

Abstract
The aim of this study is to establish and validate a rapid, selective, and sensitive ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method to determine tubeimoside I (TBMS-I) in ICR (Institute of Cancer Research) mouse whole blood and its application in the pharmacokinetics and bioavailability study. The blood samples were precipitated by acetonitrile to extract the analytes. Chromatographic separation was performed on a UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm). The mobile phase consisted of water with 0.1% formic acid and methanol (1 : 1, v/v) at a flow rate of 0.4 mL/min. The total eluting time was 4 min. The TBMS-I and ardisiacrispin A (internal standard (IS)) were quantitatively detected by a tandem mass spectrometry equipped with an electrospray ionization (ESI) in a positive mode by multiple reaction monitoring (MRM). A validation of this method was in accordance with the US Food and Drug Administration (FDA) guidelines. The lower limit of quantification (LLOQ) of TBMS-I was 2 ng/mL, and the calibration curve was linearly ranged from 2 to 2000 ng/mL (r2 ≥ 0.995). The relative standard deviation (RSD) of interday precision and intraday precision was both lower than 15%, and the accuracy was between 91.7% and 108.0%. The average recovery was >66.9%, and the matrix effects were from 104.8% to 111.0%. In this assay, a fast, highly sensitive, and reproducible quantitative method was developed and validated in mouse blood for the first time. The absolute availability of TBMS-I in the mouse was only 1%, exhibiting a poor oral absorption.
1. Introduction
Tubeimoside I (TBMS-I), a triterpenoid saponin, is derived from the traditional Chinese bulb of Bolbostemma paniculatum (Maxim.). It was often used for the treatment of poisonous snake bite and inflammation [1]. In the past couple of years, additional attention was drawn to TBMS-I as it was reported to be a potential anticancer agent and appeared to be effective against several types of cancer, such as gliomas, breast cancer, colon cancer, and non-small cell lung cancer [2–5].
Pharmacokinetic studies are important in drug research and development which can provide systemic concentrations and exposure times of the drug for predicting a diverse range of efficacy- and toxicity-related events. To systematically examine the preclinical pharmacokinetic studies of TBMS-I in a reproducible and precise manner, a sensitive, fast, and validated analytical method for the determination of TBMS-I in biological fluids is imperative. However, the research on pharmacokinetics of TBMS-I lags behind compared to its pharmacological studies. Up to January of 2018, there was only one study report that published high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) for the determination of TBMS-I, which has been applied to the pharmacokinetic study in rats in 2007 [6]. But it had several drawbacks, such as long analysis time (more than 6 min) and low sensitivity (20 ng/mL), especially requiring a large volume of plasma (100 μL), which make it unsuitable for the serial blood sampling in mice pharmacokinetic evaluation [7]. In the last couple of years, the UPLC technique has attracted more and more concern along with the development of analysis techniques [8]. Compared with LC-MS/MS, the UPLC-MS/MS method was faster, more sensitive, and with higher sample throughput [9, 10]. Meanwhile, its strong ability of isolating was more suitable for the analysis of the metabolism in vivo of the complex traditional Chinese medicine and complex compound [11, 12].
To the best of our knowledge, the profile of toxicity or pharmacokinetics of some drugs is alterable in different species [13–16], so it is not reasonable that these pharmacokinetic data are used directly in mice [17]. The mouse was chosen as the animal model to study the pharmacokinetics of TBMS-I in our study not only because of the reason mentioned above but also because the mouse is the most frequently used species for the preclinical efficacy [18, 19], toxicology [20], biodistribution [21], and pharmacokinetic [22] studies to evaluate a potential anticancer agent, particularly with a limiting drug supply or specialized animal models in the early new drug discovery stage [7, 23].
Thus, we established a rapid, sensitive, and selective UPLC-MS/MS method to quantitate the concentration of TBMS-I directly in the mouse utilizing low-volume whole blood after intravenous and oral administration in this study for the first time. A validation of this method was in accordance with the FDA guidelines. This method was successfully applied to the pharmacokinetics and bioavailability of TBMS-I in mice.
