

First-principles signal-to-noise calculations for BioSAXS have been developed and applied to evaluate the impact of energy, source brightness, window scattering and other variables on the ability to detect and distinguish protein conformational changes.
Keywords: BioSAXS, time-resolved, microfluidics, noise, X-ray damage, CHESS-U
Abstract
Biological small-angle X-ray solution scattering (BioSAXS) is now widely used to gain information on biomolecules in the solution state. Often, however, it is not obvious in advance whether a particular sample will scatter strongly enough to give useful data to draw conclusions under practically achievable solution conditions. Conformational changes that appear to be large may not always produce scattering curves that are distinguishable from each other at realistic concentrations and exposure times. Emerging technologies such as time-resolved SAXS (TR-SAXS) pose additional challenges owing to small beams and short sample path lengths. Beamline optics vary in brilliance and degree of background scatter, and major upgrades and improvements to sources promise to expand the reach of these methods. Computations are developed to estimate BioSAXS sample intensity at a more detailed level than previous approaches, taking into account flux, energy, sample thickness, window material, instrumental background, detector efficiency, solution conditions and other parameters. The results are validated with calibrated experiments using standard proteins on four different beamlines with various fluxes, energies and configurations. The ability of BioSAXS to statistically distinguish a variety of conformational movements under continuous-flow time-resolved conditions is then computed on a set of matched structure pairs drawn from the Database of Macromolecular Motions (http://molmovdb.org). The feasibility of experiments is ranked according to sample consumption, a quantity that varies by over two orders of magnitude for the set of structures. In addition to photon flux, the calculations suggest that window scattering and choice of wavelength are also important factors given the short sample path lengths common in such setups.
1. Introduction
Biological small-angle X-ray solution scattering (BioSAXS) continues to grow in popularity despite the increasing availability of high-resolution structures. This trend reflects the versatility and ease of use of the method, and also the fact that the method yields information about the behavior of molecules in solution. The method is available at many synchrotrons worldwide (Graewert & Svergun, 2013 ▸). As more routine users enter the field, there is a need for tools to help to understand the limitations of the technique and to estimate whether an experiment will be likely to yield statistically valid conclusions. Sample solubility, for example, is sometimes a limiting factor for complex multicomponent constructs, and experiments can fail to generate a strong enough signal. Buffer composition alone can alter the contrast, also reducing the signal. Size-exclusion chromatography used in tandem with BioSAXS also can introduce considerable sample dilution. Time-resolved and microfluidic SAXS experiments tend towards small beams and short sample path lengths. Radiation damage also places limits on how many scattered photons can be recovered from a finite quantity of material.
Much effort has already been invested in computing scattering profiles from atomic models, although challenges remain (Svergun et al., 1995 ▸; Schneidman-Duhovny et al., 2010 ▸). In this work, we assume that the theoretical profile is already known and we focus exclusively on modeling sources of noise and other experimental uncertainties. Our goal here is to develop a lightweight quantitative tool for estimating signal quality that is applicable to a wide range of experiments. We do not attempt to ray-trace at the level of beamline optical components (Pedersen et al., 2014 ▸; Sagan et al., 2011 ▸), but rather concentrate on validating our fit-free method against actual beamline data collected using a variety of stations and detectors. Sedlak et al. (2017 ▸) recently devised a two-parameter model fit to known scattering experiments that can yield practical noise estimates. Our goal here is to avoid fitting parameters and to develop a more detailed first-principles approach with wide applicability. Various correction factors and error sources for SAXS have been reviewed exhaustively elsewhere (Pauw, 2014 ▸; Svergun et al., 2013 ▸).
In this work, we explicitly account for buffer, contrast, window and instrumental scattering, photon flux, sample thickness, detector integration mask, sample-to-detector distance, and sensor thickness. Wavelength-dependent (or equivalently energy-dependent) terms are also correctly modeled. BioSAXS is customarily performed in the X-ray energy range of 10–12 keV (1.24–1.03 Å), but values well outside this range are of interest for several reasons. Many X-ray absorption edges of biological interest in anomalous SAXS experiments are low in energy: Zn (9.6 keV) to Ca (4.0 keV) and P (2.1 keV). Low-energy experiments are also capable of probing larger size scales. Energy is an important factor in radiation damage owing to changes in absorbed dose (Hopkins & Thorne, 2016 ▸). At higher energies, reduced absorption favors experiments that require thicker windows, such as high-pressure SAXS (Ando et al., 2008 ▸). While windows are widely acknowledged as a significant source of parasitic scatter in SAXS experiments, there appear to be no previous quantitative studies computing the magnitude of the effect. This study provides the first such calculations.
We introduce the main formula for scattering intensity and follow it with a discussion of the approximations and limitations of this implementation. Basic statistical measures are introduced to assess three common sample properties of interest to BioSAXS users: detectability, practical usability and distinguishability.
Signal-to-noise calculations are tested against protein standard data collected on a variety of beamlines at various energies, fluxes and configurations. With the advent of high-speed detection, microfluidics, time-resolved methods and high-pressure SAXS, faint signals will become more commonplace. The simulation parameters in this study are based on the actual running conditions currently achieved at MacCHESS with a prototype continuous-flow time-resolved SAXS system. To examine the ability of time-resolved BioSAXS to distinguish realistic conformational changes, we have drawn a subset of conformations from the Database of Macromolecular Motions (http://molmovdb.org; Gerstein & Krebs, 1998 ▸). The required exposure time, consumed sample volume and radiation dose are calculated under various scenarios. As synchrotron and laboratory sources continue to evolve, there are also important questions as to how increased brilliance and other beam properties will expand the range of biological problems that can be investigated. With this in mind, we use the CHESS-U1 upgrade currently in progress as an example to compare the impact of source improvements with the gains expected from the reduction of parasitic scatter and the optimization of wavelength.
2. Theory
The small-angle X-ray scattering intensity for biomolecules in solution has been expressed in parts by many authors (Kratky & Pilz, 1972 ▸; Stuhrmann, 1980 ▸; Orthaber et al., 2000 ▸; Dreiss et al., 2006 ▸; Meisburger et al., 2013 ▸):
The parameters for our implementation are defined as follows: χ(q) is the solid-angle correction (dimensionless), η(λ) is the quantum efficiency of the detector (dimensionless), T(λ) is the X-ray transmission of the sample (dimensionless), d is the thickness of the sample (cm), P is the incident photon flux (photons s−1 on the sample), r is the sample-to-detector distance (cm), M is the molecular mass (g mol−1), N A is Avogadro’s number (6.022 × 1023 mol−1), ν is the specific volume of the solute (cm3 g−1), Δρ is the excess scattering length density (cm−2), c is the concentration of the solute (g cm−3), FF(q) is the molecular form factor [dimensionless, FF(0) = 1] and SF(q) is the sample structure factor [dimensionless, SF(0) = 1].
Momentum transfer in this expression is customarily defined as q = 4πsin(θ)/λ, where 2θ is the angle of scattering and λ is the wavelength. The solid-angle correction χ(q), which is proportional to cos32θ, has been described elsewhere (Pauw, 2014 ▸).
For simplicity, our calculations assume a detector with Poisson counting statistics, zero read noise and generally idealized properties, including a uniform detector response across the surface. We do, however, account for detector quantum efficiency as well as pixel size and detector dimensions (Donath et al., 2013 ▸). Quantum efficiency, η(λ) in (1), is determined by absorption and is therefore heavily energy-dependent. For silicon-based counting detectors the efficiency is maximal for photons in the 8–11 keV range (depending on sensor thickness), but declines with increasing photon energy (Donath et al., 2013 ▸). In the energy ranges typically used for BioSAXS, the quantum-efficiency curve is dominated by the mass attenuation μ, a factor that is well modeled by a simple cubic power law in wavelength (λ) for low-Z sensor materials: μ ∝ λ3 (cm−1; Feigin & Svergun, 1987 ▸). The quantum efficiency (QE) is thus given by
where ρ is the density of a silicon sensor layer of thickness d. The constant c = 0.0048 is fitted to correct for the absorption of nonsensitive sensor layers specific to PILATUS-type detectors. (2) is accurate to better than 1% over the energy range 5–22 keV.
