

Abstract
Background
Neurodegenerative diseases such as Alzheimer's disease (AD), amyotrophic lateral sclerosis, Parkinson's disease (PD), spinal cerebellar ataxias, and spinal and bulbar muscular atrophy are described by slow and selective degeneration of neurons and axons in the central nervous system (CNS) and constitute one of the major challenges of modern medicine. Computer-aided or in silico drug design methods have matured into powerful tools for reducing the number of ligands that should be screened in experimental assays.
Methods
In the present review, the authors provide a basic background about neurodegenerative diseases and in silico techniques in the drug research. Furthermore, they review the various in silico studies reported against various targets in neurodegenerative diseases, including homology modeling, molecular docking, virtual high-throughput screening, quantitative structure activity relationship (QSAR), hologram quantitative structure activity relationship (HQSAR), 3D pharmacophore mapping, proteochemometrics modeling (PCM), fingerprints, fragment-based drug discovery, Monte Carlo simulation, molecular dynamic (MD) simulation, quantum-mechanical methods for drug design, support vector machines, and machine learning approaches.
Results
Detailed analysis of the recently reported case studies revealed that the majority of them use a sequential combination of ligand and structure-based virtual screening techniques, with particular focus on pharmacophore models and the docking approach.
Conclusion
Neurodegenerative diseases have a multifactorial pathoetiological origin, so scientists have become persuaded that a multi-target therapeutic strategy aimed at the simultaneous targeting of multiple proteins (and therefore etiologies) involved in the development of a disease is recommended in future.
Keywords: Neurodegenerative diseases, cheminformatics, chemometrics, in silico drug discovery and design, virtual screening, virtual docking, QSAR, MD
1. Introduction
Neurodegenerative diseases (NDDs), termed ‘protein-misfolding disorders’, are a heterogeneous group of disorders that are described by profound loss of neurons and distinct involvement of functional systems defining clinical presentations. Comprehensive neuropathological, molecular genetic and biochemical assessments suggested that proteins with modified physical and chemical properties are deposited in the human brain but also in peripheral organs as a fundamental phenomenon in many forms of NDDs [1]. According to this, a physiological protein triggers structural conformational changes, which can result in the loss of function or altered function, aggregation and intra- or extra- neuronal accumulation of amyloid fibrils. The ubiquitin- proteasome pathway and the autophagy-lysosome system, oxidative stress response proteins and chaperone network are protein elimination pathways that contribute to controling the quality of cellular components and serve to maintain proteostasis of the cell. These pathways have high impact on the pathogenesis of NDDs. Impaired mitochondrial function and oxidative damage, dysregulated bioenergetics and DNA oxidation, neuroinflammation, dysregulation of ion homeostasis and cellular/axonal transport defects are related to the formation of toxic forms of NDD-related proteins [2]. Classification of NDDs is based on the correlation of clinical symptoms with neuropathology, anatomical distribution of neuronal loss and cell types affected, conformationally changed proteins, and etiology. Clinical–anatomical classification of neurodegenerative disorders, which is useful mostly when clinical symptoms and signs are early diagnosed, is as follows: (1) Cognitive dysfunction as early symptom, dementia and alteration in high-order brain functions that are closely related to involvement of the hippocampus, entorhinal cortex, limbic system (amygdala, olfactory cortices, anterior cingulate cortex, subcortical structures) and neocortical areas; (2) Movement disorders in which the most important anatomical regions involved are the motor cortical areas, lower motor neurons of the spinal cord, basal ganglia, brainstem nuclei, thalamus, cerebellar cortex and nuclei; and (3) Combinations of these symptoms that form early during the clinical course [3]. Neuropathological-based classification relies on the assessment of the anatomical distribution of neuronal loss, and histopathological hallmarks like spongiform change in the neuropil, or vascular lesions, and the distinction of intracellular and