

Abstract
The adaptive cellular response to low oxygen tensions is mediated by the hypoxia‐inducible factors (HIFs), a family of heterodimeric transcription factors composed of HIF‐α and HIF‐β subunits. Prolonged HIF expression is a key contributor to cellular transformation, tumorigenesis and metastasis. As such, HIF degradation under hypoxic conditions is an essential homeostatic and tumour‐suppressive mechanism. LIMD1 complexes with PHD2 and VHL in physiological oxygen levels (normoxia) to facilitate proteasomal degradation of the HIF‐α subunit. Here, we identify LIMD1 as a HIF‐1 target gene, which mediates a previously uncharacterised, negative regulatory feedback mechanism for hypoxic HIF‐α degradation by modulating PHD2‐LIMD1‐VHL complex formation. Hypoxic induction of LIMD1 expression results in increased HIF‐α protein degradation, inhibiting HIF‐1 target gene expression, tumour growth and vascularisation. Furthermore, we report that copy number variation at the LIMD1 locus occurs in 47.1% of lung adenocarcinoma patients, correlates with enhanced expression of a HIF target gene signature and is a negative prognostic indicator. Taken together, our data open a new field of research into the aetiology, diagnosis and prognosis of LIMD1‐negative lung cancers.
Keywords: adaptive hypoxic response, HIF‐1, LIMD1, lung cancer, tumour suppressor
Subject Categories: Cancer, Vascular Biology & Angiogenesis
Introduction
The HIF family of transcription factors are heterodimeric proteins formed of a HIF‐α and HIF‐β subunit (Wang et al, 1995). HIF‐α is regulated by intracellular oxygen levels; at physiological oxygen tension (normoxia), two highly conserved proline residues within the oxygen‐dependent degradation domain of the HIF‐α subunit (P402/564 on HIF‐1α; P405/531 on HIF‐2α) are hydroxylated by prolyl hydroxylase domain (PHD) proteins. Hydroxylated HIF‐α is then recognised and ubiquitinated by the von Hippel–Lindau (VHL) E3 ubiquitin ligase complex, resulting in its degradation by the 26S proteasome (Salceda & Caro, 1997; Maxwell et al, 1999; Jaakkola et al, 2001; Foxler et al, 2012). Under low oxygen (hypoxic) conditions, the hydroxylase activity of the PHD enzymes is inhibited; HIF therefore escapes hydroxylation and degradation to initiate a transcriptional programme of cellular response and adaptation to hypoxia.
Under conditions of chronic hypoxia, a negative regulatory feedback loop is initiated whereby free oxygen from inhibited mitochondrial respiration leads to overactivation of PHDs, causing HIF‐α degradation and a desensitised hypoxic response (Ginouves et al, 2008). However, neoplastic cells survive under conditions of chronic tumour hypoxia by inhibiting the degradation of HIF (Bertout et al, 2008). This is exemplified by VHL mutations in clear cell renal carcinomas, leading to sustained HIF‐α expression and activity (Rechsteiner et al, 2011). In non‐small‐cell lung cancer (NSCLC), deregulation of the HIF negative feedback loop is far less characterised, even though HIF‐α protein expression is implicated as a poor prognostic indicator (Giatromanolaki et al, 2001; Kim et al, 2005).
The lung tumour suppressor protein LIMD1 is a member of the Zyxin family of adaptor proteins, initially characterised as signal transducers (Kadrmas & Beckerle, 2004) shuttling between the cytoplasm and nucleus. LIMD1 loss has been identified in lung, breast, head and neck squamous cell carcinomas, and adult acute leukaemia (Sharp et al, 2004, 2008; Spendlove et al, 2008; Ghosh et al, 2010b; Liao et al, 2015), and its decreased expression in diffuse large B‐cell lymphoma has clinical significance to patient prognosis and disease classification/stratification (Xu et al, 2015). Limd1‐knockout mice have increased lung tumour numbers and volume and decreased survival rate compared to Limd1‐expressing control mice when either challenged with a chemical carcinogen or cross‐bred with Kras G12D mice (Sharp et al, 2008) validating its critical role in normal cellular homeostasis. Furthermore, it has been reported that silencing of LIMD1 in multidrug‐resistant colorectal carcinoma cells increased their chemosensitivity in vitro (Chen et al, 2014).
As a scaffold protein, LIMD1 exerts multiple tumour‐suppressive functions depending on its binding partners. Basal LIMD1 gene expression is under the control of PU.1, a member of the Ets family of transcription factors (Foxler et al, 2011). LIMD1 can repress cell cycle progression through pRb‐dependent and pRb‐independent inhibition of E2F (Sharp et al, 2004) and regulates Hippo signalling by binding to LATS, causing sequestration of the Hippo kinase complex (Das Thakur et al, 2010; Codelia et al, 2014; Jagannathan et al, 2016). LIMD1 is also part of the Slug/Snail complex that regulates E‐cadherin transcription (Ayyanathan et al, 2007; Langer et al, 2008) in addition to facilitating centrosomal localisation of BRCA2 to prevent aberrant cellular proliferation (Hou et al, 2016). Our recent work has shown that LIMD1 is a critical effector of microRNA (miRNA)‐mediated gene silencing, a process generally considered to be a global tumour‐suppressive mechanism (James et al, 2010; Bridge et al, 2017).
LIMD1 forms complexes with PHD2 and VHL to post‐translationally repress HIF‐1α protein levels and therefore HIF‐1α‐mediated gene activation (Foxler et al, 2012; Zhang et al, 2015). However, the pathophysiological link between this mechanistic role of LIMD1 within the PHD‐LIMD1‐VHL HIF regulatory complex and cancer is unknown. Here, we report that LIMD1 expression is upregulated in hypoxia, through a functional HIF‐1α‐specific hypoxic response element (HRE) within the CpG island in its promoter. LIMD1 facilitates HIF‐1α protein degradation under hypoxic conditions by maintaining the PHD2/VHL/HIF‐1α degradation complex, thereby reducing HIF‐1α‐driven gene activation. Utilising an RNAi‐mediated knockdown‐rescue system, we have identified that inhibition of hypoxia‐driven increase in LIMD1 expression causes HIF‐1α protein stabilisation and HIF target gene activation. In vivo, inhibition of hypoxia‐driven LIMD1 expression results in larger and more vascularised xenograft tumours. Finally, our data provide a molecular mechanistic insight into clinico‐pathological data indicating that LIMD1 loss or haplo‐insufficiency correlated with elevated HIF‐1α‐driven gene expression within lung tumours is associated with poorer patient prognosis.
Results
LIMD1 is a HIF‐1‐responsive gene
Homeostatic signalling pathways often have in‐built self‐regulatory feedback mechanisms to attenuate their activation (Yosef & Regev, 2011). With this in mind, we hypothesised that LIMD1 might be a HIF target gene as well as a component of the degradation complex. We therefore assessed endogenous LIMD1 expression in a panel of cell lines exposed to 1% O2 (henceforth referred to as hypoxia), including transformed/immortalised lines (A549, HeLa, HEK293 and U2OS), non‐transformed small airway epithelial cells (SAEC) and primary human dermal fibroblasts (HDF). We observed an increase in LIMD1 mRNA and protein expression in all cell lines in hypoxia when compared to atmospheric oxygen (20% O2, herein referred to as normoxia) using PHD2 as a positive control and PHD1 as a hypoxia non‐responsive gene (Figs 1A–C, and EV1A and B; Stiehl et al, 2006).
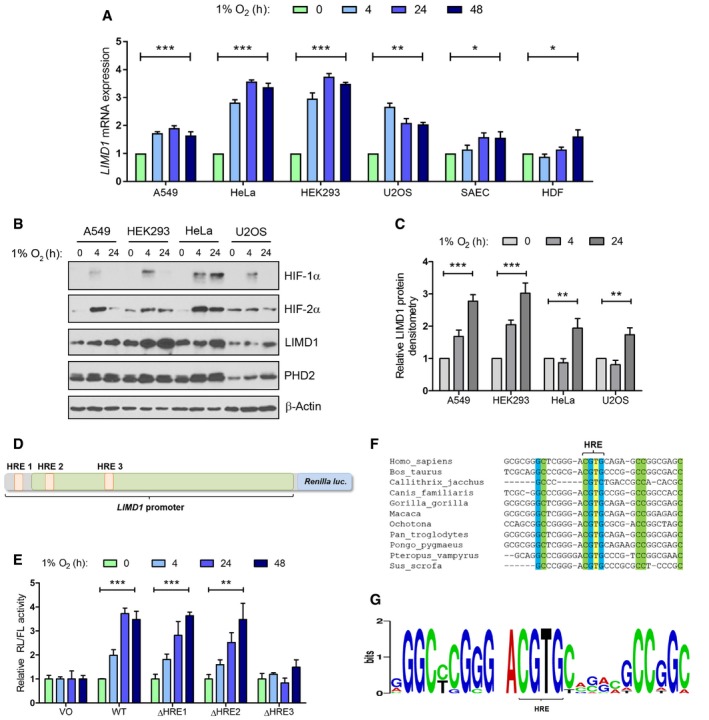
Figure 1. LIMD1 expression is regulated by hypoxia.
-
A, B(A) LIMD1 mRNA and (B) protein levels were increased following hypoxic exposure.
-
CDensitometric analysis of (B).
-
DThe LIMD1 promoter contains a hypoxic response element responsible for HIF binding and transcriptional activation of LIMD1. Three predicted HRE elements were individually deleted within the context of the wild‐type LIMD1 promoter‐driven Renilla luciferase.
-
EReporter constructs in (D) were expressed in U2OS cells and exposed to hypoxia for the indicated time‐points. Luciferase activity was then assayed and normalised to firefly control. Data are displayed normalised to the normoxic value for each construct. Deletion of the third HRE present within the LIMD1 promoter (ΔHRE3) inhibited hypoxic induction of LIMD1 transcription.
-
F, G(F) Sequence alignment and (G) sequence logo of LIMD1 promoters from the indicated species demonstrate that the HRE3 consensus sequence is highly conserved.
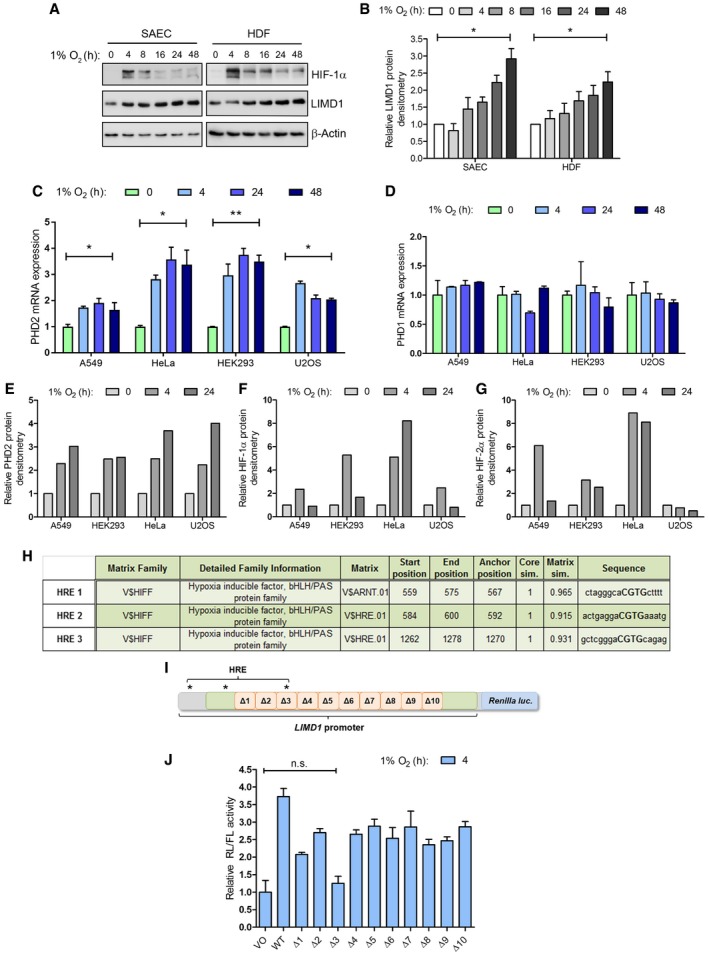
Figure EV1. LIMD1 expression is induced in hypoxia via a HIF‐1 hypoxia‐responsive element (HRE).
-
ASmall airway epithelial cells (SAEC) and human dermal fibroblasts (HDF) were incubated in hypoxia for up to 48 h, total protein extracted and analysed by Western blot.
-
BDensitometric analysis of LIMD1 protein expression in (A), normalised to β‐actin loading control and 0‐h hypoxic time‐point.
-
CPHD2 mRNA expression is upregulated by hypoxia. A549, HEK293, HeLa and U2OS cells were incubated in hypoxia for up to 48 h, RNA extracted and PHD2 mRNA expression analysed by qRT–PCR.
-
DPHD1 mRNA expression is unchanged by hypoxia. qRT–PCR analysis of PHD1 mRNA in the indicated cell lines, as in (C).
