2015). Significantly increased intestinal epithelial hypertrophy, epithelial hyperplasia, and mucosal thickness were observed in the Vil‐Ogt KO mice (Fig 3A and B). Immunohistochemistry revealed a modest increase in infiltrating neutrophils, macrophages, and CD4 T cells at the crypt base of the ileum in Vil‐Ogt KO mice (Fig 3C). Quantitative reverse transcription PCR (RT–qPCR) showed that the expression of inflammatory genes including Tnfa, Il6, Il1b, and Ifng was largely upregulated in the jejunum, ileum, and colon of Vil‐Ogt KO mice when compared to control mice (Fig 3D). Immunofluorescence of Ki‐67 showed significantly greater amounts of proliferating epithelial cells in the ileum of Vil‐Ogt KO (Fig 3E). In addition, TUNEL assay and immunostaining of Cleaved‐CASPASE3 showed increased apoptotic cells in the crypt region of ileum in Vil‐Ogt KO mice (Fig 3F and G). Consistently, cultured ileal organoids from KO mice had a profound decrease in viability, when compared to control organoids (Fig 3H). Collectively, our data illustrate that loss of protein O‐GlcNAcylation in IECs results in dysregulated proliferative/apoptotic homeostasis and susceptibility to inflammation in mice.
Figure 3. IEC‐specific loss of OGT disrupts intestinal proliferative/apoptotic homeostasis.
-
ARepresentative images of the H&E staining of ileum and colon of 10‐week‐old male wild‐type and Vil‐Ogt KO mice. Scale bars = 100 μm.
-
BCombined scores of the mucosal injury in 10‐week‐old male wild‐type and Vil‐Ogt KO mice (n = 11, ileum: P < 0.0001, colon: P = 0.0012).
-
CRepresentative images of MPO, F4/80, and CD4 immunohistochemistry in the ileum tissue of male wild‐type and Vil‐Ogt KO mice. Scale bars = 50 μm.
-
DRT–qPCR of inflammatory markers in jejunum, ileum, and colon (n = 5, jejunum: Il6 P = 0.029, Ifng P = 0.0009; ileum: Tnfa P = 0.0094, Ifng P = 0.0066; colon: Tnfa P = 0.028).
-
EImmunofluorescent staining of Ki‐67 in ileal tissues (n = 5). Scale bars = 50 μm.
-
F, GTUNEL staining (F) and immunostaining of Cleaved‐CASPASE3 (G) in the ileum. Quantification of the numbers of positive cells is shown at the right. (TUNEL WT n = 9, KO n = 6, P = 0.0003; Cleaved Cas3 WT = 8, KO = 7, P < 0.0001).
-
HNumbers of survived organoids per 1000 isolated ileal crypts (WT n = 4, KO n = 23, P < 0.0001).
Defective intestinal barrier in Vil‐Ogt KO mice
Intestinal barrier dysfunction potentiates and sustains intestinal inflammation (Turner, 2009), and we then sought to test whether the deficiency in O‐GlcNAcylation in IECs alters epithelial barrier function. Vil‐Ogt KO mice exhibited increased plasma levels of fluorescein isothiocyanate (FITC) after oral gavage of FITC‐dextran (Fig 4A) and increased fecal albumin (Fig 4B), indicating that Vil‐Ogt KO mice had increased intestinal permeability. A major component of the mucosal barrier is the junction complex between IECs (Turner, 2009). Electron microscopy of the wild‐type intestine clearly showed the tight junctions (arrows), the adherens junctions (arrowheads), and desmosomes (stars) (Fig 4C). In Vil‐Ogt KO mice, however, the density of perijunctional ring was profoundly decreased (Fig 4C). Desmosomes were reduced in number, which sometime caused open paracellular space (Fig 4C). We also observed splenomegaly (Fig EV3A) and increased levels of inflammatory gene expression in the liver (Fig EV3B), indicating that the leaky gut in Vil‐Ogt KO mice caused systemic inflammation.
Figure 4. Intestinal barrier dysfunction in Vil‐Ogt KO mice.
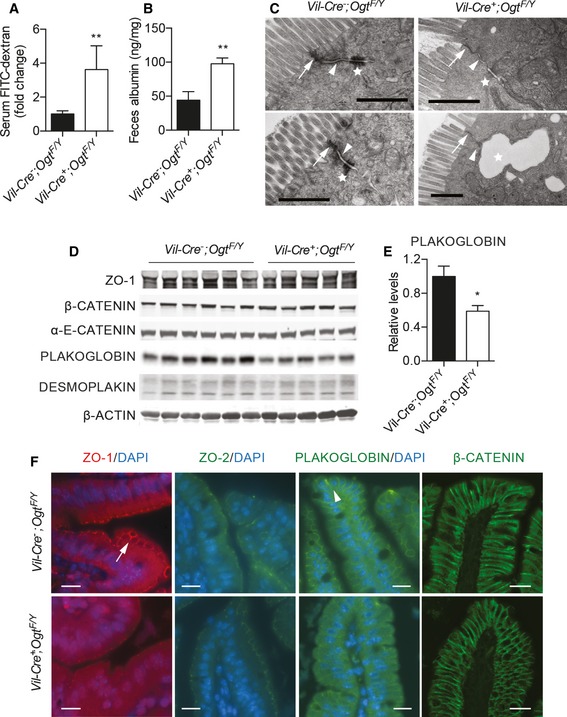
-
A, BIntestinal barrier functional assays measuring serum FITC‐dextran (A) and albumin from fecal sample (B) of male wild‐type and Vil‐Ogt KO mice. (A: WT n = 5, KO n = 6, P = 0.0015; B: WT n = 9, KO n = 6, P = 0.0053).
-
CRepresentative EM pictures of ileum epithelial cells. Arrows, arrowheads, and stars indicate tight junctions, adherens junctions, and desmosomes, respectively. Scale bars = 1 μm.
-
DImmunoblotting of protein markers of tight junction (ZO‐1), adherens junction (β‐CATENIN, α‐E‐CATENIN, and PLAKOGLOBIN), and desmosome (PLAKOGLOBIN and DESMOPLAKIN) in colon.
-
EDensitometric analysis of PLAKOGLOBIN protein levels in (D). (WT n = 6, KO n = 5, P = 0.021).
-
FRepresentative images of ZO‐1, ZO‐2, PLAKOGLOBIN, and β‐CATENIN immunofluorescent staining in the ileum tissue of male wild‐type and Vil‐Ogt KO mice. Arrow and arrowhead indicate ZO‐1 and PLAKOGLOBIN, respectively. Scale bars = 10 μm.
Figure EV3. Systemic inflammation in Vil‐Ogt KO mice.

-
A, BGross morphology of spleen (A) and RT–qPCR of inflammatory markers in liver (B) from male wild‐type and Vil‐Ogt KO mice (WT n = 4, KO n = 6, Il6 P = 0.0271).
-
CImmunoblotting of protein markers of tight junction including OCCLUDIN, CLAUDIN 3 and 5 in colon. The blots were from the same experiment as shown in Fig 7A.
We then performed Western blotting and immunofluorescence using antibodies against markers of junction complexes and found that PLAKOGLOBIN, a critical component of both desmosomes and adherens junctions that anchors transmembrane cadherins to intermediate filaments, was downregulated in Vil‐Ogt KO mice (Fig 4D–F). Even though the expression level was not changed, the tight junction protein zonula occludens‐1 (ZO‐1) remarkably lost its localization to the cell membrane in Vil‐Ogt KO mice (Fig 4D and F). We did not observe any changes in other junction complex markers including ZO‐2, β‐CATENIN, α‐E‐CATENIN, DESMOPLAKIN, CLAUDIN 3 and 5, and OCCLUDIN (Figs 4D and F, and EV3C). Taken together, these findings demonstrate that the loss of OGT in IECs impairs epithelial barrier function, which may potentiate the development of intestinal inflammation.
