

Abstract
Background
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by debilitating motor deficits, as well as autonomic problems, cognitive declines, changes in affect and sleep disturbances. Although the scientific community has performed great efforts in the study of PD, and from the most diverse points of view, the disease remains incurable. The exact mechanism underlying its progression is unclear, but oxidative stress, mitochondrial dysfunction and inflammation are thought to play major roles in the etiology.
Objective
Current pharmacological therapies for the treatment of Parkinson’s disease are mostly inadequate, and new therapeutic agents are much needed.
Methods
In this review, recent advances in computer-aided drug design for the rational design of new compounds against Parkinson disease; using methods such as Quantitative Structure-Activity Relationships (QSAR), molecular docking, molecular dynamics and pharmacophore modeling are discussed.
Results
In this review, four targets were selected: the enzyme monoamine oxidase, dopamine agonists, acetylcholine receptors, and adenosine receptors.
Conclusion
Computer aided-drug design enables the creation of theoretical models that can be used in a large database to virtually screen for and identify novel candidate molecules.
Keywords: Parkinson’s disease, QSAR, monoamine oxidase, dopamine agonists, acetylcholine receptors, and adenosine receptors
1. INTRODUCTION
Parkinson’s disease (PD) is the second most frequent neurodegenerative disorder [1, 2] after Alzheimer’s (AD). PD can cause significant disability and decreases the quality of life; clinical manifestations include tremor, rigidity, postural instability and bradykinesia [3]. As an example, PD patients carry a six-fold increased risk for dementia compared to the general population, with approximately 80% of patients developing dementia over the course of the disease [4].
Motor ability disruption in PD is due to decreased striatal dopamine levels, arising from selective and progressive loss of dopaminergic cells within the substantia nigra pars compacta and formation of α-synuclein proteinaceous intra-neuronal inclusions referred to as Lewy bodies and Lewy neurites [5, 6]. These nigrostriatal circuits are an integral part of a complex basal ganglia network and are thought to be involved in a variety of complex functions [7]. The exact mechanism underlying this process is unclear, but oxidative stress, mitochondrial dysfunction and inflammation are thought to play major roles in the etiology [8].
Non-motor symptoms can also be observed, and involve autonomic functions, sleep, cognition, mood and attention [3], these symptoms can occur across all stages of PD, and have been recognized as a key determinant factor for quality of life in PD patients [9].
Presently, there are neither medical treatments nor convincing neuroprotective agents to cure PD. Yet, there are a number of strategies that help to improve dopamine deficiency and therefore PD symptoms [7]. Treatment strategies depend on several factors, including patient disability level, age of the patient, the desire to avoid response fluctuations, potential medication side effects, and affordability [10]. Motor symptoms that result from PD may be treated with dopaminergic agents, and with functional neurosurgery, yet the currently available treatments typically fail to treat non-motor symptoms [11]. Non-motor symptoms and non-motor fluctuations can be minimized with dopaminergic treatment or with deep brain stimulation, and the dopaminergic pathophysiology of such non-motor symptoms, likely involves brain areas other than the nigrostriatal system [12].
The main strategy in the treatment of PD is dopamine replacement using carbidopa, levodopa, dopamine agonists, monoamine oxidase type B inhibitors, catechol-o-methyl-trfdansferase inhibitors, anticholinergics and amantadine [13]. Levodopa has been the therapeutic mainstay for patients with idiopathic Parkinson’s disease since the late 1960’s and continues to be the primary treatment for management of symptomatic PD [14]. On the other hand, dopamine agonists cause hallucinations, sleepiness and compulsive behaviors such as: gambling, hyper-sexuality and excessive eating [15]. Other side effects of synthetic PD medications include ankle edema, diarrhea, dry mouth, tremor, dyskinesia, cognitive impairments and urinary retraction.
When a drug therapy fails to successfully manage PD, surgical treatments are recommended. However, the surgery for PD is not devoid of risks. It has been reported that surgery may increase morbidity and mortality as a result of intracellular hemorrhages, and thermolytic lesioning of structures adjacent to the target sites [16].
Although efforts continue to study PD from the most diverse points of view, the disease remains incurable. Consequently, the major objective is to design new and more potent compounds for targets associated with PD. Many molecular modeling methods and chemical informatics techniques have been applied to differing targets in the study of PD. This review aims to examine a reasonable selection of QSAR analyses employed to develop drugs for PD, that include DA agonists, monoamine oxidase type B (MAO-B) inhibitors, levodopa or levodopa plus dopa-decarboxylase inhibitors (DDC-I) and catechol-O-methyl transferase (COMT) inhibitors.
2. DOPAMINE AGONISTS
Dopamine is an abundant neurotransmitter in the brain, and plays an important role as a regulator of many physiological functions in the central nervous system. These functions include motor activity, cognition and positive reinforcement. Additionally, in the periphery dopamine acts as a modulator of the cardiovascular and renal functions, among others [17]. In two steps that occur in the cytosol, dopamine is synthesized from the amino acid tyrosine. The first step involves hydroxylation of tyrosine to l-dihydroxyphenylanaline (l-dopa). This reaction is catalyzed by the enzyme tyrosine hydroxylase and requires oxygen. The second step is the decarboxylation of l-dopa to dopamine. This reaction is catalyzed by the aromatic amino acid enzyme decarboxylase, and generates CO2 [18].
Dopamine receptors belong to a superfamily of G-protein-coupled receptors (GPCRs) and have been subdivided into two groups based on pharmacological behavior [19]. D1 and D5 receptors are members of the D1-like family of dopamine receptors and have in common the activation of the enzyme adenylate cyclase. D2, D3 and D4 receptors are members of the D2-like family and characterized by inhibition of adenylyl cyclase [20, 21]. The D1-like group of receptors includes D1 (or D1a), D5 (D1b), D1c, and D1d [D1a (D1) and D1b (D5) these being the principal ones], while the D2-like group of receptors contains D2L, D2S, D3, and D4 (or D2aL, D2aS, D2b, and D2c) [22].
Receptors belonging to the family of GPCRs have in common a characteristic 7-transmembrane helix, each of which has 22-28 hydrophobic amino acids [20]. According to Sidhu and Niznik [23], the signal pathways involving central dopamine receptors are extremely complicated, considering that each of them can interact with more than one G protein. This leads to competitive activation in multiple directions. Dopaminergic system disorders are associated with Parkinson's disease, schizophrenia, mania and depression, among others [21, 24].
Using CoMFA (Comparative Molecular Field Analysis) [25] and CoMSIA (Comparative Molecular Similarity Indices Analysis) [26] analyses, Modi et al. [27] studied the structural requirements of D2 and D3 receptor ligands for binding affinity, and selectivity for D3 receptors (Fig. 1). The D3 receptor was chosen; considering its predominant limbic location in the central nervous system and expectations that it would cause fewer undesirable side effects [24, 28].
Fig. (1).

Molecular structures of the derivatives used in the 3D QSAR studies.
To derive the 3D QSAR models, 45 structurally diverse molecules in a dataset were selected by SAR studies focused mainly on optimization of the linker length, and the arylpiperazine moiety. After the linker length identifications and possible arylpiperazine moieties, the agonist portion of the molecule was varied. In this work, two different alignment methods, atom-based, and flexible, were tried.
Different training and test sets were used, since experimental activity varied significantly for D2, and D3; and selectivity between (D2/D3). The training sets were formed by carefully selecting 37 molecules that generated statistically significant CoMFA models. The remaining compounds (8) were used as the test set.
The best CoMFA model for D2, obtained using flexible alignment and AM1 charges, gave an rCV2 of 0.713 (4 components), a conventional r2 of 0.920, and a standard error of estimate (SEE) of 0.234. The predictive capability was rpred2 of 0.926. For the dopamine D3 receptor binding affinity, the best CoMFA model, obtained using a flexible alignment and Gasteiger–Hückel charges, gave an rcv2 of 0.453 (5 components), an rCV2 of 0.941, a SEE of 0.169 and an rpred2 of 0.710. The steric field described 41.5% and 63.6% of variance for the dopamine D2 and D3 binding affinities, respectively, and the corresponding contributions from the electrostatic field were found to be 58.5% and 36.4%, respectively. The mean rcv2 values of 0.731 and 0.472 obtained for the D2 and D3 binding affinities indicated that the derived models had good internal predictivity. The authors suggest that the greater contribution of the electrostatic field may indicate the importance of the ‘solvation–desolvation’ processes that are crucial for the observed differences in the D2/D3 receptor binding affinities.
The CoMSIA models were generated using five fields: steric, electrostatics, hydrophobic, hydrogen bond - donor and acceptor. Initially, the analyses were performed using individual fields as well as various combinations of the different fields. For D2 binding affinity, the CoMSIA model, using atom-based alignment and AM1 charges, obtained an rcv2 of 0.719 (4 components), rconv2 of 0.912, SEE of 0.245 and rpred2 of 0.911. For D3 binding affinity, the best model using flexible alignment and Gasteiger–Hückel charges obtained an rcv2 of 0.493 (6 components), an rconv2 of 0.898, a SEE of 0.227 with an rpred2 of 0.465. Removal of compound 33 (an outlier) improved the rpred2 value from 0.465 to 0.640. Once more, the derived models showed good internal predictivity considering the mean rcv2 values of 0.726 and 0.456, respectively for D2 and D3.
The CoMFA-generated plots revealed good correlation between the steric and electrostatic fields and the binding potencies at the D2, D3 receptors, and selectivity at D3 (D2/D3), with a dominating contribution made by the steric field on the electrostatic counterpart (for D3 and D2/D3), or vice versa (for D2). The models obtained revealed the importance of the carbonyl group, (which is likely involved in potential H-bonding interactions with the D3 target residues), and a biphenyl substituent, as important determinants for the D3 selectivity of the studied compounds.
Silva et al. [17] have performed 4D-QSAR analysis using 73 compounds from tetracyclic tetrahydrofuran derivatives containing a cyclic amine side chain in the 2-position (Fig. 2), and with binding affinity towards dopamine D2 receptors; related to locomotor activity [20]. From the compounds, 60 were selected from the literature [29] to be employed in the training set and the remaining 13 compounds, randomly selected from the original set, were employed as test set to validate the model. The negative logarithm of the concentration capable of inhibiting 50% of human D2L activity (pIC50 =- logIC50) was used as biological activity. The conformational ensemble profile of each ligand was constructed using molecular dynamic simulation. Calculations were done in order to model solvent effect in the absence of explicit solvent [30].
Fig. (2).

Molecular structure of the dopamine D2 receptor derivatives.
The leave-one-out cross-validation analysis of the best model resulted in a q2 value of 0.668 with a standard error of 0.263 which indicates a predictive capacity of 67% (Equation 1). The optimum number of latent variables (PLS components), used for further analysis was seven. Fig. (3) illustrates a graphic representation of the 3D-pharmacophore embedded in the 4D-QSAR model using Compound 5-((1-(4-fluorophenyl)-3-[4-[[(2R,3aR,12bS)-11-fluoro-3,3a,8,12b-tetrahydro-2H-dibenzo [3,4:6,7]cyclohepta [1,2-b]furan-2-yl]methyl]-1-piperazinyl]-1-propanone) as a reference [17].
Fig. (3).