-
E–GThe indicated cell lines were incubated in normoxia or 4‐ or 24‐h hypoxia, total protein extracted and PHD2, HIF‐1α or HIF‐2α protein expression quantified by densitometry and normalised to β‐actin loading control.
-
HThe LIMD1 promoter contains three putative HRE elements. The 2‐kb upstream region of the LIMD1 promoter was scrutinised in silico for predicted HIF binding sites using MatInspector software as previously described (Foxler et al, 2011).
-
IA 2‐kbp region of the LIMD1 promoter was cloned into a pGL4 Renilla luciferase plasmid, and a series of 10 consecutive internal deletions were constructed (Δ1–10); the positions of the three in silico HRE elements are indicated.
-
JReporter constructs in (I) were expressed in U2OS cells and exposed to hypoxia for 24 h. Data shown are normalised to vector only (VO) control. The Δ3 mutation inhibited hypoxic induction of LIMD1 transcription compared to the other nine analysed; significance values for all other constructs are omitted for clarity.
In silico analysis of the LIMD1 promoter identified three putative hypoxic response elements (HRE 1–3; Fig EV1H; Foxler et al, 2011). To assess their functionality, we used a LIMD1 promoter‐driven luciferase reporter construct, spanning 1990‐bp upstream from the LIMD1 transcriptional start site [as predicted by the RefSeq NM_014240.2, which corresponds to nucleotides 45634323‐6323 on the primary chromosome 3 ref assembly NC_000003.11 (Foxler et al, 2011)] and encompassing all three predicted HRE elements (Fig 1D). Within this construct, we created a series of ten consecutive small internal deletions within the CpG Island that have previously been identified as containing transcriptional regulatory elements (Foxler et al, 2011; Fig EV1I). These reporter constructs displayed ~ threefold induction of wild‐type LIMD1 promoter activity in hypoxia compared to normoxia. However, deletion of the 31‐bp Δ3 region that encompasses the predicted HRE3 ablated any hypoxia‐induced increase in luciferase activity (Fig EV1J). Furthermore, internal deletion of the three predicted HREs confirmed HRE3 to be the active hypoxia‐responsive element within the LIMD1 promoter (Fig 1E). The position and sequence of this HRE is also highly conserved, further supporting its functional importance (Fig 1F and G).
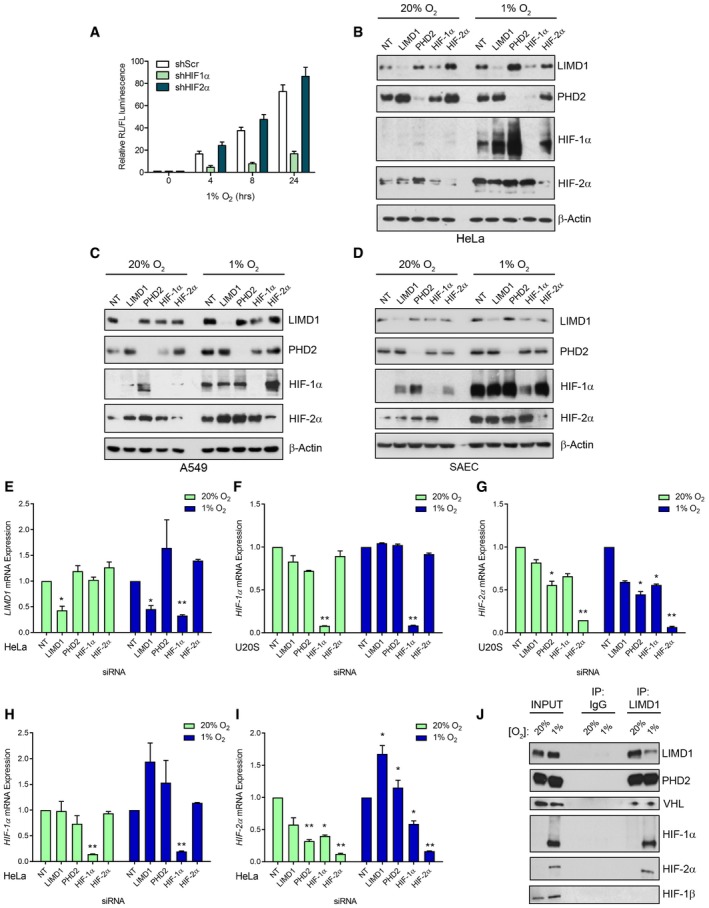
We next determined which HIF‐α paralogue was involved in LIMD1 regulation by combining the LIMD1 promoter‐driven luciferase reporters (Foxler et al, 2011) with shRNA‐mediated knockdown of HIF‐1α and HIF‐2α. Depletion of HIF‐1α, but not HIF‐2α, prevented induction of LIMD1 expression in hypoxia (Fig EV2A). This finding was corroborated by ChIP and EMSAs, which further demonstrated HIF‐1 binding to the LIMD1 promoter (Fig 2A and B). siRNA‐mediated depletion of HIF‐1α reduced LIMD1 protein and mRNA expression under hypoxic and, to a lesser extent, normoxic conditions in all cell lines examined (Figs 2C and D, and EV2B–E). LIMD1 depletion did not affect HIF1A or HIF2A mRNA expression, with the exception of an observed increase in HIF2A mRNA in HeLa cells under hypoxic conditions (Fig EV2F–I). The decrease in LIMD1 expression in normoxia following si‐HIF‐1α demonstrates that HIF‐1 activity is required for LIMD1 expression in normoxia, an observation that has been previously described for other genes (Pillai et al, 2011). Furthermore, under hypoxic conditions HIF preferentially binds to gene loci that are already transcriptionally active to further activate their expression (Xia & Kung, 2009). Thus, these data show that under hypoxic conditions, HIF‐1 binds the LIMD1 promoter to increase its expression.
Figure EV2. LIMD1 hypoxic induction occurs via HIF‐1 and facilitates formation of a hypoxic HIF‐1 degradation complex.
-
AHIF‐1 but not HIF‐2 is responsible for the hypoxic increase in LIMD1 expression. HIF‐1α or HIF‐2α was knocked down in U2OS cells using transient shRNA‐expressing plasmids. The wild‐type LIMD1 promoter‐driven firefly luciferase was co‐transfected with a Renilla normalisation plasmid into these cells and exposed to up to 24‐h hypoxia. Resultant luciferase activity was assayed and normalised to Renilla. Data are displayed normalised to the normoxic value for each shRNA knockdown line.
-
B–DHypoxic induction of LIMD1 is impaired upon knockdown of HIF‐1α. Western blot analysis for the indicated proteins from HeLa, A549 and SAEC transfected with the indicated siRNA (40 nM) for 48 h prior to exposure to hypoxia for 24 h.
-
EsiRNA‐mediated depletion of HIF‐1α but not HIF‐2α reduces LIMD1 expression in both normoxia and hypoxia. qRT–PCR analysis of LIMD1 mRNA from HeLa cells transfected with the indicated siRNA (40 nM) and maintained in normoxia (20% O2) or exposed to hypoxia (1% O2) for 24 h.
-
F–IKnockdown of HIF‐1α but not HIF‐2α significantly reduces LIMD1 mRNA expression in both normoxia and hypoxia. qRT–PCR analysis of the indicated mRNA from cellular extracts in (B) and Fig 2C.
-
JLIMD1 endogenously complexes with PHD2, VHL, HIF‐1α and HIF‐2α. Western blot analysis of endogenous LIMD1 immunoprecipitated from A549 cells in either normoxia or following 24‐h hypoxia.
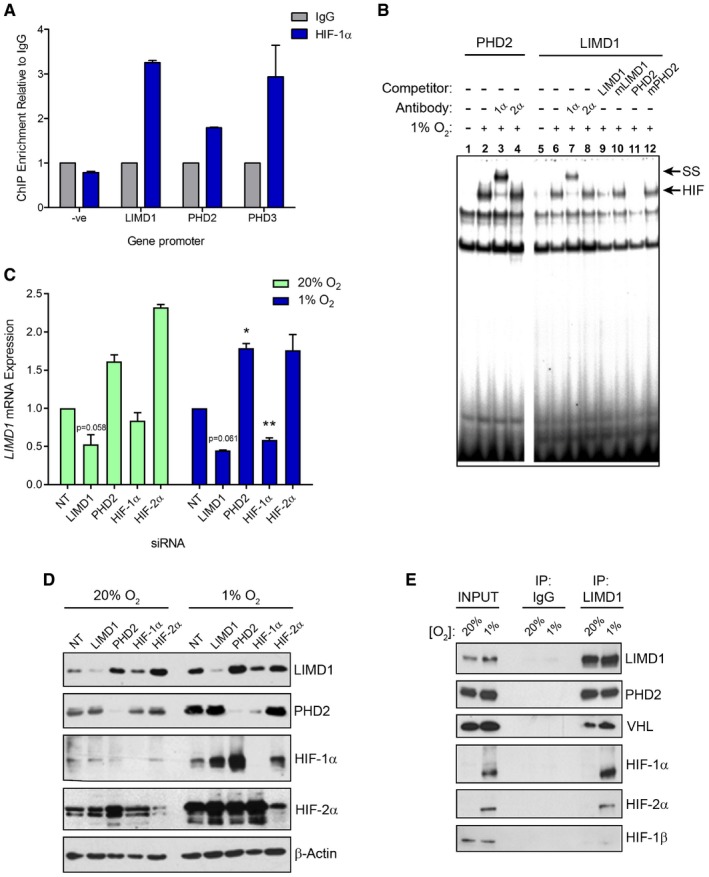
Figure 2. LIMD1 is a HIF‐1‐responsive gene.
-
AHIF‐1 binds to the LIMD1 promoter. Chromatin immunoprecipitation assay (ChIP) of endogenous HIF‐1α from paraformaldehyde cross‐linked U2OS cells exposed to 16‐h hypoxia, followed by qRT–PCR analysis of the indicated gene promoters.
-
BEMSA of nuclear extracts from U2OS cells exposed to normoxia (lanes 1 and 5) or 16‐h hypoxia identified that HIF‐1α but not HIF‐2α bound the LIMD1 HRE consensus sequence, causing a band supershift (ss). Wild‐type unlabelled oligo probes that encompass the LIMD1 or PHD2 HRE were used as controls to compete out the ss, and probes where the HRE sequences have been mutated (mLIMD1/mPHD2) were used to show specificity for HRE binding.
-
CsiRNA‐mediated depletion of HIF‐1α but not HIF‐2α reduces LIMD1 expression in both normoxia and hypoxia. qRT–PCR analysis of LIMD1 mRNA from U2OS cells transfected with the indicated siRNA (40 nM) and maintained in normoxia (20% O2) or exposed to hypoxia (1% O2) for 24 h.
-
DWestern blot analysis of protein from (C).
-
ELIMD1 endogenously complexes with PHD2, VHL, HIF‐1α and HIF‐2α. Endogenous LIMD1 was immunoprecipitated from HeLa cells in either normoxia or following 24‐h hypoxia and co‐immunoprecipitated proteins identified by immunoblot analysis.
Under normoxic conditions, LIMD1 scaffolds PHD2 and VHL to enable efficient degradation of HIF‐1α (Foxler et al, 2012). Given that LIMD1 is a hypoxia‐responsive gene, we next performed co‐immunoprecipitation assays to assess whether the PHD2‐LIMD1‐VHL complex also exists in hypoxic conditions. Indeed, LIMD1 co‐precipitated with PHD2 and VHL under hypoxia (Figs 2E and EV2J); HIF‐1α and HIF‐2α also co‐precipitated with LIMD1, which may be due to the increased protein stability of the HIF proteins under this low oxygen tension. These data demonstrate active engagement of the PHD‐LIMD1‐VHL complex with its HIF target protein in hypoxia. However, HIF‐1β did not co‐precipitate within this complex, indicating LIMD1 was facilitating HIF‐α degradation prior to heterodimerisation with the HIF‐β subunit and independent of oxygen tensions. Thus, in hypoxia, LIMD1 expression facilitates formation of an active PHD2‐LIMD1‐VHL HIF‐degradation complex.
HIF‐1‐driven LIMD1 expression is required for negative regulation of HIF in a hypoxic environment
LIMD1 protein expression has been previously shown to be significantly reduced or lost in human lung and breast cancers (Sharp et al, 2004, 2008; Spendlove et al, 2008). This led us to hypothesise that a decrease in the normal levels of LIMD1 protein expression as a result of LIMD1 loss of heterozygosity (LOH) or promoter methylation (Sharp et al, 2008) may disrupt the hypoxic PHD‐LIMD1‐VHL complex, and exacerbate HIF‐mediated gene expression and pro‐transforming effects in the context of a hypoxic tumour microenvironment.