Microbial dysbiosis in Vil‐Ogt KO mice
Microbial dysbiosis plays an important role in inflammatory bowel disease (Bevins & Salzman, 2011), and we then sought to determine whether the deficiency in intestinal epithelial O‐GlcNAcylation interferes with gut microbiota. 16S rRNA gene sequencing was used to interrogate differences in fecal microbiota between singly housed wild‐type and Vil‐Ogt KO mice. The phylogenetic diversity was comparable between control and KO mice (Fig EV4A). Principal coordinate analysis (PCoA) of the unweighted UniFrac showed that bacterial communities within the same genotype had similar bacterial composition no matter where they were housed (Fig 5A). Linear discriminant analysis (LDA) effect size (LEfSe) highlighted differentially abundant taxonomic clades (Fig EV4B). The phylum Firmicutes, the Coriobacteriia class within the Actinobacteria phylum, and Mycoplasmatales order within the Tenericutes phylum were decreased, while the Bacteriodetes family Bacteriodaceae and the Gammaproteobacteria class within the Proteobacteria phylum were enriched in Vil‐Ogt KO mice (Figs 5B and EV4C and D). Interestingly, the most consistent observations of microbial dysbiosis in IBD patients are a reduction in Firmicutes and an increase in Proteobacteria (Matsuoka & Kanai, 2015).
Figure EV4. Changes in microbial composition in Vil‐Ogt KO mice.
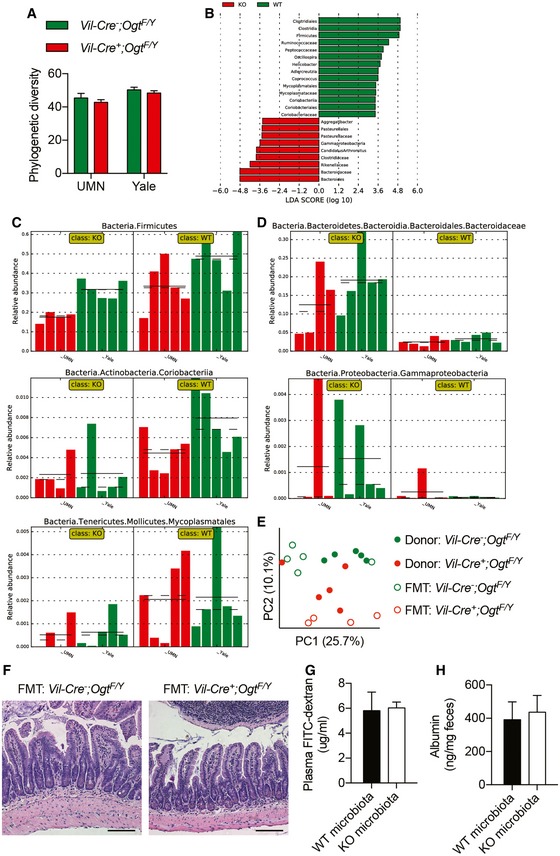
-
APhylogenetic diversity of fecal bacteria from mice that were individually housed at UMN (WT n = 5, KO n = 4) or Yale (n = 5).
-
BDifferentially abundant taxomonic clades analyzed by LEfSe.
-
C, DAbundance histogram plots of taxonomic groups detected by LEfSe that were downregulated (C) or upregulated (D) in Vil‐Ogt KO mice (n = 4–5).
-
E–HAntibiotic‐treated mice were transplanted with gut microbiota from control or Vil‐Ogt KO mice (n = 5). (E) PCoA plot of unweighted UniFrac distance of bacterial communities. (F) H&E staining of colon. Scale bars = 100 μm. (G) Levels of serum FITC‐dextran. (H) Albumin from fecal samples.
Figure 5. Microbial dysbiosis in OGT‐deficient mice.

-
APCoA plot of unweighted UniFrac distance of bacterial communities in wild‐type and Vil‐Ogt KO mice that were singly housed at Yale or UMN (UMN KO n = 4, rest n = 5).
-
BCladogram showing the most discriminative bacterial clades between wild‐type and Vil‐Ogt KO mice that were identified by LEfSe. Regions in red indicate clades that were enriched in Vil‐Ogt KO mice, while regions in green indicate clades that were enriched in wild‐type mice.
-
C–HAntibiotic‐treated C57BL/6 male mice were transplanted with fecal microbiota from wild‐type or Vil‐Ogt KO mice and then induced with DSS for colitis (WT n = 5, KO n = 7). (C) Daily changes in body weight. (D) Colitis scores. (E) Intestinal barrier functional assays measuring serum FITC‐dextran on Day 10. (F) H&E staining of colon tissues. Ulcer areas are designated by red circles. Scale bars = 200 μm. (G) Pathological scores of the mucosal injury. (H) RT–qPCR of inflammatory markers in the colon. (D: P = 0.0409; E: P = 0.0457; G: P = 0.032).
To determine whether microbial dysbiosis in Vil‐Ogt KO mice contributes to the pathogenesis of intestinal inflammation, we first performed fecal microbiota transplantation (FMT) from control and Vil‐Ogt KO mice into antibiotic‐treated wild‐type mice. Compositional differences between the microbiota from control and Vil‐Ogt KO mice generally persisted in the corresponding FMT recipients (Fig EV4E). No difference in intestinal morphology (Fig EV4F) or barrier function (Fig EV4G and H) was observed between mice transplanted with microbiota from wild‐type or Vil‐Ogt KO mice. We then induced acute colitis in these mice with dextran sodium sulfate (DSS) and found that mice receiving microbiota from Vil‐Ogt KO mice showed a tendency to lose more body weight (Fig 5C), increased colitis score (Fig 5D), and increased intestinal permeability at 5 days after DSS removal (Fig 5E). Histological examination showed severe intestinal injury and inflammation in Vil‐Ogt KO FMT mice after 5‐day recovery (Fig 5F), characterized by severer immune infiltration, more necrotic crypts, and larger size of ulcers when compared to the control group (Fig 5G). The expression of inflammatory markers including Il1b and Il6 in recipients of Vil‐Ogt KO microbiota showed an increasing tendency as compared to mice undergoing a control FMT (Fig 5H). These data together indicate that, although not sufficient to induce intestinal dysfunction, microbial dysbiosis in Vil‐Ogt KO mice exacerbates DSS‐induced colitis.
OGT regulates Paneth cell function
The Paneth cell is a type of secretory IECs found at the base of the small intestine crypt. It contains large granules high in anti‐microbial peptides (AMPs), which can alter the composition of gut microbiota and counteract enteric pathogens (Bevins & Salzman, 2011). Recent studies have demonstrated Paneth cells as a site of origin for intestinal inflammation (Adolph et al, 2013). We found that Paneth cells were missing in some crypts in the ileum of Vil‐Ogt KO mice (Figs 3A and 6A). H&E staining and electron microscopy further showed that KO Paneth cells had much smaller but more eosinophilic granules (Figs 3A and 6B). TUNEL and Cleaved‐CASPASE3 staining was striking in the crypt base where Paneth cells are located (Fig 3F and G). However, the expression of the Lzy1 gene, a marker of Paneth cells, was not changed (Fig 6C), suggesting that OGT knockout might not affect the differentiation of Paneth cells but rather promote Paneth cell death. The expression of AMP genes, including various Defensins and Ang4, was significantly downregulated in Vil‐Ogt KO ileum (Fig 6C). Mounting evidence suggests that IBD‐associated mutations in autophagy‐related genes impair Paneth cell function (Cadwell et al, 2009; Henckaerts et al, 2011; Patel & Stappenbeck, 2013; Chu et al, 2016). Consistent with our recent finding that protein O‐GlcNAcylation promotes hepatic autophagy (Ruan et al, 2017), pro‐LC3 was accumulated and failed to be cleaved and activated to form LC3‐II in the IEC of Vil‐Ogt KO mice (Fig 6D). These data indicate that loss of OGT causes Paneth cell dysfunction.
Figure 6. Paneth cell dysfunction in OGT‐deficient mice.