2. Materials and Methods
2.1. Experimental Materials
TBMS-I (purity >98%; Figure 1(a)) and ardisiacrispin A (internal standard, purity >98%; Figure 1(b)) were purchased from Chengdu Mansite Biotechnology Co., Ltd. (Chengdu, China). HPLC-grade methanol and acetonitrile were bought from Merck (Darmstadt, Germany). HPLC-grade formic acid was supplied by Tedia (Ohio, USA). A Milli-Q system (Millipore, Bedford, USA) is used for generating ultrapure water. The ICR mice (male, weight 20–22 g, n=12) obtained were from the Laboratory Animal Center of Wenzhou Medical University (Wenzhou, China).
Figure 1.

Chemical structures of tubeimoside I (a) and ardisiacrispin A (b).
2.2. UPLC and Mass Spectrometric Conditions
The determination of analytes was carried out using the ACQUITY UPLC I-Class system equipped with a triple-quadrupole mass spectrometer (Waters Corp., Milford, MA, USA). MassLynx 4.1 software (Waters Corp.) was used to collect data and control the system.
TBMS-I and IS were separated on a UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm) with a stable temperature of 40°C. The mobile phases A and B were methanol and water with 0.1% formic acid, respectively. The details of gradient elution were as follows: the percentage of methanol was kept at 10% from 0 to 0.2 min and it reached 80% within 1.3 min; then, it was kept at the same percentage for 0.5 min and subsequently it turned back to 10% for another 0.5 min, and finally it was maintained at 10% for 2.5 min. The flow rate was set at 0.4 mL/min, and the total elution time was 4.0 min.
The mass spectrometer system for analysis was equipped with an electrospray source ionization (ESI) in a positive mode. The quantitative detection was performed in a multiple reaction monitoring mode at transitions m/z 1319.7 → 1187.6 for TBMS-I (collision voltage 12 V and cone voltage 30 V) and m/z 1083.5 → 407.1 for IS (collision voltage 72 V and cone voltage 100 V). The capillary voltage was 2.3 kV. High-purity nitrogen as curtain gas and drying gas was set at 50 L/h and 800 L/h, respectively. The temperature of the ion source and dissolvent was 150°C and 400°C, respectively.
2.3. Preparation of Stock Solutions, Quality Control (QC) Samples, and Calibration Standards (CS)
TBMS-I and IS were separately dissolved in methanol at a final concentration of 1.0 mg/mL as stock solutions. The working standard solutions were diluted from the stock solution using methanol. The standard working solution of IS was diluted with acetonitrile to the concentration of 50 ng/mL. All solutions were stored at 4°C prior to analysis.
CS samples were prepared by diluting blank mouse blood into corresponding standard working solutions. A series of concentrations of standard solutions were prepared with TBMS-I stock solutions and serially diluted by using methanol. The final concentrations of TBMS-I were from 2 to 2000 ng/mL, including 2, 5, 10, 20, 50, 100, 200, 500, 1000, and 2000 ng/mL.
Low-, mid-, and high-level QC samples of TBMS-I were similarly prepared at finial concentrations of 3, 190, and 1900 ng/mL, respectively. All the solutions were stored at −20°C until processed.
2.4. Sample Preparation
A 20 μL aliquot of the mouse blood sample and 100 μL of acetonitrile containing 50 ng/mL IS were added into 1.5 mL EP tubes [24]. After vortexing for 1 min, the specimens were centrifugated (13000 rpm) for 10 min at 4°C. Then, 80 μL of the supernatant was collected; subsequently, a 2 μL of the supernatant was injected into the UPLC-MS/MS system for analysis.
2.5. Method Validation
A validation of this method was in accordance with the FDA guidelines, including selectivity, linearity, precision, accuracy, recovery, matrix effect, and stability [25].
The chromatograms of blank mouse blood, blank blood spiked with TBMS-I and IS, and the real sample from mouse after dosing were used to estimate the selectivity of the UPLC-MS/MS method.
Calibration curves were generated by analyzing different concentrations of calibration samples on three consecutive days. The linear regressions of the peak area ratios (y) of each TBMS-I to the corresponding IS versus the nominal concentration (x) of TBMS-I were fitted over the range 2–2000 ng/mL. Linearity was evaluated covering the concentration range 2–2000 ng/mL.
The interday precision, intraday precision, and accuracy were estimated by determining three concentrations of quality control samples (n=6) on the same day and on three days in a row.