Compound 5 with descriptors derived from the best QSAR-4D model.
pIC50 = +7.418 + 2.493(3,9,3,any) + 2.592(4,9,0,np) + 3.697(7,12,0,any) –1.261(5,7,4,any) + 1.323(2,5,4,any) – 3.028(4,6,3,np) – 4,285(5,12,3,any) –2.173(9,9,3,any) + 4.437(3,8,1,np) Eq (1)
n=60, r2 = 0.759, r2adj = 0.716, q2 = 0.668, q2adj = 0.616, LSE = 0.051, LOF = 0.098
The positive coefficients observed in Equation 1 for the grid cell occupancy descriptors (GCODs) (3,9,3,any), (4,9,0,np) (7,12,0,any), (2,5,4,any), (3,8,1,np) indicate favorable interactions between the substituent molecule and amino acid residues in the D2 active site. This indicates that substituents in these positions increase the potency of the compounds. On the other hand, the negative coefficients observed for (5,7,4,any), (4,6,3,np), (5,12,3,any), (9,9,3,any), indicate unfavorable interactions between the substituent molecule and amino acid residues in the D2 active site, which indicates that substituents in these positions will decrease the potency of the compounds.
Further, GCODs (2,5,4, any), (3,9,3,any), (4,6,3,np) and (5,7,4, any) situated near the cyclic amine, will implicate the group’s relative orientation as important for increasing/decreasing biological activity. GCODs (2,5,4, any) and (3,9,3, any) may represent interactions, such as salt bridges, between the piperazyl moiety and amino acid Asp114. GCODs (3,8,1,np) and (4,9,0,np) are related to nonpolar interactions and are located close to hydrogen atoms. In summary, hydrophobic substituents attached to the piperazyl ring contribute to increase biological activity and a hydrophilic substituent at (9,9,3,any) position will increase biological activity.
A set of 45 novel illoperidone analogs, 3-[[(aryloxy)alkyl]piperidinyl]-1,2-benzisoxazole (Fig. 4), for D2 antagonism, as selected from the literature [31], were studied by Dash et al. [32], using 3D-QSAR approach. In this study, the pharmacophore and 3D-QSAR modeling was carried out using PHASE software [33]. This software identifies common spatial arrangements between functional groups which are essential to biological activity, considering a set of high-affinity ligands. PHASE software provides a standard set of pharmacophore features: hydrogen-bond donor, hydrogen-bond acceptor, negatively ionizable, positively ionizable, hydrophobic group, and aromatic ring. The 1/logIC50 value for D2 inhibition was used as a biological activity parameter.
Fig. (4).

The structure of 3-[[(aryloxy)alkyl]piperidinyl]-1,2-benzis- oxazole derivatives.
In the first step, the conformational space of all the molecules was explored through a combination of Monte-Carlo multiple minimum/low mode sampling with maximum number of 2,500 conformers per structure and 100 minimization steps [34]. A pharmacophore model based on common molecular features was generated and validated by 3D-QSAR analysis. 45 compounds were divided into a training set (34 compounds), and a test set (11 compounds) for the purpose of atom-based 3D-QSAR.
The training set resulted in the following PLS factors (R2 = 0.925, SD = 0.045, F = 69.8, P = 6.281e-015), and the test set was characterized by the following PLS factors (Q2 = 0.756, RMSE = 0.107, Pearson-R = 0.907).
Resulting from the 3D-QSAR approach, the H-bond donor map suggested that the presence of a primary amine and hydroxyl group near the 2- and 5- positions of the phenyl ring have a favorable effect on biological activity. The hydrophobic volume maps suggested that hydrophobic interactions at the 4-position of the aromatic ring increase biological activity. The electron-withdrawing volume maps suggested that the presence of a methoxy group at the 2-position of the aromatic ring decreases biological activity, and the presence of a carbonyl group at the 4-position increases biological activity.
Active compounds were docked with the 3D structure of the D2 receptor using Glide XP docking. The results suggest that H-bond donor hydroxyl groups attached to the 2-position of the aromatic ring increase biological activity, and that hydrophobic benzisoxazole ring interactions occur with amino acids VAL79, ILE148, VAL154, PHE353, PHE354 and ILE358. Inhibitor piperidine rings display hydrophobic interaction with VAL75, TRP350 and PHE375, and inhibitor hydrophobic chains interact with aromatic residues of PHE74, TYR372, PHE375 and TYR380.
In the final step, an in silico screening search for novel D2 antagonists was performed considering all four pharmacophoric features, obtaining 4,171 hits in the Zinc database. The hits having fitness scores of less than 80% were discarded, and 119 compounds were further selected for cluster analysis. Taking into account the predicted value of 1/logIC50 greater than 0.411, 86 hits were chosen for Glide SP docking onto the active site of D2 receptor, and 11 hits, with XP GlideScore ≤ −8.5, were considered to be potential D2 inhibitors.
The sumary of dopamine agonist studies is presented in Table 1.
Table 1.
Main results obtained for dopamine agonists by computer-aided drug design methods.
| Authors | Compound | Principal Results |
|---|---|---|
| Modi et al. [27] | arylpiperazine derivatives | - ‘solvation–desolvation’ processes are crucial for the observed differences in the D2/D3 receptor binding affinities; - the carbonyl group and a biphenyl substituent, are determinants for the D3 selectivity. |
| Silva et al. [17] | tetracyclic tetrahydrofuran derivatives | - hydrophobic substituents attached to the piperazyl ring; - hydrophilic substituent at (9,9,3,any) position will increase biological activity. |
| Dash et al. [32] | Illoperidone derivatives | - primary amine and hydroxyl group near the 2- and 5- positions of the phenyl ring; - hydrophobic interactions at the 4-position of the aromatic ring and - the presence of a carbonyl group at the 4-position will increases biological activity. - a methoxy group at the 2-position of the aromatic ring decreases biological activity. |
3. MONOAMINE OXIDASE
Monoamine oxidase (MAO) is a flavo-protein, localized in the mitochondrial outer membrane, that catalyzes oxidative deamination of biogenic and xenobiotic amines. The enzyme has essential functions in the metabolism of neuro-active and vasoactive amines in the central nervous system and peripheral tissues [35]. MAO degrades dopamine excesses in the cytosol, catalyzing oxidative deamination of the dopamine amino group to 3,4-dihydroxyphenylace-taldehyde with concomitant formation of ammonia and hydrogen peroxide. The product of this reaction is then metabolized by the enzyme aldehyde dehydrogenase to 3,4-dihydroxyphenylacetic acid using NAD as an electron donator [18].
Two MAO isoforms can be found in all mammals, MAO-A and MAO-B. The difference between these isoforms is based on their respective substrate preferences, their sensitivities to the acetylenic inhibitors clorgyline and L-deprenyl (selegiline), and by their tissue distribution [35-37]. MAO-A has higher affinity for serotonin, dopamine, norepinephrine and epinephrine whereas high affinity substrates of MAO-B include tyramine, phenylethylamine and MPTP [38-40].
MAO-A, is found in the gastrointestinal tract [37] and plays a role maintaining low cytosolic concentrations of dopamine. It has been suggested that MAO-A plays a role in oxidative stress because the enzyme generates hydrogen peroxide [41]. MAO-B is found in the human brain, where it acts in the breakdown of dopamine and in deamination of phenylethylamine. This amine is responsible for stimulating the release of dopamine and inhibits its neuronal reuptake [42]. With aging, expression levels of MAO-B in neuronal tissue enhance 4-fold, increasing dopamine metabolism levels, and production of dopanal and hydrogen peroxide. The increase in hydrogen peroxide promotes apoptotic signaling events resulting in decreased levels of dopamine-producing cells, which play a key role in neurodegenerative diseases such as Parkinson’s and Alzheimer’s [43]. For these reasons, pharmacologists have focused attention on the development of MAO-B inhibitor drugs [44].
McNaught et al. [45] described the destruction of the dopamine-containing neurons of the substantia nigra pars compacta in PD, which may be related to the mechanism of action of the selective nigral toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Fig. 5). In brain, the toxin MPTP is metabolized by glial MAO-B to produce its active metabolite 1-methyl-4-phenylpyridinium (MPP+) [46, 47]. Prior to its energy-dependent concentration within mitochondria, MPP+ is actively accumulated by dopaminergic neurons via the dopamine reuptake system [47-49]. MPP+ induces cell death by selective inhibition of NADH ubiquinone reductase (complex I) and α-KGDH resulting in ATP depletion [50-52].
Fig. (5).

Mechanism of action of the selective nigral toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).
McNaught et al. [45] considering that isoquinoline derivatives are structurally related to MPTP or MPP+, and may be endogenous neurotoxins contributing to cell death in PD, studied substrate affinities of 14 neutral and quaternary isoquinoline derivatives for their ability to inhibit the uptake of [3H] dopamine into rat striatal synaptosomes in the dopamine reuptake system. Using the selected compounds, QSAR and 3D-QSAR produced no statistically acceptable model. However, certain favorable or unfavorable functional group regions helped to contribute to the molecular modeling analyses. Thus, a 2-methyl group in 1,2,3,4-tetrahydro-isoquinoline or a 7-methoxy group increased activity, whereas two -OH substituents in positions 6- and 7- were notably unfavorable for activity.
Twenty phenyl alkylamine derivatives (Fig. 6), taken from literature, all having four MAO inhibitory activities, [53], were studied by Hasegawa et al. [54] through QSAR analysis. The first biological activity was in vitro MAO inhibitory activity. Other three biological activities were in vivo MAO inhibitory activities within respective noradrener-gic (NA), dopaminergic (DA), and serotonergic (5-HT) neurons of the rat brain. The activity was expressed as the negative logarithm of the 50% inhibitory concentration (pIC50).
Fig. (6).
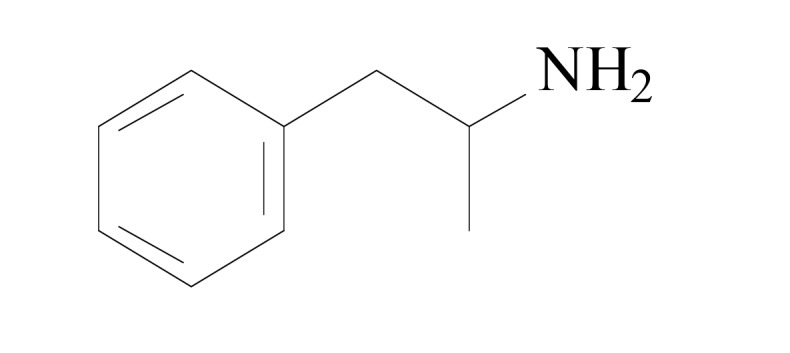
Chemical structure of phenyl alkylamines derivatives.
The relationship between MAO inhibitory activity and structural descriptors was analyzed using the nonlinear PLS method. Principal component analysis (PCA) was also employed to verify similarities and differences among the four MAO biological activities.
Two significant components were obtained when PCA analysis was employed on the dataset. The first explained 83.7% of the total variance in the conventional and cross-validated steps, and the second explained 82.1%. The loading plot of first against the second principal component showed that four biological activities were clustered into two groups, in vitro (biological variable 1) and in vivo MAO inhibitory activities (biological variables 2, 3 and 4).
Hasegawa et al. [54] performed the PLS analysis separately. The MAO in vitro activity PLS model resulted in R2 values of 0.988 and Q2 values of 0.861. The PLS models developed for the three in vivo MAO inhibitory activities resulted on R2 values of 0.863 (NA), 0.897 (DA), and 0.855 (5-HT) and Q2 values of 0.576 (NA), 0.511 (DA), and 0.595 (5-HT).
The QSAR analysis performed for the in vitro MAO inhibitory activity demonstrated that such activity is favored by large, electron-withdrawing and hydrophobic substituents at ortho positions. Meta positions were not significant. Electron-donating substituents at para positions increased biological activity. Considering the analysis performed for in vivo MAO inhibitory activity, the QSAR analyses demonstrated that electron-withdrawing substituents at ortho positions and electron-donating substituents at para positions increase biological activity. Substituents at meta positions are limited by small steric volume.
Helguera et al. [55] performed QSAR analysis to a set of over 450 different types of heterocyclic compounds, such as chromones, homo-isoflavonoids, coumarins, and their precursors (chalcones), 2-hydrazinylthiazoles, and pyrazoles. The compounds were obtained both in house [56-58], and from the literature [59-76], and then evaluated for human MAO in a single and consistent inhibition assay [77]. Considering that the data set contains stereoisomers, which cannot be distinguished by 2D descriptors, the authors discarded one. In this way, data set was formed by 449 organic compounds.
The compounds were classified into four groups considering the IC50 and pSI values, where pSI is selectivity to hMAO-B. They were divided into training and test sets to obtain validated QSAR models. The descriptors used in this work were available in the DRAGON [78], MOE [79] and MODESLAB [80] software. Linear Discriminant Analysis was employed to find classification models that best described biological activity, as a linear combination of predictor descriptors.
The three descriptor sets used in combination with the Linear Discriminant Analysis classification method revealed that DRAGON and MOE descriptors-derived models displayed higher predictive powers than those using the TOPS-MODE approach. The most frequent shape-related descriptors were associated with van der Waals volumes or areas (i.e. MATS3v, PEOE_VSA+1 and Q_VSA_FNEG), and with self-returning walk count of order 5 (SRW05). The SRW05 descriptor is related to the presence of five member rings in the chemical structure. This result is in agreement with previous SAR results that describe decreases in potency and selectivity for hMAO-B activity when simultaneous substituents are present in position 3- and 5- of the pyrazoline ring [70].
The most frequent descriptor based on the counting of atom-centered fragments was the C-019, and the descriptors based on the chemical functional groups were nArCO and nCrs, and (b_1rotN) bonds. The descriptor (C-019) describes the =CRX fragment, where R represents any group linked through carbon; X represents any electronegative atom and = represents a double bond. It was observed that this fragment is mostly included in non-selective ligands. The descriptors: nArCO representing aromatic ketones, nCrs representing the number of secondary rings C(sp3), and b_1rotN representing rotatable single bonds can be found in both selective and non-selective molecules.
The molecular descriptors: average molecular weight (AMW), Geary autocorrelation - lag 3 / weighted by atomic polarizabilities (GAT3p), and descriptors calculated from the eigenvalues of a modified distance adjacency matrix graph, weighted with partial charges (GCUT_PEOE_2) also appeared in the results with a high frequency. The authors suggest that results coming from further descriptor analyses may provide useful knowledge towards hMAO-B selective inhibitor design [81].
Pisani et al. [82] performed 3D-QSAR and docking simulations on a series of 7-metahalobenzyloxy-2H-chromen-2-one derivatives (Fig. 7), considering their rat monoamine oxidase A and B inhibition activity. The data of a series of MAO inhibitors was obtained in house [83]. The initial series, with 67 compounds, was split into a training set (58 compounds) and a test set (9 compounds), having similar coverage in terms of biological activity, range, and structural diversity. Biological activity towards MAO-B was associated via Partial Least Squares to the variation of electrostatic, steric, hydrophobic, hydrogen bond acceptor, and hydrogen bond donor fields using the Gaussian-based fields available in Phase [33].
Fig. (7).