While quantum efficiency merely scales the overall intensity of scattering (1), its effect on signal-to-noise ratios is more complex. When the probability of a successful photon-absorption event is p, the probability of I successful events is given by a binomial distribution with σ2 = Ip(1 − p). The observed detector statistics will be a convolution of these two effects, with the errors adding in quadrature as in normal Poisson statistics: σ2 det = Ip + Ip(1 − p) (Hülsen-Bollier, 2005 ▸). The contribution of absorption statistics becomes increasingly important with low quantum efficiency (high energy), but never dominates the noise. So, not only is the signal in an imperfect detector reduced by a factor of p, but the signal-to-noise ratio is also reduced by the statistics of absorption: I/σdet = [Ip/(2 − p)]1/2. The reduction factor [1/(2 − p)]1/2 is, for example, 0.95 (p = 0.9) at 10 keV, but falls significantly to 0.76 (p = 0.26) at 20 keV for a 320 µm silicon sensor.
The shape of the detector surface and any integration masks are important because they limit the number of pixels perq-space bin and consequently also influence signal to noise (Sedlak et al., 2017 ▸). Let N(R) be the number of unit-square pixels within a circle of radius R. The pixels in the ith discrete q bin are then given by the difference w(Ri) = N(Ri + δ) − N(Ri), with R i = iδ, for some bin size δ > 0. The σ associated with bin i is obtained by adding the pixel counts in quadrature so that σ(qi) = [w(qi)I(qi)]1/2, where I(qi) is the average counts per pixel in bin i. In this analysis, we compute w(qi) for a simple rectangular beamstop mask centered on the beam, the dimensions of which are specified as the number of pixels from the beam center to the edge.
X-ray transmission by the sample, which appears as T(λ) = I/I 0 = exp(−μd) in (1), is determined from the known mass attenuation coefficient μ for water (http://physics.nist.gov/PhysRefData/XrayMassCoef/ComTab/water.html) and the specified sample thickness d. The experimental mass attenuation coefficient of water fits well for energies in the range 2.5–15.5 keV (λ = 0.8–5.0 Å) by the simple expression μ/ρ (cm2 g−1) = 2.8λ3 (Feigin & Svergun, 1987 ▸). Much above 20 keV, photoabsorption is no longer the dominant absorption mechanism and μ/ρ starts to deviate from this simple power law.
In addition to transmission, contast (Δρ2 in equation 1) also modulates signal. The contrast or excess scattering density is the difference Δρ = ρsample − ρbuffer between the biomolecule and the solvent. The average electron density is customarily multiplied by the classical electron radius r 0 (2.818 × 10−13 cm) to give units of cm−2 and is referred to as ρ, the scattering length density. Given the number of electrons per dry mass of sample, ρM,protein = 3.22 × 1023 e g−1 (Orthaber et al., 2000 ▸), and the partial specific volume of the solute in buffer ν, the contrast is calculated as Δρ = ρM,sample/ν − ρbuffer. In this study we use the density of water for ρbuffer (3.34 × 1023 e cm−3). Since this is a small difference that appears in (1) as a square, variations can contribute to error. Values for the average electron density of proteins, nucleic acids and lipids have appeared elsewhere (Svergun & Koch, 2003 ▸) and software tools exist for estimating contrast (Whitten et al., 2008 ▸). In the studies here, we use ν = 0.72 cm3 g−1, which is closer to the reported values for lysozyme and glucose isomerase than the 0.7425 cm3 g−1 derived by minimizing the deviations of molecular-weight estimates (Mylonas & Svergun, 2007 ▸).
The final terms FF(q) and SF(q) in (1) contain the structural information resulting from diffraction. The form factor FF(q) describes the part of the scattering profile arising from the molecular shape alone and is given by the one-dimensional (rotationally averaged) Fourier transform of the electron density ρ of the protein. SAXS form factors have been calculated for a wide variety of geometric shapes (for example SASfit by Joachim Kohlbrecher; https://kur.web.psi.ch/sans1/SANSSoft/sasfit.pdf) as well as for fully detailed atomic models (Svergun et al., 1995 ▸). The computations here use profiles generated by FoXS (Schneidman-Duhovny et al., 2010 ▸). In the present calculations, we do not attempt to model inter-particle interference effects that might appear in the small-angle range with high sample concentrations, and the structure factor will be assumed to be constant: SF(q) = 1. Similarly, radiation damage, an effect that is most often seen at the smallest scattering angles, will not be modeled here. Approximate sample X-ray dose levels, however, are straightforward to calculate and are provided as a rough guide for acceptable sample exposure.
The quantity within the square brackets in (1) is the macroscopic differential scattering cross section dΣ/dΩ in units of cm sr−1. The remaining prefactor gives an intensity I(q) in photons s−1 cm−2 in the direction of the scattered beam. Conveniently, then, the number of photons per detector pixel at q is given by I(q) × (exposure time in seconds) × (pixel area in cm2).
In practice, experimental scattering profiles are obtained as the difference between sample and buffer. Errors in subtraction most commonly occur as a result of buffer mismatch during sample preparation, but can also happen owing to instrumental drift and normalization errors. For the purposes of this paper, we assume ideally matched buffer and accurate normalization. Formally, the counting noise from the sample–buffer difference I diff(q) = [I(q) + I b(q)] − I b(q) adds in quadrature to give σ2 diff = σ2 I(q) + 2αb 2. where I b(q) is the scattering from buffer in the sample cell, including background. Even in the case of zero sample concentration there will still be a finite noise level. Because sample concentrations are usually dilute, I b(q) dominates the counting statistics.
3. Background scattering and buffer model
Scattering from buffer, slits and imperfect X-ray optics, residual air in the beam path, trace heavy-metal fluorescence in the beampipe materials and scattering from windows are all sources that contribute to the final noise level of the data. While the scattering of water itself is nearly constant in the small-angle range 0 < q < 0.3 Å−1, instrumental and window scattering sources become increasingly important at low q and as the sample path length decreases. It is these complicating factors that make first-principles noise estimation a nontrivial task. In this section, we develop a model of buffer that explicitly accounts for energy, sample path length, and window material and thickness.
3.1. Instrumental background
Background scattering can have a major impact on the quality of SAXS intensity measurements. Direct-beam profiles are a complex superposition of multiple secondary sources such as mirror surfaces, slit edges and distortions owing to imperfect optics. Careful beamline design seeks to minimize these effects, but the low-intensity pedestals of direct beams will generally be broader and more intense than might be expected purely on the basis of reported FWHM measurements or assumptions of Gaussian shape. This is the single most unpredictable component of a model when comparing different sources and beamlines. Modeling beamline optical components, such as with ray-tracing software (Pedersen et al., 2014 ▸; Sagan et al., 2011 ▸), is beyond the scope of this simplified approach. To assess instrumental background scattering, we have taken vacuum-only exposures on various CHESS BioSAXS beamlines by removing the sample cell altogether. The vacuum levels within the approximately 1.5 m sample-to-detector path are typically held to 0.26 Pa (2 mTorr); consequently, residual air scattering is a very small effect in comparison with that produced by windows, buffer and other factors. For practical purposes in this study, we utilize a measured instrumental ‘vacuum’ background denoted I v.
3.2. Window scattering
As in the case of instrumental scattering, well designed SAXS beamlines minimize the impact of X-ray window scattering. Thin-walled glass capillaries are most commonly used, although MacCHESS has used flat glass film for some time. Glass, although high in absorption, is nearly atomically smooth. Freshly cleaved natural mica has been used historically and is used in the examples calculated here. It absorbs X-rays, but is very smooth, highly radiation resistant, chemically resistant and has mechanical rigidity. A range of materials have been evaluated by various investigators and the subject continues to be of interest (Gillilan et al., 2013 ▸; Lurio et al., 2007 ▸; Henderson, 1995 ▸; Masunaga et al., 2013 ▸; Acerbo et al., 2015 ▸). Unlike instrumental vacuum scattering, window scattering arises from well characterized materials that can be treated as beamline-independent standards once placed on an appropriate scale. We denote scattering that results entirely from windows as I w.