To directly assay the effects of hypoxia‐driven LIMD1 expression, we utilised a lentiviral shRNA‐mediated knockdown‐rescue system that concurrently expresses an shRNA and a cDNA (Foxler et al, 2012; Fig 3A schematic). We utilised this system to express an shLIMD1 to deplete cells of endogenous LIMD1, whilst simultaneously re‐expressing an RNAi‐resistant (rr) Flag epitope‐tagged LIMD1 that was under the control of the endogenous LIMD1 promoter, which we previously identified as being an active and regulated promoter sequence under both normoxic and hypoxic conditions (Fig EV1I; Foxler et al, 2011). The promoter sequence contained the wild‐type HRE motif (HREwt); we also generated a version with a mutated HRE sequence (HREmut; Figs 3A and EV3A). Ectopic LIMD1 expression in cells transduced with these vectors (where endogenous LIMD1 is repressed by the shRNA) would therefore be potentially enhanced (HREwt) or unchanged (HREmut) by hypoxia through HIF‐1. U2OS, HeLa and SAEC were transduced with the paired set of lentiviruses described and identified within this non‐clonal population, and LIMD1 controlled by the HREwt promoter had a twofold to threefold hypoxic induction of LIMD1 (Figs 3B–E and EV3B–E). In contrast, mutation of the HRE within the LIMD1 promoter (HREmut) significantly impaired hypoxic induction of LIMD1 in these lines. This was coupled with an impairment of HIF‐1α degradation under increasing exposure to hypoxia in the HREmut lines compared to HREwt (Fig 3B and F–H). Of note, the HREmut cells had increased expression of HIF1A mRNA after 24 h in hypoxia compared to the HREwt cells (Fig EV3F), which we postulate may be the result of increased HIF‐1α protein in this line further driving its own transcription (Koslowski et al, 2011). The HREmut cell lines also exhibited increased HIF‐driven luciferase activity (Figs 3I and EV3G), endogenous HIF‐1‐driven gene activation (Figs 3J and EV3H–K) and cumulative secreted VEGF‐A (Figs 3K and EV3L) when compared to the HREwt cells.
Figure 3. Induction of LIMD1 in hypoxia inhibits HIF‐1‐mediated gene expression.
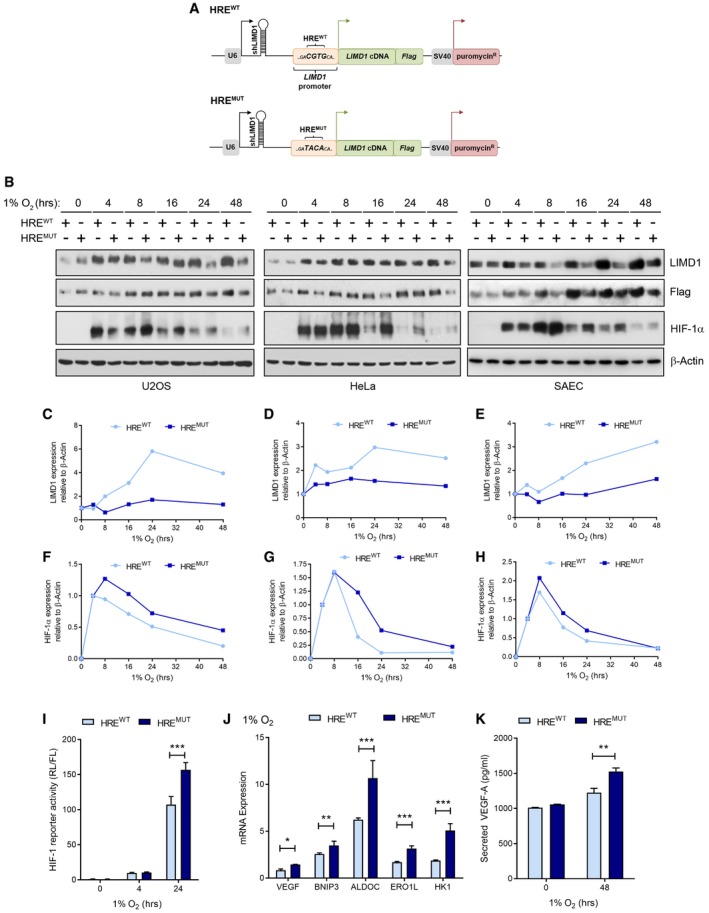
-
AA combinatorial RNAi–cDNA re‐expression lentiviral cassette was utilised to create isogenic cell lines where LIMD1 was either responsive or unresponsive to hypoxia. Endogenous LIMD1 was depleted by shRNA, whilst concurrently a Flag‐LIMD1 cDNA was expressed that was driven by the sequence of its own endogenous promoter.
-
BU2OS, HeLa and SAEC were transduced with these lentiviral cassettes to create the HREwt and HREmut paired cell lines.
-
C–EWestern blot quantification of LIMD1 relative to β‐actin and normalised to 0‐h time‐point for each cell line.
-
F–HWestern blot quantification of HIF‐1α relative to β‐actin and normalised to 4‐h time‐point for each cell line.
-
IImpaired hypoxic induction of LIMD1 induction increases HRE‐luciferase activity. U2OS isogenic cell lines were co‐transfected with a synthetic HRE‐luciferase (pNL‐HRE) and pGL3 firefly normalisation plasmid, prior to exposure to hypoxia. Luciferase activity was assayed and normalised against HRE activity in the HREwt line. After 24‐h hypoxic exposure, the HREmut line had significantly increased luciferase activity compared to the HREwt line.
-
JImpaired hypoxic induction of LIMD1 induction increases expression of HIF target genes. RNA was extracted from the U2OS isogenic cell lines following 24‐h hypoxic exposure, and a panel of HIF‐1 downstream targets were quantified by qRT–PCR. The HREmut line had significantly increased HIF‐1‐driven gene expressions compared to the HREwt line.
-
KImpaired hypoxic induction of LIMD1 induction increases VEGF‐A secretion. U2OS isogenic cell lines were incubated in hypoxia for 48 h and VEGF‐A secretion was quantified by ELISA, identifying the HREmut line as secreting a significantly increased VEGF‐A protein when compared to the HREwt line.
Figure EV3. Hypoxic induction of LIMD1 represses HIF‐1 transcriptional activity.
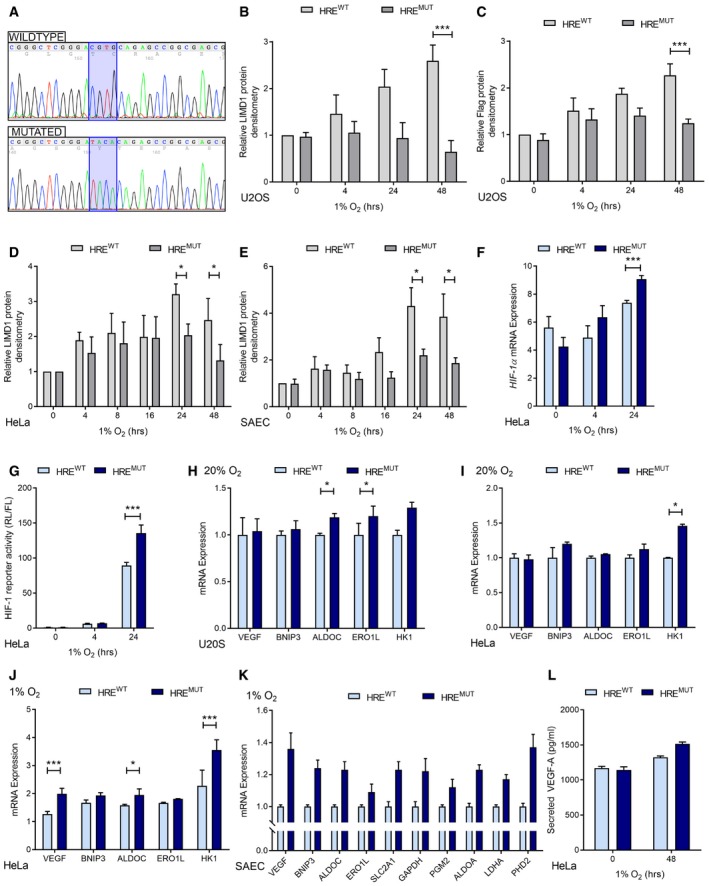
-
ASequence verification of mutation of the HRE motif within the LIMD1 promoter in the HREmut lentiviral cassette.
-
B, CDensitometric quantification of LIMD1 and Flag (LIMD1) protein expression in U2OS HRE isogenic cell lines (Fig 3B, mean ± SEM, n = 3). Expression of each protein was normalised to β‐actin loading control and the 0‐h time‐point.
-
D, EDensitometric quantification of LIMD1 protein expression in HeLa (D) and SAEC (E) HRE isogenic cell lines (Fig 3B, mean ± SEM, n = 3). Expression of each protein was normalised to β‐actin loading control and the 0‐h time‐point.
-
FHIF1A mRNA increases after 24 h hypoxia in the HREmut line. qRT–PCR analysis of RNA from HREwt and HREmut cells exposed to hypoxia for the indicated times. HIF1A mRNA expression was normalised to the RPII housekeeping gene.
-
GHypoxic LIMD1 induction decreases HRE‐luciferase activity in hypoxia. The isogenic cell lines were co‐transfected with a synthetic HRE‐luciferase (pNL‐HRE) and pGL3 Firefly normalisation plasmid, prior to exposure to hypoxia. Luciferase activity was assayed and normalised against HRE activity in the HREwt line. After 24‐h hypoxic exposure, the HREmut line had significantly increased luciferase activity compared to the HREwt line. n = 3, **P < 0.01.
-
H–KHypoxic LIMD1 induction decreases endogenous HIF‐driven gene expression in hypoxia. qRT–PCR analysis of RNA extracted from the U2OS (I), HeLa (J‐K) and SAEC (L) isogenic cell lines following normoxic or hypoxic (24 h) exposure. The HREmut line had significantly increased HIF‐1‐driven gene expressions compared to the HREwt line.
-
LHypoxic LIMD1 induction decreases VEGF‐A secretion in hypoxia. The isogenic HeLa cell lines were incubated in hypoxia for 48 h, and VEGF‐A secretion was quantified by ELISA.
We then wished to ascertain whether the observed transcriptional and phenotypic differences (Fig 3) were both HIF‐specific and due to the effects that LIMD1 expression was exerting on HIF‐1α protein turnover. Treatment with the translational inhibitor cycloheximide (Cx) to assess the degradation rate of HIF revealed the HREmut line had a significantly decreased rate of both HIF‐1α and HIF‐2α degradation compared to the HREwt line (Fig 4A and B). Furthermore, siRNA depletion of HIF‐1α ablated the differential gene expression of VEGF‐A between the lines (Fig 4C and D). Depletion of HIF‐2α also decreased VEGF‐A expression, likely due to VEGF‐A being a dual HIF‐1 and HIF‐2 target gene (Hu et al, 2003). We conclude that the differences in HIF stability and transcription observed between the HREwt and HREmut cell lines were caused by the hypoxic induction of LIMD1 via its HRE.
Figure 4. LIMD1 regulates HIF‐1α at the post‐translational level.
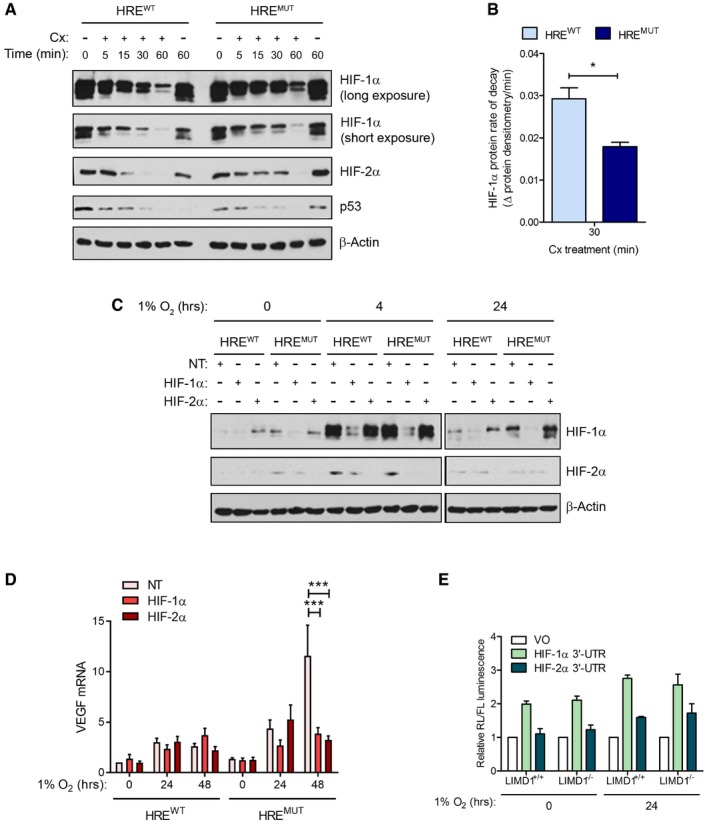
-
AThe hypoxic induction of LIMD1 facilitates HIF protein degradation. Western blot analysis of HREwt and HREmut U2OS lines exposed to hypoxia and the translation inhibitor cycloheximide (Cx) treatment for the indicated time‐points. HIF‐1α protein is degraded more efficiently in the HREwt line where LIMD1 expression is increased in hypoxia as detected by immunoblot.