Chemical structure of coumarin derivatives.
The obtained 3D-QSAR model revealed robust statistics for both sets: training (n = 58, r2 = 0.856, RMSE = 0.421, stability = 0.853, factors = 4, q2 = 0.605), and test (n = 9, r2ext = 0.794, RMSE = 0.457). The steric field contributes with 32.10% to the model; the hydrophobic field contributes 25.80%, the electrostatic contributes 23.30%, the hydrogen bond donor contributes 12.60% and the hydrogen bond acceptor contributes 6.20%. The existence of a larger forbidden steric region bound to position 4 decreases MAO-B inhibitory activity. Electron rich substituents at the meta position of the 7-benzyloxy substituent increase MAO-B activity, and the occurrence of hydrogen bond donor interactions increase MAO-B inhibition. From the computational analysis it was observed that most of the examined coumarin derivatives exhibited a very high and selective inhibition of MAO B.
Considering these characteristics, a new series of MAO inhibitors were designed and prepared. In this new series, substituents at position 4 were introduced with unhindered hydrophilic groups exhibiting hydrogen bond donor/acceptor properties. In the low nanomolar range these 4,7-substituted coumarin derivatives showed outstanding MOA-B selectivity for the MAO-A isoform, and for MAO-B inhibitory potencies.
The resume of monoamine oxidade inhibitors studies are presented in Table 2.
Table 2.
Main results obtained for monoamine oxidade inhibitors.
| Authors | Compound | Principal Results |
|---|---|---|
| McNaught et al. [45] | isoquinoline derivatives | - 2-methyl group and; -7-methoxy group will increase biological activity. -OH substituents in positions 6- and 7- will decrease biological activity. |
| Hasegawa et al. [54] | phenyl alkylamine derivatives | - at ortho positions, large, electron-withdrawing and hydrophobic substituents and; - at para positions, electron-donating substituents will increase biological activity. |
| Helguera et al. [55] | heterocyclic compounds, such as chromones, homo-isoflavonoids, coumarins, and their precursors | - van der Waals volumes or areas and five member rings are important to the biological activity. - substituents in position 3- and 5- of the pyrazoline ring will decrease biological activity. |
| Pisani et al. [82] | 7-metahalobenzyloxy-2H-chromen-2-one derivatives | - larger steric region bound to position 4 decreases MAO-B inhibitory activity. - electron rich substituents at the meta position of the 7-benzyloxy substituent increase MAO-B activity, -4,7-substituted showed outstanding MOA-B selectivity for the MAO-A isoform. |
4. ACETHYLCHOLINE RECEPTORS
The endogenous cholinergic neurotransmitter, acetylcholine, exerts its biological effect via two types of cholinergic receptors: muscarinic acetylcholine receptors (mAChRs), and nicotinic acetylcholine receptors (nAChRs). These two types of receptors are different in both structure and function.
The muscarinic acetylcholine receptors belong to a class I subfamily of hepta-helical, trans-membrane G-protein coupled receptors (GPCRs) and were discovered from their ability to bind to the alkaloid muscarine. Muscarinic receptor subtypes were initially classified pharmacologically as either M1 or M2 based on their differential sensitivity to pirenzepine, a selective antagonist to the M1 receptor [84]. Today, they are divided into five distinct subtypes, denoted as M1, M2, M3, M4 and M5 [85].
Muscarinic M1, M3 and M5 receptors couple preferentially to the Gq/11 subunit type of G-proteins, activating phos-pholipase C-β, and inducing a subsequent increase in intracellular calcium concentration [86]. On the other hand, M2 and M4 couple mainly to Gi/o G-proteins, and typically lead to adenylate cyclase inhibition, with activation of inward-rectifier potassium conductance [87].
Nicotinic acetylcholine receptors derive their name from their affinity for nicotine and are largely distributed both in the peripheral and central nervous systems [88]. Nicotine binds directly to the receptor α subunit and stimulates the opening of a nonspecific cation channel formed by various combinations of α2, β, γ, δ and ε subunits [84]. These nAChRs play a key role in signal transmission between cells at the nerve/muscle synapses [89] and in neurodegenerative pathologies [90, 91]. In the central nervous system, subunits α2–α7 and β2–β4 combine in a variety of different stoichiometries, resulting in the formation of receptors with distinct biological functions [92]. The α4β2 subtype is the most widespread heteromeric nAChR subtype in the central nervous system (CNS), involved in memory, drug addiction and excitement [93].
Reduction in nAChR activity is a dysfunction in a variety of neurological and psychiatric disorders such as Alzheimer’s, Parkinson’s, schizophrenia, hyperactivity, depression and even nociception [94-97]. Thus, pharmaceuticals that selectively target nAChRs might be valuable for the treatment of behavioral symptoms in PD [98].
In order to find out the chemical features modulating affinity to nAChRs, Nicolotti et al. [99] initially performed a 2-D QSAR analysis for the different congeneric series of nicotinic agonists separately. From the literature, 269 nAChR ligands were taken containing several different series namely: i) nicotine, phenylpyrrolidine, isonicotine and 3-aminomethylpyridine derivatives [100-104] (Fig. 8a), ii) (hetero)aryloxymethylazacyclic derivatives [105-107] (Fig. 8b), iii) arecolone and isoarecolone derivatives [108] (Fig. 8c), and iv) nitrogen polycyclic derivatives [100, 109-113] (Fig. 8d) with various core structures.
Fig. (8.

a). Chemical structures of (A) nicotinoid agonists, (B) phenylpyrrolidine derivatives, (C) isonicotine derivatives and (D) 3-aminomethylpyridine derivatives.
Fig. (8b).

Chemical structures of (A) pyridyloxymethylpyrrolidine derivatives, (B) pyridyloxymethylazetidine derivatives and (C) phenoxymethylazacyclic derivatives.
Fig. (8c).

Chemical structures of (A) arecolone and (B) isoarecolone derivatives.
Fig. (8d).

Chemical structures of nitrogen polycyclic derivatives.
The binding mode of the nAChR agonists was also investigated at the 3-D level by the standard CoMFA [25] procedure, and the GRID/GOLPE approach [114-116], with the lipophilic DRY probe was applied.
The 2-D QSAR analysis of the congeneric series of nicotinoid analogs showed that two main effects influence ligand binding at the nAChR. Steric effects are unfavorable for biological activity, and lipophilic effects are favorable to biological activity. These observations were found for all classes of examined compounds, and were related to the bulk parameter; Molar Refractivity (MR) and the STERIMOL parameter B5. In three cases the STERIMOL parameter B5 steric descriptor was correlated with better pKi values than MR.
The steric effects at positions 1, 3, 4 and 5, of nicotine ligands, can be quantitatively valued by comparing the negative coefficients with MR in equation 2.
pKi = 9.05 (0.64) - 1.17 (0.39) MR1’ - 1.81 (0.85) MR3’ - 1.06 (0.44) MR4’ - 0.97 (0.29) MR5’ Eq (2)
n=28 r2=0.731 (q2=0.572) s=0.480
The lipophilic effect, represented by π, can be observed with equation 3.
pKi = 10.32 (0.45) + 1.45 (0.26) π- 1.33(0.19)B5 Eq (3)
n=15 r =0.831 (q =0.713) s=0.538
The 3-D QSAR analyses allowed merging of all congeneric series with development a global model with good predictive ability. The arecolone and isoarecolone series, the 3-isoxazole derivatives, and the three non-pyrrolidine compounds were combined to form a unique molecular database. The full set of ligands was split into a training set, consisting of 206 compounds, and an evaluation set, consisting of 34 compounds. The good correlation between the binding affinities as calculated for the training set model, and those observed was obtained. Thus, proving the predictivity of the 3-D QSAR model generated. As observed in 2-D QSAR results, the models derived mostly showed the prevalent effects of steric features.
Nielsen et al. [117] synthesized six novel series of potent ligands with nanomolar affinity for the α4β2 nAChR subtype, which is the major subtype found in brain tissue. The affinities of the compounds for the α4β2 subtype of nAChRs have been investigated in vitro using [3H]cytisine binding to rat cerebral cortical membranes. The 3D-QSAR model was based on a training set of 25 compounds, and a test set composed of 4 compounds. All calculations were evaluated using the GRID [114, 118] and GOLPE [116, 119, 120] 3D-QSAR approach. The compounds were aligned using (R)-epibatidine and the conformationally restricted nicotinic analogue 29 as templates (Fig. 9).
Fig. (9).

Structures of (A) (R)-epibatidine, and (B) compound 29 employed as templates for the alignment step.
The GRID was used to calculate the interaction energies between the compounds and the four probes (OH2, C3, O-, and N1+) in order to mimic possible interactions with the receptor. The final model was obtained using only two probes: OH2 and C3, considering that the N1+ probe reduced the predictivity of the model dramatically, and the O- probe had coefficient plots similar to the plots for the OH2 probe.
The smart region definition (SRD) and the fractional factorial design (FFD) selection in GOLPE were applied to eliminate the noise variables. The SRD variable pre-selection reduced the number of variables from 15.155 to 2.169 without altering the quality of the model (Q2 = 0.390). The FFD variable selection reduced the number of variables to 983 with a highly significant improvement in the quality of the model, with Q2 from 0.38 to 0.81.
The coefficient plots for the OH2 probe and for the C3 probe showed some identical regions. The identical regions with highest negative values were located around the 6-position on the pyridine ring, and to a lesser extent around the 5-position. The negative coefficients indicate that bulk substituents in these positions reduce biological activity. The identical regions having the highest positive values are located around the protonated nitrogen. The introduction of substituents or bulky ring systems, which have unfavorable interactions with the C3 probe, increases biological activity. The coefficient plot for the OH2 probe differs from that for the C3 probe in the 6-position (and 5-position) of the pyridine ring. The results indicate that substituents with unfavorable electrostatic interactions with the water probe increase biological activity. Tønder et al. [121] published a pharmacophore model with similar results.
The resume of acetylcholine receptors studies are presented in Table 3.
Table 3.
Main results obtained for acetylcholine receptors.
| Authors | Compound | Principal Results |
|---|---|---|
| Nicolotti et al. [99] | nicotinic agonists | - steric effects are unfavorable for biological activity; - lipophilic effects are favorable to biological activity. |
| Nielsen et al. [117] | pyridines derivatives | - bulk substituents around the 6-position and to a lesser extent around the 5-position will decrease the biological activity. |
5. ADENOSINE RECEPTORS
Adenosine acts as an endogenous modulator in both the central and peripheral nervous systems by interacting with four transmembrane G protein coupled receptors (GPCRs) identified as adenosine receptors (ARs) A1, A2A, A2B, and A3 [122, 123] (Fig. 10). ARs (A1 and A3) are negatively coupled to adenylyl cyclase and exert an inhibitory effect on cyclic adenosine monophosphate (cAMP) production [123]. Adenosine A1 receptors inhibit adenylate cyclase activity. Activation of these receptors results in the opening of several types of potassium channels, and closing of certain calcium channels. The adenosine A3 receptors are not as well-understood as the others. Receptor stimulation leads to the formation of inositol triphosphate (IPA3), and consequently, to increased calcium concentration in the cell [124].
Fig. (10).