3.3. Buffer model
To fully parameterize a buffer measurement, we need to extract pure buffer, window and instrumental curves on an absolute scale. The procedure used in the experiments reported here is to collect a ‘vacuum’ measurement, I
v, with no cell in position. An empty cell profile is also collected, I
e, and then a cell containing buffer, I
b. The beamstop diode counts c
e, c
v and c
b are used to equalize the dose by composing normalization factors: K
e = c
e/c
b, K
v = c
v/c
b, K
b = 1. In this way, profiles can still be interpreted as photons per pixel despite the fact that the doses between sample and buffer may be slightly different owing to beam-intensity fluctuations. Standardized, parameter-free profiles, 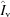 ,
, 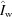 and
and 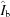 (vacuum, window and buffer, respectively) are prepared by dividing out the specific parameters under which the profiles were collected: sample-to-detector distance
(vacuum, window and buffer, respectively) are prepared by dividing out the specific parameters under which the profiles were collected: sample-to-detector distance  , exposure time
, exposure time  , detector quantum efficiency
, detector quantum efficiency 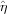 , flux
, flux 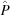 , sample and window absorption parameters
, sample and window absorption parameters 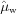 ,
, 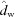 ,
, 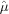 and
and 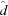 . Grouping these common constants as Ω =
. Grouping these common constants as Ω = 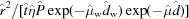 , the standardized profiles are computed as
, the standardized profiles are computed as
The same transmissions in Ω appear in all of the terms owing to the choice of K b = 1. Reassembling the standardized profiles gives the fully general parameterized model buffer,
In practice, dilute buffer profiles  closely resemble water. In particular, is constant as q → 0 and is proportional to compressibility and temperature in the case of pure water (Orthaber et al., 2000 ▸). Variable absorption and base scattering levels owing to buffer components can be expected, but departures from pure water behavior are generally small.
closely resemble water. In particular, is constant as q → 0 and is proportional to compressibility and temperature in the case of pure water (Orthaber et al., 2000 ▸). Variable absorption and base scattering levels owing to buffer components can be expected, but departures from pure water behavior are generally small.
The three components of measured buffer in the sample cell, 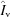 ,
, 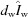 and
and  in (6), vary in their relative importance with q. Fig. 1 ▸ shows that for traditional 25 µm mica windows with a 1.5 mm sample path length, the buffer contributes 78% of the signal at the widest angles. In the region below q = 0.02 Å−1 where Guinier analysis is conducted, the windows and vacuum become more important, with vacuum eventually becoming dominant close to the direct beam. As the sample path length is made shorter, background terms will naturally play a larger role.
in (6), vary in their relative importance with q. Fig. 1 ▸ shows that for traditional 25 µm mica windows with a 1.5 mm sample path length, the buffer contributes 78% of the signal at the widest angles. In the region below q = 0.02 Å−1 where Guinier analysis is conducted, the windows and vacuum become more important, with vacuum eventually becoming dominant close to the direct beam. As the sample path length is made shorter, background terms will naturally play a larger role.
Figure 1.

Scattering sources that contribute to the buffer measured in a sample cell. (a) Total scattering produced by a 1.5 mm path length of standard lysozyme buffer between two 25 µm thick mica windows (solid line). The empty cell contains scattering from mica windows and instrumental ‘vacuum’ scatter (dashed line), while the underlying vacuum profile alone (dotted line) contributes instrumental sources of photons such as slit scatter and fluorescence. (b) Fractional contributions of buffer (solid line), windows (dashed line) and vacuum (dotted line) to the total ‘buffer in cell’ are derived from normalized differences between the profiles in (a). Buffer alone contributes 78% of the total signal at wide angles, but is linearly dependent on the sample path length. Below q = 0.02 Å−1 in the Guinier region, window and vacuum background become increasingly important components of the total signal.
4. Radiation damage and sample consumption
Biological samples in solution are sensitive to radiation damage, and consequently sample consumption is a limiting factor in data collection that should be included with any signal-to-noise estimates. For the monochromatic radiation and flux typically available at third-generation sources, damage occurs mainly in the form of induced aggregation. Calculation of the dose as the absorbed energy per unit mass (1 Gy = 1 J kg−1) is straightforward (Meisburger et al., 2013 ▸),
where P is the incident flux, E is the energy per photon, T w(E) is the single-window transmission, T s(E) is the sample transmission, ρ is the sample density and V is the illumated volume. Ideally, dose calculations should be based on mass-energy absorption, although the values do not differ significantly from the standard mass attenuation at the typical energies used in BioSAXS (Hopkins & Thorne, 2016 ▸). The concept of the critical dose, a threshold at which damage becomes evident, has been used by a number of authors, and observed critical dose limits for standard proteins have been reported over a wide range from 400 Gy to 10 kGy (Meisburger et al., 2013 ▸; Kuwamoto et al., 2004 ▸; Cho et al., 1999 ▸; Jeffries et al., 2015 ▸). Recent characterization of radiation sensitivity (the percentage change in a parameter such as R g per unit dose) suggests that damage is more a continuous process with no well defined threshold (Hopkins & Thorne, 2016 ▸). Damage and sensitivity values cannot yet be known a priori (Garman & Weik, 2017 ▸), and consequently we use dose here only as a rough indicator of when to be concerned.
Whether flowing or static, sample consumption ultimately limits the scattering signal. Protein samples can be produced in quantity with modern expression methods: often more than 1 cm3 at >10 mg ml−1 for single well behaved cytosolic proteins. Achieving high protein concentrations is risky, however, owing to possible irreversible aggregation and precipitation. Since I(0) ∝ M × c (1), commonly used concentrations of the standards lysozyme and glucose isomerase suggest that c ≃ 60/M, where c is in mg ml−1 and M is in kDa, is sufficiently dilute to reach ideality but still have a usable signal.
Flow rates in continuous-flow time-resolved configurations are dictated by the timescale being measured and the microfluidics of mixing rather than dose. Microfluidic mixer channels for TR-SAXS are typically small in at least two of the three dimensions. The X-ray sample path length could be made arbitrarily long, since it often decouples from the fluidic mixing, but in practice it is difficult to fabricate devices with high aspect ratios. Consequently, TR-SAXS systems have trended towards small path lengths (0.5 mm and less).
Because of the high flow rates, sensitivity in TR-SAXS is of paramount importance in reducing sample consumption. Graceffa and coworkers report a continuous-flow technology that requires approximately 2 mg protein per time point with a total of 90 time points (Graceffa et al., 2013 ▸). Just how much sample consumption should be regarded as ‘prohibitive’ or ‘unfeasible’ is difficult to judge and the value we choose here is unavoidably arbitrary. Since it is common for crystallographers to produce cm3 volumes of moderately concentrated protein, it is reasonable to argue that the consumption of 1–4 cm3 of sample at a sufficient concentration to obtain a modest SAXS signal (∼60/M mg ml−1) would be regarded as feasible.
5. Statistics
The statistical requirements for an experiment depend upon how the SAXS data are to be used. For this study, we define three basic statistical measures: detectability, usability and distinguishability. At the most basic level is determining whether a known component is present in the sample at all: detectability. Until recently, this statistic would have been of only theoretical interest, but the advent of SAXS-coupled size-exclusion chromatography (SEC–SAXS) has made SAXS, for the first time, a means of basic detection.
In practice, reliable σ values are not always available in experiments, so a number of methods to detect differences have evolved that utilize only measured counts of sample 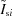 and buffer
and buffer 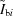 . Of particular note is the CORMAP statistic, which has been compared in detail with the standard χ2 results (Franke et al., 2015 ▸). Since the calculations here provide smooth theoretical I
si, I
bi curves with true σ values, there is no need to generate random deviates to simulate noise. We can confine our calculations to the classical χ2 statistic χ2
sb,
. Of particular note is the CORMAP statistic, which has been compared in detail with the standard χ2 results (Franke et al., 2015 ▸). Since the calculations here provide smooth theoretical I
si, I
bi curves with true σ values, there is no need to generate random deviates to simulate noise. We can confine our calculations to the classical χ2 statistic χ2
sb,
where p is the number of parameters if either of the curves is a parameterized model fit to the data. The extra ‘1’ in (8) arises from our use of smooth curves and is derived in Appendix A .