-
BHIF‐1α rate of decay (ROD) is significantly slower in HREmut U20S compared to HREwt. Densitometric analysis of HIF‐1α protein, double normalised to β‐actin and 0 min Cx treatment in each line, was used to calculate ROD (Δ relative protein densitometry per minute) of HIF‐1α protein between 0 and 30 min of Cx treatment.
-
C, D(C) The increase in HIF target gene expression in the HREmut lines is due to HIF protein expression. siRNA (40 nM) targeting HIF‐1α and HIF‐2α was transfected into the HREwt and HREmut cell lines, and protein depletion was confirmed by immunoblot, which resulted in decreased expression of VEGF mRNA as analysed by qRT–PCR from the simultaneous extraction of RNA (D).
-
ELIMD1 expression does not affect miRNA silencing of HIF‐1α and HIF‐2α 3′UTR in normoxia or hypoxia. The 3′UTR of either HIF‐1α or HIF‐2α was cloned into the psiCheck2 luciferase vector. These were then transfected into LIMD1‐expressing control (LIMD1+/+) or LIMD1‐knockout (LIMD1−/−) HeLa cells and luciferase activity assayed. Expression of LIMD1 did not affect the stability or silencing of either the HIF‐1α or HIF‐2α 3′UTR‐containing reporters.
To exclude the possibility that the miRNA‐silencing function of LIMD1 (James et al, 2010) was complicit in this observed effect, we utilised luciferase reporter constructs containing the HIF‐1/2α 3′UTRs (as defined from RefSeq identifiers NM_001530.3 and NM_001430.4, respectively). There were no differences in HIF‐α 3′ UTR regulation in LIMD1‐expressing or null cells, regardless of oxygen tension (Fig 4E), indicating that LIMD1 was only affecting HIF‐α levels post‐translationally and not post‐transcriptionally in this experimental context of subtle but significant change in LIMD1 protein induction [twofold to threefold increase (Fig 3B–E)]. Together, these data reveal that inhibition of the HIF‐1/LIMD1 feedback loop causes an increased cellular hypoxic HIF phenotype in vitro, demonstrating that increased LIMD1 protein expression in hypoxia is necessary for correct modulation of HIF‐1 expression and signalling in a hypoxic environment.
Ablation of HIF‐driven LIMD1 expression promotes tumour growth
We next investigated the physiological relevance of this newly identified hypoxic HIF‐1–LIMD1 negative feedback loop using in vivo xenograft tumour growth as a model system. LIMD1 is a lung tumour suppressor, with decreased mRNA and protein expression shown to occur in a large proportion of lung adenocarcinomas (Sharp et al, 2008). For our xenograft model, we therefore utilised the A549 lung adenocarcinoma cell line. A549 LIMD1‐HREwt and A549 LIMD1‐HREmut cell lines were created as described above. The transduced cell lines were validated in vitro, where ablation of the hypoxic induction in LIMD1 expression in the HREmut line increased synthetic HIF‐1‐driven luciferase activity, HIF‐1‐responsive genes and secreted VEGF (Fig 5A–C). These findings also corroborated the results obtained in U2OS and HeLa cells (Figs 3 and EV3).
Figure 5. Increased LIMD1 expression in hypoxia inhibits tumour growth and vascularisation.
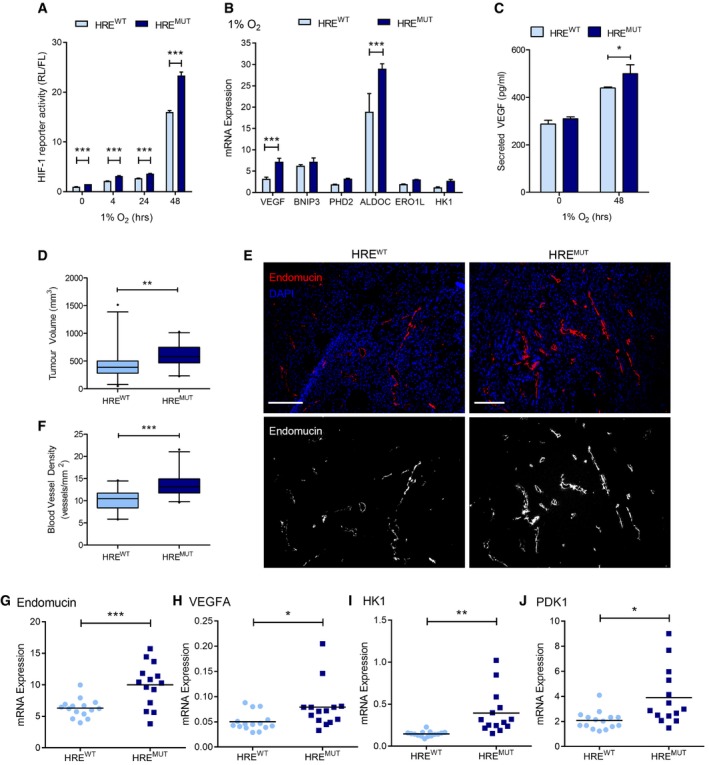
-
AImpaired hypoxic induction of LIMD1 induction increases HRE‐luciferase activity in lung adenocarcinoma cells. Isogenic HREwt and HREmut A549 cell lines were co‐transfected with a synthetic HIF‐1‐driven luciferase (pNL‐HRE) and pGL3 firefly normalisation plasmid, prior to exposure to hypoxia. Luciferase activity was assayed and normalised against HRE activity in the HREwt line.
-
BImpaired hypoxic induction of LIMD1 induction increases endogenous HIF‐mediated gene expression. RNA was extracted from the isogenic cell lines following 24‐h hypoxic exposure, and the expression of a panel of HIF‐1 target genes was quantified by qRT–PCR. The HREmut line had significantly increased HIF‐1‐driven gene expression compared to the HREwt line.
-
CImpaired hypoxic induction of LIMD1 induction increases VEGF‐A secretion in lung adenocarcinoma cells. The isogenic cell lines were incubated in hypoxia for 48 h, and VEGF secretion was quantified by ELISA, identifying the HREmut line as secreting a significantly increased level of VEGF‐A protein when compared to the HREwt line.
-
DImpaired hypoxic induction of LIMD1 induction increases 3D tumour growth in vivo. Eight‐ to 12‐week‐old female SCID/beige mice were injected subcutaneously with 5 × 109 A549 HREwt or HREmut cells and subsequent xenograft growth measured. HREwt‐derived xenografts were smaller in volume compared to the HREmut where LIMD1 expression was unresponsive to hypoxia.
-
EHREwt‐derived xenografts have lower blood vessel density compared to HREmut‐derived xenografts. Xenografts were snap frozen in liquid nitrogen, sectioned and stained with endomucin (red) and DAPI (blue) (upper panels). Lower panels show endomucin staining in white for visual clarity. Scale bar, 100 μm.
-
FBlood vessels were manually counted throughout the entire section and xenograft cross‐sectional area calculated.
-
G–J(G) HREwt‐derived xenografts have lower expression of the blood vessel marker endomucin. RNA was extracted from snap‐frozen xenografts and analysed by qRT–PCR. In addition, HREwt xenografts also had lower expression of the HIF‐driven genes (H) VEGF‐A, (I) HK1 and (J) PDK1. n = 15 (HREwt) and n = 14 (HREmut).
Subcutaneous xenografts were established on the flanks of SCID/beige mice from either the A549 LIMD1 HREwt or HREmut cell lines. Xenografts from A549 HREmut cells (which have an impaired HIF–LIMD1 negative feedback loop) had increased age‐matched endpoint tumour volumes compared to A459 HREwt cells (which have an intact HIF–LIMD1 negative feedback loop; Fig 5D). The effect on in vivo tumour growth was not due to intrinsic differences in proliferation rates as HREwt and HREmut cells had similar growth rates in in vitro proliferation assays and in colony formation assays under either normoxia or hypoxia (Fig EV4A and B). Endomucin staining, as a marker of blood vessels, revealed increased vasculature in HREmut A549‐derived xenografts (Fig 5E and F) and was associated with increased endomucin mRNA expression (Fig 5G) along with an increased HIF‐1‐mediated gene expression profile of pro‐angiogenic and glycolytic genes (Figs 5H–J and EV4C–O). HIF1A mRNA expression was not altered upon LIMD1 loss (Fig EV3F), demonstrating that the increase in HIF‐1‐driven gene expression was not due to increased HIF‐1α transcription. Together, these data indicate that ablation of this HIF‐1–LIMD1 negative regulatory feedback mechanism in vivo increases tumour growth and vascularisation.
Figure EV4. LIMD1 hypoxic induction represses HIF‐1 target gene expression in vivo .
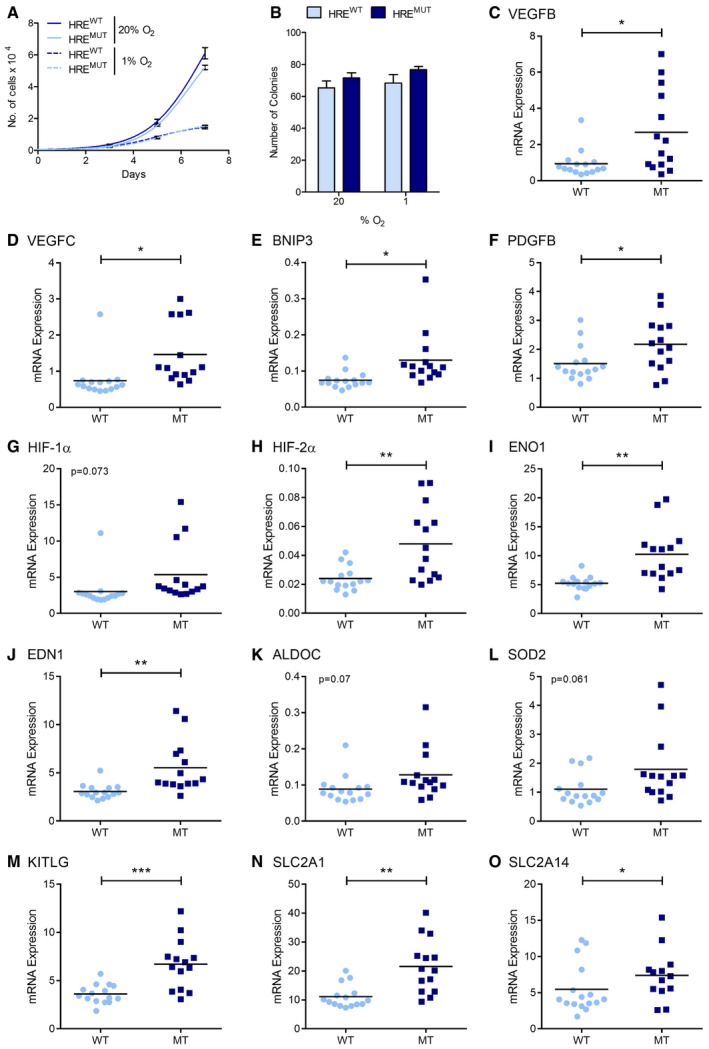
-
AIncreased hypoxic LIMD1 expression does not affect 2D growth rates. A549 isogenic cell lines were seeded into 6‐well plates and counted every 2 days to determine their proliferation rate in both normoxia and hypoxia.
-
BIncreased hypoxic LIMD1 expression does not affect colony formation ability. A549 isogenic cell lines were seeded into 6‐well plates at 100 cells/well and colony formation allowed to proceed for 10 days in either normoxia or hypoxia. Cells were then fixed and stained with crystal violet for counting.
-
C–OqRT–PCR analysis of the indicated HIF‐responsive genes from HREwt‐ and HREmut‐derived xenografts. Eight‐ to 12‐week‐old female SCID/beige mice were injected subcutaneously with 5 × 109 HREwt or HREmut A549 cells and subsequent xenograft growth measured until the first measurement exceeded the legal limit of 1.44 cm2. Xenografts were removed and snap frozen in TriPure. Xenografts were homogenised and followed by aqueous phase RNA extraction. RNA expression was analysed by qRT–PCR and normalised to the RPII housekeeping gene. n = 15 (HREwt) and n = 14 (HREmut).
LIMD1 is a prognostic indicator in NSCLC
Finally, to investigate the clinical relevance and significance of our in vitro and in vivo findings in NSCLC, we examined LIMD1 protein expression in a tissue microarray of 276 lung cancer patients and investigated correlation with patient outcome (representative staining Fig 6A; marker distribution Fig 6B). In agreement with previous studies, LIMD1 protein expression was detected in both nuclear and cytoplasmic compartments (Sharp et al, 2008; Spendlove et al, 2008). Kaplan–Meier survival analysis demonstrated that patients exhibiting low LIMD1 expression within this cohort had significantly worse overall survival compared to those with high LIMD1 expression (P = 0.045; Fig 6C). Immunohistochemical analysis of HIF‐1α and downstream angiogenic marker VEGF‐A was not significantly correlated with LIMD1 expression in this cohort; however, high VEGF expression was correlated with poor patient prognosis (P = 0.045; Appendix Fig S1, Fig EV5A–C).