-
ANumbers of Paneth cells in the ileum of wild‐type and Vil‐Ogt KO mice (n = 6, P = 0.0048).
-
BRepresentative electron microscopic images of Paneth cells. Scale bars = 1 μm.
-
CExpression of AMP genes in ileum tissues of wild‐type and Vil‐Ogt KO mice (n = 6, Pan Defesin P = 0.031, Defa3 P = 0.049, Defa4 P = 0.013, Defa5 P = 0.023, Defa6 P = 0.034, Ang4 P = 0.031).
-
DImmunoblotting of LC3 in IEC cells isolated from WT and Vil‐Ogt KO mice.
-
ERepresentative images of O‐GlcNAc staining in Paneth cells of male wild‐type and Defa6‐Ogt KO mice. Stars indicate lysozyme‐positive Paneth cells.
-
FMorphology and numbers of Paneth cells in the ileum of wild‐type and Defa6‐Ogt KO mice (n = 5, P = 0.024). Arrow indicates Paneth cell. Scale bars = 10 μm.
-
GExpression of AMP genes in the ileum of wild‐type and Defa6‐Ogt KO mice (n = 5 Pan Defesin P = 0.0291, Defa3 P = 0.0462).
-
HRepresentative images of cultured organoid on Day 3.
-
IExpression of inflammatory markers in the ileum of wild‐type and Defa6‐Ogt KO mice (n = 5).
-
J–LAntibiotic‐treated wild‐type and Defa6‐Ogt KO male mice were transplanted with fecal microbiota from Vil‐Ogt KO mice and then induced with DSS for colitis. Daily changes in body weight (J), colitis scores (K), and RT–qPCR of inflammatory genes in the ileum (L) are shown (n = 4–5). (L: P = 0.049).
To determine whether O‐GlcNAcylation autonomously controls Paneth cell function, we generated Paneth cell‐specific OGT (Defa6‐Ogt) KO mice by crossing the Ogt‐floxed mice to the Defa6‐iCre line (Adolph et al, 2013). Immunofluorescent staining demonstrated that O‐GlcNAcylation was enriched in the nucleus of Paneth cells in WT ileum, and it was specifically and efficiently depleted in Paneth cells of Defa6‐Ogt KO mice (Fig 6E). H&E staining showed that these mutant mice had fewer Paneth cells and granules were more eosinophilic, similar to the morphological changes observed in Vil‐Ogt KO mice (Fig 6F). The reduction in Paneth cell number was confirmed by immunofluorescent staining using an anti‐lysozyme antibody (Fig EV5). The expression of AMP genes was also downregulated (Fig 6G). Paneth cell is an important source of niche factors to support intestinal stem cells in culture (Barker, 2014). We found that the majority of cultured ileal organoids from Defa6‐Ogt KO mice could not survive (Fig 6H). These data suggest that protein O‐GlcNAcylation is indispensable for the survival and function of Paneth cells.
Figure EV5. Specific knockout of OGT in Paneth cells.
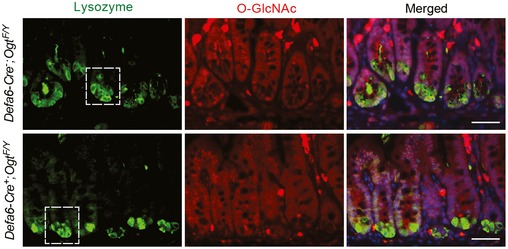
Immunostaining of protein O‐GlcNAcylation in Paneth cells (marked by lysozyme staining) from wild‐type and Defa6‐Ogt KO mice, showing the knockout specificity/efficiency of OGT and the reduction of Paneth cell numbers. Zoom‐in view of the regions indicated by the rectangles was shown in Fig 6E. Scale bars = 50 μm.
However, we did not observe any intestinal inflammation in Defa6‐Ogt KO mice (Fig 6I). Intestinal structure and barrier were largely intact in Defa6‐Ogt KO mice, indicating that Paneth cell dysfunction alone is not sufficient to precipitate intestinal inflammation. Recent evidence suggests that a second “hit” is required for Paneth cell dysfunction to induce intestinal inflammation (Kaser et al, 2008). We then asked whether alterations in gut microbiota together with the disruption of Paneth cell function instigate a pro‐inflammatory response. Control and Defa6‐Ogt KO mice were treated with an antibiotic cocktail and then both received FMT from Vil‐Ogt KO mice. When colitis was induced by DSS, Defa6‐Ogt KO mice receiving the dysbiotic microbiota derived from Vil‐Ogt KO mice had a tendency to lose more body weight (Fig 6J), showed more severe colitis (Fig 6K), and expressed higher levels of inflammatory genes including Ifng and Il1b (Fig 6L), compared to control mice receiving the same FMT. Collectively, these data demonstrate that intestinal epithelial OGT regulates multiple homeostatic modules, including gut microbiota and Paneth cell function, to prevent from spontaneous and induced intestinal inflammation.
Defective O‐GlcNAcylation of STAT proteins in Vil‐Ogt KO mice
We then sought to determine the molecular mechanisms by which OGT controls intestinal homeostasis. Key regulators in intestinal inflammation such as NF‐κB and STAT3 can be modified and regulated by O‐GlyNAcylation (Yang et al, 2015; Li et al, 2017); however, we did not observe any changes in the phosphorylation of NF‐κB or STAT3 in the colon of Vil‐Ogt KO mice (Fig 7A). To identify other potential molecules and pathways modulated by O‐GlcNAcylation, we performed RNA sequencing of isolated IECs from the ileum and colon of control and Vil‐Ogt KO mice. The sample correlation matrix and hierarchical clustering based on the correlation showed that IECs from control and Vil‐Ogt KO mice clustered separately (Fig 7B). We identified 3,247 and 2,031 genes that significantly show more than twofold changes in expression in ileal and colonic IECs, respectively (Fig 7C). These differentially expressed genes were then subjected for Ingenuity Pathway Analysis (IPA). Many inflammatory pathways were enriched, such as atherosclerosis signaling, IL‐10 signaling, Th1 and Th2 pathways, and T‐cell receptor signaling (Fig 7D). To find potential upstream regulators, a bioinformatics program, distant regulatory elements of coregulated genes (DiRE; Gotea & Ovcharenko, 2008), was used to predict common regulatory elements for these differentially expressed genes. STAT proteins including STAT1 were predicted as top candidate transcription factors (Fig 7E). It has been shown that many STAT proteins can be modified and regulated by O‐GlcNAcylation (Gewinner et al, 2004; Freund et al, 2017), and loss of STAT proteins potentiates the intestinal injury and mucosal inflammation (Gilbert et al, 2012; Willson et al, 2013; Chiriac et al, 2017). We found that in KO IECs, levels of total and O‐GlcNAcylated STAT1 were reduced (Fig 7F), indicating that O‐GlcNAcylation may control the stability of STAT1 proteins (Ruan et al, 2013a).
Figure 7. O‐GlcNAc regulation of STAT1 in IECs.

-
AExpression of phosphorylated and total STAT3 and NF‐κB p65 in the colon of wild‐type and Vil‐Ogt KO mice.
-
BIleum and colon IECs from 10‐week‐old wild‐type and Vil‐Ogt KO mice were isolated for RNA‐sequencing analyses. The heatmaps of the sample‐to‐sample distances are shown.
-
CVolcano plots of gene expression changes. The x‐axis specifies the fold changes (FC), and the y‐axis specifies the negative logarithm to the base 10 of the P‐values adjusted for multiple testing with the Benjamini–Hochberg procedure. FC > 2 and P < 0.05 genes are shown in solid dots.
-
DMost significantly enriched canonical signaling pathways in differentially expressed genes, determined by the Comparison Analysis module in the IPA program.
-
ETop candidate transcription factors controlling the observed differentially expressed genes, predicted by DiRE.
-
FLevels of STAT1 protein in IECs determined by immunoblotting and O‐GlcNAc modification of STAT1 determined by immunoprecipitation using the O‐GlcNAc antibody followed by Immunoblotting.