extracellular protein accumulations. The conformationally changed proteins involved in the majority of sporadic and genetic adult-onset NDDs are as follows: α-synuclein (α-syn), an abundant brain protein of 140 residues that belongs to a family of three closely related proteins (α-, β- and γ-syn); transactive-response (TAR) DNA-binding protein 43 (TDP-43), a nuclear protein with 414 aa which is ubiquitously expressed in non-central nervous system in nearly all tissues; the microtubule-associated protein tau (MAPT) with pivotal role for the assembly of tubulin into microtubules and stabilization of microtubules; amyloid β-peptide (Aβ); PrP, a 253 aa protein involved in prion diseases or transmissible spongiform encephalopathies; the fused-in-sarcoma (FUS), Ewing’s sarcoma RNA-binding protein 1 (EWSR1) and TATA-binding protein-associated factor 15 (TAF15) proteins, belong to the FET (FUS, EWS and TAF15) family of DNA/RNA binding proteins [4]. Protein aggregation in the nervous system can be deposited intracellularly including tau, α-syn, TDP-43, FUS/FET proteins, and those related to trinucleotide repeat expansion or rare hereditary diseases and extracellularly consisting mainly of Aβ or PrP. Alzheimer's disease (AD) is described by the existence of extracellular amyloid plaques and by the intraneuronal aggregates of hyperphosphorylated and misfolded tau protein. Lewy body (LB)-associated disorders, including Parkinson disease (PD) and dementia with Lewy bodies (DLB) exhibit intraneuronal cytoplasmic and neuritic inclusions, whereas multiple system atrophy (MSA), a sporadic, adult-onset degenerative movement disorder of unknown cause, is described by α-syn–positive glial cytoplasmic and rare neuronal inclusions. Tauopathies are a spectrum of neurodegenerative disorders which are classified into three categories (neuronal, mixed neuronal/ glial and glial predominant) in view of the cellular distribution of tau pathology. Neuronal tau deposition is an important feature of AD, Pick disease (PiD), neurofibrillary tangle (NFT)-dementia or primary age-related tauopathy (PART) and frontotemporal lobar degenerations (FTLD). Argyrophilic grain disease (AGD), progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) show both neuronal and glial tau aggregates, while globular glial tauopathies (GGT) are characterized by the presence of glial tau pathologies [5, 6]. Prion diseases make up a group of rare fatal neurological disorders with various etiologies, including sporadic (Creutzfeldt-Jakob disease (CJD)), genetic (genetic CJD, Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI)), and acquired (kuru, variant CJD, and iatrogenic CJD) forms [7]. TDP-43 is a major protein component of the ubiquitin-immunoreactive inclusions in the pathologic condition of sporadic and familial FTLD with ubiquitin-positive and sporadic amyotrophic lateral sclerosis (ALS). FUS is a multifunctional RNA-binding protein whose mutations in FUS could cause familial ALS and a rare group of diseases with FTLD. ALS and spinal muscular atrophy (SMA) are among the group of neurodegenerative disorders that essentially influence the spinal cord, in which the most severe lesions are observed in the anterior portion of the spinal cord. Non-neuronal deposits are also seen in the posterior part of the spinal cord in Friedreich ataxia (FRDA), the most frequent hereditary ataxia, which is described by the early loss of the large sensory neurons of the spinal cord and cardiomyopathy [8, 9]. Besides these obvious NDDs, there is one group of neurological disorders such as Tourette syndrome, essential tremor, torsion dystonia, and schizophrenia that are often viewed as neurodegenerative in light of their chronic course and obscure etiopathogenesis, but they do not show any apparent structural abnormalities. All neurodegenerative disorders of cerebellum are related to ataxia and can readily be classified into three main types based on the loci of pathology: pontocerebellar atrophy, FRDA, and cerebellar cortical atrophy. Huntingtons disease (HD), spinocerebellar ataxias, FRDA, Kennedy´s disease (SBMA), dentirubral-pallidoluysian atrophy are a group of genetic diseases caused by expansion mutations of trinucleotide repeats. PD, DLB, MSA (P and C) fall into the category of the synucleinopathies. GSS, FFI, sporadic fatal insomnia (FSI), Kuru, CJD - sporadic, iatrogenic, familial, and new variant assigned to the class of the prion diseases. FTLD and Parkinsonism dementia, PSP, CBD, PiD, and AGD fit into the spectrum of the tauopathies [10].