The largest contributions to the statistic come from regions of the scattering curve that are well separated from buffer relative to the total scattering level. This region tends to be at the smallest angles except as limited by parasitic scattering, which dominates both sample and buffer near the beamstop. It can thus be expected that parasitic scatter may strongly influence the ability to detect species. As conventionally used, the condition 〈χ2 sb〉 ≥ 1.1 would favor rejecting the null hypothesis that the profiles are the same. Differences this small are difficult to discern visually on a typical plot and, considering that other systematic errors may come into play that are not accounted for here (such as buffer mismatch and instrumental drift), we have opted for a more definitive criterion of 〈χ2 sb〉 ≥ 1.5. The summation in 〈χ2 sb〉 is linear in flux P and exposure time t, but not in concentration c. The denominator in the sum is dominated by the buffer and will be nearly constant for dilute samples, with the result that P, t ∝ c2 when 〈χ2 sb〉 is constant.
In addition to detecting when the sample signal rises above buffer, the χ2 statistic can generally serve to distinguish between different subtracted profiles 1 and 2. This is the case, for example, when determining which of two possible conformational changes best matches the data. A difference between two profiles of the same molecular weight will vanish at small angles, but differ at wider angles, where conformational changes alter the profile shapes. For illustration, we have selected a subset of structure pairs from the Database of Macromolecular Movements (Gerstein & Krebs, 1998 ▸). As in the case of detectability, we opt for the criterion 〈χ2 12〉 ≥ 1.5 as a definitive measure of difference. This measure does not tell us whether the difference in profiles is sufficient to make structural interpretations about what is happening, but it does imply that algorithms using the profiles will show some difference.
Finally, it is desirable to have some measure of what constitutes a ‘useable’ profile. This depends on the use, of course, so there are many possible choices. In practice, the AutoRg program is widely used for automated Guinier analysis of experimental data. The error estimates provided by this program are based on standard error modified to account for possible systematic deviations owing to the choice of the q range for analysis (Franke et al., 2017 ▸). As a point of reference for what constitutes a reasonably ‘normal’ BioSAXS data set, we use the criterion error(R g)/R g ≃ 0.05.
We have now defined three statistical conditions that can serve as indicators for how beam characteristics, parasitic scatter and other conditions impact the range of biological problems that can be investigated: detectability (〈χ2 sb〉 ≥ 1.5), usability [error(R g)/R g ≃ 0.05] and distinguishability (〈χ2 12〉 ≥ 1.5).
6. Experimental methods
Scattering measurements were conducted on the CHESS G1, A1 and F1 beamlines using an experimental BioSAXS setup as previously documented (Acerbo et al., 2015 ▸). PILATUS 100K detectors (Dectris, Baden, Switzerland) were used to acquire SAXS images (except where noted). All detectors used silicon-type detection with sensor thicknesses of 0.320, 0.45 or 1.00 mm. Data were also collected on the PETRA III P12 beamline as part of a special radiation-damage and multilayer-testing experiment and therefore do not represent typical running conditions for that beamline. Station energies, bandwidths, fluxes and other parameters are listed in Table 1 ▸. Incident beam flux for the A1, F1 and G1 setups was measured by temporarily moving the in vacuo beamstop out of the way and replacing the sample cell with a windowless spacer. The end flightpath vacuum window, made of 51 µm (2 mil) Mylar film, has a measured X-ray absorption of 2.6%. Since this window is in place during flux and sample measurements, its effect can be neglected. A standard 6 cm long parallel-plate N2 ion chamber was placed in front of the detector. As a crosscheck, a comparison of the standard ion-chamber readings was previously made with a commercial calibrated PIN diode (Forvis Technologies Inc., Santa Barbara, California, USA) at 11.213 keV using an attenuated beam (4 × 108 photons s−1). Measurements gave 10% agreement between the two methods. For two of the validation experiments, we also employed a novel diamond flux monitor that has been shown to have a linear response over 11 orders of magnitude (Bohon et al., 2010 ▸). The flux at P12 was measured with a 65 µm thick diamond flux monitor, moving the sample capillary out of the beam and creating a temporary 7 cm air gap using a 12.7 µm (½ mil) Kapton window to hold the vacuum. The total transmission (air + Kapton) is 0.976. The transmission of diamond at density 3.5 g cm−3 is T = 0.9512 at 10 keV. The flux was calculated from the current using 13.3 eV as the electron–hole pair-creation energy in diamond. The beam diameter was 299 µm (horizontal) × 100 µm (vertical).
Table 1. Beamline characteristics.
| A1, CHESS | F1, CHESS | G1, CHESS | P12, PETRA III | |
|---|---|---|---|---|
| Source | Undulator | Wiggler | Undulator | Undulator |
| Optics | Diamond mono | Silicon mono | Multilayer | Multilayer |
| Energy (keV) | 19.845, 32.428 | 12.688 | 9.962, 11.166 | 10.0 |
| Beam size (horizontal × vertical) (µm) | 250 × 250 | 100 × 250 | 250 × 250 | 300 × 100 |
| Flux (photons s−1) | 2.3 × 1011, 3.4 × 1010 | 5.1 × 1010 | 8.4 × 1011, 4.1 × 1011 | 3.4 × 1014 |
| Detector distance (cm) | 168.38 | 155.15 | 150.47 | 163.73 |
| Bandwidth (%) | 0.13 | 0.014 | 1.5 | 1.5 |
| Detector sensor type (µm) | PILATUS 100K, 1000† | PILATUS 100K, 1000† | PILATUS 100K, 320 | EIGER 4M, 450 |
The same detector was used for A1 and F1.
With the exception of experiments conducted on P12 at PETRA III, sample cells were constructed as described by Acerbo et al. (2015 ▸). Scratch-free ruby mica windows (25 µm) were originally purchased from Attwater Group, Preston, England. The ultrathin glass ribbon (5 µm) used for the windows was obtained from Nippon Electric Glass America, Schaumburg, Illinois, USA. Lysozyme (EMD Millipore, Billerica, Massachusetts, USA) was prepared in 50 mM NaCl, 40 mM sodium acetate pH 4.5 in 1% glycerol. The concentration was measured (in triplicate) by the A 280 method. Glucose isomerase (Hampton Research, Aliso Viejo, California, USA) was prepared in 10 mM HEPES pH 7.0 with 1 mM MgCl2.
Data reduction was preformed using the BioXTAS RAW software.
7. Results and discussion
To understand the level of accuracy to expect from the calculations presented here, we measured two well known protein standards, glucose isomerase and lysozyme, on four different beamlines under multiple carefully characterized conditions of flux, energy and sample-cell type. Once the method had been validated, we examined how the choice of X-ray energy influences the basic sensitivity of a scattering experiment. Finally, the statistical measures presented here were applied to a diverse set of protein conformational changes to understand how choices of energy, flux and windows determine the kinds of changes that can be practically distinguished given realistic limitations on sample consumption.