-
G, HCaco‐2 cells were treated with 100 uM Ac45S‐GlcNAc for 24 h, and levels of STAT1 and global O‐GlcNAcylation (G) and STAT1 O‐GlcNAcylation (H) were determined.
-
IsiRNAs against scrambled sequence or human OGT were transfected in 293T cells, and STAT1 levels were determined 48 h later.
-
JThe pGAS‐luc Cis‐Reporter plasmid was cotransfected with siRNAs in 293T cells for luciferase assay to determine the transcriptional activity of STAT1 (n = 4, P = 0.0003).
To test the regulation of STAT1 protein levels by O‐GlcNAcylation, we first treated Caco‐2 cells with an inhibitor of OGT, Ac45S‐GlcNAc (Gloster et al, 2011). Global O‐GlcNAcylation was dramatically downregulated by Ac45S‐GlcNAc, as well as the levels of STAT1 total protein (Fig 7G) and STAT1 O‐GlcNAcylation (Fig 7H). In 293T cells, knocking down OGT also reduced the levels of STAT1 protein (Fig 7I). More importantly, OGT knockdown significantly blunted the transcription activity of STAT1 (Fig 7J). These data suggest that defective STAT1 signaling caused by the loss of O‐GlcNAcylation may contribute to intestinal defects observed in Vil‐Ogt KO mice.
Inhibition of OGA diminished DSS‐induced intestinal inflammation
We have shown that O‐GlcNAc signaling is defective in IBD patients and Ogt knockout in IECs leads to intestinal damage and inflammation in mice. To determine whether pharmacologically elevated protein O‐GlcNAcylation strengthens barrier function and protects mice from chemical‐induced acute colitis, we orally administered water or Thiamet‐G (TMG, an OGA inhibitor) to C57BL/6 mice (n = 5) for 2 weeks. The mice were then treated with 2.5% DSS in drinking water for 5 days followed by 4 days of water only. TMG‐treated mice lost less body weight (Fig 8A) and had lower colitis score (Fig 8B) measured as described in Alenghat et al (2013). In vivo barrier functional assay showed that TMG treatment reduced intestinal permeability before and after DSS induction (Fig 8C and D). TMG‐treated mice also had longer colon length (Fig 8E), accelerated mucosal recovery (Fig 8F), and lower levels of inflammatory genes such as Tnfa and Il1b (Fig 8G). These data demonstrate that augmenting global protein O‐GlcNAcylation by TMG pretreatment ameliorates chemical‐induced acute colitis.
Figure 8. Elevation of protein O‐GlcNAcylation diminishes DSS‐induced intestinal inflammation.

-
A, BWild‐type mice were treated with H2O or TMG for 2 weeks before the induction of colitis by DSS. Daily changes in body weight (A) and colitis score (B; n = 5). (B: Day 6 P = 0.0171, Day 7 P = 0.0445)
-
CIntestinal barrier functional assays measuring serum FITC‐dextran before DSS treatment (n = 5, P = 0.0117).
-
D–GAlbumin levels in feces (D), colon length (E), H&E staining of colon tissues (F), and RT–qPCR of inflammatory genes in the colon (G) on Day 4 of recovery (n = 5). Scale bars = 100 μm. (E: P = 0.0474; G: Tnfa P = 0.0190, Il1b P = 0.0301).
Discussion
Genomewide association studies (GWASs) have identified more than 160 IBD‐associated gene loci (Khor et al, 2011; Cleynen & Vermeire, 2015); however, the heterogeneity of IBD and the low disease penetrance in individuals carrying disease‐susceptibility alleles suggest that complex environment–host interactions cause IBD (Kayama & Takeda, 2012; Kamada et al, 2013; Knights et al, 2013; Sonnenberg & Artis, 2015). A multi‐hit model of IBD has thus been proposed (Maloy & Powrie, 2011). The induction and perpetuation of intestinal inflammation require the convergence of several abnormalities that affect overlapping layers of regulatory modules, including genetic susceptibility, barrier defects, microbial dysbiosis, and sustained innate immunity. Humans or animals with defects in one layer of these modules are only predisposed to but not sufficient to develop IBD.
Here, we report that protein post‐translational O‐GlcNAc modification in the intestinal epithelium is compromised in human IBD. IEC‐specific knockout of OGT leads to intestinal damage and inflammation in mice. The early‐onset spontaneous intestinal damages observed in Vil‐Ogt KO mice suggests that protein O‐GlcNAcylation is a regulator of multiple homeostatic modules in the epithelium. We demonstrated that O‐GlcNAc deficiency results in disruptive epithelial barrier, Paneth cell dysfunction, and microbial dysbiosis. Inducing Paneth cell disorder by knocking out the Ogt gene specifically only in Defa6‐expressing cells or transplanting fecal microbiota from Vil‐Ogt KO mice into antibiotic‐treated wild‐type mice did not cause intestinal inflammation, again supporting the multi‐hit model of IBD (Maloy & Powrie, 2011). Nevertheless, microbial dysbiosis and Paneth cell dysfunction together may potentiate chemical‐induced inflammation.
Many genetically engineered mouse models have been established to understand the molecular mechanisms and to develop therapeutic strategies for IBD (Mizoguchi, 2012; Kiesler et al, 2015). However, only a handful of IEC‐specific genetic models were shown to develop spontaneous intestinal inflammation; these include NEMO IEC‐KO, IKK1/2 IEC‐KO, TAK1 IEC‐KO, XBP1 IEC‐KO, and ATG16L1 IEC‐KO mice (Nenci et al, 2007; Kajino‐Sakamoto et al, 2008; Kaser et al, 2008; Tschurtschenthaler et al, 2017). NEMO, IKK1 and 2, and TAK1 are all required for the activation of nuclear factor kappa B (NF‐κB), suggesting that IEC‐specific NF‐κB signaling is protective in intestinal inflammation. Abnormal autophagy and unresolved endoplasmic reticulum (ER) stress are common features of IBD (Cadwell et al, 2008; Kaser et al, 2008). Dysfunctions in either unfolded protein response factor, X‐box binding protein 1 (XBP1), or autophagy (ATG16L1 or ATG7) in IECs result in the reciprocal compensatory, and severe spontaneous ileitis develops if both mechanisms are defective (Adolph et al, 2013).
O‐GlcNAcylation has been associated with both pro‐ and anti‐inflammatory effects in various conditions (Baudoin & Issad, 2014; Zheng et al, 2017; Thi Do et al, 2018). The NF‐κB complex is modified by O‐GlcNAc; however, O‐GlcNAcylation has both positive (James et al, 2002; Yang et al, 2008; Allison et al, 2012; Ramakrishnan et al, 2013; Zhang et al, 2015) and negative (Zou et al, 2009; Xing et al, 2011) effects on NF‐κB activity and inflammation based on reported results. Here, we did not observe any changes in NF‐κB activity in Vil‐Ogt KO mice, suggesting intestinal epithelial O‐GlcNAcylation is not a major driver of the NF‐κB pathway. In contrast to our findings, a recent work showed that OGA+/− knockout mice are susceptible to DSS‐induced colitis, suggesting that hyper‐O‐GlcNAcylation may promote intestinal inflammation by activating NF‐κB signaling (Yang et al, 2015). However, in the whole‐body heterozygous knockout mouse model used by Yang et al, it is unclear what cell types are important in mediating the function of OGA in DSS‐induced colitis. Given the protective role of NF‐κB in IEC (Nenci et al, 2007; Kajino‐Sakamoto et al, 2008), it is unlikely that the elevation of protein O‐GlcNAcylation in IECs contributes to the phenotype. In addition, Li et al (2017) showed that inhibition of OGT in macrophage by myeloid‐derived cullin 3 protects against intestinal inflammation. They found that the E3 ubiquitin ligase cullin 3 downregulated the expression of OGT gene, thereby inhibiting STAT3 O‐GlcNAcylation. Since both NF‐κB and STAT3 are critical components in immune activation, we propose that protein hyper‐O‐GlcNAcylation in the immune system may promote immune activation and intestinal inflammation.
