

Abstract
Coeluting peptides are still a major challenge for the identification and validation of MS/MS spectra, but carry great potential. To tackle these problems, we have developed the here presented CharmeRT workflow, combining a chimeric spectra identification strategy implemented as part of the MS Amanda algorithm with the validation system Elutator, which incorporates a highly accurate retention time prediction algorithm. For high-resolution data sets this workflow identifies 38–64% chimeric spectra, which results in up to 63% more unique peptides compared to a conventional single search strategy.
Keywords: tandem mass spectrometry, MS/MS, database search, chimeric spectra, mixed spectra, retention time prediction, validation
Introduction
Advancements in mass spectrometer instrument precision and acquisition time1,2 made mass spectrometry the primary instrument in proteomics analyses. The interpretation of the measured spectra is often performed using a database search algorithm.3−6 Most database search algorithms stick to the “one-spectrum-one-peptide” paradigm, although the occurrence of coeluting peptides and the accompanied challenges of chimeric spectra have been widely studied.7−9 Even though several solutions for processing chimeric spectra already exist,10−14 they are still often not used in an everyday proteomics workflow. In addition, the validation of more than one peptide match per spectrum (here called mPSM) is an important task,15 as the confidence score for the most abundant peptide in a spectrum is not easily comparable to the score of a second coeluting peptide also present in the spectrum. However, through ignoring this valuable information a large amount of unique peptides remains unidentified, as recent studies show that about 50% of all spectra contain more than one peptide.7,15
In general, the dynamic range of proteins is a big challenge in proteomics experiments.16 Detecting highly abundant proteins is a lot simpler than identifying the least abundant part of the proteome.16,17 Many approaches have been conducted to increase proteome coverage and enable deep proteome analysis,18−25 being more or less straightforward and affordable techniques for an everyday proteomics workflow.
We here propose a combination of identifying chimeric spectra and validating detected mPSMs using retention time prediction, jointly leading to a significant increase in validated unique peptides for each data set accompanied by higher coverage of low abundant proteins: the CharmeRT workflow.
Methods
CharmeRT Workflow
The first part of the CharmeRT workflow identifies chimeric spectra using a second search approach in our database search engine MS Amanda.26 The second part of CharmeRT validates the identified PSMs of first and second searches using Elutator, a newly developed tool based on the principles of Percolator,27 featuring a new approach for retention time prediction. An overview of the workflow can be seen in Figure 1.
Figure 1.
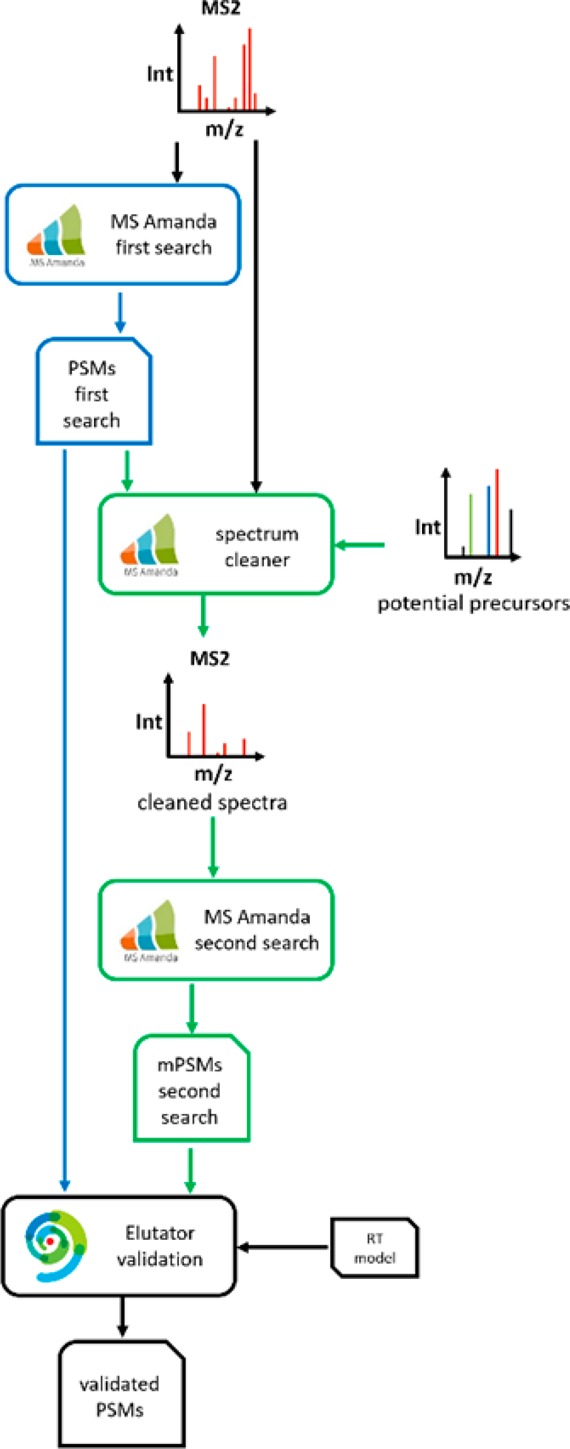
Overview of the CharmeRT workflow. After a first search round with MS Amanda, spectra are cleaned, potential interfering precursors are identified, and spectra are submitted to a second search round. Resulting PSMs of the first and the second search are validated by Elutator using a retention time model.