7.1. Validation
The primary goal here is to assess how well the first-principles calculations match the experimental I(0) and I/σ values. Table 2 ▸ lists a series of experiments conducted on four different beamlines using the well characterized standard proteins lysozyme and glucose isomerase. Energy, flux, exposure times, window materials and detector sensor thickness vary while concentrations are essentially fixed. Sample scattering is based on the specified structures (PDB codes) as calculated by FoXS (Schneidman-Duhovny et al., 2010 ▸). Actual measured buffer curves were used as the buffer model for noise calculations. Most I(0) measurements fall within a 10% error range similar to that expected for molecular-weight estimates, where the overall error is dominated by errors in concentration measurements (Mylonas & Svergun, 2007 ▸). The notable exceptions are the highest energy experiment and the highest flux experiment. The highest energy experiment at 32 keV was problematic for multiple reasons: excessive parasitic scattering, reduced low-q range, the uncertain accuracy inherent in a 6 cm ion-chamber measurement and the breakdown of power-law assumptions for absorption calculations. The highest flux measurements at PETRA III were special nonflowing sample conditions shot at very high speed (1.35 ms exposure) as part of another experiment. Both the glucose isomerase and lysozyme intensities are underestimated by more than 10%, suggesting some systematic error beyond concentration measurement. Although subtle radiation damage cannot be ruled out in these cases, Guinier analysis does not reveal an obvious nonlinearity. The R g value for lysozyme, in fact, was slightly below normal (R g = 13.8 versus 14.3 Å), possibly as a result of concentration effects owing to a higher than normal concentration (5.4 mg ml−1 versus a typical 3–4 mg ml−1). The glucose isomerase data were also collected at a higher than normal concentration (1.1 mg ml−1 versus 0.3–0.4 mg ml−1) but gave a slightly low, but reasonable, radius of gyration (R g = 32.2 Å).
Table 2. Experimental tests of the signal-to-noise calculation.
The protein was either lysozyme (LYS) or glucose isomerase (GI). Concentration (Conc) is given in mg ml−1, energy in keV, flux in photons s−1, sensor thickness in centimeters, single-window thickness (Window) in micrometres and sample path length (Path) in centimetres. Time is the exposure time and Model is the PDB code used in the simulation. Experimental I(0) [Exp. I(0)] is determined from the data by Guinier analysis, and calculated I(0) [Cal. I(0)] is derived from the model; both values are in photons per pixel. Rel err is the relative error in I(0) of the simulation with respect to the experiment. The relative scale required to overlay experimental and computed I/σ curves by least squares is denoted I/σ scl (see text). Relative standard deviation (I/σ RSD) is the standard deviation of overlaid experimental and calculated I/σ curves normalized by the mean difference (see text).
| Protein | Conc | Beamine | Energy | Flux | Sensor | Window | Path | Time | Model | Exp. I(0) | Cal. I(0) | Rel err | I/σ scl | I/σ RSD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LYS | 4.53 | G1 | 9.962 | 8.71 × 1011 | 0.032 | 25 µm mica | 0.15 | 1 s | 6lyz | 28.60 | 29.296 | 0.02 | 0.036 | 0.088 |
| GI | 0.47 | G1 | 9.962 | 8.71 × 1011 | 0.032 | 25 µm mica | 0.15 | 1 s | 1oad | 36.40 | 33.035 | −0.09 | 0.034 | 0.183 |
| LYS | 4.53 | G1 | 9.962 | 8.71 × 1011 | 0.032 | 5 µm glass | 0.15 | 1 s | 1oad | 34.30 | 37.539 | 0.09 | −0.051 | 0.083 |
| LYS | 4.68 | A1 | 19.845 | 3.23 × 1011 | 0.1 | 5 µm glass | 0.15 | 10 s | 6lyz | 150.00 | 164.18 | 0.10 | −0.101 | 0.192 |
| GI | 0.31 | A1 | 19.845 | 3.23 × 1011 | 0.1 | 5 µm glass | 0.15 | 10 s | 1oad | 139.06 | 131.57 | −0.05 | −0.157 | 0.274 |
| LYS | 4.68 | A1 | 32.428 | 3.40 × 1010 | 0.1 | 5 µm glass | 0.15 | 10 s | 6lyz | 29.97 | 18.83 | −0.37 | −0.078 | 0.391 |
| LYS | 4.4 | F1 | 12.688 | 5.12 × 1010 | 0.1 | 5 µm glass | 0.15 | 10 s | 6lyz | 34.70 | 35.391 | 0.02 | 0.019 | 0.147 |
| LYS | 4.64 | G1 | 11.166 | 4.05 × 1011 | 0.032 | 5 µm glass | 0.15 | 2 s | 6lyz | 52.10 | 43.529 | −0.16 | 0.233 | 0.110 |
| LYS | 5.4 | P12 | 10.0 | 3.40 × 1014 | 0.045 | 50 µm glass | 0.17 | 1.35 ms | 6lyz | 3.91 | 3.31 | −0.15 | 0.450 | 0.097 |
| GI | 1.1 | P12 | 10.0 | 3.40 × 1014 | 0.045 | 50 µm glass | 0.17 | 1.35 ms | 1oad | 10.09 | 8.16 | −0.19 | −0.023 | 0.144 |
As an overall measure of how well experimental noise is reproduced by the calculations, we report two parameters. The first measure indicates by what fraction the calculated I/σ curve must be scaled to superimpose the experimental data. Minimizing
with respect to α, ‘I/σ scl’ = 1 − α in Table 2 ▸. The second measure indicates how well the shape of the I/σ curve matches the experiment. In Table 2 ▸, this is ‘I/σ RSD’ = SD/mean(I/σ), the relative standard deviation. When the relative error in I(0) (the ‘Rel err’ column in Table 2 ▸) is high, one of these two measures of I/σ error also tends to be high.
Figs. 2 ▸(a) and 2 ▸(c) give example simulated (dashed lines with error bars) and experimental (dots) scattering-curve pairs for lysozyme and glucose isomerase taken from the first two entries in Table 2 ▸. Error bars are plotted as ±σ; consequently, 68% of the experimental data points are expected to fall within the error bars. The bars and the data points appear asymmetrical owing to the semi-log scale of the plot. Since the point of the simulation is to reproduce the true scale of scattering as well as the noise level, the curves have not been scaled to superimpose. As a result, a small but systematic shift between data and simulation is expected. The calculated signal-to-noise ratio (I/σ) is also plotted (Figs. 2 ▸ b and 2 ▸ d) and precisely reproduces the experimental ratio derived from the standard deviation of the pixel counts in each q bin. As the simulated I/σ for glucose isomerase falls below 1.0 near q = 0.1 Å−1, the experimental curve contains no more useful information and breaks up on the log scale as negative noise fluctuations become frequent.
Figure 2.

Simulated lysozyme and glucose isomerase scattering profiles and signal-to-noise ratios compared with experimental data. (a) A noise-free theoretical profile (dashed line) for lysozyme generated from PDB entry 6lyz was calculated using FoXS. The computed noise level is shown as ±σ error bars (every fifth point) compared with the experimental profile (dots). Error bars are asymmetrical owing to the semi-log scale. (b) The simulated signal-to-noise ratio I/σ of lysozyme (dashed line) overlays the equivalent experimental result (solid line). (c) The glucose isomerase theoretical profile generated from PDB entry 1oad (dashed line) is also given with ±σ error bars (every fifth point) and compared with the experimental profile (dots). (d) The computed signal-to-noise ratio for glucose isomerase (dashed line) falls more rapidly with q than the smaller lysozyme molecule. The experimental signal-to-noise curve (solid line) breaks up (becomes negative) on the semi-log scale for I/σ < 1, indicating that no useful information is contained beyond q = 0.1 Å−1.
7.2. Energy dependence
The energy dependence of scattering arises from the transmission T(λ) and detector-efficiency η(λ) terms in (1). A fair comparison of scattering profiles at different energies (E 0, E 1) should be constrained to the same q ranges. This constraint introduces an additional energy dependence on the sample-to-detector distance r (r 1 = r 0 E 1/E 0 for small angles). The concentration of (flowing) lysozyme for which a 1 s exposure gives χ2 sb = 1.5 is shown as a function of energy in Fig. 3 ▸(a). The q-space range (q max = 0.3 Å−1) has been held fixed in this comparison by varying the sample-to-detector distance from 800 mm (5 keV) to 4520 mm (30 keV). The sensitivity of detection declines for energies above 10 keV (solid line). This is true even for an ideal 100% efficient detector (dashed line). Where samples are exposed at a maximum allowable dose (7 kGy for this flowing sample) rather than a constant time, and detection is also 100% efficient, the sensitivity does improve modestly at high energies (dashed–dotted line). Above 20 keV, the convenient ∼λ3 power law for absorption used in these calculations begins to deteriorate and results should be considered only to be approximate.
Figure 3.