Adenosine receptors.
ARs (A2A and A2B) stimulate adenylyl cyclase activity, inducing cAMP level increases in cells [125]. Both subtypes differ in location and pharmacological properties. In the CNS, adenosine A2B receptor is widely spread, yet adenosine A2A receptors are found only in dopaminergic regions of the brain [124]. Adenosine A1 and A2A receptors are characterized by high affinity for adenosine, while A2B and A3 receptors show significantly lower affinity for adenosine.
Adenosine A2A receptors are primarily expressed in dopamine rich areas of the CNS [126], and are located on the bodies of indirect pathway medium spiny striatal neurons and dopamine terminals. Currently, connections between A2A and D2 receptors are of great interest for Parkinson’s disease (PD) treatment, which involves a decrease in dopamine levels [127].
Antagonism of AR (A2A) reduces adenosine signaling, enhances the sensitivity of the dopaminergic neurons, and restores balance to the signaling pathway controlling muscle movement. Thus, an A2A receptor antagonist may be a beneficial monotherapy for the treatment of PD and could be a very interesting target in new drug design. There has been a significant effort over the past decade to synthesize novel and selective A2A receptor antagonists, and as result, istradefylline (KW-6002) was launched under the name Nouriast® as the first antiparkinsonian agent based on A2A receptor antagonism [128].
Khanfar et al. [129] employed a genetic function algorithm (GFA) to build predictive QSAR models for a collection of 188 Adenosine A2A antagonists in order to generate differing pharmacophore binding hypotheses. The GFA method was employed to select differing combinations of pharmacophores and molecular descriptors. The phar-macophoric space of Adenosine A2A antagonists was explored through eight HYPOGEN automatic runs that were performed on seven training subsets. Compounds in the training subsets were selected considering structural diversity and a wide range of bioactivities. The training subsets were chosen considering that differences in Adenosine A2A bioactivity primarily results from the presence or absence of pharmacophoric features.
In this work, Khanfar et al. [129] implemented the genetic function algorithm as a tool for selecting differing combinations of pharmacophores and molecular descriptors. The ability of the resulting pharmacophore(s)/descriptor(s) combinations to explain biological activity variations was explored using two methodologies: (a) multiple linear regression (MLR) analysis, and (b) kNN regression.
Unfortunately, the QSAR predictive models obtained by using GFA/MLR-based QSAR analyses were statistically insignificant. In order to improve the results, kNN-based QSAR analysis was employed. This approach relies on a distance learning methodology, where the activity of an unknown member is predicted from the activity of a certain number (k) of nearest neighbors (kNNs) in the training subset. To validate the kNN-QSAR selected pharmacophores, operating characteristic curve (ROC) analysis was employed. Such analysis makes it possible to assess the ability to selectively capture diverse Adenosine A2A antagonists from a large list of decoys [130].
The successful pharmacophores were complemented with exclusion spheres to improve their ROC receiver profiles. The best QSAR models were used as 3D search queries to perform a virtual screen in the National Cancer Institute structural database, to identify novel Adenosine A2A antagonist leads. The most potent hit yielded an IC50 value of 545.7 nM.
Using 3D-QSAR, molecular dynamics, and thermodynamic analysis, Zhang et al. [131] studied the interactions of 278 monocyclic and bicyclic pyrimidine derivatives with the human A2A adenosine receptor. The compounds were classified and separated artificially into three sets, i.e., Training set I: pyrimidine and triazine derivatives (97 compounds); Training set II: pyrazolo [3,4-d]pyrimidines, pyrrolo [2,3- d]pyrimidines, triazolo [4,5-d]pyrimidines and 6-arylpurines (120 compounds); and Training set III: thieno [3,2-d]pyrimidines (61 compounds) (Fig. 11). Two kinds of alignment were performed: i) the most active compound in each dataset was considered as template and the Align-Database function in Sybyl [25] was executed, and ii) the bioactive conformations of all compounds were at once derived from docking, and then processed, using the initial method.
Fig. (11).

Structures of (A) pyrimidine and triazine derivatives (X=C/N), model I; (B) pyrazolo[3,4-d]pyrimidines, pyrrolo[2,3- d]pyrimidines, triazolo[4,5-d]pyrimidines and 6-arylpurines derivatives (X=C/N and Y=C/N), model II and (C) thieno[3,2-d]pyrimidines derivatives, model III.
Docking analysis was performed to verify binding sites of wild AR [A2A (PDB code 3PWH.pdb)] with certain mutations (PDB code 3EMS.pdb). To mimic the impacts of receptor flexibility, and water solvation effects on the ligand-receptor complex, a dynamic simulation was carried out. Analysis of the docking results showed that the binding poses for the three kinds of the derivatives maintained similar binding modes within the AR (A2A).
Interactions between ligands and the active AR (A2A) site involves polar interactions with GLU169 and ASN253 side chains, non-polar interactions with VAL84, LEU249, MET270 and ILE274, and π-stacking between aromatic moieties of the ligands and the conserved PHE168 side chain of the receptor. The docking results showed that ASN253 is capable of forming stable H-bonding with ligands. This indicates that the residue is fundamental in maintaining binding poses with different heterocyclic compounds. After docking, the energetically favorable conformations from among the compounds were selected for CoMFA and CoMSIA modeling.
Several molecular descriptors were included in the PLS analyses to derive more reasonable QSAR models. All of the statistical parameters obtained using the CoMFA and CoMSIA approaches were reasonably high, which confirms the stability and predictability of the models. Three models were generated.
The maps of model I show that bulky groups at C4 and C6 positions on the pyrimidine ring increase binding affinity. Bulky groups near substituents at C2, C5 and C6 positions decrease binding affinity. The upper region of the group at C4 is favorable for hydrophobic interaction. Interactions between the ligands may occur with ALA63, ILE66 and ILE274. Hydrophilic groups at C2 and C4 increase binding affinities. At C2, a small, electronegative and hydrophilic substituent would increase binding activity. To increase the binding affinity, a limitedly bulky group at C4 should bear both electronegativity and hydrophilic interactions but not in the role of H-bond donor. Non-H groups at C5 such as methyl would decrease inhibitory activity.
The maps of model II indicate that at C6 of the pyrimidine ring, a minor electronegative group should play a role as H-bond acceptor and increase biological activity. At C2 of the pyrimidine ring, an H-bond donating group increases biological activity as can be observed for compounds substituted with an amino group at this position. At Nitrogen at position 3 of ring B, N3, a limitedly bulky group as H-bond acceptor increases biological activity. An aromatic ring attached to the nitrogen is essential for both affinity and selectivity for A2A antagonists.
In the maps of model III at position C6 of the pyrimidine ring, electronegative, hydrophilic and limited bulky groups as H-bond donors increase inhibitory activity. At position C2 of the pyrimidine ring, a small and electronegative group as H-bond donor is well tolerated and would functionally deliver good potency. At position C2, a small lipophilic group plays an important role in AR (A2A) affinity and selectivity over AR (A1).
A series of 4-arylthieno [3, 2-d] pyrimidine derivatives was studied by Ahmed et al. [132] through QSAR analysis in order to evaluate antagonist activity towards both adenosine A1 and adenosine A2A targets (Fig. 11). Biological data of adenosine A1 and adenosine A2A for QSAR analysis was obtained from the literature [133]. The structures of 4-arylthieno [3, 2-d] pyrimidine derivatives, Fig. (12), were built using INSIGHT-II (Accelrys Software Inc., US) software. The Cerius2 package was used to calculate the molecular descriptors, which included: 2D topological, thermodynamic, structural descriptors and charge dependent descriptors.
Fig. (12).