Chimeric Spectra Search in MS Amanda
To identify multiple peptides per spectrum, a second search approach was implemented in the database search engine MS Amanda. For each spectrum, all peaks of the highest scoring peptide identified in the first search are removed. Basis of this removal are the selected fragment ions in the search, additionally neutral loss ions can be removed as well. As interfering peptides may have the same c- or n-terminal amino acid due to the used enzyme, leading to a shared y1/b1 ion in a mixed spectrum, y1 ions can optionally be kept, and b1 ions are not considered at all by MS Amanda. Tests showed that all other potentially shared peaks can be neglected, as they are very unlikely. We identified an average overlap of 0.7%, see Supplemental Table S2. Corresponding MS1 spectra are investigated and potential interfering precursors are determined, optionally performing a preceding deisotoping of the MS1 spectrum. There are several ways to treat precursor peaks where the charge state cannot be determined: not considering them, testing various selectable charge states, or only testing the most abundant ones of them at different charge states. All spectra are submitted to a further search lap testing each of the identified precursors with the option to research the original precursor. For each spectrum, multiple second search hits, i.e., the best n PSMs for the top m precursors, are reported.
mPSM Validation in Elutator
The second part of the CharmeRT workflow is realized by Elutator, a new tool for validating identified mPSMs. Elutator is based on the principles of Percolator27 and validates mPSMs using a set of features optimized for the analysis of MS Amanda results. A complete list of all used features is given in the supplemental data (Supplemental Table S1), including the deviation of an estimated peptide elution retention time (RT) from the actual value, as well as recalibrated masses for precursor and fragment ions. The most important features are explained in the next sections.
Elutator Retention Time Prediction Model
An important factor in the context of validating mPSMs is the difference between predicted and measured retention times. Several approaches already exist to construct RT prediction models.28−30 However, the use of these models for validation is often limited due to specific requirements, such as, a significant amount of training data and correct handling of chemical modifications. We have therefore developed a new retention time prediction algorithm: Elutator’s RT model is based on the SSRCalc30 model and estimates the hydrophobicity index of peptides based on their sequences and chemical modifications, which can be linearly mapped to retention time. It was significantly redesigned and extended for better performance but preserves most of the features and ideas of the original SSRCalc algorithm. The features used for predicting the model include peptide length, certain properties for special amino acids (e.g., Proline), the isoelectric charge, properties for short peptides, or parameters for hydrophobic amino acid patterns likely forming helices, and are similar to the features described by Krokhin.31
An important improvement compared to the original model of SSRCalc for retention time prediction is the consideration of neighboring effects of amino residuals being not restricted to nearest neighbors only. Experiments showed a statistically significant effect of amino residual interactions even for residuals separated by several positions in the polypeptide chain. A detailed description on how we model these interactions is given in the Supporting Information.
The described features are used in an optimized nonlinear retention time model implemented in Elutator. The original formulation of the model was given by Krokhin for SSRCalc.31 The parameters (coefficients) of the used model are optimized using Newton’s method of minimizing the sum of squares of retention time deviations for all peptides in the training sets. A detailed information on the model calculation is given in the supplemental data. The optimization procedure assumes simultaneous training over several different data sets measured under similar elution conditions (gradient duration, chemical composition of eluents, column temperature, etc.). To avoid overfitting, the retention time model has been trained using 94122 highly reliable PSMs (FDR threshold was 0.001) corresponding to 44 271 unique sequences obtained from in-house measured data sets of different organisms: trypsin digested human (HeLa), mouse, yeast, B. subtilis, E. coli, phosphorylated peptides from TiO2 enriched human cell lysate, and chymotrypsin digested human data set. After a preliminary optimization, we removed 0.1% of the outliers, corresponding to the number of expected false matches, and repeated the optimization. By considering additional peptide properties, such as the interactions of neighboring amino residuals in the peptide chain, we considerably increased the RT prediction accuracy (Figure 2). A similar accuracy of retention time prediction was achieved for phosphorylated and unmodified peptides (see Supplemental Figure S1).
Figure 2.

Comparison of elution retention time prediction models: (a) Elutator,
(b) BioLCCC,29 (c) SSRCalc,30 and (d) Elude.28 Depending
upon the model design the output is either an absolute retention time
or a relative hydrophobicity index, which can be linearly mapped to
the retention time in a particular data set. We here compare the correlation
of predicted and measured retention times of data set I, which is
important for validation. R2 is the coefficient
of determination, and 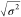 is the dispersion of the error in minutes.
As Elude cannot be trained on multiple raw files, we here used 50%
randomly chosen PSMs over all raw files for training and the others
for testing.
is the dispersion of the error in minutes.
As Elude cannot be trained on multiple raw files, we here used 50%
randomly chosen PSMs over all raw files for training and the others
for testing.
For practical usage, the applicability of the trained model on data sets measured under a different chromatographic setup is of high interest. Elutator maps the predicted hydrophobicity index to the observed retention time by applying a linear fitting for all peptides in a single HPLC run. This allows for an application to data sets with different setups. We investigated this using a publicly available externally measured HeLa data set.32 The accuracy of the retention time prediction is lower for the external data set, as can be seen through the correlation coefficient R2. Nevertheless, as demonstrated in Supplemental Figure S2, using retention time prediction also here leads to a higher number of PSMs. Smaller retention time dispersion for the external data set can be explained by the shorter gradient (90 min versus 180 min for the in-house data set). The smaller gradient duration leads to a proportional decrease of retention time deviations. Alternatively, a new model can be easily trained for specific elution conditions using the Elutator RT Trainer (see Availability).