The minimum detectable concentration (χ2 = 1.5) of a flowing lysozyme sample as a function of energy. (a) A fixed 1 s exposure at 8 × 1011 photons s−1 for a typical SAXS cell with path length 1.5 mm, 2 × 25 µm mica windows and a counting detector with 320 µm silicon sensor thickness (solid line). Sample-to-detector distances have been scaled so as to maintain a fixed q range as the energy varies. The combination of fixed sample thickness and fixed q range diminishes the sensitivity at high energies even when a hypothetical 100% efficient detector (dashed curve) is used. Where the exposure time is adjusted according to expected radiation damage, the sensitivity shows a modest improvement with increasing energy but no longer has a minimum (dashed–dotted curve). (b) Better sensitivity can be achieved at high energies with thicker sample path lengths, but at the expense of high sample-cell volume (blue solid line). The highest sensitivity at microfluidic scale path lengths (0.5 mm and smaller) is achieved at the lowest energies (red dashed–dotted line). In the absence of windows [d w = 0 in equation (6)] the short-path experiment can detect a 35% lower concentration (cyan solid line), a level comparable to the standard 1.5 mm with windows (green dashed line).
Varying the sample path length moves the point of highest sensitivity in Fig. 3 ▸(b). The minima for these curves (highest sensitivity) roughly correspond to the energy at which the path length would be optimal according to the classical 1/μ formula (Glatter & Kratky, 1982 ▸). For a prohibitively long sample path length of 5.0 mm, the optimum sensitivity is above 15 keV (solid blue line). For the short path length of 0.5 mm used in our time-resolved microfluidic mixer chip the optimum sensitivity falls close to 7.5 keV (red dashed–dotted line). The complete removal of window absorption and scattering pushes the optimum sensitivity to near 6 keV (red solid line) and results in the ability to detect a 35% more dilute sample. A hypothetical windowless cell thus brings the performance of a 0.5 mm path-length sample cell back to the level of a standard 1.5 mm cell (green dashed line).
7.3. Distinguishing conformational states
Both SEC-SAXS and TR-SAXS experiments attempt to distinguish between different species. Distinguishing between monomers and higher oligomeric states is usually straightforward owing to large differences in molecular weight. However, when species differ only in conformation, differentiation can be challenging. To assess how microfluidic flow cells perform under various conditions, we have drawn a variety of matched structures from the Database of Macromolecular Motions, a curated list of known characterized molecular motions (http://molmovdb.org; Gerstein & Krebs, 1998 ▸). For fair comparison, we opted to remove any loops that are resolved in one structure but not in another. Fig. 4 ▸ is a composite of predicted scattering profiles from 12 matched structure pairs. The area between matched profile curves is color-coded to the list of PDB codes in the legend. All profiles are shown on a realistic intensity scale, where the exposure time has been chosen so that χ2 ab = 1.5. Pairs such as 1c9k–1cbu show separation over a wide range of angles and therefore require only a short exposure time to resolve. Because profile intensities fall rapidly with q, differences that occur only at wide angles require longer exposures to resolve (1clb–4icb, for example) and appear highest on the plot.
Figure 4.

Calculated scattering profiles for 12 matched structure pairs taken from the Database of Macromolecular Motions, a curated list of known characterized molecular motions. Colors between curves show the differences in scattering caused by the motion. Each set of profiles is scaled according to the exposure necessary to statistically differentiate between the two curves. Curves that are separated over large ranges of q space (1c9k–1cbu) require shorter exposure times than curves that are separated only at the widest angles.
The ability to resolve conformational changes is connected to how much sample is available for consumption. Continuous-flow time-resolved microfluidic systems flow sample in order to probe timescales. Consequently, sample flow rates are dictated by the physics of mixing and the timescales to be achieved. The length of exposure time necessary to resolve conformational changes directly determines how much sample is consumed. The microfluidic time-resolved SAXS chip currently used by MacCHESS has a 0.5 mm X-ray path length, a 0.4 mm channel width and a total window length of 1 cm. To collect different exposure times, the beam is positioned at different points along the 1 cm length. At its fastest flow speed and highest time resolution (11.3 µl s−1, 2.29 ms) the goal is to generate up to 90 time points. The rationale for the large number of time points is that it permits more sophisticated data analysis for separating species. In actual practice, multiple flow rates are possible, but this simple one-speed, 90-point test gives a worst-case sample-consumption rate. The compound refractive lenses (CRL; RXOPTICS, Monschau, Germany) used for preliminary tests of the actual setup produced 3.8 × 1011 photons s−1 in an 30 µm (vertical) × 50 µm (horizontal) beam.
Table 3 ▸ reports simulations for four different cases illustrating the effects of energy, windows and flux on the ability to distinguish molecular conformational changes. In each case the sample is at a concentration of 60/M mg ml−1 (column 1). The exposure time necessary to reach each of the statistical criteria is calculated. The exposure time for detection (t
det) of a single component to χ2
sb = 1.5 is generally very short in all cases (only reported for the first case). The exposure time necessary to obtain a ‘reasonable’ Guinier plot (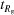 ) with error(R
g)/R
g = 0.05 is very similar across all species here because of the choice of a concentration of 60/M mg ml−1. The exposure times necessary to distinguish between conformations (t
com) vary widely over more than two orders of magnitude. In case 1 the conditions for the current mixing chip and CRL beam are used: energy = 10 keV and flux = 3.8 × 1011 photons s−1. While our current chip design uses thin Kapton windows, these simulations use the thicker traditional mica windows to evaluate the impact of window choice on sensitivity. Under these conditions, the volume consumed (volTR) for 90 time points is well beyond our 1–4 cm3 criterion for all but the first two structure pairs in the database. The dose reported in Table 3 ▸ is the X-ray dose in gray received by any illuminated volume during the flow. In all of these cases the dose is acceptable for even a sensitive protein such as lysozyme. In case 2 we remove the mica windows on the cell to find that the volume consumption is cut nearly in half, more than doubling the number of feasible proteins. In case 3 we have left the windows off and moved the energy down to 6 keV, the optimum energy for a windowless cell of thickness 0.05 cm. The sample-to-detector distance has been reduced for this calculation to maintain an equivalent q-space range. Energy optimization in this case yields only a modest reduction in sample consumption (∼10%). The final case receives a tenfold boost in flux with full windows present, as expected for some of the beamlines currently under construction at CHESS-U. Not surprisingly, the required exposure time and consumed volume scale linearly with X-ray flux, moving nine out of 12 structure pairs into the feasible category
) with error(R
g)/R
g = 0.05 is very similar across all species here because of the choice of a concentration of 60/M mg ml−1. The exposure times necessary to distinguish between conformations (t
com) vary widely over more than two orders of magnitude. In case 1 the conditions for the current mixing chip and CRL beam are used: energy = 10 keV and flux = 3.8 × 1011 photons s−1. While our current chip design uses thin Kapton windows, these simulations use the thicker traditional mica windows to evaluate the impact of window choice on sensitivity. Under these conditions, the volume consumed (volTR) for 90 time points is well beyond our 1–4 cm3 criterion for all but the first two structure pairs in the database. The dose reported in Table 3 ▸ is the X-ray dose in gray received by any illuminated volume during the flow. In all of these cases the dose is acceptable for even a sensitive protein such as lysozyme. In case 2 we remove the mica windows on the cell to find that the volume consumption is cut nearly in half, more than doubling the number of feasible proteins. In case 3 we have left the windows off and moved the energy down to 6 keV, the optimum energy for a windowless cell of thickness 0.05 cm. The sample-to-detector distance has been reduced for this calculation to maintain an equivalent q-space range. Energy optimization in this case yields only a modest reduction in sample consumption (∼10%). The final case receives a tenfold boost in flux with full windows present, as expected for some of the beamlines currently under construction at CHESS-U. Not surprisingly, the required exposure time and consumed volume scale linearly with X-ray flux, moving nine out of 12 structure pairs into the feasible category
Table 3. Distinguishing molecular conformations with continuous-flow time-resolved SAXS.