Figure 6. Loss of LIMD1 expression is a poor prognostic indicator in lung cancer.
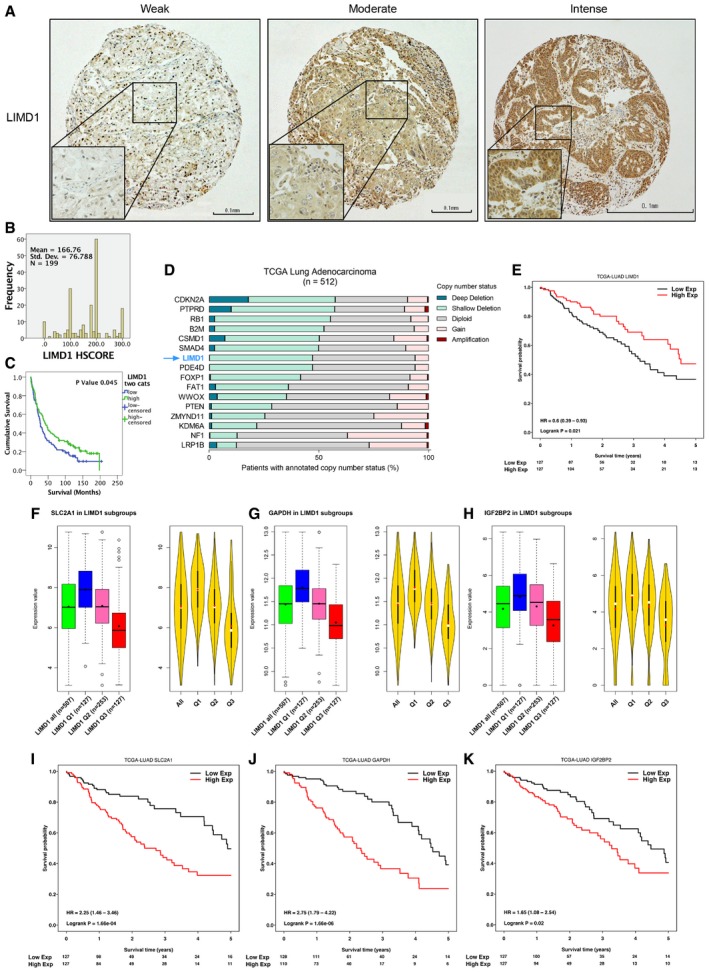
-
A, BRepresentative immunohistochemistry (IHC) staining of cores for LIMD1 that were scored in the 276 patient sample TMA to ascertain relative expression (H‐score) within the cohort (B). Scale bar 100 μm; 20 μm on zoom panel.
-
CKaplan–Meier analysis identified that patients stratified as having low LIMD1 expression (weak staining) exhibit poorer overall survival compared to high (intense staining).
-
DCopy number alterations of LIMD1 and other validated tumour suppressor genes from a lung adenocarcinoma (LUAD) cohort, publically available from TCGA.
-
EStratification of TCGA LUAD cohort into quartiles based on LIMD1 mRNA expression (where Q1 = lowest expression quartile, Q3 = highest expression quartile) demonstrates worse overall survival for patients within the lowest LIMD1‐expressing quartile (Low Exp) compared to the highest LIMD1‐expressing quartile (High Exp).
-
F–H(F) Correlation analysis of LIMD1 mRNA expression in patients from (D) identified a significant inverse correlation between LIMD1 and HIF target genes SLC2A1, (G) GAPDH and (H) IGF2BP2. The violin plots show the local density of data: black bars represent the IQR, red dot is the mean and white dot is the median.
-
I–K(I) Stratification of TCGA LUAD cohort into quartiles based on SLC2A1, (J) GAPDH and (K) IGF2BP2 expression demonstrates worse overall survival for patients within the highest expressing quartile (High Exp) compared to the lowest expressing quartile (Low Exp) for each gene.
Figure EV5. HIF target gene VEGF‐A expression is a poor prognostic indicator in lung adenocarcinoma and squamous cell carcinoma, where LIMD1 is frequently lost.
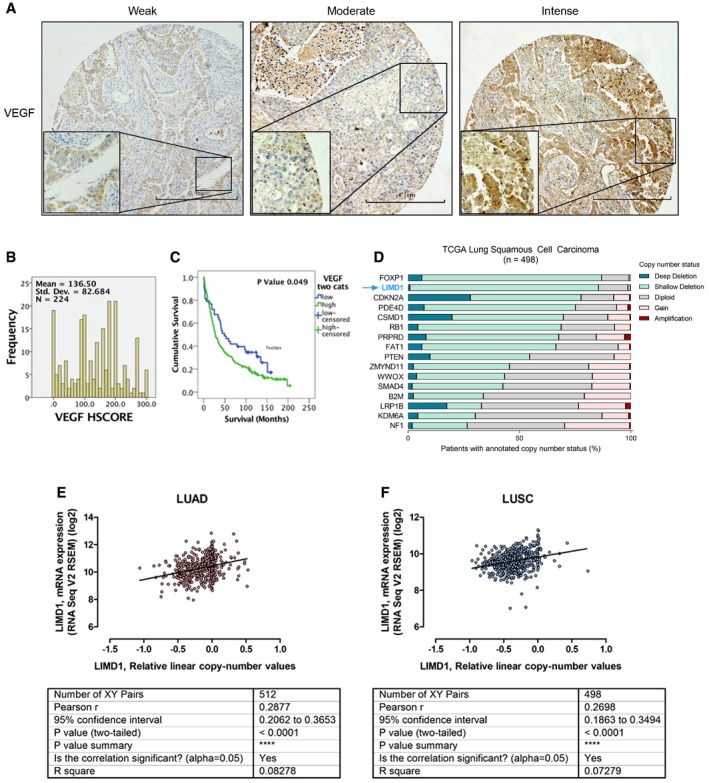
-
A, B(A) Representative immunohistochemistry (IHC) staining of cores for VEGF‐A that were scored in the 276 patient sample TMA to ascertain relative expression (H‐score) within the cohort (B). Scale bar 100 μm; 20 μm on zoom panel.
-
CKaplan–Meier analysis identifies patients stratified as having high VEGF‐A expression (intense staining) exhibit poorer overall survival compared to low (weak staining).
-
DCopy number alterations of LIMD1 and other validated tumour suppressor genes from a lung squamous cell carcinoma (LUSC) cohort (n = 498), publically available from TCGA.
-
E, F(E) Regression analysis performed upon a lung adenocarcinoma (LUAD) and (F) squamous cell carcinoma (LUSC) cohort from TCGA demonstrates significant correlation between LIMD1 copy number and mRNA expression.
We next interrogated The Cancer Genome Atlas (TCGA) datasets to assess LIMD1 loss in a much larger cohort of NSCLC. First, gene copy number analysis of LIMD1 and a number of other well‐characterised tumour suppressor genes in lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) cohorts (n = 512 and n = 498, respectively) demonstrated that single (shallow) or bi‐allele (Ghosh et al, 2008) deletion of the LIMD1 gene occurred in 47.1% (LUAD) and 85.4% of patients (LUSC; Figs 6D and EV5D). Regression analysis demonstrated correlation between LIMD1 copy number and reduced mRNA expression (Fig EV5E and F); therefore, lung adenocarcinoma patients were stratified into risk groups (quartiles) based on mRNA abundance intensities, and patient survival was determined using a Cox proportional hazards model. We determined that patients in the risk group exhibiting high LIMD1 expression (High Exp) had increased overall survival, whereas patients exhibiting low LIMD1 expression (Low Exp) had reduced overall survival (log‐rank P = 0.021, HR 0.6; Fig 6E).
To assess the impact of LIMD1 loss on HIF regulation and outcome in these patients, we analysed the TCGA LUAD cohort to identify hypoxia/HIF signature genes correlated with low LIMD1 expression. We identified a strong inverse correlation between LIMD1 and HIF target genes SLC2A1, GAPDH and IGF2BP2 mRNA expression (Fig 6F–H). Kaplan–Meier analysis of survival of patients stratified by expression of these genes revealed that patients with the highest expression of these genes have significantly worse overall survival compared to patients who demonstrate the lowest expression (Fig 6I–K).
To complement the bioinformatic analysis of patient tumours, we performed cell line‐based in vitro analysis of the gene expression changes that occur in a primary lung epithelial cell background following a reduction in LIMD1 expression. We used siRNA to knock down LIMD1 in primary human lung bronchial epithelial cells (HBEC) and performed micro‐array analysis to identify the resultant gene ontology changes. Due to the cell type used in this analysis, this models the role of LIMD1 loss in lung SCC. Differentially expressed genes and associated pathways following depletion of LIMD1 were analysed by ingenuity pathway analysis (IPA), and this identified upregulation of HIF‐1α and a network of HIF‐1 interactions following LIMD1 loss (Appendix Figs S2 and S3, and Dataset EV1).
In summary, these findings reveal the existence of a previously uncharacterised tumour‐suppressive, negative regulatory feedback loop in which LIMD1 facilitates HIF degradation in hypoxia (Fig 7). These findings add another level to the rapidly growing number of pathways and processes that regulate HIF. This is the first example of the main scaffold protein LIMD1 within the regulatory PHD2‐LIMD1‐VHL complex being itself regulated by HIF and therefore providing this regulatory triad with an intrinsic homeostatic negative regulatory functionality, which when deregulated contributes to lung adenocarcinoma tumour growth.
Figure 7. Proposed model of the HIF‐1–LIMD1 negative regulatory feedback mechanism.
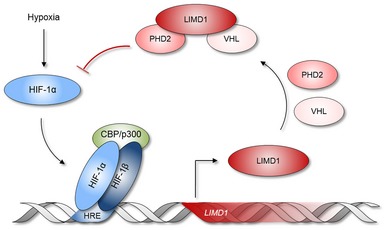
HIF‐1α is stabilised in hypoxia and heterodimerises with HIF‐1β, forming an active HIF‐1 transcription factor complex with CBP/p300. HIF‐1 binds to hypoxic response elements (HRE) within the promoters of genes that are required for hypoxic survival and adaptation, increasing their gene expression. Classically, these include genes required for metabolic adaptation and angiogenesis. Here, we identify that the tumour suppressor gene LIMD1 is also a HIF‐1‐responsive gene, and in hypoxia, its expression is increased at both the mRNA and protein levels. This increased expression facilitates formation of a hypoxic PHD2‐LIMD1‐VHL degradation complex, facilitating hypoxic HIF‐1α degradation. In turn, this attenuates the tumorigenic cellular adaption to hypoxia and subsequent tumorigenesis, thus identifying a new level of complexity of a tumour‐suppressive mechanism of LIMD1.
Discussion
We have previously shown that the scaffold protein LIMD1 is a critical component of the HIF‐degradation complex (Foxler et al, 2012). In this study, we have demonstrated that LIMD1 expression is induced by HIF‐1 under hypoxic conditions, forming a negative feedback loop to degrade HIF. HIF‐1‐driven LIMD1 expression enables the cell to degrade HIF‐1α under conditions of chronic hypoxia to prevent protracted HIF‐1 activation, frequently associated with an oncogenic phenotype. In a xenograft model for tumorigenesis, ablation of the hypoxic inducibility of LIMD1 expression and subsequent loss of hypoxic HIF‐1α protein regulation caused increased tumour vasculature and growth. From a clinical perspective, decreased LIMD1 expression correlates with increased expression of HIF target genes SLC2A1, GAPDH and IGF2BP2, each of which correlates with significantly worse survival outcomes for patients.
By virtue of its scaffold function, the cellular processes and pathways that LIMD1 regulates are dependent on its protein partners. HIF‐α is post‐transcriptionally regulated (Gorospe et al, 2011), including by microRNA‐20b, microRNA‐199a and microRNA‐424 (Rane et al, 2009; Cascio et al, 2010; Ghosh et al, 2010a). Therefore to rule out the possibility that the miRNA‐silencing function of LIMD1 (James et al, 2010) was contributing to the observations made, we identified that loss of LIMD1 does not affect the stability nor silencing of HIF1A/HIF2A mRNA. LIMD1's multiple tumour‐suppressive functions and its discrimination between binding partners are likely to be regulated by different signalling cascades, with multiple phosphorylation events on LIMD1 having already been identified (Huggins & Andrulis, 2008; Sun & Irvine, 2013). However, any post‐translational modification events that promote LIMD1 to function preferentially in HIF regulation are yet to be elucidated, but may stem from a hypoxia‐/HIF‐activated signalling cascade (Wouters & Koritzinsky, 2008; Lee et al, 2012; Xu et al, 2017).