On the other hand, both NF‐κB and STAT signaling pathways are indispensable for intestinal epithelial homeostasis, as IEC‐specific deficiencies in either the NF‐κB complex or STAT family proteins cause or predispose to intestinal inflammation (Nenci et al, 2007; Kajino‐Sakamoto et al, 2008; Gilbert et al, 2012; Willson et al, 2013; Chiriac et al, 2017). By RNA sequencing of IECs, we predicted STAT proteins, particularly STAT1, as a downstream target of protein O‐GlcNAcylation and mediator of intestinal damage in Vil‐Ogt KO mice. Several STAT proteins including STAT1, STAT3, STAT5A, STAT5B, and STAT6 can be O‐GlcNAc‐modified (Gewinner et al, 2004; Freund et al, 2017). We confirmed that STAT1 in IECs was O‐GlcNAcylated, and loss of OGT reduced STAT1 expression and transcription activity. In the intestinal epithelium, deficiencies in STAT1, STAT3, or STAT5A did not lead to spontaneous inflammation, even though these mice were prone to chemical‐induced colitis (Gilbert et al, 2012; Willson et al, 2013; Chiriac et al, 2017). Redundancy and compensation between different STAT proteins may attenuate the detrimental effects caused by the loss of individual STAT proteins. However, in Vil‐Ogt KO mice, impairment in O‐GlcNAc signaling is likely to dysregulate multiple STAT proteins to elicit profound inflammation in the epithelium. Further studies on the O‐GlcNAcylation of specific STAT proteins are warranted to delineate the precise control and function of protein O‐GlcNAcylation in intestinal homeostasis and inflammation.
In human IBD, levels of OGT protein expression and global O‐GlcNAcylation in epithelial cells are greatly decreased. Linear regression analysis indicated an inverse correlation between disease severity and OGT/O‐GlcNAc levels. Statistical significance was reached even with limited numbers of samples, emphasizing the clinical importance of epithelial O‐GlcNAcylation in intestinal pathology. However, we could not observe such correlations within disease‐only groups (data not shown), probably due to the small sample size and the lack of a broad spectrum of disease severity. A larger cohort, with controlled age, gender, diet, disease status, and treatment, is needed in the future to elucidate the relationship between the reduction in OGT and the progress of disease. Nevertheless, no mutations in or polymorphisms associated with the OGT gene have be reported, probably because the OGT gene is resided in the chromosome X, which is commonly excluded from GWAS analyses. Further investigations are required to determine the cause of deficiencies in protein O‐GlcNAcylation in IBD.
Currently, there is no cure for IBD. Patients rely on diet and lifestyle changes, drug therapy, or surgery to relieve symptoms and induce remission. A remarkable observation of this study is that pretreatment with an OGA inhibitor to elevate the intestinal epithelial O‐GlcNAc modification protects mice from chemical‐induced colitis. It is warranted to test whether genetic and chemical approaches to increase O‐GlcNAc levels also have therapeutic effects on chronic intestinal inflammation. Our study will shed light on the future design of novel preventions and therapeutics for IBD.
Materials and Methods
Human samples
De‐identified intestine slides from UC and CD patients were purchased from the Biological Materials Procurement Network (BioNet) at the University of Minnesota. Normal colon tissues adjacent to tumor from age‐matched subjects were used as controls. The Institutional Review Board of the University of Minnesota has reviewed our study and determined that it did not meet the regulatory definition of human subject research. Informed consent was obtained from all Chinese subjects enrolled in the study for biopsy sampling per protocol approved by the Institutional Review Board at the Fourth Military Medical University. The intensity of OGT and O‐GlcNAc staining was semi‐quantified by visually scoring. 10–20 crypts per sample were randomly selected and ranked 1–4 based on the staining intensity (1 being weakest and 4 being strongest). Average scores were used for comparison between control, UC, and CD patients. For the correlation between the disease severity and the level of O‐GlcNAcylation, slides from another cohort of 10 controls, 10 UCs, and 9 CDs were obtained from BioNet (Appendix Table S2). Tissue inflammation and damage were evaluated by the Geboes scoring system for UC and the global histological activity (GHA) scoring system for CD (Dataset EV1; Levesque et al, 2015). Lineage regression analysis was performed to determine the correlation.
Animal
Ogt‐floxed mice on the C57BL/6 background (Shafi et al, 2000) were kindly provided by Dr. Xiaoyong Yang (Yale University). Villin‐Cre mice (stock no. 004586) were purchased from the Jackson Laboratory. Defa6‐iCre mice on the C57BL/6 background were generated and reported earlier (Adolph et al, 2013). Homozygous floxed Ogt F/F mice were bred to Villin‐Cre or Defa6‐iCre mice to generate Vil‐Ogt KO and Defa6‐Ogt KO mice, respectively. 10‐ to 15‐week‐old male mice were used for experiments unless otherwise mentioned. All animals were kept on a 14‐h: 10‐h light: dark cycle in the animal facility at the University of Minnesota or on a 12‐h: 12‐h light: dark cycle at Yale University. Mice were group‐housed unless otherwise mentioned, with free access to water and standard chow diet. All procedures involving animals were conducted within IACUC guidelines under approved protocols.
In vivo intestinal barrier function assays
Mice with free access to water were fasted for 2‐h and orally gavaged with fluorescein isothiocyanate (FITC)‐dextran (average molecular weight: 3,000–5,000, 0.6 mg/g; Sigma) diluted in PBS. Fluorescence intensity of plasma samples was measured (excitation 492 nm/emission 520 nm) 4 h after the gavage. For fecal albumin assays, fecal pellets were weighed and homogenized in diluent (PBS, 1% BSA, 0.05% Tween 20). Albumin levels in fecal homogenates were measured by ELISA (Bethyl Laboratories) according to the manufacturer's protocol.
Fecal microbiota colonization
Donor mice were euthanized, and ceca were aseptically removed immediately. The content was diluted 1:10 in a 50% glycerol/PBS solution and frozen at −80°C. On the day of inoculation, the cecal content was further diluted 1:5 in autoclaved PBS prior to oral gavage of 0.15 ml per mouse. Prior to inoculation, recipient animals were treated with a cocktail of broad‐spectrum antibiotics to deplete gut microbiota (Hill et al, 2010; Reikvam et al, 2011). Specifically, animals having free access to autoclaved food and water were subjected to oral gavage daily for 14 days with 100 μl of autoclaved water supplemented with ampicillin (2 mg/ml), gentamicin (2 mg/ml), metronidazole (2 mg/ml), neomycin (2 mg/ml), and vancomycin (1 mg/ml).
Murine colitis model
36–50 kDa colitis‐grade dextran sodium sulfate (DSS; MP Biomedicals) was added to drinking water at 2.0 or 2.5% weight/volume for 5–6 days and then removed for the recovery. Thiamet‐G (TMG; CarboSynth) at a dose that sufficiently increases O‐GlcNAc level (0.2 g/kg BW; Yuzwa et al, 2008) was gavaged daily from 2 weeks before the DSS treatment until the end of experiment. Disease was scored as described in Alenghat et al (2013): (i) weight loss (no loss = 0; < 5% = 1; 5–10% = 2; 10–20% = 3; > 20% = 4); (ii) stool (normal = 0; soft, watery =1; very soft, semi‐formed = 2; liquid, sticky, or unable to defecate = 3); (iii) blood (no blood = 0; visible blood in rectum = 1; visible blood on fur = 2); and (iv) general appearance (normal = 0; piloerection = 1; lethargy and piloerection = 2; motionless = 4). Histological injury and inflammation were scored as described in Gilbert et al (2012). Scoring parameters included edema (scale: 1–4), erosion/ulceration of the epithelial monolayer (scale: 1–4), crypt loss/damage (scale: 1–4), and infiltration of immune cells into the mucosa (scale: 1–4).