Combined Retention Time Score
Besides the deviation of the predicted RT to the measured RT, Elutator also uses the combined retention time score as feature for mPSM validation. It includes the PSM score of the search engine and the retention time deviation obtained from the retention time model. To calculate a combined score, the MS Amanda score is recalibrated on the posterior error using linear regression to define coefficients a and b using the model
where f(A) is the probability for a match with score A to be false (i.e., local FDR), and A is the MS Amanda score.
After this calibration, the combined score is calculated using the following scoring function:
where σ is the dispersion
of the predicted
retention time, calculated considering highly reliable matches (FDR
= 0.001), T is the duration of the linear part of
the gradient, erf is the Gauss error function, and ε is defined
as 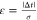 , where Δt is the
retention time deviation from the predicted value for the scored peptide.
, where Δt is the
retention time deviation from the predicted value for the scored peptide.
Calibration of Mass Differences
The aim of calibrating mass differences is to eliminate constant biases in mass measurements for precursors and fragments to enhance the mass resolution and is included as additional feature for mPSM validation. In Elutator this calibration is based on theoretically known masses of highly reliable matches of the first search (FDR = 0.001, calculated on MS Amanda score).
Recalibration can be done for measured deviations of m/z values, 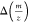 , as well as for relative mass deviations, Δmppm. Elutator uses the following approximation
of mass deviations over retention time t and m/z to determine the calibration coefficients a, b, and c:
, as well as for relative mass deviations, Δmppm. Elutator uses the following approximation
of mass deviations over retention time t and m/z to determine the calibration coefficients a, b, and c:
Results of mass recalibration for a human data set32 are presented in Supplemental Figure S3. This data set was analyzed with lock mass disabled (available in Q Exactive instruments, Thermo Fisher Scientific). Constant bias and variable error seemed to be similar in this case. Activating the lock mass option partly eliminates a constant bias, but increases a variable error because it is based on measuring the mass of known ions present in the spectrum. Therefore, we suggest that disabling the lock mass is preferable for better mass resolution when PSM validation by Elutator is used.
Longest Consecutive Series A + B + Y
We introduce a combined consecutive sequence of N- and C-terminal ions as additional feature for validation, namely the sequence of a, b, and y ions, which typically constitute HCD/CID spectra. PSMs with scores close to the FDR threshold contain relatively few matched fragment peaks; therefore, y ions are likely not able to form any consecutive sequence. However, longer sequences can be potentially constructed by taking into account a and b ions, which fill gaps between y fragments (see Supplemental Figure S4).
Experiments
In House Data Generation
Samples were reduced and alkylated using dithiothreotiol (1 μg DTT per 20 μg protein) and iodacetamide (5 μg per 20 μg protein). Proteins were predigested with Lys-C at 30 °C for 2 h (1 μg Lys-C per 50 μg protein in 6 M urea and 12 mM Triethylammonium bicarbonate buffer (100 mM Ammonium bicarbonate (ABC) buffer for mouse samples)) and digested overnight with trypsin (Promega, Trypsin Gold, Mass spectrometry grade) at 37 °C (1 μg trypsin per 30 μg protein, 0.8 M urea in 45 mM Triethylammonium bicarbonate buffer (mouse: 2 M urea with 100 mM ABC buffer)); digestion was stopped by adding concentrated TFA to a pH of approximately 2. Phosphorylated peptides were enriched following the in-house TiO2 enrichment protocol,33 HeLa peptides were obtained following the in-house HeLa protocol.34
The HPLC system used was an UltiMate 3000 HPLC RSLC nano system coupled to an Q Exactive mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), equipped with a Proxeon nanospray source (Proxeon, Odense, Denmark). Peptides were loaded onto a trap column (Thermo Fisher Scientific, Bremen, Germany, PepMap C18, 5 mm × 300 μm ID, 5 μm particles, 100 Å pore size) at a flow rate of 25 μL/min using 0.1% TFA as mobile phase. After 10 minutes the trap column was switched in line with the analytical column (Thermo Fisher Scientific, Bremen, Germany, PepMap C18, 500 mm × 75 μm ID, 3 μm, 100 Å). Peptides were eluted using a flow rate of 230 nL/min. The eluting peptides were directly analyzed using hybrid quadrupole-orbitrap mass spectrometers (Q Exactive or Q Exactive Hybrid, Thermo Fisher). The Q Exactive mass spectrometer was operated in data-dependent mode using a full scan (m/z range 350–1650Th, nominal resolution of 70 000, target value 1E6) followed by MS/MS scans of the 12 most abundant ions. MS/MS spectra were acquired at a resolution of 17 500 using normalized collision energy 30%, isolation widths of 2, 4, or 8, and the target value was set to 5E4. Precursor ions selected for fragmentation (charge state 2 and higher) were put on a dynamic exclusion list for 10 s. Additionally, the underfill ratio was set to 20%, resulting in an intensity threshold of 2E4.