The recovery was calculated by comparison of the peak areas of TBMS-I and IS in the extracted low (3 ng/mL), middle (190 ng/mL), and high (1900 ng/mL) concentrations of QC samples with those of the extracted blank blood spiked with TBMS-I and IS at corresponding concentrations.
Matrix effects were tested by comparison of the peak areas of these new working solutions with those of the corresponding standard solutions diluted with methanol : 0.1% formic acid (1 : 1, v/v) at equivalent concentrations, and this peak area ratio is defined as the matrix effect.
The stability of TBMS-I was tested under four conditions: storage in an autosampler at 4°C, storage at room temperature for 2 hours, storage at −20°C for a month, and three complete freeze-thaw cycles (from −20°C to room temperature).
The stability of TBMS-I in mouse blood was obtained by comparing the areas of the newly configured QC samples with the corresponding three concentrations (3, 190, and 1900 ng/mL) of standard samples.
2.6. Pharmacokinetic Study
Twelve mice were randomly and equally divided into two groups (A and B). Mice in the group A were injected sublingually with 5 mg/kg TBMS-I, and mice in the group B were given TBMS-I orally at a final concentration of 20 mg/kg. The study protocol was approved by the Animal Care and Use Committee of Wenzhou Medical University. Mice were allowed to receive standard food and water ad libitum in a temperature-controlled room (25°C) with a 12-hour on and 12-hour off light cycle before the experiment.
Blood samples (20 μL) were obtained from an individual mouse by tail vein bleeding in 1.5 mL tubes at 0 (prior to dosing), 0.0833, 0.5, 1, 1.5, 2, 3, 4, 8, 12, and 24 h after dosing. Six separate mice were used for sample collection and analysis at each time point. All the blood samples were directly stored at −20°C until analysis. DSA 2.0 pharmacokinetic software (China Pharmaceutical University, China) was used to calculate the main pharmacokinetic parameters, including the area under the time-concentration curve (AUC), half-life (t1/2), the maximum of blood concentration(Cmax), blood clearance rate (CL), apparent volume of distribution (V), and mean retention time (MRT). Bioavailability was calculated by absolute bioavailability = 100% × AUCpo · Div/(AUCiv · Dpo), where AUCiv and AUCpo are the AUC of the drug from (0–∞) after intravenous and oral administration, and Div and Dpo are the single dosage of TBMS-I for the intravenous and oral administration, respectively.
3. Results and Discussion
3.1. Method Optimization
The mode of electronic source ionization (positive- or negative-ion mode) selection was often tested in a methodological study [26–30]. In this study, we chose the positive mode for the higher response achieved. According to the optimized results of mass spectrometric conditions, we can see that the daughter ions at m/z 1187.8 and m/z 407.3 were the strongest and the most stable among abundant fragment ions produced, respectively, by TBMS-I and IS, which was presented in Figure 2. Thus, we selected m/z 1319.7 → 1187.8 and m/z 1083.4 → 407.3 for TBMS-I and IS, respectively.
Figure 2.

Mass spectrum of tubeimoside I (a) and ardisiacrispin A (b).
In order to wash out the endogenous compounds as much as possible and avoid endogenous interference, the mobile phase was optimized [31, 32]. Several mobile phases were investigated on the ACQUITY BEH C18 column to obtain a perfect separation and a more symmetrical peak shape [33], including acetonitrile and water with 0.1% formic acid, acetonitrile and 10 mmol/L ammonium acetate solution (0.1% formic acid), methanol and water (0.1% formic acid), and methanol and 10 mmol/L ammonium acetate solution (0.1% formic acid). Among this, the mobile phase containing the mixture of methanol and water (including 0.1% formic acid) was chosen in this study for the best mass spectrometry peak and retention time using gradient eluting.