Colonoscopy
Mice were anaesthetized using intraperitoneal injection of ketamine, xylazine, and acepromazine. The “Coloview system” (Karl Storz) was used for colonoscopy as described in Becker et al (2005). The endoscopic procedure was viewed on a color monitor, and pictures were digitally recorded.
Ileal organoid and Caco‐2 cell culture
Isolated ileal crypts were counted and cultured in Matrigel following a published protocol (Mahe et al, 2013). Advanced DMEM/F12 was supplemented by EGF (50 ng/ml), Noggin (100 ng/ml), and R‐spondin conditional media. Caco‐2 cells were cultured as described previously (Natoli et al, 2012). Differentiated Caco‐2 cells were treated with 100 μM Ac45S‐GlcNAc for 24 h and subjected to protein extraction.
Antibodies
Anti‐O‐GlcNAc (RL2, ab2739) was from Abcam. Anti‐ZO‐1 (617300) was from Life technologies. Anti‐ZO‐2 (18900‐1‐AP), anti‐CLAUDIN 3 (16456), and anti‐OCCLUDIN (13409) were from proteintech. Anti‐Ki67 (550609) was from BD Biosciences. Anti‐Myeloperoxidase (MPO, PA5‐16672) was from Thermo Fisher. Anti‐F4/80 (123102) and Anti‐CD4 (100506) were from Biolegend. Anti‐OGT (#24083), anti‐Cleaved CASPASE 3 (#9661), anti‐LC3B (#2775), anti‐α‐E‐CATENIN (#3042), anti‐β‐CATENIN (#8480), anti‐PLAKOGLOBIN (#2309), anti‐STAT1 (#9172), anti‐STAT3 (#4904), anti‐phospho‐STAT3 (Tyr705, #9145), anti‐STAT5 (#9363), anti‐NF‐κB p65 (#8242), and anti‐phospho‐NF‐κB p65 (Ser536, #3033) were from Cell Signaling Technology. Anti‐lysozyme (CSB‐PA02769A0Rb) was from Cusabio. Anti‐DESMOPLAKIN I/II (sc‐33555) was from Santa Cruz Biotechnology. Anti‐β‐ACTIN (A5441) and anti‐CLAUDIN 5 (ABT45) were from Millipore Sigma.
Histology, immunohistochemistry, immunofluorescence, and pathological analysis
Tissues were fixed in 10% neutral buffered formalin. Paraffin sections of intestine tissues were stained with hematoxylin and eosin (H&E) and alcian blue (Sigma) according to standard procedures. Immunohistochemistry was carried out using Histostain‐Plus 3rd Gen IHC Detection Kit (Life technologies) following manufacturer's instruction. Antigen retrieval was performed in citric buffer using a 2100 Retriever (Aptum Biologics). For immunofluorescence, tissue slides were blocked with 3% BSA, 0.2% TWEEN 20 in PBS, incubated with primary antibodies (1:100 dilution) overnight, and secondary antibodies (Alexa Fluor 488 anti‐Rabbit IgG, Alexa Fluor 594 anti‐Rabbit IgG, and Alexa Fluor 594 anti‐Mouse IgG, 1:400) for 1 h. TUNEL assay was carried out using In Situ Cell Death Detection Kit (Roche), following manufacturer's instruction. A Nikon system was used for fluorescence detection. A modified histopathologic scoring system was used to analyze pathological alterations in the intestinal mucosa (Gilbert et al, 2015). Briefly, scores of epithelial hyperplasia (scale: 1–4), epithelial hypertrophy (scale: 1–4), crypt elongation (scale: 1–4), villus length (scale: 1–4), cell apoptosis (scale: 1–4), immune cell infiltration (scale: 1–4), and mucosal thickness (scale: 1–4) were combined.
Immunoprecipitation and Western blot
Tissues were lysed in RIPA or NP‐40 buffer containing proteinase inhibitors, protein phosphatase inhibitors, and an OGA inhibitor. For immunoprecipitation, whole‐cell lysates were mixed with various antibodies as specified in text and precipitated by Protein A/G agarose beads (Santa Cruz). Equal amounts of whole lysates or immunoprecipitation samples were electrophoresed on TGX precast gels (Bio‐Rad) and transferred to nitrocellulose membrane. Primary antibodies (1:500 dilution for antibody from Santa Cruz Biotechnology and 1–1,000 dilution for the rest) were incubated at 4°C for overnight. Western blotting was visualized by using IRDye secondary antibodies and the Odyssey imaging system (LI‐COR Biosciences).
RNA and real‐time PCR
Total RNA was extracted from mouse tissues and cells using TRIzol reagent (Invitrogen). cDNA was reverse‐transcribed (Bio‐Rad) and amplified with SYBR Green Supermix (Bio‐Rad) using a C1000 Thermal Cycler (Bio‐Rad). All data were normalized to the expression of the Rplp0 gene. Primer sequences are as follows: Ang4 for 5′‐TCTCCAGGAGCACACAGCTA‐3′; Ang4 rev 5′‐ACAACAAAGGACATGGGCTC‐3′; Defa3 for 5′‐GTCCAGGCTGATCCTATCCA‐3′; Defa3 rev 5′‐AGAGCCTTCTGGGTCTCCA‐3′; Defa4 for 5′‐GTCCAGGCTGATCCTATCCA‐3′; Defa4 rev 5′‐TGGCCTCCAAAGGAGATAGA‐3′; Defa5 for 5′‐TCCAGGCTGATCCTATCCAC‐3′; Defa5 rev 5′‐TGGCCTCCAAAGGAGATAGA‐3′; Defa6 for 5′‐GGACCAGGCTGTGTCTGTCT‐3′; Defa6 rev 5′‐TTGCAGCCTCTTGCTCTACA‐3′; Ifng for 5′‐TCAAGTGGCATAGATGTGGAAGAA‐3′; Ifng rev 5′‐TGGCTCTGCAGGATTTTCATG‐3′; Il1b for 5′‐TGTGAAATGCCACCTTTTGA‐3′; Il1b rev 5′‐GGTCAAAGGTTTGGAAGCAG‐3′; Il6 for 5′‐GAGGATACCACTCCCAACAGACC‐3′; Il6 rev 5′‐AAGTGCATCATCGTTGTTCATACA‐3′; Lyz1 for 5′‐CTGTGGGATCAATTGCAGTG‐3′; Lyz1 rev 5′‐GAATGCCTTGGGGATCTCTC‐3′; Ogt (human) for 5′‐CTGTCACCCTTGACCCAAAC‐3′; Ogt (human) rev 5′‐CTCTGGGAAGACTTCTAATGC‐3′; Pan Defensin for 5′‐GGTGATCATCAGACCCCAGCATCAGT‐3′; Pan Defensin rev 5′‐AAGAGACTAAAACTGAGGAGCAGC‐3′; Rplp0 for 5′‐AGATGCAGCAGATCCGCAT‐3′; Rplp0 rev 5′‐GTTCTTGCCCATCAGCACC‐3′; Tnfa for 5′‐CATCTTCTCAAAATTCGAGTGACAA‐3′; Tnfa rev 5′‐TGGGAGTAGACAAGGTACAACCC‐3′.
Electron microscopy
Mice were perfused with PBS followed by 4% PFA. Intestinal tissues were cut into less than 1 × 1×1 mm3 cubes for post‐fixation overnight in 2.5% gluteraldehyde and 2% PFA in 0.1 M sodium cacodylate buffer. Embedding, sectioning, and observation were carried out at the Electron Microscopy Core at Yale School of Medicine.
16S rRNA gene sequencing
Total DNA in stool and cecal contents was extracted using the PowerFecal DNA Isolation Kit (MoBio). The V4 region of the bacterial 16S rRNA gene was amplified by triplicate PCR (F515/R806) using barcoded fusion primers. Samples were pooled in sets with a maximum of 96 samples in equal quantities. Paired‐end sequencing of the amplicon library was performed on the Illumina MiSeq 300‐bp paired‐end platform at the University of Minnesota Genomics Center (Gohl et al, 2016). A multi‐step bioinformatics analysis was performed using the QIIME 1.9.1 software, including filtering raw fastq files for primer and adapter dimer sequences, removing contaminating host sequences and chimeric sequences, clustering sequences into operational taxonomic units (OTUs) using the open‐reference OTU calling method with the greengenes 16S reference, and calculating alpha and beta diversity metrics. Linear discriminant analysis (LDA) effect size (LEfSe) method was used for microbial biomarker discovery (Segata et al, 2011).