Data Set Description
To assess the quality of the CharmeRT workflow, we applied it to several different data sets (3 replicates each, measured on Thermo Q Exactive or Q Exactive Hybrid): several in-house HeLa tryptic digests with different isolation widths and different gradient times (data sets A-F, I), an in-house phospho-enriched HeLa tryptic digest (data set G), and an external HeLa tryptic digest32 (data set H).
-
(A, B)
HeLa tryptic digest, in-house measurement (Thermo Q Exactive Hybrid, 1 h gradient (A) and 3 h gradient (B), 2 m/z isolation width, 1 μg, Figure 3).
-
(C, D)
HeLa tryptic digest, in-house measurement (Thermo Q Exactive Hybrid, 1 h gradient (C) and 3 h gradient (D), 4 m/z isolation width, 1 μg, Figure 3).
-
(E, F)
HeLa tryptic digest, in-house measurement (Thermo Q Exactive Hybrid, 1 h gradient (E) and 3 h gradient (F), 8 m/z isolation width, 1 μg, Figure 3).
-
(G)
HeLa tryptic digest, in-house measurement, phospho enrichment (Thermo Q Exactive, 3 h gradient, 2 m/z isolation width, 100 ng, Figure 4 and Figure S1, TiO2 enrichment of phosphorylated peptides).
-
(H)
HeLa tryptic digest, external measurement32 (Thermo Q Exactive, 90 min gradient, 4 m/z isolation width, 5 μg, Figure 4 and Figure S2).
-
(I)
HeLa tryptic digest, in-house measurement (Thermo Q Exactive, 3 h gradient, 2 m/z isolation width, 100 ng, Figures 2 and S2)
Figure 3.

Comparison of identification results of HeLa data sets measured with various isolation widths and gradient times analyzed with the CharmeRT workflow. We analyzed triplicates of tryptic HeLa samples for 2 m/z, 4 m/z, and 8 m/z isolation width, each either at a gradient time of 1 h or 3 h. Results are given for 1% FDR calculated at peptide level, showing the (a) number of identified PSMs in the first and in the second search and (b) number of unique peptides identified only in the first, only in the second, and in both searches.
Figure 4.

Comparison of MS Amanda and Elutator with other scoring methods and validation tools. Comparison was performed using (a) an external HeLa data set obtained from Michalski et al.32 (data set H) and (b) an in-house data set of human HeLa after TiO2 enrichment of phosphorylated peptides (data set G). The FDR threshold of 1% was calculated at PSM level for consistency between different search tools, which typically operate at PSM level. In cases where several high confident matches were reported for the same spectrum, the match with best q-value was selected such that the number of PSMs corresponds to the number of confidently identified spectra. Elutator includes features derived from a peptide elution retention time prediction model. Model training was performed on in–house data sets, the same model was applied to in-house and external data sets.
Database Search Parameters
When possible, runs have been performed in Proteome Discoverer 1.4, using Mascot version 2.2.7, MS Amanda v 1.4.14.9288, and Elutator v 1.14.1.236. For results obtained with pParse, all raw files have been preprocessed with pParse version 2.0.8 and resulting files submitted to PD 1.4. MaxQuant results were obtained with version 1.5.5.1, and all settings were set to default values as this lead to the best performance.
The following parameter settings have been used for MS Amanda, Mascot, and MaxQuant: swissprot database 2016–06 (human/mouse) including the “cRAP” contaminants database; trypsin as enzyme; 2 missed cleavages; Carbamidomethyl(C) as fixed PTM; Oxidation(M) and (for the phosphorylated data set) Phospho(S,T) as variable modifications. For MS Amanda and Mascot 10 ppm precursor mass tolerance and 0.02 Da fragment mass tolerance were used.
We applied the following additional settings specific for MS Amanda, where second search has been enabled: MS1 spectrum deisotoping set to false; keep y1 ion, remove water losses, remove ammonia losses, and exclude first precursor set to true; top 5 results per precursor in Figures 3 and 4/top 10 results per precursor for Supplemental Figure S7.
For Mascot we set the peptide cutoff score to 0.
The Elutator FDR threshold was set to 1% on peptide level for results in Figure 3 and on PSM level for the experiments in Figure 4. For results in Figure 4, the match with the best q-value was selected in a case when several high confident matches were reported for the same spectrum, such that the number of PSMs corresponds to the number of confidently identified spectra. For all results obtained using Percolator, numbers were obtained applying an extra Proteome Discoverer node “Multi-confident PSMs fix”, available at http://ms.imp.ac.at/?goto=charmert. MaxQuant results were filtered manually.
Results
CharmeRT Performance
To demonstrate the performance of the CharmeRT workflow, we analyzed HeLa samples using different isolation widths during acquisition. In standard mass spectrometry experiments, very narrow isolation widths (≤2 m/z) are applied to decrease the probability of coeluting peptides. However, being able to reliably identify multiple coeluting peptides per spectra reveals new possibilities for peptide identification and acquisition. By using broader isolation widths, we were able to considerably increase the numbers of identified peptides at a constant FDR (Figure 3).