Proteins and other potential interference would affect the analysis of the mass spectrometry system [34, 35]. Therefore, an effective and simple sample preparation was a key point for establishing the UPLC-MS/MS method of TBMS-I. Liquid-liquid extraction (LLE) has the advantages of the high extraction rate and low limit of quantification [36], but this method required a long time for evaporation of the extracting solvent and large sample volumes. The number of blood samples, which can be taken from a mouse (∼20 g), is limited. Thus, it is not easy to get enough plasma after centrifuging for liquid-liquid extraction at each point (10 total time points in 24 h) by tail vein transection bleeding. Taking these factors into consideration, only 20 μL of blood samples was collected at different time points, and a one-step protein precipitation procedure for whole blood was chosen in our study following the example of the previous literature [37]. The small sample volume requirement further supports a serial blood sampling and enables entire pharmacokinetics from a single mouse which significantly reduces the numbers of mice used and inaccuracy of the pharmacokinetics because of individual differences [7, 23]. Our method provides a simple, direct, and high-throughput assay for measuring TBMS-I because of the simple sample processing. The following precipitating solvents and their mixtures in different combinations and ratios were tested: methanol, acetonitrile, and acetonitrile-methanol. The results indicated that acetonitrile was a good precipitating reagent for the best recoveries for the analytes. Considering that blood samples are more complex than plasma, 20 μL of the blood sample was mixed with 5 volumes of acetonitrile, which can not only provide higher recoveries and less matrix effect but also provide a sufficient supernatant volume for multiple injections for analysis. The level of TBMS-I in the supernatant obtained from the blood after protein precipitation and centrifugation is high enough to be detected by UPLC-MS/MS because the LLOD is 0.7 ng/mL and LLOQ is 2 ng/mL for TBMS-I, which will contribute to the assay of lower concentration of TBMS-I at the last time point for sample collection.
Internal standard was also an important task for establishing this method [38–40]. Tubeimoside I and ardisiacrispin A had a similar structure, so the retention time and the way of ionization of them are similar. In addition, ardisiacrispin A was a good choice for IS in our study because of its robustness, stability, absence of matrix effects, and reproducible extraction.
3.2. Method Validation
3.2.1. Selectivity
Figure 3 presents the ion chromatogram of a blank extract, a blank extract with TBMS-I and IS, and a blood sample from the caudal vein spiked with IS. The peaks of TBMS-I and IS appeared at 2.62 and 2.52 min, respectively. No interfering peaks were found at or close by the retention times of TBMS-I and IS. The total runtime was 4.0 minutes.
Figure 3.

Spectrogram of the TBMS-I and IS: (a) a blank extract, (b) a blank extract with tubeimoside I and IS, and (c) the blood samples after administration spiked with IS.
3.2.2. Linearity
The regression equation of the calibration curve of TBMS-I was y = 0.00027776x + 0.0000866688 (y represents the value of the peak area ratio of TBMS-I and IS and x represents the concentrations of TBMS-I in blood). The correlation coefficient r2 was 0.9976, which showed a good linearity. The LLOQ was 2 ng/mL with the signal-to-noise ratio (S/N ratio) of 10 for the determination of TBMS-I in mouse blood, and the lower limit of detection was 0.7 ng/mL with the S/N ratio of 3.
3.2.3. Precision, Accuracy, Recovery, and Matrix Effect
Table 1 shows the results of the precision, accuracy, recovery, and matrix effect. The RSD of interday precision and intraday precision was no more than 14% and 15%, respectively. The accuracy was in the range of 91.7% to 108.0% at each QC level. All of the recoveries were above 66.9%, and matrix effects were between 104.8% and 111.0%. These data suggest that this method was satisfied with the pharmacokinetic study of TBMS-I.
Table 1.
Accuracy, precision, matrix effect, and recovery of the TBMS-I in mouse blood (n=6).
| Concentration (ng/mL) | Precision (RSD %) | Accuracy (%) | Matrix effect (%) | Recovery (%) | ||
|---|---|---|---|---|---|---|
| Intraday | Interday | Intraday | Interday | |||
| 3 | 13.4 | 14.5 | 105.8 | 108.0 | 105.2 | 78.4 |
| 190 | 11.2 | 10.6 | 94.4 | 91.7 | 104.8 | 68.6 |
| 1900 | 5.3 | 8.4 | 101.3 | 104.1 | 111.0 | 66.9 |
3.2.4. Stability
The blood samples under the different storage conditions mentioned above (n=3) were carried out the stability experiment (results shown in Table 2). In this study, the variations of each condition were within 14% and RSD was under 15%, which indicated a reliable stability behavior of TBMS-I under the different storage conditions.
Table 2.