LIMD1 has few reported coding sequence mutations and none that correlate with loss of function (Huggins et al, 2007; Ghosh et al, 2008). Such data suggest that gene dosage and thus small but significant changes in protein levels may be important in disease aetiology. Loss of total gene expression is frequently observed, where reduced LIMD1 gene copy number and mRNA expression occur in a significant proportion of lung carcinomas (Sharp et al, 2008). In this study, we have identified that a reduction in LIMD1 expression (through ablation of the hypoxic responsiveness of LIMD1 promoter) is sufficient to cause a HIF‐mediated pathological transcriptome and phenotype in the form of increased tumour size and vasculature.
The regulation of HIF under normoxic conditions is well characterised (Salceda & Caro, 1997; Maxwell et al, 1999); however, the mechanism of HIF degradation under long‐term hypoxic conditions is still poorly defined at the molecular level, even less so with respect to the physiological relevance. However, our new data show that discrete regulatory processes such as the HIF/LIMD1 negative feedback loop described here can modulate HIF activity under chronic hypoxic conditions. Biological pathways often have in‐built negative feedback loops whereby a transcription factor induces the expression of an upstream negative regulator (Yosef & Regev, 2011); such a negative feedback loop exists for the HIF proteins and the hypoxic response (Marxsen et al, 2004; Stiehl et al, 2006; Tan et al, 2008; Nakayama et al, 2009). PHD2 depletion under hypoxia results in stabilisation of HIF‐1α (Stiehl et al, 2006), demonstrating HIFs can be actively degraded and regulated independent of oxygen tensions. Indeed, the complexity of HIF regulation via the hydroxylation–ubiquitination degradation pathway is becoming increasingly clear, with the identification of a plethora of HIF regulators, more recently including Siah2, SHARP1 and RHOBTB3 (Nakayama et al, 2009; Montagner et al, 2012; Zhang et al, 2015).
Expression of HIF‐α mRNA has historically been described as constitutive; however, a growing number of studies are demonstrating the existence of factors that regulate HIF‐α mRNA (Uchida et al, 2004; Chamboredon et al, 2011; Hamidian et al, 2015). From our investigations, the LIMD1 HREmut‐derived A549 xenografts had an increase in HIF2A mRNA expression compared to the HREwt controls. We propose that inhibition of hypoxia‐driven LIMD1 expression, resulting in enhanced HIF‐α protein stability, drives HIF2A mRNA expression. Stabilisation of HIF‐α protein and hypoxia has previously been described as inducers of HIF2A mRNA expression (Hamidian et al, 2015; Mohlin et al, 2015).
Many HIF‐activated genes have been identified as prognostic and diagnostic markers. The oncogenic properties of HIF in cancer and disease have resulted in an abundance of potential therapeutics in development and clinical trials that target HIF at the transcriptional, translational, post‐translational and functional levels (Masoud & Li, 2015; Nakazawa et al, 2016). Indeed, the hypoxic HIF signalling pathway is already a therapeutic target with multiple drugs currently in clinical trials (Wilson & Hay, 2011; Bryant et al, 2014; Masoud & Li, 2015). VEGF expression is a marker of poor prognosis in NSCLC (Kaya et al, 2004); as such, VEGF‐targeted therapies are widely used to target VEGF‐mediated angiogenesis, although the details of how they exert their effects are not yet clearly defined (Ellis & Hicklin, 2008). In some cases, vasculature promotion and normalisation have been demonstrated to yield greater therapeutic efficacy (Wong et al, 2015, 2016). In our small cohort of 276 NSCLC, high VEGF expression was correlated with poor survival, with 80% of the cohort demonstrating high VEGF expression.
Analysis of a TCGA lung adenocarcinoma cohort determined an inverse correlation between expression of LIMD1 and HIF target genes SLC2A1, GAPDH and IGF2BP2. Overexpression of glucose transporter 1 (GLUT‐1), the uniporter protein encoded by the SLC2A1 gene, is correlated with poor survival in most solid tumours (Wang et al, 2017), and selective GLUT‐1 inhibition has been demonstrated to inhibit glucose uptake and reduce viability of lung adenocarcinoma and squamous cell carcinoma cell lines in vitro and in vivo (Goodwin et al, 2017). Correlative studies have also determined upregulation or de novo expression of the IGF2BP family of oncofetal proteins across a number of solid tumours to be associated with tumour aggressiveness, metastasis and poorer overall survival (Bell et al, 2013). GAPDH overexpression is also associated with reduced patient survival (Guo et al, 2013), and therapeutic targeting of GAPDH has been demonstrated to have clinical application in both hepatocellular and colorectal cancers (Ganapathy‐Kanniappan et al, 2012; Yun et al, 2015). Our data correlate LIMD1 loss with overexpression of each of these HIF target genes, and as such hold potential for both stratification of patients based on a LIMD1low, SLC2A1high/IGF2BP2high/GAPDHhigh genetic profile and targeted therapies based upon this HIF target gene signature.
Taken together, our findings hold significant impact for the aetiology of LIMD1‐negative lung cancers and hold the potential for advances in the diagnosis and prognosis of such cancers with respect to deregulated HIF regulation and associated oncogenic phenotypes, and subsequently hypoxia‐targeted therapies.
Materials and Methods
Bioinformatic analysis
As a reference point for referring to positions within the LIMD1 promoter, the unconfirmed transcriptional start site (TSS) was assigned according to the NCBI reference sequence NM_014240.2. This corresponds to nucleotide 45636323 on the primary chromosome 3 ref assembly NC_000003.11 and is 49‐bp upstream from the AUG translation initiation codon. The human LIMD1 promoter, which was preliminarily designated as 2.5‐kbp upstream of the ATG translation initiation codon, was scrutinised using the Ensembl Genome Browser (http://www.genome.ucsc.edu) for the presence of CpG Islands, utilising the default software thresholds. The in silico screen for transcription factor binding motifs within the promoter was performed using the MatInspector software programme (http://genomatix.de) using the Matrix Family Library Version 8.1. HRE3 multiple sequence alignment was performed using LIMD1 promoter sequences from the UCSC Genome Browser and aligned using Clustal Omega. HRE3 sequence logo was generated using WebLogo.
Copy number analysis (CBioportal)
Provisional TCGA LUAD and LUSC datasets were accessed and downloaded from cbioportal.org. The datasets included 512 (LUAD) and 498 (LUSC) patients. Focally deleted genes altered in lung adenocarcinoma and squamous cell carcinoma were selected from a study by Campbell et al (2016) and used, along with LIMD1, to query LUAD and LUSC provisional datasets (via http://www.cbioportal.org). Putative copy number alterations from GISTIC were used to identify the following copy number categories: amplification, gain, diploid, shallow deletion and deep deletion for each gene. The percentage of samples displaying each category was then calculated for each gene. For each sample, linear LIMD1 copy number values were plotted against LIMD1 mRNA expression values (log2‐transformed RNA Seq V2 RSEM). Pearson's r correlation analysis was performed using GraphPad Prism version 7.04.
The Cancer Genome Atlas (TCGA) analysis
All analyses were conducted in the R statistical environment (v 3.3.2).
Data
Level‐3 RNA‐Seq data for human lung adenocarcinoma (LUAD) primary tumours (n = 517) were downloaded from the TCGA using the TCGAbiolinks R package (Cancer Genome Atlas Research Network, 2014; Colaprico et al, 2016).
Correlation
Pearson's product‐moment correlation coefficient and P‐value of pairwise comparisons between the mRNA abundance values of LIMD1 and hallmark hypoxia gene set (GSEA) were computed.
Survival
Patients were stratified into three risk groups based on the quartile values of mRNA abundance densities. The prognostic values of SLC2A1, GAPDH and IGF2BP2 in lower quartile (n = 127) and upper quartile (n = 127) risk groups were assessed using a Cox proportional hazards regression model, with P‐values estimated using log‐rank test. The survival modelling and Kaplan–Meier analyses were conducted using the survival package (v 2.41‐3; Therneau & Grambsch, 2000). All analyses were conducted for a 5‐ and 10‐year survival timeframe.
Cell culture
U2OS, HEK293T, HeLa and A549 cells were maintained in D‐MEM (Sigma) supplemented with 10% FCS and 1% pen/strep solution in a humidified 37°C incubator and 5% CO2. SAEC were maintained in complete small airway epithelial cell growth medium (Lonza). Cells were regularly tested for mycoplasma. Hypoxic incubations were carried out at 1% O2 within a humidified ProOx110 controller chamber (BioSpherix Ltd, New York, USA). Cells were transfected using Viafect (Promega E4981) with a 3:1 ratio of Viafect:DNA.
Promoter cloning
Site‐directed mutagenesis reactions were performed using QuikChange XL Site‐Directed Mutagenesis Kit (Stratagene #200517) as per the supplied protocol as confirmed by sequencing (Source Bioscience UK Limited, Nottingham). Primer sequences are supplied in Appendix Table S10.
shRNA plasmids generation and transduction
shRNA sequences (supplied in Appendix Table S10) were annealed and ligated into psiRNA‐DUO plasmid (Invivogen # ksirna4‐gz3) with Acc65I and HindIII (NEB #R0599S and #R0104S).
The knockdown‐rescue shRNA lentiviral system was a kind gift from Greg Longmore (Feng et al, 2010). The lentiviral system allows simultaneous shRNA‐mediated knockdown of an endogenous target with concurrent rescue of the same RNAi‐resistant target. Lentiviral plasmids containing a LIMD1 targeting shRNA with concurrent RNAi‐resistant LIMD1 cDNA expression was modified so that expression of the cDNA was driven by the endogenous LIMD1 promoter. A 2‐kb region upstream of the LIMD1 ATG translation initiation codon was amplified using the primers ggagcgGTCGACCAGGCACTTGGCATACAGATATGGTC (SalI forward) and cgctccGAATTCGCTGCAGACAGGTGTCCGGGCCTAG (EcoR1 reverse). The ubiquitin C promoter from the pFlRu plasmid that drives the rescue expression was then replaced with the amplified LIMD1 promoter. To create a rescue plasmid with a mutated HRE, site‐directed mutagenesis with the already described ΔHRE3 primers was used. Lentivirally transduced cell lines were created as previously described (Foxler et al, 2012).
Luciferase assays
The HRE‐pGL3 luciferase construct containing three copies of the HRE from the phosphoglycerate kinase promoter was a kind gift from Thilo Hagen (Department of Biochemistry, National University of Singapore). The HRE element was subcloned into the pNL1.1 vector (Promega). Cells were co‐transfected with 50 ng of HRE reporter vector and 5 ng firefly normalisation reporter plasmid DNA per well of a 12‐well plate and lysed 36 h post‐transfection in 1× Passive Lysis Buffer (Promega E1941). Luciferase activity assayed using a Nano‐Glo Dual‐Luciferase Reporter Assay System (Promega N1610). The 3′UTR sequences for HIF‐1α and HIF‐2α were PCR cloned from a HeLa cell cDNA and into the psiCheck2 vector using 5′XhoI and 3′NotI sites incorporated into the PCR primers. Primer sequences are supplied in Appendix Table S10.
qRT–PCR
All qRT–PCR was performed using a 1‐step RT–qPCR method (Promega A6020). All reactions were performed in triplicate with 20 ng of RNA 200 nM forward and reverse primers in a 25 μl reaction volume run on an ABI7000 instrument (Applied Biosystems). Gene‐specific primers were designed to span an exon boundary and data normalised to the housekeeping genes RNA polymerase II. RNA extractions from cells were performed using a column‐based purification (Promega Z6011) and from xenograft tissue following homogenisation in Tripure (Roche Applied Science 11667157001) and aqueous phase extraction. The list of primers used appear in Appendix Table S10.
VEGF‐A ELISA
ELISAs were performed on cell supernatants following 48‐h hypoxia utilising the Human VEGF‐A Quantikine ELISA Kit (R&D Systems DVE00).