Applying the second search approach increased the number of reliable identifications for all tested isolation widths and gradient times. Even for narrow isolation widths (2 m/z) and small gradient times (1 h) we observed a considerable number of validated chimeric spectra, which increased the number of identified unique peptides by 41% (5360 unique peptides). As expected, the amount of reliably identified PSMs and peptides in the first search decreases by 2–15% for broad isolation widths (8 m/z, 14219 PSMs (1 h)/23138 PSMs (3 h)) compared to narrow isolation widths (2 m/z, 14 506 PSMs (1 h)/27 340 PSMs (3 h)), as spectra complexity increases. This is alleviated by the chimeric approach, which identified almost the same number of unique peptides (20 438 (1 h)/28 550 (3 h) unique peptides) compared to the 2 m/z isolation width runs (18 566 (1 h)/31 346 (3 h) unique peptides). In our tests an isolation width of 4 m/z combined with a longer gradient resulted in the highest number of identified peptides (33 138 unique peptides) and the deepest insight into the investigated sample. This results not only in further evidence for already identified proteins, but also in additional proteins unidentified before (Supplemental Figure S6). Similar results can be achieved for an external data set:35 analyzing label-free data acquired at 1.4 m/z isolation width we see an average increase in PSMs of 75%, whereas for a TMT data set measured at a very narrow isolation width of 0.4 m/z only a small amount of chimeric spectra can be identified (see Supplemental Figure S5).
On average, 38% of the reliably identified spectra at 2 m/z isolation width (1 h gradient) were chimeric spectra (Supplemental Figure S7). This number increases to 53% at an isolation width of 4 m/z (3 h gradient). Additionally, on average, almost 20% of all reliably identified spectra at 4 m/z contain more than two peptides. Several examples of randomly drawn identified chimeric spectra of data set D are given in Supplemental Figures S11–S18.
Comparison to State of the Art Approaches
The combination of chimeric spectra identification and mPSM validation using the power of accurate retention time prediction increased the number of identified PSMs (38373 PSMs (HeLa)/5463 PSMs (enriched phospho data set)) by up to 129% and considerably outperformed all other methods (Figure 4, Supplemental Table S3). Compared to the widely used combination of Mascot and Percolator (17 916 PSMs (HeLa)/4088 PSMs (enriched phospho data set)), CharmeRT was able to identify 34–114% more PSMs and 25–62% more unique peptides. Mascot and Percolator can be additionally improved by using pParse,36 which enables the detection of mixed spectra (23 841 PSMs (HeLa)/4488 PSMs (enriched phospho data set)). Still, CharmeRT identified 22–61% more PSMs than this combination.
Compared to a single search strategy, the CharmeRT approach was able to identify 52–90% more PSMs and 23–45% more peptides. In addition, 29–36% of all validated peptides identified in the first search could be confirmed using the second search. The efficacy of Elutator was much higher for matches identified in the second search, as the spectrum quality for coeluting peptides is lower and therefore the effect of including auxiliary information used in Elutator is higher: the increase in PSMs was 17–51% for the first search and 106–149% for the second search (see Supplemental Table S3 and Supplemental Figure S8). The overall positive effect of retention time prediction appeared to be 8–15%. Notably, the RT prediction model was applied to the externally measured data sets without any additional training.
Only a minor amount of mixed spectra can be identified when the second search approach is used on phosphorylated sample. The validation through Elutator leads to 25% additionally identified PSMs in this case for the conventional single search compared to Mascot + Percolator. Chemical modifications hamper spectrum identification due to an increased combinatorial search space. However, only a small number of mixed spectra is expected in this case, as the enrichment of phosphorylated peptides with, for example, titanium dioxide (TiO2) reduces the overall complexity of the sample.
We hypothesized that the additional peptides identified in the second search correspond to lower abundant proteins, which typically are difficult to be identified in standard shotgun workflows.16,17 If this hypothesis could be confirmed, the dynamic range of mass spectrometry measurements could effectively be expanded. To validate our assumption, we used publicly available RNA expression profiles of HeLa proteins.37 High reliable peptides identified in a single raw file (data set D) with a global peptide level FDR of 1% from first and second search were used to infer 4696 protein groups (Proteome Discoverer 1.4, no additional filters).
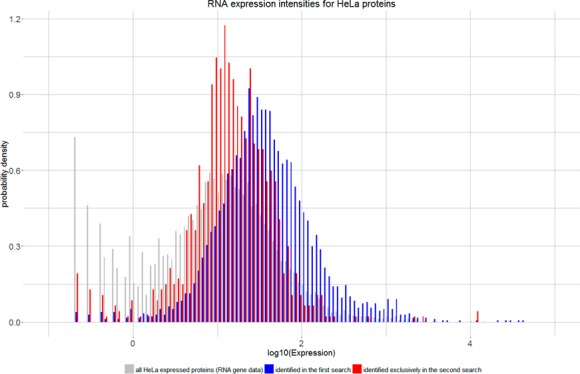
For 4435 (94%) proteins, nonzero HeLa RNA expressions were found. The remaining proteins mainly correspond to contaminant proteins or proteins absent in the RNA expression database (Supplemental Table S4). Of the expressed proteins, 885 (20%) were identified exclusively in the second search. The statistical distributions of expression levels of proteins identified in the first search and second search strongly indicate that activating second search shifts the sensitivity toward lower abundant proteins (Figure 5 and Supplemental Figure S9). As the correlation between protein and RNA abundance is only about 40%,38,39 we support this finding by additionally analyzing a publicly available spike in data set40 (see Supplemental Figure S10).