The concentration c is in mg ml−1 and PDB1 and PDB2 are the PDB codes of the matched structures used in the calculation. Molecular weight (mwt) is given in kilodaltons, while the radius of gyration R
g is in angstroms. All exposure times are given in seconds, with t
det being the minimum exposure necessary for detection, being the exposure necessary to achieve 5% error in R
g and t
com being the exposure necessary to distinguish between matched conformations. The volume volTR, given in microlitres, is the amount of sample that flows through the cell in time t
com. The , t
com and volTR values are given for four calculations labelled according to the dose received (Gy), whether or not windows are present, the X-ray energy and flux. Experiments that are deemed to be ‘feasible’ by virtue of a sufficiently low sample consumption (≤4000 µl) are shown in bold (see text).
| Dose = 38.3 Gy (windows) 10 keV 3.8 × 1011 photons s−1 | Dose = 44.0 Gy (no windows) 10 keV 3.8 × 1011 photons s−1 | Dose = 79.5 Gy (no windows) 6 keV 3.8 × 1011 photons s−1 | Dose = 382.9 Gy (windows) 10 keV 3.8 × 1012 photons s−1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c | PDB1 | PDB2 | mwt | R g | t det |
|
t com | volTR |
|
t com | volTR |
|
t com | volTR |
|
t com | volTR |
| 1.01 | 1c9k | 1cbu | 59.276 | 24.3 | 0.01412 | 14.5 | 3.51 | 3569 | 10.8 | 1.98 | 2017 | 6.6 | 1.8 | 1823 | 1.17 | 0.35 | 359 |
| 3.01 | 1e0s | 2j5x | 19.951 | 16.1 | 0.00757 | 10.3 | 3.97 | 4035 | 12.9 | 2.29 | 2328 | 4.4 | 2.1 | 2095 | 0.89 | 0.40 | 403 |
| 3.19 | 1jkn | 1f3y | 18.815 | 15.5 | 0.00718 | 9.1 | 4.81 | 4888 | 6.2 | 2.82 | 2871 | 7.7 | 2.5 | 2561 | 0.68 | 0.49 | 495 |
| 6.58 | 1hdn | 1pfh | 9.119 | 12.2 | 0.00521 | 7.5 | 5.04 | 5121 | 4.4 | 2.90 | 2949 | 7.2 | 2.6 | 2638 | 0.84 | 0.50 | 504 |
| 10.41 | 4ins | 2hiu | 5.766 | 10.6 | 0.00445 | 9.7 | 7.02 | 7138 | 5.0 | 4.12 | 4190 | 5.0 | 3.7 | 3724 | 0.72 | 0.71 | 718 |
| 1.70 | 2ran | 1axn | 35.383 | 22.1 | 0.01187 | 24.4 | 7.48 | 7604 | 15.9 | 4.43 | 4500 | 5.9 | 4.0 | 4035 | 1.47 | 0.75 | 766 |
| 2.25 | 1brd | 2brd | 26.673 | 16.1 | 0.00743 | 13.7 | 11.60 | 11794 | 6.0 | 6.71 | 6828 | 6.1 | 6.0 | 6052 | 0.92 | 1.14 | 1164 |
| 2.96 | 1a03 | 1cnp | 20.290 | 17.5 | 0.00840 | 11.1 | 21.36 | 21726 | 6.0 | 12.21 | 12415 | 5.7 | 11.0 | 11173 | 1.03 | 2.14 | 2173 |
| 4.40 | 1dc7 | 1dc8 | 13.624 | 14.3 | 0.00643 | 12.9 | 25.63 | 26071 | 5.5 | 14.65 | 14898 | 5.7 | 13.1 | 13346 | 0.92 | 2.59 | 2638 |
| 6.98 | 1clb | 4icb | 8.592 | 12.0 | 0.00521 | 6.9 | 161.13 | 163872 | 6.4 | 92.77 | 94351 | 4.0 | 83.0 | 84419 | 0.62 | 16.17 | 16449 |
| 1.64 | 1beb | 1b0o | 36.601 | 21.6 | 0.01119 | 12.5 | 380.86 | 387334 | 6.9 | 214.84 | 218496 | 8.7 | 195.3 | 198633 | 0.87 | 37.84 | 38485 |
| 3.33 | 1rx2 | 1rx7 | 18.000 | 16.0 | 0.00763 | 8.9 | 507.81 | 516445 | 8.1 | 292.97 | 297949 | 6.6 | 258.8 | 263189 | 0.95 | 50.66 | 51520 |
8. Conclusions
We have developed a methodology for simulating signal-to-noise levels in biological small-angle solution scattering that explicitly accounts for buffer, contrast, window and instrumental scattering, photon flux, sample thickness, detector integration mask, sample-to-detector distance, and sensor thickness. Energy-dependent terms are also correctly modeled. The method has been validated by comparing predictions with experimental results for two standard proteins collected on four different synchrotron beamlines. The estimation of absolute scattering intensity for BioSAXS experiments can be expected to be about as accurate as molecular-weight estimates derived from SAXS data. Noise estimates based on assumptions of basic counting statistics can give signal-to-noise curves that are in excellent agreement with experiment when the background scatter is correctly modeled. Instrumental background is inherently difficult to model in general given the variety of beamline optics possible, but window and buffer scattering can be parameterized based on standardized measured profiles that are valid on any beamline or source. Window and instrumental ‘vacuum’ scattering begins to overtake buffer signal at small angles and the effect is exacerbated for short sample path lengths.
Sample consumption is an important limiting factor in BioSAXS. Improvements in the sensitivity of detection directly translate into less sample consumption. While detection limits and the quality of Guinier-derived parameters depend mainly on sample molecular weight, the ability of BioSAXS to distinguish between alternate protein conformations varies widely over more than two orders of magnitude in exposure time for our sampling of structures and depends upon specific profile shape in regions that fall rapidly with q. Window scattering and absorption in microfluidic (time-resolved) SAXS can significantly degrade this ability to distinguish species. Lower X-ray energies are favorable for data collection in narrow microfluidic channels, but the overall effect is small in comparison to windows. Distinguishability scales linearly with beam flux, so the tenfold flux improvement expected from the CHESS-U upgrade significantly expands the number of feasible structures, but the magnitude of window scattering is not insignificant in comparison. Consequently, improvements in source technology should always include improvements in background reduction.
We have demonstrated some basic uses for the accurate estimation of BioSAXS signal quality. Such calculations can be readily applied to more advanced data-processing techniques such as distance distribution functions, shape reconstruction, model refinement and analysis of solution ensembles. When approximate models are available, these kinds of calculations should also be valuable in experiment planning prior to synchrotron visits, although negative results in these simulations should not necessarily rule out experiments. The Python code and data used in this paper are available from the author (REG) upon request. A simplified web-based interface to the code can be run at https://www.classe.cornell.edu/wsgi/macchess/Web_SAXS/parsing_post.wsgi.
Acknowledgments
Thanks to Jesse Hopkins for contributing in numerous ways to BioSAXS at CHESS, particularly for his discussions on radiation damage and time-resolved SAXS. Also, to thanks to Steve Meisburger for valuable insights and to Tilman Donath for providing information on detector performance. The following people helped in configuring the CHESS A1 station for high-energy BioSAXS: Irina Kriksunov, Oluwasina Okunoye, Phil Sorenson, Bill Miller, Tom Krawczyk, Zack Brown and Jesse Hopkins. CHESS F1 was configured for BioSAXS with help from Manjie Huang, Melanie MacMullan, Bill Miller and Scott Smith. Thanks to Haydyn Mertens, Clement Blanchet, Cy Jeffries and Dmitri Svergun (EMBL, Hamburg) for access and assistance during the P12 experiments.