Chromatin immunoprecipitation (ChIP)
1 × 107 cells were stimulated overnight in hypoxia. Formaldehyde (1% v/v) was added to cross‐link protein–DNA for 10 min at 37°C and was quenched with ice‐cold 0.125 M glycine/PBS. Cells were harvested in 1 ml harvesting buffer (0.125 M glycine, 1 mM EDTA and protease inhibitors in PBS) and pelleted by centrifugation at 3,500 g for 10 min at 4°C. The cell pellet was resuspended in 100 μl of lysis buffer (50 mM Tris–HCl pH8.0, 1% SDS, 10 mM EDTA plus protease inhibitors) and incubated on ice for 10 min. 50 μl of dilution buffer (20 mM Tris–HCl pH8.0, 1% Triton X‐100, 2 mM EDTA, 150 mM NaCl plus protease inhibitors) was added and lysates sonicated on ice to shear the DNA to 200–600 bp. Lysates were cleared of insoluble material by centrifugation at 13,000 g for 10 min at 4°C. An input sample was taken, and the remaining soluble chromatin containing supernatant diluted to 1 ml with dilution buffer and added to an antibody‐conjugated IP matrix and incubated overnight at 4°C with rotation. IP matrix beads were washed 6 × 1 ml RIPA and protein–DNA complexes eluted in 2 × 75 μl elution buffer (1% SDS, 0.1 M NaHCO3) for 15 min at room temperature with rotation. Cross‐links were then reversed for 6 h at 65°C with NaCl (0.2 M), followed by incubation with 20 μg proteinase K, 40 mM Tris–HCl pH 6.5, 10 mM EDTA. DNA was then purified using the Qiagen PCR purification kit.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays were performed using the method described in Foxler et al (2011) following incubation of cells at 1% O2 for 24 h, prior to nuclear extraction. MG132 was included in all buffers to prevent degradation of HIF‐1α. 5 μg of nuclear extracts was incubated in a total volume of 20 μl HIF binding buffer (50 mM KCl, 10 mM Tris pH 7.7, 5 mM DTT, 1 mM EDTA, 2 mM MgCl2, 5% glycerol, 0.03% NP‐40 and 400 ng salmon testes DNA) with 1 μl of HIF‐1α or HIF‐2α antibodies. Binding reactions were pre‐incubated on ice for 30 min prior to addition of 32P‐labelled probe with an overnight incubation at 4°C. Complexes were then resolved on 5% polyacrylamide gels (acrylamide: bisacrylamide 30:0.8) at room temperature. Gels were dried and developed using a Fuji‐film LAS‐3000 phosphor‐imager. For specificity of the HIF binding site and for competition assays, probes with the HIF site mutated were also used. Probes were synthesised by Sigma‐Aldrich and listed in Appendix Table S10.
Immunoblotting
Protein lysates were analysed using SDS–PAGE using standard Western blotting protocols. The list of antibodies used appear in Appendix Table S11.
Cycloheximide (Cx) treatment
Cells were plated 24 h prior to treatment with cycloheximide (200 μg/ml; sc‐3508). Media containing 400 μg/ml Cx were incubated in 1% O2 for 24 h to allow de‐oxygenation prior to addition to an equal volume of cell media. Lysates were taken immediately prior to Cx addition (start‐point) and following the last time‐point without Cx added (endpoint) to disseminate between drug‐induced and endogenous HIF‐α protein turnover. Cx was added so that all drug treatment times finished at the same time. HIF‐α protein expression was assayed by Western blot and quantified with ImageJ software and normalised to β‐actin loading control. The rate of turnover was calculated from the gradient of log[HIF‐α protein] against time.
Animal studies
All animal experiments conformed to the British Home Office Regulations (Animal Scientific Procedures Act 1986; Project License PPL 70/7263 to Prof Nick Lemoine). Trial experiments and experiments done previously were used to determine sample size with adequate statistical power. Twenty‐five mice for each group were studied in total in two independent experiments. Eight‐ to 12‐week‐old female SCID/beige mice (Harlan Laboratories) were given a subcutaneous injection of 5 × 106 transduced A549 cells in 100 μl PBS into the right flank for subcutaneous tumour growth. Calliper measurements were taken over time, and the experiment reached an endpoint when the first tumour measurement of maximum length x maximum breadth exceeded the maximum size dictated by the Project licence. Mice were killed by Schedule 1 cervical dislocation. Xenografts were then immediately excised prior to the onset of rigour mortis. Xenografts were bisected and half flash frozen in liquid N2 or frozen on dry ice in 0.8 ml Tripure for subsequent RNA extractions. Snap‐freezing of fresh subcutaneous tumours is the best recognised method for subsequent effective blood vessel immunostaining. This method avoids many of the limitations of prefixing the tissue that can actually reduce antigenicity when it comes to immunostaining for blood vessels.
Xenograft analysis
Snap‐frozen xenograft samples were sectioned into 4–6 μM sections (Pathology Department, Bart's Cancer Institute, Queen Mary University of London). Sections were fixed in ice‐cold acetone for 10 min and stained overnight with the endothelial blood vessel marker endomucin (1/100, Santa Cruz V7C7) and AlexaFlour 546‐conjugated secondary antibody and mounted in Prolong Gold anti‐fade reagent with DAPI (Invitrogen, Paisley, UK). Stained sections were visualised on a Zeiss Axioplan microscope, and blood vessel number and tumour section area systematically counted and measured for the whole section area. For each xenograft, blood vessel density was calculated across a midline tumour section using the formula (Σ number of blood vessels)/(Σ section area). RNA was extracted using the manufacturer's recommended protocol following homogenisation in Tripure reagent.
In vitro clonogenic and proliferation assays
A549 HRE wild‐type and mutant cells were seeded into 6‐cm plates at 5 × 104 cells per plate. Twenty‐four hours after seeding plates were placed into 1% O2. Forty‐eight hours postseeding, three plates for each condition were trypsinised and counted using a TC20TM BioRad Cell Counter. Each plate was counted in duplicate and an average cell count calculated. Plates were counted 48, 72, 120 and 168 h postseeding, and growth curves generated. Four biological repeats of this experiment were conducted.
Microarray analysis
RNA was extracted from HBEC treated for 72 h with 80 nM scrambled or LIMD1 targeting siRNA in quadruplet. Microarray analysis was performed by the Genome Centre, Bart's Cancer Institute using a HT12v4.0 Illumina array. Raw data were normalised and fold‐change gene expression calculated from the average expression value for each condition. Genes with a q value cut‐off of < 0.15 were analysed by ingenuity pathway analysis (IPA) [Qiagen] software using fold‐change values for each mRNA: siRNA LIMD1 versus siRNA scrambled. Gene ontology was collected from the Bio Functions read‐out of IPA results where activation was > +1 or < −1 (Dataset EV1), and categories were collapsed into similar overall functions, e.g. apoptosis and cell survival, and directionality of function was inferred from the activation z‐score in Dataset EV1. The HIF‐1α transcription factor was indicated as an activated upstream regulator by IPA (HBEC activation score 3.114 and P‐value of overlap 1.76E‐04); therefore, this was also represented in the heat map. These categories were then applied to the heat map for functional clustering; only genes that were placed in these enriched categories are shown. IPA upstream analysis also produced a network of HIF‐1α interactions for HBEC siRNA LIMD1 versus siRNA scrambled treated. These interactions and the predicted activation state of downstream effectors are inferred from the total gene set submitted for IPA (q < 0.15).
Patient cohort and immunohistochemistry
The overall number of patients within the cohort was 276 of which 276 were valid for immunohistochemistry staining. The cohort consisted of 150 males and 126 females with an age range of 36–91 years. Ninety‐three percentage of cases were adenocarcinoma, 3% small‐cell carcinoma and 4% other types, and all 276 tumour cores were of primary lung tumour origin. Informed consent was obtained from all subjects, and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. IHC was carried out as previously described (Sharp et al, 2008; Spendlove et al, 2008). Briefly, slides were heated to 60°C on a hotplate (Leica, HI1220) for 10 min, allowed to cool for 5 min before being dewaxed in a Leica autostainer. Antigen retrieval was then performed via microwave for 20 min in citrate buffer pH 6.0 for VEGF sections. Antigen retrieval for LIMD1 was carried out in a water bath Epitope retrieval solution 2 pH 9.0 (Bond) at 95°C for 35 min. Slides were peroxidase treated (Novolink) for 5 min in Sequenza trays, washed with TBS twice for 5 min and blocked (Novolink) for 5 min. The working dilutions of the antibodies were then made up in antibody diluent (Bond leica); LIMD1 1/200, and VEGF‐A Pre‐diluted (SP28, Abcam). Beta‐2‐microglobulin 1/2,000 (Dako, A0072) was used as a reference positive control, and negative controls were without primary antibody. Each antibody at its chosen dilution was incubated for 60 min before being washed (×2). Secondary and tertiary reagents (Novolink postprimary and polymer) were incubated on the slides for 30 min each with a washing step between. Slides were developed with DAB solution (Novolink) and counterstained with haematoxylin (Novolink) for 6 min. Slides were then dehydrated and mounted in DPX prior to observation and laser scanning.
Statistics
Statistical analyses were performed using R 3.4.4 and are described in each figure legend. Where systematic differences existed between experimental runs, data were analysed with mixed‐model ANOVA allowing a separate intercept and effect of time for each run. Homoscedasticity and normality of all model residuals were evaluated graphically. Where residuals were non‐normal or heteroscedastic, the model was refit to the log10‐transformed dependent variable. If model assumptions were still not met, nonparametric tests or Welch‐corrected Student's t‐tests were used instead. Where data are normalised to a group, this group was excluded from the analysis. One‐sample Student's t‐tests were used when comparing data to a standardised group (theoretical value of 1).
Data availability
Microarray data from this publication have been deposited to Gene Expression Omnibus and assigned the identifier accession number https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114692.
Author contributions
DEF, KSB, JGF, PG, SC, KMS, KMD, AN, EG, HIR, PTK, MAH, T‐YC, PES, LER and TVS designed and performed experiments and analysed the data. TRM, H‐WW, PSR, MJP, DL, NRL, PR, TAG, CC, KMH‐D and IS provided reagents, experimental advice and design. All authors contributed to editing and proofreading the manuscript. DEF, KSB and TVS wrote the manuscript. TVS supervised and managed all research.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Within solid tumours, including lung cancer, inadequate oxygen levels (hypoxia) create an environment that is a driving force of cancer progression and form a resistance mechanism to all forms of therapy such as standard chemotherapies and ionising radiation. In chronic hypoxia, the transcriptional regulators of hypoxia (HIFs) are degraded through negative feedback loops; however, neoplastic cells evade this to survive in this harsh microenvironment. How this occurs in non‐small‐cell lung cancer is poorly understood and serves as an area of significant interest for cancer biology and potential hypoxia‐based targeted therapies.
Results
We identified that the tumour suppressor LIMD1, which facilitates efficient degradation of the transcriptional regulator of hypoxia (Foxler et al, 2012), is itself a HIF target gene. This creates a negative feedback loop whereby the activity of HIF is limited under prolonged hypoxic exposure, and mitigates pro‐tumorigenic effects of hypoxia. Subcutaneous implantation of cells lacking this feedback mechanism formed larger and more vasculature in vitro. Furthermore, in silico analysis of TCGA data shows that LIMD1 is lost in 47% of lung adenocarcinoma and serves as an independent prognostic marker. Deeper analysis of this dataset reveals a negative correlation between LIMD1 and a hypoxic gene set which further correlates with patient outcome.
To conclude, we have identified a novel LIMD1‐mediated negative feedback loop of HIF regulation that effects tumour growth, highlighting the functional importance of LIMD1 expression in normal lung homeostasis and the tumorigenic advantage its loss/deregulation gives to the hypoxic tumour microenvironment.
Impact
Our findings open a new field of research into the aetiology, diagnosis and prognosis of LIMD1‐negative lung cancers and hold the potential for advances in the stratification of patients with respect to deregulated HIF regulation and associated phenotypes. This holds the potential for development of chemotherapeutic drugs that target LIMD1 loss which could be used in combination with hypoxia‐targeted therapies.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by RCUK funding awarded to TVS from the BBSRC (grant BB/L027755/1), MRC (grant MR/N009185/1) and CRUK (grant CRUK‐A12733). PR is funded by a joint Royal College of Surgeons/Cancer Research UK Clinician Scientist Fellowship in Surgery (C19198/A15339). PR and JGF are both supported by the Barts Charity and the Orchid Charity (0000 0003 1861 7984) (to NRL). MJP is a BBSRC New Investigator (BB/N018818/1). DL is a Medical Research Council New Investigator Research Grant holder (MR/L008505/1).