Figure 5.

Comparison of protein expression values. Proteins identified in the second search (red) correspond in a higher proportion to low abundant proteins compared to proteins already identified in the first search (blue). Overall expression values for HeLa cells (gray) have been taken from ProteinAtlas.37
Discussion
We have shown that already in experiments with narrow isolation widths (2 m/z, 1 h and 3 h gradient) a large number of chimeric spectra exists (39%), indicating that coeluting peptides are a common issue in tandem mass spectra identification. Still, chimeric spectra generally remain unconsidered, as standard peptide identification workflows stick to the one-peptide-one-spectrum approach. By combining chimeric spectra identification and appropriate validation with retention time prediction, this challenge can be turned into a major chance. We are able to identify almost up to three-times as many PSMs as compared to a standard workflow, leading to an increase of identified unique peptides of up to 63% at 1% FDR (peptide level). The CharmeRT workflow allows the use of wider isolation widths, which enable a deeper insight into measured samples. This indicates a possible expansion suitable for data-independent measurements (DIA). More importantly, CharmeRT increases the proteome coverage at unaltered acquisition time, enabling the identification of low abundant proteins at no extra cost, except for algorithmic runtime. As proteins with regulatory functions often occur at low abundance,41 identifying them is essentially important for understanding and investigating cell mechanisms. By applying CharmeRT, we are able to expand the sensitivity range of mass spectrum analysis.
Availability
CharmeRT is freely available at http://ms.imp.ac.at/?goto=charmert for Proteome Discoverer 1.4 and 2.2. A version for Proteome Discoverer 2.3 and a standalone version are currently in progress and will be available soon. In addition, a tool for training RT models on user specific in-house columns is provided.
Acknowledgments
The authors want to thank all colleagues of the Protein Chemistry Group and the Bioinformatics Research Group for their help, especially Gerhard Dürnberger for the input, Marina Knögler for the support on the software, and Georg Pirklbauer for the help on the visualization of the results. Special thanks shall be given to Johannes Stadlmann for carefully reading the manuscript and for the fruitful discussions. We also want to thank Johannes Griss for the comments on the manuscript and the Proteome Discoverer Team of Thermo Fisher Scientific, especially Carmen Paschke, Kai Fritzemeier, Torsten Ueckert, and Bernard Delanghe for their help on the PD software. This work was supported by the Austrian Science Fund (FWF) (TRP 308-N15).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00836.
All features used in Elutator to validate PSMs and peptides; correlation of theoretically calculated hydrophobicity index to the measured retention time for high confident matches of in-house HeLa and TiO2 enriched data sets; correlation of theoretically calculated hydrophobicity index to measured retention time for high confident matches of in-house and external HeLa data set; histogram of mass deviations for highly reliable identifications before and after recalibrationl longest consecutive series A + B + Y; shared ions between first and second peptides; results for data of O’Connell et al.35; protein evidence origin; presence of chimeric spectra in data sets with different isolation widths and gradient times; score distributions of MS Amanda scores for target and decoy peptides; RNA abundance of HeLa proteins; proportion of second search PSMs for spike-in data; identified PSMs and unique peptides at 1% FDR; mapping grouped proteins identified in first and second searches to RNA HeLa protein expression data; chimeric spectrum examples (PDF)
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Notes
All in-house data sets have been deposited to the ProteomeXchange Consortium via the PRIDE42 partner repository with the data set identifier PXD007750.
Supplementary Material
References
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4 (9), 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Andrews G. L.; Simons B. L.; Young J. B.; Hawkridge A. M.; Muddiman D. C. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal. Chem. 2011, 83 (13), 5442–5446. 10.1021/ac200812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20 (9), 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Kim S.; Mischerikow N.; Bandeira N.; Navarro J. D.; Wich L.; Mohammed S.; Heck A. J. R.; Pevzner P. a. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 2010, 9 (12), 2840–2852. 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3 (5), 958–964. 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 2011, 10 (4), 1785–1793. 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- Houel S.; Abernathy R.; Renganathan K.; Meyer-Arendt K.; Ahn N. G.; Old W. M. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 2010, 9 (8), 4152–4160. 