Structures of 4-arylthieno [3, 2-d] pyrimidine derivatives.
The physicochemical screening of 4-arylthieno [3, 2-d] pyrimidine derivatives was executed using FAF-Drugs [134]. This tool performs various physicochemical calculations, identifies key functional groups, certain toxic, and unstable molecules or functional groups.
The QSAR model was generated using respectively 19 and 21 4-arylthieno [3, 2-d] pyrimidine derivative compounds as training sets for A1 and A2A inhibitors. The best models are presented in equations 4 and 5. The QSAR models were generated using the genetic function approximation (GFA). This algorithm is a useful technique for a database with a large number of descriptors and a small number of molecules. In this step, only 37 compounds were analyzed, considering their poor scalability.
A1 = 23.4694 + 0.701334 ∗ (DIPOLE MAG) − 2.7753 ∗ (CHI-V-3-P) Eq (4)
n=19, r2 =0.77, r2adj=0.752, LOF=0.808, q2=0.662.
A2A = 73.649 − 1.2009 ∗ (SC − 2) − 0.248049 ∗ (AREA) + 0.10049 ∗ (WIENER) − 0.0014297 ∗ (PHI-MAG); Eq (5)
n=21, r2 =0.936, r2adj=0.913, LOF=0.668, q2=0.881.
The predictive ability of the QSAR model was further validated with the test set containing 12 compounds for A1, and 12 compounds for A2A inhibitors. The r2 values of the A1 and A2A antagonists were above 0.7, indicating a good percentage of total variance in biological activity. The q2 > 0.6 suggested that the models will be useful in the future for meaningful predictions. Validation was done employing test sets that contained 12 compounds of A1 and A2A inhibitors. The predictive power of the model was reasonably good with predictive r2 values (0.961, 0.914), and cross validated r2 being respectively (0.912, and 0.781).
For equation 7, the DIPOLE MAG descriptor suggests that the strength and behavior of the molecule’s orientation will increase A1 inhibitory activity. The molecular connectivity index CHI-V-3-P suggests that molecular bonds, clusters, rings and flexibility are less favored for A1 inhibitory activity.
For equation 8, the topological descriptor SC-2 suggests unfavorable molecular branching for inhibition activity. The molecular surface area (AREA) describes binding, transport, and solubility for a molecule; and the negative weight suggests a less favorable inhibition of the A2A receptor. The Wiener graph–theoretical descriptor represents the sum of the chemical bonds existing between all pairs of heavy atoms in the molecule. The positive value suggests that increases in the number of heavy atom pairs in the molecules increases inhibitory potency. Finally, the negative weight of the PHI-MAG descriptor suggests less flexibility for 4-arylthieno [3, 2-d] pyrimidine derivatives in enhanced A2A receptor inhibition.
The resume of adenosine receptors studies are presented in Table 4.
Table 4.
Main results obtained for adenosine receptors by computer-aided drug design methods.
| Authors | Compound | Principal Results |
|---|---|---|
| Zhang et al. [131] | monocyclic and bicyclic pyrimidine derivatives |
Model I - at C4 on the pyrimidine ring hydrophilic and bulky groups and; - at C6 bulky groups will increase binding affinity; - upper region of the group C4 hydrophobic interaction are favorable; - at C2, a small, electronegative and hydrophilic substituent will increase binding activity. - at C5 and C6 bulky groups will decrease binding affinity; - at C5 non-H groups will decrease inhibitory activity. Model II - at C6 a minor electronegative group should play a role as H-bond acceptor and iwill ncrease biological activity; - at N3 of ring B a limitedly bulky group as H-bond acceptor will increase biological activity; - an aromatic ring attached to the nitrogen is essential for both affinity and selectivity for A2A antagonists. Model III - at C6 electronegative, hydrophilic and limited bulky groups as H-bond donors will increase inhibitory activity; - at C2 a small lipophilic group plays an important role in AR (A2A) affinity and selectivity over AR (A1). |
| Ahmed et al. [132] | 4-arylthieno[3, 2-d] pyrimidine derivatives | - the strength and behavior of the molecule’s orientation will increase A1 inhibitory activity; - molecular bonds, clusters, rings and flexibility are less favored for A1 inhibitory activity; - molecular branching will decrease the biological activity; - binding, transport, and solubility and the negative weight suggests a less favorable inhibition of the A2A receptor; - the increase in the number of heavy atom pairs will increase the biological activity; - less flexibility for the compounds will enhance A2A receptor inhibition. |
CONCLUSION
Application of computational methods is of great importance to drug discovery, and methods such as molecular docking, QSAR, pharmacophore modeling and molecular dynamics are being broadly applied in drug development in order to cure Parkinson disease. Computer aided-drug design enables the creation of theoretical models that can be used in a large database to virtually screen for and identify novel candidate molecules. The models obtained using congeneric series allow a more in-depth understanding of binding sites, and permit modifications in ligand structures that enhance receptor binding. For the 3D-QSAR the alignment step is crucuial to perform a correct study. Analysis, understanding and improvement of a pharmacophoric group can also aid in the development of new binders. However, the limited set of substituents and the not very good quality for uncommon functional groups can injure the QSAR models. Computational methods provide benefits to the drug discovery process, expanding and guiding all stages. The results presented in this review may help the development of new drugs against Parkinson's disease and promote its cure.
Consent for Publication
Not applicable.
Acknowledgements
Declared none.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.de Lau L.M.L., Breteler M.M.B. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5(6):525–535. doi: 10.1016/S1474-4422(06)70471-9. [http://dx.doi.org/ 10.1016/S1474-4422(06)70471-9]. [PMID: 16713924]. [DOI] [PubMed] [Google Scholar]
- 2.Yao S.C., Hart A.D., Terzella M.J. An evidence-based osteopathic approach to Parkinson disease. OFP. 2013;5:96–101. [Google Scholar]
- 3.Chaudhuri K.R., Healy D.G., Schapira A.H. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5(3):235–245. doi: 10.1016/S1474-4422(06)70373-8. [http://dx.doi.org/10.1016/ S1474-4422(06)70373-8]. [PMID: 16488379]. [DOI] [PubMed] [Google Scholar]
- 4.Hely M.A., Reid W.G.J., Adena M.A., Halliday G.M., Morris J.G.L. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov. Disord. 2008;23(6):837–844. doi: 10.1002/mds.21956. [http://dx.doi.org/10.1002/mds.21956]. [PMID: 18307261]. [DOI] [PubMed] [Google Scholar]
- 5.Samii A., Nutt J.G., Ransom B.R. Parkinson’s disease. Lancet. 2004;363(9423):1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [http://dx.doi.org/10.1016/S0140-6736(04)16305-8]. [PMID: 15172778]. [DOI] [PubMed] [Google Scholar]
- 6.Tan E-K., Skipper L.M. Pathogenic mutations in Parkinson disease. Hum. Mutat. 2007;28(7):641–653. doi: 10.1002/humu.20507. [http://dx.doi.org/10. 1002/humu.20507]. [PMID: 17385668]. [DOI] [PubMed] [Google Scholar]
- 7.Almeida Q.J., Hyson H.C. The evolution of pharmacological treatment for Parkinson’s disease. Recent Patents CNS Drug Discov. 2008;3(1):50–54. doi: 10.2174/157488908783421500. [http://dx.doi.org/10.2174/157488908783421500]. [PMID: 18221241]. [DOI] [PubMed] [Google Scholar]
- 8.Lim K.L., Zhang C.W. Molecular events underlying Parkinson’s disease - an interwoven tapestry. Front. Neurol. 2013;4:33. doi: 10.3389/fneur.2013.00033. [http://dx.doi.org/10.3389/fneur.2013.00033]. [PMID: 23580245]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhuri K.R., Schapira A.H. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8(5):464–474. doi: 10.1016/S1474-4422(09)70068-7. [http://dx.doi.org/10.1016/ S1474-4422(09)70068-7]. [PMID: 19375664]. [DOI] [PubMed] [Google Scholar]
- 10.Guttman M., Kish S.J., Furukawa Y. Current concepts in the diagnosis and management of Parkinson’s disease. CMAJ. 2003;168(3):293–301. [PMID: 12566335]. [PMC free article] [PubMed] [Google Scholar]
- 11.Tolosa E., Gaig C., Acevedo L. Non-motor symptom management in parkinson's. 2013.
- 12.Chung S.J., Lee J.J., Ham J.H., Ye B.S., Lee P.H., Sohn Y.H. Striatal dopamine depletion patterns and early non-motor burden in parkinsons disease. PLoS One. 2016;11(8):e0161316. doi: 10.1371/journal.pone.0161316. [http:// dx.doi.org/10.1371/journal.pone.0161316]. [PMID: 27529171]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz N.L., Waters C.H. Current strategies in the treatment of Parkinson’s disease and a personalized approach to management. Expert Rev. Neurother. 2009;9(12):1781–1789. doi: 10.1586/ern.09.117. [http://dx.doi.org/ 10.1586/ern.09.117]. [PMID: 19951137]. [DOI] [PubMed] [Google Scholar]
- 14.Müller T., Hefter H., Hueber R., Jost W.H., Leenders K.L., Odin P., Schwarz J. Is levodopa toxic? 2004. [DOI] [PubMed]
- 15.Tamminga C.A. Partial dopamine agonists in the treatment of psychosis. J. Neural Transm. (Vienna) 2002;109(3):411–420. doi: 10.1007/s007020200033. [http://dx.doi.org/10.1007/s007020200033]. [PMID: 11956961]. [DOI] [PubMed] [Google Scholar]
- 16.Obeso J.A., Guridi J., Obeso J.A., DeLong M. Surgery for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry. 1997;62(1):2–8. doi: 10.1136/jnnp.62.1.2. [http://dx.doi.org/10.1136/jnnp.62.1.2]. [PMID: 9010390]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva D.R., Ramalho T.C., Cunha E.F.F. 4D-QSAR model for compounds with binding affinity towards dopamine D2 receptors. Lett. Drug Des. Discov. 2014;11:649–664. [http://dx.doi.org/ 10.2174/1570180810666131113213448]. [Google Scholar]
- 18.Segura-Aguilar J., Paris I., Muñoz P., Ferrari E., Zecca L., Zucca F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014;129(6):898–915. doi: 10.1111/jnc.12686. [http://dx.doi.org/ 10.1111/jnc.12686]. [PMID: 24548101]. [DOI] [PubMed] [Google Scholar]
- 19.Kalani M.Y.S., Vaidehi N., Hall S.E., Trabanino R.J. Fred- dolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; God- dard III, W.A. The predicted 3D structure of the human D2 dopa- mine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA. 2004;101:3815–3820. doi: 10.1073/pnas.0400100101. [http://dx.doi.org/10.1073/pnas.0400100101]. [PMID: 14999101]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estevinho M.F., Fortunato J.S. Dopamine e receptoes. Rev. Portug Psicossomát. 2003;5:21–31. [Google Scholar]
- 21.Zhang A., Neumeyer J.L., Baldessarini R.J. Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2007;107(1):274–302. doi: 10.1021/cr050263h. [http://dx.doi.org/10.1021/ cr050263h]. [PMID: 17212477]. [DOI] [PubMed] [Google Scholar]
- 22.Weed M.R., Woolverton W.L., Paul I.A. Dopamine D1 and D2 receptor selectivities of phenyl-benzazepines in rhesus monkey striata. Eur. J. Pharmacol. 1998;361(1):129–142. doi: 10.1016/s0014-2999(98)00669-4. [http://dx.doi. org/10.1016/S0014-2999(98)00669-4]. [PMID: 9851550]. [DOI] [PubMed] [Google Scholar]