RNA sequencing
IECs from ileum and colon were isolated by shaking intestinal tissue in PBS with 10 mM EDTA at 37°C for 30 min. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer's instruction. RNA concentration and integrity of each sample were measured on an Agilent Bioanalyzer. Equal amounts of RNA from each sample were pooled for cDNA library construction and sequencing on an Illumina HiSeq instrument with 1 × 75 bp reads at the Yale Center for Genome Analysis. Sequencing analysis was performed on the Galaxy server hosted by the Minnesota Supercomputing Institute. FastQ files were trimmed using Trimmomatic. Quality control checks on raw sequence data for each sample were performed with FastQC. Read mapping was performed via TopHat using the UCSC mouse genome (mm 10) as reference. DESeq2 was used for differential gene expression analysis. The Ingenuity Pathway Analysis (IPA) tool from Qiagen was used for pathway analysis and upstream regulator analysis of significant genes that showed more than twofold of differential expression. Finally, these coregulated genes were subjected to the Distant Regulatory Elements (DiRE) server to predict common regulatory elements (Gotea & Ovcharenko, 2008).
Luciferase assay
HEK 293T cells (ATCC) were seeded at 48‐well plates while scramble and OGT siRNA was transfected using Lipofectamine 2000 (Invitrogen). On the second day, cells were transfected with pGAS‐Luc (IFN‐γ‐activated sequence) plasmid (Agilent) with pcDNA or STAT1α plasmid (Addgene). The reporter activity was measured 36 h after pGAS‐Luc transfection with the Dual‐Luciferase® Reporter Assay System (Promega) following manufacturer's instruction.
Statistical analyses
Mice receiving FMT or drug treatment were randomly assigned to control or treatment groups. The histological scores were provided by a professional pathologist without any information of the treatment group. Otherwise, no blinding was done, due to the obvious size difference between WT and KO mice for most measurements. Results are shown as mean ± SEM. Shapiro–Wilk normality test was used to assess normal distribution. The comparisons were carried out using two‐tailed unpaired Student's t‐test, one‐way ANOVA with the Dunnett post hoc test, and two‐way ANOVA followed by post hoc comparisons using Tukey or Bonferroni corrections. The prevalence of rectal prolapse was compared between WT and Vil‐Ogt KO mice using the Log‐rank (Mantel–Cox) test in Prism.
Data availability
RNA‐Seq data: Gene Expression Omnibus GSE100473.
Author contributions
MZ, with the help from XX, KR, MC, CS, and ZH, designed, performed, and analyzed most experiments. BX, KW, and YN performed experiments using Chinese human biopsies. XH performed pathological analyses. RSB provided the Defa6‐iCre mouse strain. H‐BR conceived, designed, and performed experiments. MZ and H‐BR with contributions from XH and RSB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
It is generally accepted that IBD requires the convergence of several abnormalities that affect overlapping layers of regulatory modules including genetic mutations, permeable barriers, bacterial alterations, and immune over‐activation. However, we still do not fully understand genes and pathways that contribute to such multi‐layer defects.
Results
Here, we report a post‐translational modification on proteins, termed O‐GlcNAcylation, in the gut epithelium controls multiple regulatory mechanisms including epithelial barrier, autophagy, and gut bacteria to maintain intestinal homeostasis. We found that levels of protein O‐GlcNAcylation were reduced in both UC and CD patients and negatively correlated with disease severity. In mouse models, the deficiency of O‐GlcNAcylation in intestinal epithelial cells broke down the gut barrier, disrupted Paneth cell function, and caused changes in microbial composition to eventually elicit intestinal inflammation. On the other hand, a drug that increases O‐GlcNAcylation alleviated chemical‐induced colitis in mice.
Impact
Our data reveal that protein O‐GlcNAcylation controls multiple layers of regulatory mechanisms in intestinal epithelial cells. Drugs specifically enhancing O‐GlcNAcylation in the gut epithelium may prevent and treat IBD.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4D
Source Data for Figure 6D
Source Data for Figure 7
Acknowledgements
We thank Dr. Xiaoyong Yang for providing the Ogt‐floxed mouse line, Dr. David Vocadlo for providing Ac45S‐GlcNAc, and Dr. Daniel Vallera for providing Caco‐2 cells. We thank Dr. Tim Starr for providing the Karl Storz Coloview system and Patrick Blaney for assisting with the colonoscopy. We thank Dr. Dan Knights and Trevor Gould for assisting the analysis of 16S sequencing data. We thank Dr. Weiqi He and Huashan Li for providing the technical assistance in organoid culture. This work was supported by National Natural and Science Foundation of China (81770543), American Heart Association Scientist Development Grant (14SDG20120052), Mizutani Foundation for Glycoscience Grant (170133), and University of Minnesota Medical School 2017 Innovation Grant to H.‐B.R, Key Science and Technology Project of Henan Province (182102310107) to X. X, Natural Science Foundation of Jiangsu Province (BK20150687) to Z. H, NIH DK088199 to R.S.B, and Crohn's Colitis Foundation of America Senior Research Award (426234) to X.H.
EMBO Mol Med (2018) 10: e8736
References
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez‐Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S et al (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503: 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky‐Dolfi M et al (2013) Histone deacetylase 3 coordinates commensal‐bacteria‐dependent intestinal homeostasis. Nature 504: 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, Jones DR, Mayo MW (2012) Modification of RelA by O‐linked N‐acetylglucosamine links glucose metabolism to NF‐kappaB acetylation and transcription. Proc Natl Acad Sci USA 109: 16888–16893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N (2014) Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 15: 19–33 [DOI] [PubMed] [Google Scholar]
- Baudoin L, Issad T (2014) O‐GlcNAcylation and inflammation: a vast territory to explore. Front Endocrinol 5: 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Wirtz S, Nikolaev A, Kiesslich R, Lehr HA, Galle PR, Neurath MF (2005) In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 54: 950–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins CL, Salzman NH (2011) Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9: 356–368 [DOI] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S et al (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456: 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Stappenbeck TS, Virgin HW (2009) Role of autophagy and autophagy genes in inflammatory bowel disease. Curr Top Microbiol Immunol 335: 141–167 [DOI] [PubMed] [Google Scholar]
- Chiriac MT, Buchen B, Wandersee A, Hundorfean G, Gunther C, Bourjau Y, Doyle SE, Frey B, Ekici AB, Buttner C et al (2017) Activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology 153: 123–138 e128 [DOI] [PubMed] [Google Scholar]
- Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, Mayer AE, Shen Y, Wu WL, Kambal A et al (2016) Gene‐microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 352: 1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen I, Vermeire S (2015) The genetic architecture of inflammatory bowel disease: past, present and future. Curr Opin Gastroenterol 31: 456–463 [DOI] [PubMed] [Google Scholar]
- Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, Zeitz M, Siegmund B, Kuhl AA (2014) A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol 7: 4557–4576 [PMC free article] [PubMed] [Google Scholar]
- Freund P, Kerenyi MA, Hager M, Wagner T, Wingelhofer B, Pham HT, Elabd M, Han X, Valent P, Gouilleux F et al (2017) O‐GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5‐dependent malignancies. Leukemia 31: 2132–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewinner C, Hart G, Zachara N, Cole R, Beisenherz‐Huss C, Groner B (2004) The coactivator of transcription CREB‐binding protein interacts preferentially with the glycosylated form of Stat5. J Biol Chem 279: 3563–3572 [DOI] [PubMed] [Google Scholar]
- Gilbert S, Zhang R, Denson L, Moriggl R, Steinbrecher K, Shroyer N, Lin J, Han X (2012) Enterocyte STAT5 promotes mucosal wound healing via suppression of myosin light chain kinase‐mediated loss of barrier function and inflammation. EMBO Mol Med 4: 109–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S, Nivarthi H, Mayhew CN, Lo YH, Noah TK, Vallance J, Rulicke T, Muller M, Jegga AG, Tang W et al (2015) Activated STAT5 confers resistance to intestinal injury by increasing intestinal stem cell proliferation and regeneration. Stem Cell Rep 4: 209–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ (2011) Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7: 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R et al (2016) Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol 34: 942–949 [DOI] [PubMed] [Google Scholar]