10.1021/pr1003856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves G.; Ogurtsov A. Y.; Kwok S.; Wu W. W.; Wang G.; Shen R.-F.; Yu Y.-K. Detection of co-eluted peptides using database search methods. Biol. Direct 2008, 3, 27. 10.1186/1745-6150-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Pirmoradian M.; Chernobrovkin A.; Zubarev R. a. DeMix Workflow for Efficient Identification of Co-fragmented Peptides in High Resolution Data-dependent Tandem Mass Spectrometry. Mol. Cell. Proteomics 2014, 13 (11), 3211–3223. 10.1074/mcp.O114.038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D.; Mendoza L.; Hoopmann M. R.; Sun Z.; Schmidt F.; Deutsch E. W.; Moritz R. L. ReSpect: Software for Identification of High and Low Abundance Ion Species in Chimeric Tandem Mass Spectra. J. Am. Soc. Mass Spectrom. 2015, 26 (11), 1837–1847. 10.1007/s13361-015-1252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10 (4), 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Wang J.; Bourne P. E.; Bandeira N. Peptide identification by database search of mixture tandem mass spectra. Mol. Cell. Proteomics 2011, 10 (12), M111.010017. 10.1074/mcp.M111.010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Li X. J.; Ye M.; Pan S.; Schwikowski B.; Aebersold R. ProbIDtree: An automated software program capable of identifying multiple peptides from a single collision-induced dissociation spectrum collected by a tandem mass spectrometer. Proteomics 2005, 5 (16), 4096–4106. 10.1002/pmic.200401260. [DOI] [PubMed] [Google Scholar]
- Wang J.; Bourne P. E.; Bandeira N. MixGF: Spectral Probabilities for Mixture Spectra from more than One Peptide. Mol. Cell. Proteomics 2014, 13 (12), 3688–3697. 10.1074/mcp.O113.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M. C.; Yates J. R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 2013, 13 (5), 723–726. 10.1002/pmic.201200451. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen D. B.; Kelstrup C. D.; Batth T. S.; Larsen S. C.; Haldrup C.; Bramsen J. B.; Sørensen K. D.; Høyer S.; Ørntoft T. F.; Andersen C. L.; Nielsen M. L.; Olsen J. V. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4 (6), 587–599.e4. 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.-S. S.; Pinto S. M.; Getnet D.; Nirujogi R. S.; Manda S. S.; Chaerkady R.; Madugundu A. K.; Kelkar D. S.; Isserlin R.; Jain S.; Thomas J. K.; Muthusamy B.; Pamela L.-R.; Kumar P.; Sahasrabuddhe N. A.; Balakrishnan L.; Advani J.; George B.; Renuse S.; Selvan L. D.; Patil A. H.; Nanjappa V.; Radhakrishnan A.; Prasad S.; Subbannayya T.; Raju R.; Kumar M.; Sreenivasamurthy S. K.; Marimuthu A.; Sathe G. J.; Chavan S.; Datta K. K.; Subbannayya Y.; Sahu A.; Yelamanchi S. D.; Jayaram S.; Rajagopalan P.; Sharma J.; Murthy K. R.; Syed N.; Goel R.; Khan A. A.; Ahmad S.; Dey G.; Mudgal K.; Chatterjee A.; Huang T.-C. C.; Zhong J.; Wu X.; Shaw P. G.; Freed D.; Zahari M. S.; Mukherjee K. K.; Shankar S. S. K.; Mahadevan A.; Lam H.; Mitchell C. J.; Shankar S. S. K.; Satishchandra P.; Schroeder J. T.; Sirdeshmukh R.; Maitra A.; Leach S. D.; Drake C. G.; Halushka M. K.; Prasad T. S.; Hruban R. H.; Kerr C. L.; Bader G. D.; Christine I.-D.; Gowda H.; Pandey A.; Leal-Rojas P.; Kumar P.; Sahasrabuddhe N. A.; Balakrishnan L.; Advani J.; George B.; Renuse S.; Selvan L. D.; Patil A. H.; Nanjappa V.; Radhakrishnan A.; Prasad S.; Subbannayya T.; Raju R.; Kumar M.; Sreenivasamurthy S. K.; Marimuthu A.; Sathe G. J.; Chavan S.; Datta K. K.; Subbannayya Y.; Sahu A.; Yelamanchi S. D.; Jayaram S.; Rajagopalan P.; Sharma J.; Murthy K. R.; Syed N.; Goel R.; Khan A. A.; Ahmad S.; Dey G.; Mudgal K.; Chatterjee A.; Huang T.-C. C.; Zhong J.; Wu X.; Shaw P. G.; Freed D.; Zahari M. S.; Mukherjee K. K.; Shankar S. S. K.; Mahadevan A.; Lam H.; Mitchell C. J.; Shankar S. S. K.; Satishchandra P.; Schroeder J. T.; Sirdeshmukh R.; Maitra A.; Leach S. D.; Drake C. G.; Halushka M. K.; Prasad T. S.; Hruban R. H.; Kerr C. L.; Bader G. D.; Iacobuzio-Donahue C. A.; Gowda H.; Pandey A. A draft map of the human proteome. Nature 2014, 509 (7502), 575–581. 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M.; Schlegl J.; Hahne H.; Moghaddas Gholami A.; Lieberenz M.; Savitski M. M.; Ziegler E.; Butzmann L.; Gessulat S.; Marx H.; Mathieson T.; Lemeer S.; Schnatbaum K.; Reimer U.; Wenschuh H.; Mollenhauer M.; Slotta-Huspenina J.; Boese J.-H.; Bantscheff M.; Gerstmair A.; Faerber F.; Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509 (7502), 582–587. 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- Iwasaki M.; Sugiyama N.; Tanaka N.; Ishihama Y. Human proteome analysis by using reversed phase monolithic silica capillary columns with enhanced sensitivity. J. Chromatogr. A 2012, 1228, 292–297. 10.1016/j.chroma.2011.10.059. [DOI] [PubMed] [Google Scholar]