The stability of TBMS-I under various storage conditions (n=3).
| Concentration (ng/mL) | Autosampler (4°C, 12 h) | Ambient 2 h | −20°C 30 d | Freeze-thaw | ||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | RSD | Accuracy | RSD | Accuracy | RSD | Accuracy | RSD | |
| 3 | 96.0 | 3.8 | 107.5 | 5.1 | 92.6 | 14.2 | 113.5 | 8.8 |
| 190 | 106.2 | 6.7 | 108.0 | 4.8 | 109.9 | 9.4 | 111.0 | 11.2 |
| 1900 | 104.1 | 5.5 | 95.8 | 4.4 | 91.7 | 6.0 | 90.3 | 7.5 |
3.3. Pharmacokinetic Studies
Time-concentration curve of TBMS-I after oral and intravenous administration is shown in Figure 4. The pharmacokinetic parameters were calculated according to the noncompartment model (results are presented in Table 3). The t1/2z was 2.3 ± 0.5 h for oral administration and 6.8 ± 5.6 h for intravenous administration, respectively. The Tmax was 1.8 ± 1.3 h after oral administration. The absolute availability was only 1.0%. These results in mice were similar to that of rats described by Liang et al. [6].
Figure 4.
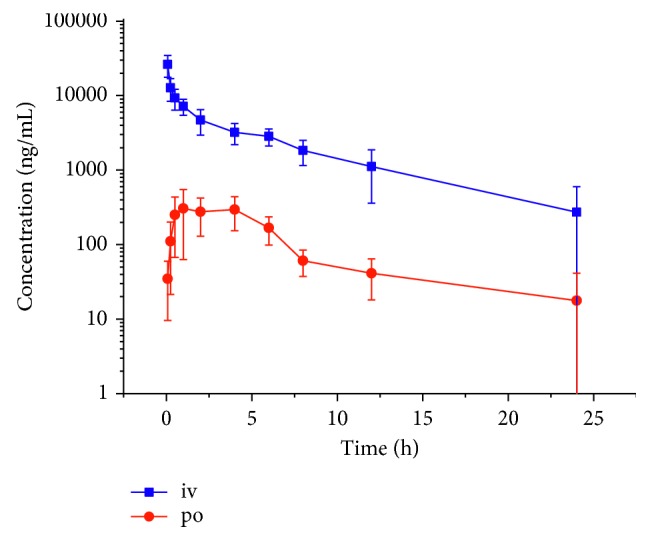
Mean blood concentration of TBMS-I after sublingual intravenous administration at the dose of 3 mg/kg and gavage of 15 mg/kg.
Table 3.
The pharmacokinetic parameters of TBMS-I after oral and intravenous administration (n=6).
| Parameters | Unit | iv (5 mg/kg) | po (20 mg/kg) |
|---|---|---|---|
| AUC (0–t) | ng/mL·h | 51205.8 ± 13134.0 | 2023.9 ± 1145.5 |
| AUC (0–∞) | ng/mL·h | 59370.3 ± 21468.0 | 2051.8 ± 1106.5 |
| MRT (0–t) | h | 5.1 ± 1.8 | 4.6 ± 2.4 |
| MRT (0–∞) | h | 8.9 ± 7.0 | 4.9 ± 2.0 |
| t 1/2z | h | 6.8 ± 5.6 | 2.3 ± 0.5 |
| CLz/F | L/h/kg | 0.1 | 16.3 ± 17.4 |
| T max | H | — | 1.8 ± 1.3 |
| V z/F | L/kg | 0.4 | 53.9 ± 56.9 |
| C max | ng/mL | 26192.5 ± 8491.9 | 429.6 ± 164.9 |
4. Conclusions
A novel UPLC-MS/MS method for the quantitative measurement of TBMS-I in mouse blood has been developed and validated. The application of this method for the determination of TBMS-I extracted from only 20 μL of whole blood using a simple one-step protein precipitation procedure within 4 min was more sensitive, more convenient, and faster than traditional and commonly used analytical techniques. It is clear, in addition, that there are potentially large savings in the amount of compound and number of animals required for early pharmacokinetic studies. In the present study, the UPLC/MS/MS method has been successfully applied to the pharmacokinetic investigations of TBMS-I in mice after sublingual intravenous and intragastric administration. The oral bioavailability of tubeimoside I in mice is 1%, which indicates that tubeimoside I is not easily absorbed into blood circulatory system through the gastrointestinal tract.