- 23.Sidhu A., Niznik H.B. Coupling of dopamine receptor subtypes to multiple and diverse G proteins. Int. J. Dev. Neurosci. 2000;18(7):669–677. doi: 10.1016/s0736-5748(00)00033-2. [http://dx.doi.org/10.1016/S0736-5748(00)00033-2]. [PMID: 10978845]. [DOI] [PubMed] [Google Scholar]
- 24.Missale C., Nash S.R., Robinson S.W., Jaber M., Caron M.G. Dopamine receptors: from structure to function. Physiol. Rev. 1998;78(1):189–225. doi: 10.1152/physrev.1998.78.1.189. [http://dx.doi.org/10.1152/physrev.1998.78. 1.189]. [PMID: 9457173]. [DOI] [PubMed] [Google Scholar]
- 25.Cramer R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110(18):5959–5967. doi: 10.1021/ja00226a005. [http://dx.doi.org/10.1021/ja00226a005]. [PMID: 22148765]. [DOI] [PubMed] [Google Scholar]
- 26.Klebe G., Abraham U., Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37(24):4130–4146. doi: 10.1021/jm00050a010. [http://dx.doi.org/10.1021/jm00050a010]. [PMID: 7990113]. [DOI] [PubMed] [Google Scholar]
- 27.Modi G., Sharma H., Kharkar P.S., Aloke K., Dutta A.K. Understanding the structural requirements of hybrid (S)-6-((2-(4-phenylpiperazin-1-yl)-ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-1-ol and its analogs as D2/D3 receptor ligands: a 3D QSAR investigation. MedChemComm. 2014;5:1384–1399. doi: 10.1039/C4MD00159A. [http://dx. doi.org/10.1039/C4MD00159A]. [PMID: 25221669]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emilien G., Maloteaux J.M., Geurts M., Hoogenberg K., Cragg S. Dopamine receptors--physiological understanding to therapeutic intervention potential. Pharmacol. Ther. 1999;84(2):133–156. doi: 10.1016/s0163-7258(99)00029-7. [http:// dx.doi.org/10.1016/S0163-7258(99)00029-7]. [PMID: 10596903]. [DOI] [PubMed] [Google Scholar]
- 29.Cid-Nunez J.M., Trabanco-Suarez A.A. 2008.
- 30.da Cunha E.F.F., Martins R.C.A., Albuquerque M.G., Alencastro R.B. LIV-3D-QSAR model for estrogen receptor ligands. J. Mol. Model. 2004;10:297–304. [http://dx.doi.org/10.1007/s00894-004-0198-5]. [Google Scholar]
- 31.Strupczewski J.T., Bordeau K.J., Chiang Y., Glamkowski E.J., Conway P.G., Corbett R., Hartman H.B., Szewczak M.R., Wilmot C.A., Helsley G.C. 3-[[(Aryloxy)alkyl]piperidinyl]-1,2-benzisoxazoles as D2/5-HT2 antagonists with potential atypical antipsychotic activity: antipsychotic profile of iloperidone (HP 873). J. Med. Chem. 1995;38(7):1119–1131. doi: 10.1021/jm00007a009. [http://dx.doi.org/10. 1021/jm00007a009]. [PMID: 7707315]. [DOI] [PubMed] [Google Scholar]
- 32.Dash R.C., Bhosale S.H., Shelke S.M., Suryawanshi M.R., Kanhed A.M., Mahadik K.R. Scaffold hopping for identification of novel D(2) antagonist based on 3D pharmacophore modelling of illoperidone analogs. Mol. Divers. 2012;16(2):367–375. doi: 10.1007/s11030-011-9349-7. [http://dx.doi.org/10.1007/s11030-011-9349-7]. [PMID: 22161148]. [DOI] [PubMed] [Google Scholar]
- 33.Phase, Version 3.1. New York, NY: Schrodinger, LLC; 2009. [Google Scholar]
- 34.Kolossvary I., Guida W.C. Low mode search: an efficient, automated computational method for conformational analysis- application to cyclic and acyclic alkanes and cyclic peptides. J. Am. Chem. Soc. 1996;118:5011–5019. [http://dx.doi.org/10.1021/ ja952478m]. [Google Scholar]
- 35.Elmer L.W., Bertoni J.M. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin. Pharmacother. 2008;9(16):2759–2772. doi: 10.1517/14656566.9.16.2759. [http://dx.doi.org/ 10.1517/14656566.9.16.2759]. [PMID: 18937611]. [DOI] [PubMed] [Google Scholar]
- 36.Youdim M.B., Edmondson D., Tipton K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006;7(4):295–309. doi: 10.1038/nrn1883. [http://dx.doi.org/10.1038/nrn1883]. [PMID: 16552415]. [DOI] [PubMed] [Google Scholar]
- 37.Chen J.J., Swope D.M., Dashtipour K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007;29(9):1825–1849. doi: 10.1016/j.clinthera.2007.09.021. [http://dx.doi.org/10.1016/j.clinthera.2007.09.021]. [PMID: 18035186]. [DOI] [PubMed] [Google Scholar]
- 38.Shih J.C. Molecular basis of human MAO A and B. Neuropsychopharmacology. 1991;4(1):1–7. [PMID: 2003865]. [PubMed] [Google Scholar]
- 39.Strolin B.M., Dostert P., Tipton K.F. Developmental aspects of the monoamine-degrading enzyme monoamine oxidase. Dev. Pharmacol. Ther. 1992;18(3-4):191–200. [PMID: 1306808]. [PubMed] [Google Scholar]
- 40.Cases O., Seif I., Grimsby J., Gaspar P., Chen K., Pournin S., Müller U., Aguet M., Babinet C., Shih J.C., De-Maeyer E. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268(5218):1763–1766. doi: 10.1126/science.7792602. [http://dx.doi.org/10.1126/science.7792602]. [PMID: 7792602]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinreb O., Amit T., Riederer P., Youdim M.B., Mandel S.A. Neuroprotective profile of the multitarget drug rasagiline in Parkinson’s disease. Int. Rev. Neurobiol. 2011;100:127–149. doi: 10.1016/B978-0-12-386467-3.00007-8. [http://dx.doi.org/10.1016/B978-0-12-386467-3.00007-8]. [PMID: 21971006]. [DOI] [PubMed] [Google Scholar]
- 42.Chen J.J., Swope D.M. Pharmacotherapy for Parkinson’s disease. Pharmacotherapy. 2007;27(12 Pt 2):161S–173S. doi: 10.1592/phco.27.12part2.161S. [http://dx. doi.org/10.1592/phco.27.12part2.161S]. [PMID: 18041936]. [DOI] [PubMed] [Google Scholar]
- 43.Mallajosyula J.K., Kaur D., Chinta S.J., Rajagopalan S., Rane A., Nicholls D.G., Di Monte D.A., Macarthur H., Andersen J.K. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One. 2008;3(2):e1616. doi: 10.1371/journal.pone.0001616. [http://dx.doi.org/10. 1371/journal.pone.0001616]. [PMID: 18286173]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solís-García del Pozo J., Mínguez-Mínguez S., de Groot P.W., Jordán J. Rasagiline meta-analysis: a spotlight on clinical safety and adverse events when treating Parkinson’s disease. Expert Opin. Drug Saf. 2013;12(4):479–486. doi: 10.1517/14740338.2013.790956. [http://dx.doi.org/10.1517/ 14740338.2013.790956]. [PMID: 23634791]. [DOI] [PubMed] [Google Scholar]
- 45.McNaught K.S., Thull U., Carrupt P.A., Altomare C., Cellamare S., Carotti A., Testa B., Jenner P., Marsden C.D. Inhibition of [3H]dopamine uptake into striatal synaptosomes by isoquinoline derivatives structurally related to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem. Pharmacol. 1996;52(1):29–34. doi: 10.1016/0006-2952(96)00133-5. [http://dx.doi.org/10.1016/0006-2952(96)00133-5]. [PMID: 8678905]. [DOI] [PubMed] [Google Scholar]
- 46.Chiba K., Trevor A., Castagnoli N., Jr Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem. Biophys. Res. Commun. 1984;120(2):574–578. doi: 10.1016/0006-291x(84)91293-2. [http:// dx.doi.org/10.1016/0006-291X(84)91293-2]. [PMID: 6428396]. [DOI] [PubMed] [Google Scholar]
- 47.Przedborski S., Vila M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003;991:189–198. [http://dx.doi.org/10.1111/j.1749-6632.2003.tb07476.x]. [PMID: 12846987]. [PubMed] [Google Scholar]
- 48.Javitch J.A., D’Amato R.J., Strittmatter S.M., Snyder S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA. 1985;82(7):2173–2177. doi: 10.1073/pnas.82.7.2173. [http://dx.doi. org/10.1073/pnas.82.7.2173]. [PMID: 3872460]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramsay R.R., Salach J.I., Dadgar J., Singer T.P. Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism. Biochem. Biophys. Res. Commun. 1986;135(1):269–275. doi: 10.1016/0006-291x(86)90972-1. [http:// dx.doi.org/10.1016/0006-291X(86)90972-1]. [PMID: 3485428]. [DOI] [PubMed] [Google Scholar]
- 50.Di Monte D., Jewel S.A., Ekstrom G. Sandy, M.S.; Smith, M.T. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 1-methyl-4-phenylpyridine (MPP’) causes A TP depletion in isolated hepatocytes. Biochem. Biophys. Res. Commun. 1986;137:310–315. doi: 10.1016/0006-291x(86)91211-8. [http://dx.doi.org/10.1016/0006-291X(86)91211-8]. [PMID: 3487319]. [DOI] [PubMed] [Google Scholar]
- 51.Mizuno Y., Saitoh T., Sone N. Inhibition of mitochondrial alpha-ketoglutarate dehydrogenase by 1-methyl-4-phenylpyridinium ion. Biochem. Biophys. Res. Commun. 1987;143(3):971–976. doi: 10.1016/0006-291x(87)90346-9. [http:// dx.doi.org/10.1016/0006-291X(87)90346-9]. [PMID: 3494449]. [DOI] [PubMed] [Google Scholar]
- 52.Nicklas W.J., Vyas I., Heikkila R.E. Inhibition of NADH- linked oxidation in brain mitochondria by 1-methyl-4-phenylpyridine, a metabolite of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [http://dx.doi.org/10.1016/0024-3205 (85)90146-8]. [PMID: 2861548]. [DOI] [PubMed] [Google Scholar]
- 53.Norinder U., Florvall L., Ross S.B. A PLS quantitative structure- activity relationship study of some monoamine oxidase inhibitors of the phenyl alkylamine type. Eur. J. Med. Chem. 1994;29:191–195. [http://dx.doi.org/10.1016/0223-5234(94)90037-X]. [Google Scholar]
- 54.Hasegawa K., Kimura T., Miyashita Y., Funatsu K. Nonlinear partial least squares modeling of phenyl alkylamines with the monoamine oxidase inhibitory activities. J. Chem. Inf. Comput. Sci. 1996;36(5):1025–1029. doi: 10.1021/ci960362j. [http://dx.doi.org/10.1021/ci960362j]. [PMID: 8831142]. [DOI] [PubMed] [Google Scholar]
- 55.Helguera A.M., Pérez-Garrido A., Gaspar A., Reis J., Cagide F., Vina D., Cordeiro M.N.D.S., Borges F. Combining QSAR classification models for predictive modeling of human monoamine oxidase inhibitors. Eur. J. Med. Chem. 2013;59:75–90. doi: 10.1016/j.ejmech.2012.10.035. [http:// dx.doi.org/10.1016/j.ejmech.2012.10.035]. [PMID: 23207409]. [DOI] [PubMed] [Google Scholar]
- 56.Alcaro S., Gaspar A., Ortuso F., Milhazes N., Orallo F., Uriarte E., Yáñez M., Borges F. Chromone-2- and -3-carboxylic acids inhibit differently monoamine oxidases A and B. Bioorg. Med. Chem. Lett. 2010;20(9):2709–2712. doi: 10.1016/j.bmcl.2010.03.081. [http://dx.doi.org/10.1016/j. bmcl.2010.03.081]. [PMID: 20382016]. [DOI] [PubMed] [Google Scholar]
- 57.Gaspar A., Reis J., Fonseca A., Milhazes N., Viña D., Uriarte E., Borges F. Chromone 3-phenylcarboxamides as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2011;21(2):707–709. doi: 10.1016/j.bmcl.2010.11.128. [http://dx.doi.org/10.1016/j.bmcl.2010.11.128]. [PMID: 21194943]. [DOI] [PubMed] [Google Scholar]
- 58.Gaspar A., Silva T., Yáñez M., Vina D., Orallo F., Ortuso F., Uriarte E., Alcaro S., Borges F. Chromone, a privileged scaffold for the development of monoamine oxidase inhibitors. J. Med. Chem. 2011;54(14):5165–5173. doi: 10.1021/jm2004267. [http://dx.doi.org/10.1021/ jm2004267]. [PMID: 21696156]. [DOI] [PubMed] [Google Scholar]
- 59.Santana L., González-Díaz H., Quezada E., Uriarte E., Yáñez M., Viña D., Orallo F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J. Med. Chem. 2008;51(21):6740–6751. doi: 10.1021/jm800656v. [http://dx.doi. org/10.1021/jm800656v]. [PMID: 18834112]. [DOI] [PubMed] [Google Scholar]
- 60.Matos M.J., Viña D., Picciau C., Orallo F., Santana L., Uriarte E. Synthesis and evaluation of 6-methyl-3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009;19(17):5053–5055. doi: 10.1016/j.bmcl.2009.07.039. [http://dx.doi.org/10.1016/j.bmcl.2009. 07.039]. [PMID: 19628387]. [DOI] [PubMed] [Google Scholar]