EMBO Mol Med (2018) 10: e8304
References
- Ayyanathan K, Peng H, Hou Z, Fredericks WJ, Goyal RK, Langer EM, Longmore GD, Rauscher FJ (2007) The Ajuba LIM domain protein is a corepressor for SNAG domain mediated repression and participates in nucleocytoplasmic shuttling. Can Res 67: 9097–9106 [DOI] [PubMed] [Google Scholar]
- Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, Huttelmaier S (2013) Insulin‐like growth factor 2 mRNA‐binding proteins (IGF2BPs): post‐transcriptional drivers of cancer progression? Cell Mol Life Sci 70: 2657–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertout JA, Patel SA, Simon MC (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8: 967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge KS, Shah KM, Li Y, Foxler DE, Wong SCK, Miller DC, Davidson KM, Foster JG, Rose R, Hodgkinson MR et al (2017) Argonaute utilization for miRNA silencing is determined by phosphorylation‐dependent recruitment of LIM‐domain‐containing proteins. Cell Rep 20: 173–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant JL, Meredith SL, Williams KJ, White A (2014) Targeting hypoxia in the treatment of small cell lung cancer. Lung Cancer 86: 126–132 [DOI] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA et al (2016) Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48: 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio S, D'Andrea A, Ferla R, Surmacz E, Gulotta E, Amodeo V, Bazan V, Gebbia N, Russo A (2010) miR‐20b modulates VEGF expression by targeting HIF‐1 alpha and STAT3 in MCF‐7 breast cancer cells. J Cell Physiol 224: 242–249 [DOI] [PubMed] [Google Scholar]
- Chamboredon S, Ciais D, Desroches‐Castan A, Savi P, Bono F, Feige JJ, Cherradi N (2011) Hypoxia‐inducible factor‐1alpha mRNA: a new target for destabilization by tristetraprolin in endothelial cells. Mol Biol Cell 22: 3366–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhu X, Xie T, Xie J, Quo K, Liu X (2014) Drug resistance reversed by silencing LIM domain‐containing protein 1 expression in colorectal carcinoma. Oncol Lett 8: 795–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codelia VA, Sun G, Irvine KD (2014) Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol 24: 2012–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I et al (2016) TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44: e71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Feng Y, Jagannathan R, Seppa MJ, Skeath JB, Longmore GD (2010) Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr Biol 20: 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis LM, Hicklin DJ (2008) VEGF‐targeted therapy: mechanisms of anti‐tumour activity. Nat Rev Cancer 8: 579–591 [DOI] [PubMed] [Google Scholar]
- Feng Y, Nie L, Thakur MD, Su Q, Chi Z, Zhao Y, Longmore GD (2010) A multifunctional lentiviral‐based gene knockdown with concurrent rescue that controls for off‐target effects of RNAi. Genom Proteomics Bioinform 8: 238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxler DE, James V, Shelton SJ, Vallim TQ, Shaw PE, Sharp TV (2011) PU.1 is a major transcriptional activator of the tumour suppressor gene LIMD1. FEBS Lett 585: 1089–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxler DE, Bridge KS, James V, Webb TM, Mee M, Wong SC, Feng Y, Constantin‐Teodosiu D, Petursdottir TE, Bjornsson J et al (2012) The LIMD1 protein bridges an association between the prolyl hydroxylases and VHL to repress HIF‐1 activity. Nat Cell Biol 14: 201–208 [DOI] [PubMed] [Google Scholar]
- Ganapathy‐Kanniappan S, Kunjithapatham R, Geschwind JF (2012) Glyceraldehyde‐3‐phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 3: 940–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ghosh A, Maiti GP, Alam N, Roy A, Roy B, Roychoudhury S, Panda CK (2008) Alterations of 3p21.31 tumor suppressor genes in head and neck squamous cell carcinoma: correlation with progression and prognosis. Int J Cancer 123: 2594–2604 [DOI] [PubMed] [Google Scholar]
- Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y et al (2010a) Hypoxia‐induced microRNA‐424 expression in human endothelial cells regulates HIF‐alpha isoforms and promotes angiogenesis. J Clin Invest 120: 4141–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ghosh A, Maiti GP, Mukherjee N, Dutta S, Roy A, Roychoudhury S, Panda CK (2010b) LIMD1 is more frequently altered than RB1 in head and neck squamous cell carcinoma: clinical and prognostic implications. Mol Cancer 9: 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, Gatter KC, Harris AL (2001) Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non‐small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer 85: 881–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E (2008) PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci USA 105: 4745–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin J, Neugent ML, Lee SY, Choe JH, Choi H, Jenkins DMR, Ruthenborg RJ, Robinson MW, Jeong JY, Wake M et al (2017) The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat Commun 8: 15503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorospe M, Tominaga K, Wu X, Fahling M, Ivan M (2011) Post‐transcriptional control of the hypoxic response by RNA‐binding proteins and microRNAs. Front Mol Neurosci 4: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Liu S, Sun MZ (2013) Novel insight into the role of GAPDH playing in tumor. Clin Transl Oncol 15: 167–172 [DOI] [PubMed] [Google Scholar]
- Hamidian A, von Stedingk K, Munksgaard Thoren M, Mohlin S, Pahlman S (2015) Differential regulation of HIF‐1alpha and HIF‐2alpha in neuroblastoma: estrogen‐related receptor alpha (ERRalpha) regulates HIF2A transcription and correlates to poor outcome. Biochem Biophys Res Comm 461: 560–567 [DOI] [PubMed] [Google Scholar]
- Hou X, Li T, Ren Z, Liu Y (2016) Novel BRCA2‐interacting protein, LIMD1, is essential for the centrosome localization of BRCA2 in esophageal cancer cell. Oncol Res 24: 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia‐inducible factor 1alpha (HIF‐1alpha) and HIF‐2alpha in hypoxic gene regulation. Mol Cell Biol 23: 9361–9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins CJ, Gill M, Andrulis IL (2007) Identification of rare variants in the hLIMD1 gene in breast cancer. Cancer Genet Cytogenet 178: 36–41 [DOI] [PubMed] [Google Scholar]
- Huggins CJ, Andrulis IL (2008) Cell cycle regulated phosphorylation of LIMD1 in cell lines and expression in human breast cancers. Cancer Lett 267: 55–66 [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ et al (2001) Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 292: 468–472 [DOI] [PubMed] [Google Scholar]
- Jagannathan R, Schimizzi GV, Zhang K, Loza AJ, Yabuta N, Nojima H, Longmore GD (2016) AJUBA LIM proteins limit hippo activity in proliferating cells by sequestering the hippo core kinase complex in the cytosol. Mol Cell Biol 36: 2526–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James V, Zhang Y, Foxler DE, de Moor CH, Kong YW, Webb TM, Self TJ, Feng Y, Lagos D, Chu C‐Y et al (2010) LIM‐domain proteins, LIMD1, Ajuba, and WTIP are required for microRNA‐mediated gene silencing. Proc Natl Acad Sci USA 107: 12499–12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadrmas JL, Beckerle MC (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5: 920–931 [DOI] [PubMed] [Google Scholar]
- Kaya A, Ciledag A, Gulbay BE, Poyraz BM, Celik G, Sen E, Savas H, Savas I (2004) The prognostic significance of vascular endothelial growth factor levels in sera of non‐small cell lung cancer patients. Respir Med 98: 632–636 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Rabbani ZN, Dewhirst MW, Vujaskovic Z, Vollmer RT, Schreiber EG, Oosterwijk E, Kelley MJ (2005) Expression of HIF‐1alpha, CA IX, VEGF, and MMP‐9 in surgically resected non‐small cell lung cancer. Lung Cancer 49: 325–335 [DOI] [PubMed] [Google Scholar]
- Koslowski M, Luxemburger U, Tureci O, Sahin U (2011) Tumor‐associated CpG demethylation augments hypoxia‐induced effects by positive autoregulation of HIF‐1α. Oncogene 30: 876–882 [DOI] [PubMed] [Google Scholar]
- Langer EM, Feng Y, Zhaoyuan H, Rauscher FJ III, Kroll KL, Longmore GD (2008) Ajuba LIM proteins are snail/slug corepressors required for neural crest development in Xenopus . Dev Cell 14: 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Jin Y, He G, Zeng SX, Wang YV, Wahl GM, Lu H (2012) Hypoxia activates tumor suppressor p53 by inducing ATR‐Chk1 kinase cascade‐mediated phosphorylation and consequent 14‐3‐3gamma inactivation of MDMX protein. J Biol Chem 287: 20898–20903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao ZL, Liu L, Wang Y, Fan H (2015) [Expression and clinical significance of LIMD‐1 gene in adult patients with acute leukemia]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 23: 34–38 [DOI] [PubMed] [Google Scholar]
- Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E (2004) Hypoxia‐inducible factor‐1 (HIF‐1) promotes its degradation by induction of HIF‐alpha‐prolyl‐4‐hydroxylases. Biochem J 381: 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud GN, Li W (2015) HIF‐1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5: 378–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399: 271–275 [DOI] [PubMed] [Google Scholar]
- Mohlin S, Hamidian A, von Stedingk K, Bridges E, Wigerup C, Bexell D, Pahlman S (2015) PI3K‐mTORC2 but not PI3K‐mTORC1 regulates transcription of HIF2A/EPAS1 and vascularization in neuroblastoma. Can Res 75: 4617–4628 [DOI] [PubMed] [Google Scholar]
- Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, Basso G, Leo G, Rosato A, Bicciato S et al (2012) SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia‐inducible factors. Nature 487: 380–384 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Qi J, Ronai Z (2009) The ubiquitin ligase Siah2 and the hypoxia response. Mol Cancer Res 7: 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa MS, Keith B, Simon MC (2016) Oxygen availability and metabolic adaptations. Nat Rev Cancer 16: 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai R, Huypens P, Huang M, Schaefer S, Sheinin T, Wettig SD, Joseph JW (2011) Aryl hydrocarbon receptor nuclear translocator/hypoxia‐inducible factor‐1{beta} plays a critical role in maintaining glucose‐stimulated anaplerosis and insulin release from pancreatic {beta}‐cells. J Biol Chem 286: 1014–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M (2009) Downregulation of miR‐199a derepresses hypoxia‐inducible factor‐1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res 104: 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P, Moch H (2011) VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Can Res 71: 5500–5511 [DOI] [PubMed] [Google Scholar]
- Salceda S, Caro J (1997) Hypoxia‐inducible factor 1alpha (HIF‐1alpha) protein is rapidly degraded by the ubiquitin‐proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox‐induced changes. J Biol Chem 272: 22642–22647 [DOI] [PubMed] [Google Scholar]
- Sharp TV, Munoz F, Bourboulia D, Presneau N, Darai E, Wang H‐W, Cannon M, Butcher DN, Nicholson AG, Klein G et al (2004) LIM domains‐containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F‐driven transcription. Proc Natl Acad Sci USA 101: 16531–16536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Al‐Attar A, Foxler DE, Ding L, de A Vallim TQ, Zhang Y, Nijmeh HS, Webb TM, Nicholson AG, Zhang Q et al (2008) The chromosome 3p21.3‐encoded gene, LIMD1, is a critical tumor suppressor involved in human lung cancer development. Proc Natl Acad Sci USA 105: 19932–19937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spendlove I, Al‐Attar A, Watherstone O, Webb TM, Ellis IO, Longmore GD, Sharp TV (2008) Differential subcellular localisation of the tumour suppressor protein LIMD1 in breast cancer correlates with patient survival. Int J Cancer 123: 2247–2253 [DOI] [PubMed] [Google Scholar]
- Stiehl DP, Wirthner R, Koditz J, Spielmann P, Camenisch G, Wenger RH (2006) Increased prolyl 4‐hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen‐sensing system. J Biol Chem 281: 23482–23491 [DOI] [PubMed] [Google Scholar]
- Sun G, Irvine KD (2013) Ajuba family proteins link JNK to Hippo signaling. Sci Signal 6: ra81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Gu Q, He H, Pamarthy D, Semenza GL, Sun Y (2008) SAG/ROC2/RBX2 is a HIF‐1 target gene that promotes HIF‐1 alpha ubiquitination and degradation. Oncogene 27: 1404–1411 [DOI] [PubMed] [Google Scholar]
- Therneau TM, Grambsch PM (2000) Modeling survival data: extending the Cox model. New York, NY: Springer‐Verlag; [Google Scholar]
- Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E, Clerici C (2004) Prolonged hypoxia differentially regulates hypoxia‐inducible factor (HIF)‐1alpha and HIF‐2alpha expression in lung epithelial cells: implication of natural antisense HIF‐1alpha. J Biol Chem 279: 14871–14878 [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92: 5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J, Chen Y, Zheng S, Wang L (2017) Glucose transporter GLUT1 expression and clinical outcome in solid tumors: a systematic review and meta‐analysis. Oncotarget 8: 16875–16886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11: 393–410 [DOI] [PubMed] [Google Scholar]
- Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, Cereser B, Crnogorac‐Jurcevic T, McDonald S, Elia G et al (2015) Dual‐action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 27: 123–137 [DOI] [PubMed] [Google Scholar]
- Wong PP, Bodrug N, Hodivala‐Dilke KM (2016) Exploring novel methods for modulating tumor blood vessels in cancer treatment. Curr Biol 26: R1161–R1166 [DOI] [PubMed] [Google Scholar]
- Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8: 851–864 [DOI] [PubMed] [Google Scholar]
- Xia X, Kung AL (2009) Preferential binding of HIF‐1 to transcriptionally active loci determines cell‐type specific response to hypoxia. Genome Biol 10: R113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tan C, Ni S, Wang Q, Wu F, Liu F, Ye X, Meng X, Sheng W, Du X (2015) Identification and validation of a two‐gene expression index for subtype classification and prognosis in Diffuse Large B‐Cell Lymphoma. Sci Rep 5: 10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Zhou W, Cheng M, Wang J, Liu Z, He S, Luo X, Huang W, Chen T, Yan W et al (2017) Hypoxia activates Wnt/beta‐catenin signaling by regulating the expression of BCL9 in human hepatocellular carcinoma. Sci Rep 7: 40446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Regev A (2011) Impulse control: temporal dynamics in gene transcription. Cell 144: 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C et al (2015) Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350: 1391–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Liu Q, Li M, Lin SY, Peng Y, Wu D, Li TY, Fu Q, Jia W, Wang X et al (2015) RHOBTB3 promotes proteasomal degradation of HIFalpha through facilitating hydroxylation and suppresses the Warburg effect. Cell Res 25: 1025–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
Microarray data from this publication have been deposited to Gene Expression Omnibus and assigned the identifier accession number https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114692.