Acknowledgments
This research was supported by the Science and Technology Foundation of Wenzhou (Y20160540 and Y20150183) and a grant from the National Natural Science Foundation of China (81702388). The authors thank Wenzhou Medical University Analytical and Testing Center for excellent technical support.
Contributor Information
Xueliang Zhang, Email: 854867181@qq.com.
Yunfang Zhou, Email: zyf2808@126.com.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Yu T. X., Ma R. D., Yu L. J. Structure-activity relationship of tubeimosides in anti-inflammatory, antitumor, and antitumor-promoting effects. Acta Pharmacologica Sinica. 2001;22(5):463–468. [PubMed] [Google Scholar]
- 2.Gu Y., Korbel C., Scheuer C., Nenicu A., Menger M. D., Laschke M. W. Tubeimoside-1 suppresses tumor angiogenesis by stimulation of proteasomal VEGFR2 and Tie2 degradation in a non-small cell lung cancer xenograft model. Oncotarget. 2016;7(5):5258–5272. doi: 10.18632/oncotarget.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bian Q., Liu P., Gu J., Song B. Tubeimoside-1 inhibits the growth and invasion of colorectal cancer cells through the Wnt/beta-catenin signaling pathway. International Journal of Clinical and Experimental Pathology. 2015;8(10):12517–12524. [PMC free article] [PubMed] [Google Scholar]
- 4.Jia G., Wang Q., Wang R., et al. Tubeimoside-1 induces glioma apoptosis through regulation of Bax/Bcl-2 and the ROS/Cytochrome C/Caspase-3 pathway. OncoTargets and Therapy. 2015;8:303–311. doi: 10.2147/OTT.S76063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng Y., Zhong Y., Li G. Tubeimoside-1 suppresses breast cancer metastasis through downregulation of CXCR4 chemokine receptor expression. BMB Reports. 2016;49(9):502–507. doi: 10.5483/bmbrep.2016.49.9.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang M. J., Zhang W. D., Zhang C., et al. Quantitative determination of the anticancer agent tubeimoside I in rat plasma by liquid chromatography coupled with mass spectrometry. Journal of Chromatography B. 2007;845(1):84–89. doi: 10.1016/j.jchromb.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 7.Kim J., Min J. S., Kim D., et al. A simple and sensitive liquid chromatography-tandem mass spectrometry method for trans-epsilon-viniferin quantification in mouse plasma and its application to a pharmacokinetic study in mice. Journal of Pharmaceutical and Biomedical Analysis. 2017;134:116–121. doi: 10.1016/j.jpba.2016.11.044. [DOI] [PubMed] [Google Scholar]
- 8.Xiong J. H., Ye H., Lin Y. X., et al. Determining concentrations of icotinib in plasma of rat by UPLC method with ultraviolet detection: applications for pharmacokinetic studies. Current Pharmaceutical Analysis. 2017;13(4):340–344. doi: 10.2174/1573412912666160815115244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L. G., Yang S. P., Liu Z. Z., et al. Pharmacokinetic study of macitentan in rat plasma by ultra performance liquid chromatography-tandem mass spectrometry. Latin American Journal of Pharmacy. 2015;34(7):1411–1416. [Google Scholar]
- 10.Chen L. G., Wang Z., Wang S., Li T., Pan Y., Lai X. Determination of apremilast in rat plasma by UPLC-MS-MS and its application to a pharmacokinetic study. Journal of Chromatographic Science. 2016;54(8):1336–1340. doi: 10.1093/chromsci/bmw072. [DOI] [PubMed] [Google Scholar]
- 11.Yuan T. F., Wang S. T., Li Y. Quantification of menadione from plasma and urine by a novel cysteamine-derivatization based UPLC-MS/MS method. Journal of Chromatography B. 2017;1063:107–111. doi: 10.1016/j.jchromb.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar M., Grossman R. G., Toups E. G., Chow D. S. UPLC-MS/MS assay of riluzole in human plasma and cerebrospinal fluid (CSF): application in samples from spinal cord injured patients. Journal of Pharmaceutical and Biomedical Analysis. 2017;146:334–340. doi: 10.1016/j.jpba.2017.08.045. [DOI] [PubMed] [Google Scholar]