- Gotea V, Ovcharenko I (2008) DiRE: identifying distant regulatory elements of co‐expressed genes. Nucleic Acids Res 36: W133–W139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, Love DC (2012) Bittersweet memories: linking metabolism to epigenetics through O‐GlcNAcylation. Nat Rev Mol Cell Biol 13: 312–321 [DOI] [PubMed] [Google Scholar]
- Hart GW, Housley MP, Slawson C (2007) Cycling of O‐linked beta‐N‐acetylglucosamine on nucleocytoplasmic proteins. Nature 446: 1017–1022 [DOI] [PubMed] [Google Scholar]
- Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, Vermeire S (2011) Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflamm Bowel Dis 17: 1392–1397 [DOI] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D (2010) Metagenomic analyses reveal antibiotic‐induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3: 148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LR, Tang D, Ingram A, Ly H, Thai K, Cai L, Scholey JW (2002) Flux through the hexosamine pathway is a determinant of nuclear factor kappaB‐ dependent promoter activation. Diabetes 51: 1146–1156 [DOI] [PubMed] [Google Scholar]
- Kajino‐Sakamoto R, Inagaki M, Lippert E, Akira S, Robine S, Matsumoto K, Jobin C, Ninomiya‐Tsuji J (2008) Enterocyte‐derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J Immunol 181: 1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, Nunez G (2013) Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13: 321–335 [DOI] [PubMed] [Google Scholar]
- Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH et al (2008) XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134: 743–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayama H, Takeda K (2012) Regulation of intestinal homeostasis by innate and adaptive immunity. Int Immunol 24: 673–680 [DOI] [PubMed] [Google Scholar]
- Khor B, Gardet A, Xavier RJ (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474: 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesler P, Fuss IJ, Strober W (2015) Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol 1: 154–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Lassen KG, Xavier RJ (2013) Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 62: 1505–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque BG, Sandborn WJ, Ruel J, Feagan BG, Sands BE, Colombel JF (2015) Converging goals of treatment of inflammatory bowel disease from clinical trials and practice. Gastroenterology 148: 37–51 e31 [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, Herring LE, Asara JM, Singer JD, Wen H (2017) Myeloid‐derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med 214: 1093–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, Coffer PJ (2014) Post‐translational modification networks regulating FOXP3 function. Trends Immunol 35: 368–378 [DOI] [PubMed] [Google Scholar]
- Mahe MM, Aihara E, Schumacher MA, Zavros Y, Montrose MH, Helmrath MA, Sato T, Shroyer NF (2013) Establishment of gastrointestinal epithelial organoids. Curr Protoc Mouse Biol 3: 217–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F (2011) Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474: 298–306 [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Kanai T (2015) The gut microbiota and inflammatory bowel disease. Semin Immunopathol 37: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi A (2012) Animal models of inflammatory bowel disease. Prog Mol Biol Transl Sci 105: 263–320 [DOI] [PubMed] [Google Scholar]
- Natoli M, Leoni BD, D'Agnano I, Zucco F, Felsani A (2012) Good Caco‐2 cell culture practices. Toxicol In Vitro 26: 1243–1246 [DOI] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B et al (2007) Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446: 557–561 [DOI] [PubMed] [Google Scholar]
- Patel KK, Stappenbeck TS (2013) Autophagy and intestinal homeostasis. Annu Rev Physiol 75: 241–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153 [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh‐Wilson LC, Baltimore D (2013) Activation of the transcriptional function of the NF‐kappaB protein c‐Rel by O‐GlcNAc glycosylation. Sci Signal 6: ra75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, McCoy KD, Macpherson AJ, Meza‐Zepeda LA, Johansen FE (2011) Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE 6: e17996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, Zhao L, Bennett AM, Samuel VT, Wu J et al (2012) O‐GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC‐1alpha stability. Cell Metab 16: 226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Nie Y, Yang X (2013a) Regulation of protein degradation by O‐GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics 12: 3489–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Singh JP, Li MD, Wu J, Yang X (2013b) Cracking the O‐GlcNAc code in metabolism. Trends Endocrinol Metab 24: 301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL et al (2014) O‐GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159: 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Ma Y, Torres S, Zhang B, Feriod C, Heck RM, Qian K, Fu M, Li X, Nathanson MH et al (2017) Calcium‐dependent O‐GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev 31: 1655–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD (2000) The O‐GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA 97: 5735–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Artis D (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21: 698–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Do T, Phoomak C, Champattanachai V, Silsirivanit A, Chaiyarit P (2018) New evidence of connections between increased O‐GlcNAcylation and inflammasome in the oral mucosa of patients with oral lichen planus. Clin Exp Immunol 192: 129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres CR, Hart GW (1984) Topography and polypeptide distribution of terminal N‐acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O‐linked GlcNAc. J Biol Chem 259: 3308–3317 [PubMed] [Google Scholar]
- Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, Bhattacharyya J, Flak MB, Shih DQ, Fuhler GM et al (2017) Defective ATG16L1‐mediated removal of IRE1alpha drives Crohn's disease‐like ileitis. J Exp Med 214: 401–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9: 799–809 [DOI] [PubMed] [Google Scholar]
- Willson TA, Jurickova I, Collins M, Denson LA (2013) Deletion of intestinal epithelial cell STAT3 promotes T‐lymphocyte STAT3 activation and chronic colitis following acute dextran sodium sulfate injury in mice. Inflamm Bowel Dis 19: 512–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, Oparil S (2011) O‐GlcNAc modification of NFkappaB p65 inhibits TNF‐alpha‐induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS ONE 6: e24021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, Kim YS, Lee HC, Kim KS, Cho JW (2008) NFkappaB activation is associated with its O‐GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci USA 105: 17345–17350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YR, Kim DH, Seo YK, Park D, Jang HJ, Choi SY, Lee YH, Lee GH, Nakajima K, Taniguchi N et al (2015) Elevated O‐GlcNAcylation promotes colonic inflammation and tumorigenesis by modulating NF‐kappaB signaling. Oncotarget 6: 12529–12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XY, Qian KV (2017) Protein O‐GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18: 452–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ et al (2008) A potent mechanism‐inspired O‐GlcNAcase inhibitor that blocks phosphorylation of tau in vivo . Nat Chem Biol 4: 483–490 [DOI] [PubMed] [Google Scholar]
- Zhang D, Cai Y, Chen M, Gao L, Shen Y, Huang Z (2015) OGT‐mediated O‐GlcNAcylation promotes NF‐kappaB activation and inflammation in acute pancreatitis. Inflamm Res 64: 943–952 [DOI] [PubMed] [Google Scholar]
- Zheng J, Jiao S, Li Q, Jia P, Yin H, Zhao X, Du Y, Liu H (2017) Antrodia cinnamomea oligosaccharides suppress lipopolysaccharide‐induced inflammation through promoting O‐GlcNAcylation and repressing p38/Akt phosphorylation. Molecules 23: pii: E51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, Chatham JC (2009) Glucosamine improves cardiac function following trauma‐hemorrhage by increased protein O‐GlcNAcylation and attenuation of NF‐{kappa}B signaling. Am J Physiol Heart Circ Physiol 296: H515–H523 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4D
Source Data for Figure 6D
Source Data for Figure 7
Data Availability Statement
RNA‐Seq data: Gene Expression Omnibus GSE100473.