- 61.Matos M.J., Viña D., Quezada E., Picciau C., Delogu G., Orallo F., Santana L., Uriarte E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009;19(12):3268–3270. doi: 10.1016/j.bmcl.2009.04.085. [http://dx.doi. org/10.1016/j.bmcl.2009.04.085]. [PMID: 19423346]. [DOI] [PubMed] [Google Scholar]
- 62.Chimenti F., Fioravanti R., Bolasco A., Chimenti P., Secci D., Rossi F., Yáñez M., Orallo F., Ortuso F., Alcaro S. Chalcones: a valid scaffold for monoamine oxidases inhibitors. J. Med. Chem. 2009;52(9):2818–2824. doi: 10.1021/jm801590u. [http://dx.doi.org/10.1021/jm801590u]. [PMID: 19378991]. [DOI] [PubMed] [Google Scholar]
- 63.Chimenti F., Secci D., Bolasco A., Chimenti P., Bizzarri B., Granese A., Carradori S., Yáñez M., Orallo F., Ortuso F., Alcaro S. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009;52(7):1935–1942. doi: 10.1021/jm801496u. [http://dx.doi.org/10.1021/jm801496u]. [PMID: 19267475]. [DOI] [PubMed] [Google Scholar]
- 64.Chimenti F., Carradori S., Secci D., Bolasco A., Chimenti P., Granese A., Bizzarri B. Synthesis and biological evaluation of novel conjugated coumarin-thiazole systems. J. Heterocycl. Chem. 2009;46:575–578. [http://dx.doi.org/10.1002/jhet.110]. [Google Scholar]
- 65.Chimenti F., Secci D., Bolasco A., Chimenti P., Granese A., Carradori S., Maccioni E., Cardia M.C., Yáñez M., Orallo F., Alcaro S., Ortuso F., Cirilli R., Ferretti R., Distinto S., Kirchmair J., Langer T. Synthesis, semipreparative HPLC separation, biological evaluation, and 3D-QSAR of hydrazothiazole derivatives as human monoamine oxidase B inhibitors. Bioorg. Med. Chem. 2010;18(14):5063–5070. doi: 10.1016/j.bmc.2010.05.070. [http://dx.doi.org/10.1016/ j.bmc.2010.05.070]. [PMID: 20579890]. [DOI] [PubMed] [Google Scholar]
- 66.Chimenti F., Secci D., Bolasco A., Chimenti P., Granese A., Carradori S., Yáñez M., Orallo F., Sanna M.L., Gallinella B., Cirilli R. Synthesis, stereochemical separation, and biological evaluation of selective inhibitors of human MAO-B: 1-(4-arylthiazol-2-yl)-2-(3-methylcyclohexylidene)hydrazines. J. Med. Chem. 2010;53(17):6516–6520. doi: 10.1021/jm100120s. [http://dx.doi.org/10.1021/ jm100120s]. [PMID: 20715818]. [DOI] [PubMed] [Google Scholar]
- 67.Chimenti F., Secci D., Bolasco A., Chimenti P., Granese A., Carradori S., D’Ascenzio M., Yáñez M., Orallo F. Synthesis and selective inhibition of human monoamine oxidases of a large scaffold of (4,5-substituted-thiazol-2-yl)hydrazones. MedChemComm. 2010;1:61–72. [http://dx.doi.org/10.1039/c0md00014k]. [Google Scholar]
- 68.Chimenti F., Bolasco A., Secci D., Chimenti P., Granese A., Carradori S., Yáñez M., Orallo F., Ortuso F., Alcaro S. Investigations on the 2-thiazolylhydrazyne scaffold: synthesis and molecular modeling of selective human monoamine oxidase inhibitors. Bioorg. Med. Chem. 2010;18(15):5715–5723. doi: 10.1016/j.bmc.2010.06.007. [http:// dx.doi.org/10.1016/j.bmc.2010.06.007]. [PMID: 20615716]. [DOI] [PubMed] [Google Scholar]
- 69.Chimenti F., Carradori S., Secci D., Bolasco A., Bizzarri B., Chimenti P., Granese A., Yáñez M., Orallo F. Synthesis and inhibitory activity against human monoamine oxidase of N1-thiocarbamoyl-3,5-di(hetero)aryl-4,5-dihydro-(1H)-pyrazole derivatives. Eur. J. Med. Chem. 2010;45(2):800–804. doi: 10.1016/j.ejmech.2009.11.003. [http://dx. doi.org/10.1016/j.ejmech.2009.11.003]. [PMID: 19926363]. [DOI] [PubMed] [Google Scholar]
- 70.Chimenti F., Fioravanti R., Bolasco A., Chimenti P., Secci D., Rossi F., Yáñez M., Orallo F., Ortuso F., Alcaro S., Cirilli R., Ferretti R., Sanna M.L. A new series of flavones, thioflavones, and flavanones as selective monoamine oxidase-B inhibitors. Bioorg. Med. Chem. 2010;18(3):1273–1279. doi: 10.1016/j.bmc.2009.12.029. [http://dx.doi.org/ 10.1016/j.bmc.2009.12.029]. [PMID: 20045650]. [DOI] [PubMed] [Google Scholar]
- 71.Fioravanti R., Bolasco A., Manna F., Rossi F., Orallo F., Yáñez M., Vitali A., Ortuso F., Alcaro S. Synthesis and molecular modelling studies of prenylated pyrazolines as MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2010;20(22):6479–6482. doi: 10.1016/j.bmcl.2010.09.061. [http://dx. doi.org/10.1016/j.bmcl.2010.09.061]. [PMID: 20934874]. [DOI] [PubMed] [Google Scholar]
- 72.Maccioni E., Alcaro S., Orallo F., Cardia M.C., Distinto S., Costa G., Yanez M., Sanna M.L., Vigo S., Meleddu R., Secci D. Synthesis of new 3-aryl-4,5-dihydropyrazole-1-carbothioamide derivatives. An investigation on their ability to inhibit monoamine oxidase. Eur. J. Med. Chem. 2010;45(10):4490–4498. doi: 10.1016/j.ejmech.2010.07.009. [http://dx. doi.org/10.1016/j.ejmech.2010.07.009]. [PMID: 20702005]. [DOI] [PubMed] [Google Scholar]
- 73.Matos M.J., Viña D., Janeiro P., Borges F., Santana L., Uriarte E. New halogenated 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2010;20(17):5157–5160. doi: 10.1016/j.bmcl.2010.07.013. [http://dx.doi.org/10.1016/j.bmcl.2010.07.013]. [PMID: 20659799]. [DOI] [PubMed] [Google Scholar]
- 74.Vergel N.E., López J.L., Orallo F., Viña D., Buitrago D.M., del Olmo E., Mico J.A., Guerrero M.F. Antidepressant-like profile and MAO-A inhibitory activity of 4-propyl-2H-benzo[h]-chromen-2-one. Life Sci. 2010;86(21-22):819–824. doi: 10.1016/j.lfs.2010.04.001. [http://dx.doi.org/ 10.1016/j.lfs.2010.04.001]. [PMID: 20394758]. [DOI] [PubMed] [Google Scholar]
- 75.Delogu G., Picciau C., Ferino G., Quezada E., Podda G., Uriarte E., Viña D. Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur. J. Med. Chem. 2011;46(4):1147–1152. doi: 10.1016/j.ejmech.2011.01.033. [http://dx.doi.org/10.1016/j.ejmech.2011.01.033]. [PMID: 21316817]. [DOI] [PubMed] [Google Scholar]
- 76.Desideri N., Bolasco A., Fioravanti R., Monaco L.P., Orallo F., Yáñez M., Ortuso F., Alcaro S. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J. Med. Chem. 2011;54(7):2155–2164. doi: 10.1021/jm1013709. [http://dx. doi.org/10.1021/jm1013709]. [PMID: 21405131]. [DOI] [PubMed] [Google Scholar]
- 77.Yáñez M., Fraiz N., Cano E., Orallo F. Inhibitory effects of cis- and trans-resveratrol on noradrenaline and 5-hydroxytryptamine uptake and on monoamine oxidase activity. Biochem. Biophys. Res. Commun. 2006;344(2):688–695. doi: 10.1016/j.bbrc.2006.03.190. [http://dx.doi.org/10.1016/ j.bbrc.2006.03.190]. [PMID: 16631124]. [DOI] [PubMed] [Google Scholar]
- 78.2006 www.talete.mi.it
- 79.Chemical Computing Group . 2008. [Google Scholar]
- 80.Gutierrez Y.E.E. 2002. [Google Scholar]
- 81.Helguera A.M., Combes R.D., González M.P., Cordeiro M.N. Applications of 2D descriptors in drug design: a DRAGON tale. Curr. Top. Med. Chem. 2008;8(18):1628–1655. doi: 10.2174/156802608786786598. [http://dx.doi.org/ 10.2174/156802608786786598]. [PMID: 19075771]. [DOI] [PubMed] [Google Scholar]
- 82.Pisani L., Farina R., Nicolotti O., Gadaleta D., Soto-Otero R., Catto M., Di Braccio M., Mendez-Alvarez E., Carotti A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015;89:98–105. doi: 10.1016/j.ejmech.2014.10.029. [http://dx.doi.org/10.1016/j.ejmech.2014.10.029]. [PMID: 25462230]. [DOI] [PubMed] [Google Scholar]
- 83.Pisani L., Catto M., Nicolotti O., Grossi G., Di Braccio M., Soto-Otero R., Mendez-Alvarez E., Stefanachi A., Gadaleta D., Carotti A. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2013;70:723–739. doi: 10.1016/j.ejmech.2013.09.034. [http://dx.doi.org/10.1016/j.ejmech.2013.09.034]. [PMID: 24231308]. [DOI] [PubMed] [Google Scholar]
- 84.Felder C.C. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 1995;9(8):619–625. [http:// dx.doi.org/10.1096/fasebj.9.8.7768353]. [PMID: 7768353]. [PubMed] [Google Scholar]
- 85.Ishii M., Kurachi Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006;12(28):3573–3581. doi: 10.2174/138161206778522056. [http://dx.doi.org/10.2174/ 138161206778522056]. [PMID: 17073660]. [DOI] [PubMed] [Google Scholar]
- 86.Nahorski S.R., Tobin A.B., Willars G.B. Muscarinic M3 receptor coupling and regulation. Life Sci. 1997;60(13-14):1039–1045. doi: 10.1016/s0024-3205(97)00045-3. [http:// dx.doi.org/10.1016/S0024-3205(97)00045-3]. [PMID: 9121345]. [DOI] [PubMed] [Google Scholar]
- 87.Volpicelli L.A., Levey A.I. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. 2004. [DOI] [PubMed] [Google Scholar]
- 88.Holladay M.W., Lebold S.A., Lin N-H. Drug Dev. Res. 1995;35:191. [http://dx.doi.org/10.1002/ddr.430350402]. [Google Scholar]
- 89.McDonald I.A., Cosford N., Vernier J.M. Nicotinic acetylcholine receptors: Molecular biology, chemistry and pharmacology. Annu. Rep. Med. Chem. 1995;5:41–50. [http://dx.doi.org/10.1016/S0065-7743(08)60918-5]. [Google Scholar]
- 90.Shacka J.J., Robinson S.E. Central and peripheral anatomy of nicotinic sites. Med. Chem. Res. 1996;6:444–464. [Google Scholar]
- 91.Zhang J., Xue F., Liu Y., Yang H., Wang X. The structural mechanism of the Cys-loop receptor desensitization. Mol. Neurobiol. 2013;48(1):97–108. doi: 10.1007/s12035-013-8420-z. [http://dx.doi.org/10.1007/s12035-013-8420-z]. [PMID: 23397136]. [DOI] [PubMed] [Google Scholar]
- 92.Tønder J.E., Olesen P.H. Agonists at the α4β2 nicotinic acetylcholine receptors relationships and molecular modeling. Curr. Med. Chem. 2001;8:651–674. doi: 10.2174/0929867013373165. [http://dx.doi.org/10.2174/ 0929867013373165]. [PMID: 11281847]. [DOI] [PubMed] [Google Scholar]
- 93.Mazzo F., Pistillo F., Grazioso G., Clementi F., Borgese N., Gotti C., Colombo S.F. Nicotine-modulated subunit stoichiometry affects stability and trafficking of α3β4 nicotinic receptor. J. Neurosci. 2013;33(30):12316–12328. doi: 10.1523/JNEUROSCI.2393-13.2013. [http://dx.doi.org/10.1523/ JNEUROSCI.2393-13.2013]. [PMID: 23884938]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sirviö J., Rinne J.O., Valjakka A., Rinne U.K., Riekkinen P.J., Paljärvi L. Different forms of brain acetylcholinesterase and muscarinic binding in Parkinson’s disease. J. Neurol. Sci. 1989;90(1):23–32. doi: 10.1016/0022-510x(89)90042-7. [http://dx.doi.org/10.1016/0022-510X(89)90042-7]. [PMID: 2723671]. [DOI] [PubMed] [Google Scholar]
- 95.Gohlke H., Schwarz S., Gündisch D., Tilotta M.C., Weber A., Wegge T., Seitz G. 3D QSAR analyses-guided rational design of novel ligands for the (α4)2(β2)3 nicotinic acetylcholine receptor. J. Med. Chem. 2003;46(11):2031–2048. doi: 10.1021/jm020859m. [http://dx.doi.org/10.1021/ jm020859m]. [PMID: 12747776]. [DOI] [PubMed] [Google Scholar]
- 96.Cassels B.K., Bermúdez I., Dajas F., Abin-Carriquiry J.A., Wonnacott S. From ligand design to therapeutic efficacy: the challenge for nicotinic receptor research. Drug Discov. Today. 2005;10(23-24):1657–1665. doi: 10.1016/S1359-6446(05)03665-2. [http://dx.doi.org/10.1016/S1359-6446(05)03665-2]. [PMID: 16376826]. [DOI] [PubMed] [Google Scholar]
- 97.Osswald J., Rellán S., Gago A., Vasconcelos V. Toxicology and detection methods of the alkaloid neurotoxin produced by cyanobacteria, anatoxin-a. Environ. Int. 2007;33(8):1070–1089. doi: 10.1016/j.envint.2007.06.003. [http://dx.doi.org/10.1016/j.envint.2007.06.003]. [PMID: 17673293]. [DOI] [PubMed] [Google Scholar]