- 13.Mueller M., Maldonado-Adrian C., Yuan J., McCann U. D., Ricaurte G. A. Studies of (+/-)-3,4-methylenedioxymethamphetamine (MDMA) metabolism and disposition in rats and mice: relationship to neuroprotection and neurotoxicity profile. Journal of Pharmacology and Experimental Therapeutics. 2013;344(2):479–488. doi: 10.1124/jpet.112.201699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatum-Gibbs K., Wambaugh J. F., Das K. P., et al. Comparative pharmacokinetics of perfluorononanoic acid in rat and mouse. Toxicology. 2011;281(1–3):48–55. doi: 10.1016/j.tox.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Toutain P. L., Ferran A., Bousquet-Melou A. Handbook of Experimental Pharmacology. Berlin, Germany: Springer; 2010. Species differences in pharmacokinetics and pharmacodynamics; pp. 19–48. [DOI] [PubMed] [Google Scholar]
- 16.Hu Y., Smith D. E. Species differences in the pharmacokinetics of cefadroxil as determined in wildtype and humanized PepT1 mice. Biochemical Pharmacology. 2016;107:81–90. doi: 10.1016/j.bcp.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z., Chen C., Ai D., et al. Pharmacokinetics and tissue residues of hydrochloric acid albendazole sulfoxide and its metabolites in crucian carp (Carassius auratus) after oral administration. Environmental Toxicology and Pharmacology. 2012;33(2):197–204. doi: 10.1016/j.etap.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Butler S. M., Wallig M. A., Nho C. W., et al. A polyacetylene-rich extract from Gymnaster koraiensis strongly inhibits colitis-associated colon cancer in mice. Food and Chemical Toxicology. 2013;53:235–239. doi: 10.1016/j.fct.2012.11.049. [DOI] [PubMed] [Google Scholar]
- 19.Park K. C., Kim S. W., Park J. H., et al. Potential anti-cancer activity of N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), a histone deacetylase inhibitor, against breast cancer both in vitro and in vivo. Cancer Science. 2011;102(2):343–350. doi: 10.1111/j.1349-7006.2010.01798.x. [DOI] [PubMed] [Google Scholar]
- 20.Yamazaki H., Suemizu H., Mitsui M., Shimizu M., Guengerich F. P. Combining chimeric mice with humanized liver, mass spectrometry, and physiologically-based pharmacokinetic modeling in toxicology. Chemical Research in Toxicology. 2016;29(12):1903–1911. doi: 10.1021/acs.chemrestox.6b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gota V., Goda J. S., Doshi K., et al. Biodistribution and pharmacokinetic study of 3,3′ diseleno dipropionic acid (DSePA), a synthetic radioprotector, in mice. European Journal of Drug Metabolism and Pharmacokinetics. 2016;41(6):839–844. doi: 10.1007/s13318-015-0301-6. [DOI] [PubMed] [Google Scholar]
- 22.Kumar R., Suresh P. S., Rudresh G., et al. Determination of ulixertinib in mice plasma by LC-MS/MS and its application to a pharmacokinetic study in mice. Journal of Pharmaceutical and Biomedical Analysis. 2016;125:140–144. doi: 10.1016/j.jpba.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe A., Watari R., Ogawa K., et al. Using improved serial blood sampling method of mice to study pharmacokinetics and drug-drug interaction. Journal of Pharmaceutical Sciences. 2015;104(3):955–961. doi: 10.1002/jps.24236. [DOI] [PubMed] [Google Scholar]
- 24.Gao S., Yang Z., Yin T., You M., Hu M. Validated LC-MS/MS method for the determination of maackiain and its sulfate and glucuronide in blood: application to pharmacokinetic and disposition studies. Journal of Pharmaceutical and Biomedical Analysis. 2011;55(2):288–293. doi: 10.1016/j.jpba.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.US Department of Health and Human Services. Guidance for Industry: Bioanalytical Method Validation. Washington, DC, USA: F.a.D.A. US Department of Health and Human Services; 2013. [Google Scholar]