Appendix A. Derivation of equation (8)
The classical χ2 statistic for simulated scattering data can be calculated without resorting to generating random deviates, but the formula has an extra term. Let I si be the true scattering intensity of the ‘sample’ in the ith of N discrete bins. Similarly, let I bi represent the true ‘buffer’ scattering intensity. An actual experimental sample measurement is the noisy profile Í si = I si + δI si, where each δI si is a random deviate drawn from a Poisson distribution having population mean 〈δI si〉 = 0 and population standard deviation σsi. Similarly, Í bi = I bi + δI bi with 〈δI bi〉 = 0 and standard deviation σbi. A single reduced χ2 statistic for comparisons between the measured curves s and b is
where p is the number of parameters if either of the curves is a parameterized model fit to the data. Here, 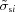 and
and 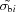 are estimates of the population standard deviations usually obtained from the detector pixels that fall within the ith q bin. The sum of squares in the denominator of (9) is a consequence of how errors owing to Poission distributions add in quadrature. Multiple independent measurements of the same profiles would produce a range of χ2
sb values that follow the well known χ2 distribution. The χ2
sb could consequently be used to make rigorous probability statements. Utilizing the properties that 〈δI
si〉 = 0, SD(I
si) = σsi, 〈δI
bi〉 = 0, SD(δI
bi) = σbi, and the definitions of Í
si and Í
bi above, the mean value of the χ2 statistic over multiple random samplings expands to give
are estimates of the population standard deviations usually obtained from the detector pixels that fall within the ith q bin. The sum of squares in the denominator of (9) is a consequence of how errors owing to Poission distributions add in quadrature. Multiple independent measurements of the same profiles would produce a range of χ2
sb values that follow the well known χ2 distribution. The χ2
sb could consequently be used to make rigorous probability statements. Utilizing the properties that 〈δI
si〉 = 0, SD(I
si) = σsi, 〈δI
bi〉 = 0, SD(δI
bi) = σbi, and the definitions of Í
si and Í
bi above, the mean value of the χ2 statistic over multiple random samplings expands to give
Funding Statement
This work was funded by National Institute of General Medical Sciences grant GM-103485. National Science Foundation, Directorate for Mathematical and Physical Sciences grant DMR-1332208.
Footnotes
CHESS-U is a major upgrade to the Cornell High Energy Synchrotron Source. With the transition to single-beam running at 6 GeV and 200 mA, CHESS-U will become a world-class high-flux density, high-energy source complementary to fourth-generation rings, which are optimized mainly for coherence. New beamlines will leverage the minimized emittance and high bunch charge with state-of-the-art undulators. Completion is expected in early 2019.
References
- Acerbo, A. S., Cook, M. J. & Gillilan, R. E. (2015). J. Synchrotron Rad. 22, 180–186. [DOI] [PMC free article] [PubMed]
- Ando, N., Chenevier, P., Novak, M., Tate, M. W. & Gruner, S. M. (2008). J. Appl. Cryst. 41, 167–175.
- Bohon, J., Muller, E. & Smedley, J. (2010). J. Synchrotron Rad. 17, 711–718. [DOI] [PMC free article] [PubMed]
- Cho, Y., Yang, J. S. & Song, K. B. (1999). Food. Res. Int. 32, 515–519.
- Donath, T., Brandstetter, S., Cibik, L., Commichau, S., Hofer, P., Krumrey, M., Lüthi, B., Marggraf, S., Müller, P., Schneebeli, M., Schulze-Briese, C. & Wernecke, J. (2013). J. Phys. Conf. Ser. 425, 062001.
- Dreiss, C. A., Jack, K. S. & Parker, A. P. (2006). J. Appl. Cryst. 39, 32–38.
- Feigin, L. A. & Svergun, D. I. (1987). Structure Analysis by Small-Angle X-ray and Neutron Scattering. New York: Springer.
- Franke, D., Jeffries, C. M. & Svergun, D. I. (2015). Nature Methods, 12, 419–422. [DOI] [PubMed]
- Franke, D., Petoukhov, M. V., Konarev, P. V., Panjkovich, A., Tuukkanen, A., Mertens, H. D. T., Kikhney, A. G., Hajizadeh, N. R., Franklin, J. M., Jeffries, C. M. & Svergun, D. I. (2017). J. Appl. Cryst. 50, 1212–1225. [DOI] [PMC free article] [PubMed]
- Garman, E. F. & Weik, M. (2017). J. Synchrotron Rad. 24, 1–6. [DOI] [PubMed]
- Gerstein, M. & Krebs, W. (1998). Nucleic Acids Res. 26, 4280–4290. [DOI] [PMC free article] [PubMed]
- Gillilan, R. C. M., Temnykh, G., Møller, M. & Nielsen, S. (2013). Trans. Am. Crystallogr. Assoc. 44, 40–50.
- Glatter, O. & Kratky, O. (1982). Small-Angle X-ray Scattering. London: Academic Press.
- Graceffa, R., Nobrega, R. P., Barrea, R. A., Kathuria, S. V., Chakravarthy, S., Bilsel, O. & Irving, T. C. (2013). J. Synchrotron Rad. 20, 820–825. [DOI] [PMC free article] [PubMed]
- Graewert, M. A. & Svergun, D. I. (2013). Curr. Opin. Struct. Biol. 23, 748–754. [DOI] [PubMed]
- Henderson, S. J. (1995). J. Appl. Cryst. 28, 820–826.
- Hopkins, J. B. & Thorne, R. E. (2016). J. Appl. Cryst. 49, 880–890. [DOI] [PMC free article] [PubMed]
- Hülsen-Bollier, G. (2005). PhD thesis, University of Erlangen-Nürnberg, Germany.
- Jeffries, C. M., Graewert, M. A., Svergun, D. I. & Blanchet, C. E. (2015). J. Synchrotron Rad. 22, 273–279. [DOI] [PubMed]
- Kratky, O. & Pilz, I. (1972). Q. Rev. Biophys. 5, 481–537. [DOI] [PubMed]
- Kuwamoto, S., Akiyama, S. & Fujisawa, T. (2004). J. Synchrotron Rad. 11, 462–468. [DOI] [PubMed]
- Lurio, L., Mulders, N., Paetkau, M., Jemian, P. R., Narayanan, S. & Sandy, A. (2007). J. Synchrotron Rad. 14, 527–531. [DOI] [PubMed]
- Masunaga, H., Sakurai, K., Akiba, I., Ito, K. & Takata, M. (2013). J. Appl. Cryst. 46, 577–579.
- Meisburger, S. P., Warkentin, M., Chen, H., Hopkins, J. B., Gillilan, R. E., Pollack, L. & Thorne, R. E. (2013). Biophys. J. 104, 227–236. [DOI] [PMC free article] [PubMed]
- Mylonas, E. & Svergun, D. I. (2007). J. Appl. Cryst. 40, s245–s249.
- Orthaber, D., Bergmann, A. & Glatter, O. (2000). J. Appl. Cryst. 33, 218–225.
- Pauw, B. R. (2014). J. Phys. Condens. Matter, 26, 239501. [DOI] [PubMed]
- Pedersen, M. C., Hansen, S. L., Markussen, B., Arleth, L. & Mortensen, K. (2014). J. Appl. Cryst. 47, 2000–2010.
- Sagan, D., Chee, J. Y., Finkelstein, K. & Hoffstaetter, G. (2011). Proc. SPIE, 8141, 81410Y.
- Schneidman-Duhovny, D., Hammel, M. & Sali, A. (2010). Nucleic Acids Res. 38, W540–W544. [DOI] [PMC free article] [PubMed]
- Sedlak, S. M., Bruetzel, L. K. & Lipfert, J. (2017). J. Appl. Cryst. 50, 621–630. [DOI] [PMC free article] [PubMed]
- Stuhrmann, H. B. (1980). Synchrotron Radiation Research, edited by H. Winick & S. Doniach, pp. 513–531. New York: Plenum Press.
- Svergun, D., Barberato, C. & Koch, M. H. J. (1995). J. Appl. Cryst. 28, 768–773.
- Svergun, D. I. & Koch, M. H. J. (2003). Rep. Prog. Phys. 66, 1735–1782.
- Svergun, D. I., Koch, M. H. J., Timmins, P. A. & May, R. P. (2013). Small Angle X-ray and Neutron Scattering from Solutions of Biological Macromolecules. Oxford University Press.
- Whitten, A. E., Cai, S. & Trewhella, J. (2008). J. Appl. Cryst. 41, 222–226.