- Nagaraj N.; Kulak N. A.; Cox J.; Neuhauser N.; Mayr K.; Hoerning O.; Vorm O.; Mann M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 2012, 11 (3), M111.013722. 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P.; Mani D. R.; Ruggles K. V.; Gillette M. A.; Clauser K. R.; Wang P.; Wang X.; Qiao J. W.; Cao S.; Petralia F.; Kawaler E.; Mundt F.; Krug K.; Tu Z.; Lei J. T.; Gatza M. L.; Wilkerson M.; Perou C. M.; Yellapantula V.; Huang K.; Lin C.; McLellan M. D.; Yan P.; Davies S. R.; Townsend R. R.; Skates S. J.; Wang J.; Zhang B.; Kinsinger C. R.; Mesri M.; Rodriguez H.; Ding L.; Paulovich A. G.; Fenyö D.; Ellis M. J.; Carr S. A.; Nci C. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534 (7605), 55–62. 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yang F.; Gritsenko M. A.; Wang Y.; Clauss T.; Liu T.; Shen Y.; Monroe M. E.; Lopez-Ferrer D.; Reno T.; Moore R. J.; Klemke R. L.; Camp D. G.; Smith R. D. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11 (10), 2019–2026. 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S.; Charles P. D.; He L.; Mowlds P.; Kessler B. M.; Fischer R. Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J. Proteome Res. 2017, 16 (3), 1288–1299. 10.1021/acs.jproteome.6b00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfer V.; Pichler P.; Stranzl T.; Stadlmann J.; Taus T.; Winkler S.; Mechtler K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13 (8), 3679–3684. 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L.; Canterbury J. D.; Weston J.; Noble W. S.; MacCoss M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4 (11), 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Moruz L.; Tomazela D.; Käll L. Training, selection, and robust calibration of retention time models for targeted proteomics. J. Proteome Res. 2010, 9 (10), 5209–5216. 10.1021/pr1005058. [DOI] [PubMed] [Google Scholar]
- Krokhin O. V.; Ying S.; Cortens J. P.; Ghosh D.; Spicer V.; Ens W.; Standing K. G.; Beavis R. C.; Wilkins J. A. Use of peptide retention time prediction for protein identification by off-line reversed-phase HPLC-MALDI MS/MS. Anal. Chem. 2006, 78 (17), 6265–6269. 10.1021/ac060251b. [DOI] [PubMed] [Google Scholar]
- Gorshkov A. V.; Tarasova I. A.; Evreinov V. V.; Savitski M. M.; Nielsen M. L.; Zubarev R. A.; Gorshkov M. V. Liquid chromatography at critical conditions: Comprehensive approach to sequence-dependent retention time prediction. Anal. Chem. 2006, 78 (22), 7770–7777. 10.1021/ac060913x. [DOI] [PubMed] [Google Scholar]
- Krokhin O. V. Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: Application to 300- and 100-A pore size C18 sorbents. Anal. Chem. 2006, 78 (22), 7785–7795. 10.1021/ac060777w. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Damoc E.; Hauschild J.-P.; Lange O.; Wieghaus A.; Makarov A.; Nagaraj N.; Cox J.; Mann M.; Horning S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Mol. Cell. Proteomics 2011, 10 (9), M111.011015. 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitinger E.; Hofer M.; Köcher T.; Pichler P.; Novatchkova M.; Yang J.; Schlögelhofer P.; Mechtler K. Quantitative Phosphoproteomics of the ATM and ATR dependent DNA damage response. Mol. Cell. Proteomics 2015, 14 (3), M114.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köcher T.; Pichler P.; Swart R.; Mechtler K. Preparation of HeLa peptides for LC-MS. Protoc. Exch. 2012, 1, 2–5. 10.1038/protex.2012.001. [DOI] [Google Scholar]
- O’Connell J. D.; Paulo J. A.; O’Brien J. J.; Gygi S. P. Proteome-wide evaluation of two common protein quantification methods. J. Proteome Res. 2018, 17 (5), 1934. 10.1021/acs.jproteome.8b00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z. F.; Liu C.; Wang H. P.; Sun R. X.; Fu Y.; Zhang J. F.; Wang L. H.; Chi H.; Li Y.; Xiu L. Y.; Wang W. P.; He S. M. pParse: A method for accurate determination of monoisotopic peaks in high-resolution mass spectra. Proteomics 2012, 12 (2), 226–235. 10.1002/pmic.201100081. [DOI] [PubMed] [Google Scholar]
- Uhlén M.; Fagerberg L.; Hallström B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson Å.; Kampf C.; Sjöstedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.-K.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P.-H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Pontén F. Tissue-based map of the human proteome. Science (Washington, DC, U. S.) 2015, 347 (6220), 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vogel C.; Marcotte E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13 (4), 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa Abreu R.; Penalva L. O.; Marcotte E. M.; Vogel C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5 (12), 1512–1526. 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramus C.; Hovasse A.; Marcellin M.; Hesse A.-M.; Mouton-Barbosa E.; Bouyssié D.; Vaca S.; Carapito C.; Chaoui K.; Bruley C.; Garin J.; Cianférani S.; Ferro M.; Van Dorssaeler A.; Burlet-Schiltz O.; Schaeffer C.; Couté Y.; Gonzalez de Peredo A. Benchmarking quantitative label-free LC–MS data processing workflows using a complex spiked proteomic standard dataset. J. Proteomics 2016, 132, 51–62. 10.1016/j.jprot.2015.11.011. [DOI] [PubMed] [Google Scholar]
- Beck M.; Schmidt A.; Malmstroem J.; Claassen M.; Ori A.; Szymborska A.; Herzog F.; Rinner O.; Ellenberg J.; Aebersold R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q. W.; Wang R.; Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44 (D1), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.