- 26.Ye W., Chen R., Sun W., et al. Determination and pharmacokinetics of engeletin in rat plasma by ultra-high performance liquid chromatography with tandem mass spectrometry. Journal of Chromatography B. 2017;1060:144–149. doi: 10.1016/j.jchromb.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Lin G. Y., Hu L. F., Yang X. Z., Pan X. J., Wang X. Q. Determination of gemcitabine in rabbit plasma by LC-ESI-MS using an allure PFP propyl column. Latin American Journal of Pharmacy. 2011;30(3):571–575. [Google Scholar]
- 28.Ding C., Dong W., Yang F. F., et al. Determination of ibudilast in rabbit plasma by liquid chromatography-mass spectrometry and its application. Latin American Journal of Pharmacy. 2011;30(10):2065–2069. [Google Scholar]
- 29.Wang S. H., Lin Z. X., Su K., et al. Effect of curcumin and pirfenidone on toxicokinetics of paraquat in rat by UPLC-MS/MS. Acta Chromatographica. 2018;30(1):26–30. doi: 10.1556/1326.2017.00175. [DOI] [Google Scholar]
- 30.Chen L. G., You W. W., Chen D. W., et al. Pharmacokinetic interaction study of ketamine and rhynchophylline in rat plasma by ultra-performance liquid chromatography tandem mass spectrometry. BioMed Research International. 2018;2018:8. doi: 10.1155/2018/6562309.6562309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang M. L., Zhang J., Wan L. Y., Wu X. L., Wen C. C., Wang X. Q. A simple and selective high performance liquid chromatography developed for determination diphenoxylate in rat plasma and tissues. Latin American Journal of Pharmacy. 2016;35(10):2327–2330. [Google Scholar]
- 32.Guo J. Y., Xu Q. Q., Tong S. H., et al. The effect of transmetil on pharmacokinetics of MS-275 in rats. Latin American Journal of Pharmacy. 2014;33(9):1567–1570. [Google Scholar]
- 33.Wang S., Wu H., Huang X., et al. Determination of N-methylcytisine in rat plasma by UPLC-MS/MS and its application to pharmacokinetic study. Journal of Chromatography B. 2015;990:118–124. doi: 10.1016/j.jchromb.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 34.Fang B. M., Bao S. H., Wang S. H., et al. Pharmacokinetic study of ardisiacrispin A in rat plasma after intravenous administration by UPLC-MS/MS. Biomedical Chromatography. 2017;31(3) doi: 10.1002/bmc.3826.e3826 [DOI] [PubMed] [Google Scholar]
- 35.Wang S., Wu H., Geng P., et al. Pharmacokinetic study of dendrobine in rat plasma by ultra-performance liquid chromatography tandem mass spectrometry. Biomedical Chromatography. 2016;30(7):1145–1149. doi: 10.1002/bmc.3641. [DOI] [PubMed] [Google Scholar]
- 36.Eliassen E., Kristoffersen L. Quantitative determination of zopiclone and zolpidem in whole blood by liquid-liquid extraction and UHPLC-MS/MS. Journal of Chromatography B. 2014;971:72–80. doi: 10.1016/j.jchromb.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen M. K., Johansen S. S. Determination of olanzapine in whole blood using simple protein precipitation and liquid chromatography-tandem mass spectrometry. Journal of Analytical Toxicology. 2009;33(4):212–217. doi: 10.1093/jat/33.4.212. [DOI] [PubMed] [Google Scholar]
- 38.Wen C. C., Wang S. H., Huang X. L., et al. Determination and validation of hupehenine in rat plasma by UPLC-MS/MS and its application to pharmacokinetic study. Biomedical Chromatography. 2015;29(12):1805–1810. doi: 10.1002/bmc.3499. [DOI] [PubMed] [Google Scholar]
- 39.Wen C., Zhang Q., He Y., Deng M., Wang X., Ma J. Gradient elution LC-MS determination of dasatinib in rat plasma and its pharmacokinetic study. Acta Chromatographica. 2015;27(1):81–91. doi: 10.1556/achrom.27.2015.1.7. [DOI] [Google Scholar]
- 40.Tian W. Q., Cai J. Z., Xu Y. Y., et al. Determination of xanthotoxin using a liquid chromatography-mass spectrometry and its application to pharmacokinetics and tissue distribution model in rat. International Journal of Clinical and Experimental Medicine. 2015;8(9):15164–15172. [PMC free article] [PubMed] [Google Scholar]