- 98.Aarsland D., Hutchinson M., Larsen J.P. Cognitive, psychiatric and motor response to galantamine in Parkinson’s disease with dementia. Int. J. Geriatr. Psychiatry. 2003;18(10):937–941. doi: 10.1002/gps.949. [http:// dx.doi.org/10.1002/gps.949]. [PMID: 14533126]. [DOI] [PubMed] [Google Scholar]
- 99.Nicolotti O., Pellegrini-Calace M., Altomar C., Carotti A., Carrieri A., Sanz F. Ligands of neuronal nicotinic acetylcholine receptor (nAChR): inferences from the Hansch and 3-D quantitative structure-activity relationship (QSAR) Models. Curr. Med. Chem. 2002;9(1):1–29. doi: 10.2174/0929867023371463. [http://dx.doi.org/10.2174/ 0929867023371463]. [PMID: 11864064]. [DOI] [PubMed] [Google Scholar]
- 100.Damaj M.I., Glassco W., Dukat M., May E.L., Glennon R.A., Martin B.R. Pharmacology of novel nicotinic analogs. Drug Dev. Res. 1996;38:177–187. [http://dx.doi.org/10.1002/(SICI)1098-2299(199607/08)38:3/4<177:AID-DDR6>3.0.CO;2-J]. [Google Scholar]
- 101.Glennon R.A., Herndon J.L., Dukat M. Epibatidine-aided studies toward definition of a nicotine receptor pharmacophore. Med. Chem. Res. 1994;4:461–473. [Google Scholar]
- 102.Lin N-H., Carrera G.M., Jr, Anderson D.J. Synthesis and evaluation of nicotine analogs as neuronal nicotinic acetylcholine receptor ligands. J. Med. Chem. 1994;37(21):3542–3553. doi: 10.1021/jm00047a012. [http:// dx.doi.org/10.1021/jm00047a012]. [PMID: 7932582]. [DOI] [PubMed] [Google Scholar]
- 103.Cosford N.D.P., Bleicher L., Herbaut A., McCallum J.S., Vernier J.M., Dawson H., Whitten J.P., Adams P., Chavez-Noriega L., Correa L.D., Crona J.H., Mahaffy L.S., Menzaghi F., Rao T.S., Reid R., Sacaan A.I., Santori E., Stauderman K.A., Whelan K., Lloyd G.K., McDonald I.A. (S)-(-)-5-ethynyl-3-(1-methyl-2-pyrrolidinyl)pyridine maleate (SIB-1508Y): a novel anti-parkinsonian agent with selectivity for neuronal nicotinic acetylcholine receptors. J. Med. Chem. 1996;39(17):3235–3237. doi: 10.1021/jm960328w. [http://dx.doi.org/10.1021/jm960328w]. [PMID: 8765504]. [DOI] [PubMed] [Google Scholar]
- 104.Dukat M., Dowd M., Damaj M.I., Martin B., El-Zahabi M.A., Glennon R.A. Synthesis, receptor binding and QSAR studies on 6-substituted nicotine derivatives as cholinergic ligands. Eur. J. Med. Chem. 1999;34:31–40. [http://dx.doi.org/10.1016/S0223-5234(99) 80038-5]. [Google Scholar]
- 105.Lin N.H., Gunn D.E., Li Y., He Y., Bai H., Ryther K.B., Kuntzweiler T., Donnelly-Roberts D.L., Anderson D.J., Campbell J.E., Sullivan J.P., Arneric S.P., Holladay M.W., Holladay M.W. Synthesis and structure-activity relationships of pyridine-modified analogs of 3-[2-((S)-pyrrolidinyl)methoxy] pyridine, A-84543, a potent nicotinic acetylcholine receptor agonist. Bioorg. Med. Chem. Lett. 1998;8(3):249–254. doi: 10.1016/s0960-894x(98)00019-5. [http://dx. doi.org/10.1016/S0960-894X(98)00019-5]. [PMID: 9871663]. [DOI] [PubMed] [Google Scholar]
- 106.Holladay M.W., Bai H., Li Y., Lin N-H., Daanen J.F., Ryther K.B., Wasicak J.T., Kincaid J.F., He Y., Hettinger A.M., Huang P., Anderson D.J., Bannon A.W., Buckley M.J., Campbell J.E., Donnelly-Roberts D.L., Gunther K.L., Kim D.J.B., Kuntzweiler T.A., Sullivan J.P., Decker M.W., Arneric S.P. Structure-activity studies related to ABT-594, a potent nonopioid analgesic agent: effect of pyridine and azetidine ring substitutions on nicotinic acetylcholine receptor binding affinity and analgesic activity in mice. Bioorg. Med. Chem. Lett. 1998;8(19):2797–2802. doi: 10.1016/s0960-894x(98)00504-6. [http://dx. doi.org/10.1016/S0960-894X(98)00504-6]. [PMID: 9873625]. [DOI] [PubMed] [Google Scholar]
- 107.Elliott R.L., Kopecka H., Gunn D.E., Lin N., Garvey D.S., Ryther K.B., Holladay M.W., Anderson D.J., Campbell J.E., Sullivan J.P., Buckley M.J., Gunther K.L., O’Neill A.B., Decker M.W., Arneric S.P. 2-(Aryloxymethyl) azacyclic analogues as novel nicotinic acetylcholine receptor (neuronal nicotinic AChR) ligands. Bioorg. Med. Chem. Lett. 1996;6:2283–2288. [http://dx.doi.org/10.1016/0960-894X(96)00416-7]. [Google Scholar]
- 108.Ward J.S., Merritt L., Bymaster F.P., Calligaro D.O. Isoarecolone and arecolones - selective central nicotinic agonists that cross the blood-brain barrier. Bioorg. Med. Chem. Lett. 1994;4:573–578. [http://dx.doi.org/10.1016/S0960-894X(01)80157-8]. [Google Scholar]
- 109.Thomas P., Brough P.A., Gallagher T., Wonnacott S. Alkyl-modified side-chain variants of anatoxin-a - a series of potent nicotinic agonists. Drug Dev. Res. 1994;31:147–156. [http://dx. doi.org/10.1002/ddr.430310210]. [Google Scholar]
- 110.Kanne D.B., Abood L.G. Synthesis and biological characterization of pyridohomotropanes. Structure-activity relationships of conformationally restricted nicotinoids. J. Med. Chem. 1988;31(3):506–509. doi: 10.1021/jm00398a004. [http://dx.doi.org/10.1021/jm00398a004]. [PMID: 3346870]. [DOI] [PubMed] [Google Scholar]
- 111.Canu B.C., Sparatore F. Synthesis and preliminary phar-macological evaluation of some cytisine derivatives. Farmaco. 1999;54(7):438–451. doi: 10.1016/s0014-827x(99)00049-x. [http://dx.doi.org/10.1016/S0014-827X(99) 00049-X]. [PMID: 10486911]. [DOI] [PubMed] [Google Scholar]
- 112.Dukat M., Fiedler M., Dumas D., Damaj I., Martin B.R., Rosecrans J.A., James J.R., Glennon R.A. Pyrrolidine-modified and 6-substituted analogs of nicotine: A structure-affinity investigation. Eur. J. Med. Chem. 1996;31:875–888. [http://dx.doi.org/10.1016/ S0223-5234(97)89850-9]. [Google Scholar]
- 113.Badio B., Daly J.W. Epibatidine, a potent analgetic and nicotinic agonist. Mol. Pharmacol. 1994;45(4):563–569. [PMID: 8183234]. [PubMed] [Google Scholar]
- 114.Goodford P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985;28(7):849–857. doi: 10.1021/jm00145a002. [http://dx. doi.org/10.1021/jm00145a002]. [PMID: 3892003]. [DOI] [PubMed] [Google Scholar]
- 115.Goodford P. 1998. [Google Scholar]
- 116.Cruciani G., Watson K.A. Comparative molecular field analysis using GRID force-field and GOLPE variable selection methods in a study of inhibitors of glycogen phosphorylase b. J. Med. Chem. 1994;37(16):2589–2601. doi: 10.1021/jm00042a012. [http://dx.doi.org/10.1021/jm00042a012]. [PMID: 8057302]. [DOI] [PubMed] [Google Scholar]
- 117.Nielsen S.F., Nielsen E.Ø., Olsen G.M., Liljefors T., Peters D. Novel potent ligands for the central nicotinic acetylcholine receptor: synthesis, receptor binding, and 3D-QSAR analysis. J. Med. Chem. 2000;43(11):2217–2226. doi: 10.1021/jm990973d. [http://dx.doi.org/10.1021/ jm990973d]. [PMID: 10841800]. [DOI] [PubMed] [Google Scholar]
- 118.GRID Molecular Discovery Ltd. Oxford, England: 1998. [Google Scholar]
- 119.Baroni M., Constantino G., Cruciani G., Riganelli D., Valigi R., Clementi S. Generating Optimal Linear PLS Estimations (GOLPE): An Advanced Chemometric Tool for Handling 3D- QSAR Problems. Quant. Struct.-. Act. Relat. 1993;12:9–20. [http://dx.doi.org/10.1002/qsar.19930120103]. [Google Scholar]
- 120.1999.
- 121.Tønder J.E., Hansen J.B., Begtrup M., Pettersson I., Rimvall K., Christensen B., Ehrbar U., Olesen P.H. Improving the nicotinic pharmacophore with a series of (Isoxazole)methylene-1-azacyclic compounds: synthesis, structure-activity relationship, and molecular modeling. J. Med. Chem. 1999;42(24):4970–4980. doi: 10.1021/jm9910627. [http://dx.doi.org/10.1021/jm9910627]. [PMID: 10585207]. [DOI] [PubMed] [Google Scholar]
- 122.Klinger M., Freissmuth M., Nanoff C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell. Signal. 2002;14(2):99–108. doi: 10.1016/s0898-6568(01)00235-2. [http://dx.doi.org/10.1016/S0898-6568(01)00235-2]. [PMID: 11781133]. [DOI] [PubMed] [Google Scholar]
- 123.Fredholm B.B.A.P. I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol. Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [http://dx.doi.org/10.1124/pr.110. 003285]. [PMID: 21303899]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Romanowska M., Komoszyński M. Adenosine--neurotransmitter and neuromodulator in the central nervous system. Postepy Biochem. 2002;48(3):230–238. [PMID: 12625251]. [PubMed] [Google Scholar]
- 125.Chen J.F., Eltzschig H.K., Fredholm B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013;12(4):265–286. doi: 10.1038/nrd3955. [http://dx.doi.org/10.1038/nrd3955]. [PMID: 23535933]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schiffmann S.N., Fisone G., Moresco R., Cunha R.A., Ferré S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007;83(5):277–292. doi: 10.1016/j.pneurobio.2007.05.001. [http://dx.doi.org/10.1016/ j.pneurobio.2007.05.001]. [PMID: 17646043]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Antonini A., Poewe W. Adenosine A2A receptor antagonists in Parkinson’s disease: still in the running. Lancet Neurol. 2014;13(8):748–749. doi: 10.1016/S1474-4422(14)70153-X. [http://dx.doi.org/10.1016/S1474-4422(14)70153-X]. [PMID: 25008550]. [DOI] [PubMed] [Google Scholar]
- 128.LeWitt P.A., Guttman M., Tetrud J.W., Tuite P.J., Mori A., Chaikin P., Sussman N.M. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008;63(3):295–302. doi: 10.1002/ana.21315. [http://dx. doi.org/10.1002/ana.21315]. [PMID: 18306243]. [DOI] [PubMed] [Google Scholar]
- 129.Khanfar M.A., Al-Qtaishat S., Habash M., Taha M.O. Discovery of potent adenosine A2a antagonists as potential anti-Parkinson disease agents. Non-linear QSAR analyses integrated with pharmacophore modeling. Chem. Biol. Interact. 2016;254:93–101. doi: 10.1016/j.cbi.2016.05.023. [http://dx.doi.org/10.1016/j.cbi.2016.05.023]. [PMID: 27216633]. [DOI] [PubMed] [Google Scholar]
- 130.Verdonk M.L., Berdini V., Hartshorn M.J., Mooij W.T., Murray C.W., Taylor R.D., Watson P. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004;44(3):793–806. doi: 10.1021/ci034289q. [http://dx.doi.org/10.1021/ ci034289q]. [PMID: 15154744]. [DOI] [PubMed] [Google Scholar]
- 131.Zhang L., Liu T., Wang X., Wang J., Li G., Li Y., Yang L., Wang Y. Insight into the binding mode and the structural features of the pyrimidine derivatives as human A2A adenosine receptor antagonists. Biosystems. 2014;115:13–22. doi: 10.1016/j.biosystems.2013.04.003. [http://dx.doi.org/ 10.1016/j.biosystems.2013.04.003]. [PMID: 23665268]. [DOI] [PubMed] [Google Scholar]
- 132.Ahmed S.S.S.J., Ahameethunisa A., Santosh W. QSAR and pharmacophore modeling of 4-arylthieno[3, 2-d] pyrimidine derivatives agaianst adenosine receptor of parkinson’s disease. J. Theor. Comput. Chem. 2010;9:975–991. [http://dx.doi.org/10.1142/ S0219633610006146]. [Google Scholar]
- 133.Roger J., Ian A., Claire E., Colin T., Paul R., Allan M.R., Gaur K.S., Paul R., Allan M., Antony R., Lawrence A., Lerpiniere J., Misra A., Robert M., Richard S., Upton R., Scott M., Douglas S. Bioorg. Antagonists of the human adenosine A2A receptor. Part 2: Design and synthesis of 4-arylthieno[3,2-d]pyrimidine derivatives. Med. Chem. Lett. 2008;18:2920–2923. doi: 10.1016/j.bmcl.2008.03.076. [http://dx.doi.org/10.1016/ j.bmcl.2008.03.076]. [DOI] [PubMed] [Google Scholar]
- 134.Lagorce D., Sperandio O., Galons H., Miteva M.A., Villoutreix B.O. FAF-Drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinformatics. 2008;9:396. doi: 10.1186/1471-2105-9-396. [http://dx.doi.org/10.1186/1471-2105-9-396]. [PMID: 18816385]. [DOI] [PMC free article] [PubMed] [Google Scholar]