It is challenging to develop new drugs because drug discovery process is very time and resources consuming processes. However, the processes of drug discovery have been changed with the assistance of powerful computers and information technology to expedite drug discovery, lead optimization, drug development and design. In biomedical field, computer-aided or in silico design which uses computational techniques in drug discovery process is being used to streamline and accelerate hit identification and hit-to-lead optimization process [11]. Methods employed in computer-aided drug design (CADD) can be broadly break down into two general categories: structure-based and ligand-based. When the target protein's structure is known, structure-based CADD is generally favored especially for soluble proteins that can readily be crystallized. However, when there is no information on the structure of the target, ligand-based CADD is applied based on information of known active and inactive compounds through chemical similarity searches or construction of predictive, quantitative structure-activity relationship (QSAR) models [12, 13]. Several computational techniques have been proposed to recognize and select therapeutic relevant targets, study the molecular basis of drug-receptor complexes interactions, structurally characterize ligand binding sites on biological targets, design de novo target-specific compound libraries, predict target protein structure, identify hit compound by ligand- and structure-based virtual screening, estimate binding free energy between a ligand and receptor, and optimize high-affinity ligands, all of which can be used to rationalize and improve the productivity, speed, and cost-efficiency of the drug discovery process [14].
2. Protein Structure Determination
In order to use structure-based drug design tools, structural information about the target can be determined by experimental techniques including X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. In the situations where an experimental structure of a target is not available, computational methods like homology modeling can be utilized to anticipate the three-dimensional (3D) structures of targets [15]. Homology modeling, also known as comparative modeling of protein, is a popular computational structure prediction method for modeling of 3D structure of a protein using the known structure of other proteins as a template that possess sequences similar to the amino acid sequences of a target protein. Since amino acid sequence specifies protein unique 3D shape and structure manages biochemical function, structures that share sequence identity to the target sequence of interest, can contribute to determining the target structure, function and even possible binding and functional sites of the structure [16]. In bioinformatics, NCBI Basic Local Alignment Search Tool (BLAST) is an algorithm that performs comparisons between pairs of sequences, searching for regions of local similarity. The model building of a target structure is performed by comparative modeling algorithms in light of the comparison with the information derived from homologous sequences with known structures, and then the models built are evaluated and refined [17, 18]. In this review, we provided an overview of several successful instances of reported homology modeling studies used to predict target structures that are essential in neurodegenerative disorders.
Lee and Kim [19] investigated human Catecholamine-O-methyltransferase (COMT) for designing anti-PD drug by using the ligand docking and comparative homology modeling. COMT is an S-adenosylmethionine (SAM, AdoMet) dependent methyltransferase, which is associated with the functions of dopamine and epinephrine in several mental processes, including PD. 3D structure of human COMT (hCOMT) was built by comparative modeling approach using MODELLER based on x-ray crystal structure of rat COMT (rCOMT) as a template protein for homology modeling. Ligand docking study was then performed using AutoDock for complex of hCOMT and fifteen molecules which were collected as COMT inhibitors from world patent. AutoDock revealed that among the fifteen inhibitors which included catechol ring, nine inhibitor binding models were energetically favorable (-6.3 to -8.2 kcal.mol-1). From the analysis of binding model, authors deduced that Arg201 and Cys173 on hCOMT play critical roles in the interaction with COMT inhibitors.
Homology modeling using Geno3D, SWISS-MODEL, and MODELLER 9v7 was applied by Dhanavade et al. [20] to constructed a 3D structure of cysteine protease from bacterial source Xanthomonas campestris. The model comparison between cysteine protease from X. campestris structure and human cathepsin B (CB) showed that active site pocket forming residues of cysteine protease are almost identical to the active site residues of human CB. The predicted cysteine protease structure was then utilized for docking of the patch of Aβ peptide using AutoDock 4.2. The results revealed that the hydrogen atom of sulfhydryl group of active site residue Cys17 of cysteine protease enzyme forms hydrogen bonding interactions with backbone carboxyl oxygen atoms of Lys16 and Leu17 of Aβ peptide, hence authors concluded that it might play a role in Aβ peptide cleavage as a new therapeutic strategy for the treatment of AD patients. Then, molecular dynamics simulations studies were implemented to confirm the stable behavior of the complex of cysteine protease and patch of Aβ peptide over the entire simulation period.
Conforti et al. [21] conducted a structure-based virtual screening (SBVS) study to identify huntingtin (HTT) mimetics, a group of small organic molecules that can disrupt the complex formation between paired amphipathic helix 1 (PAH1) hydrophobic cleft of mSIN3 molecules and repressor element 1 silencing transcription factor (REST). The structure of REST in complex with mSIN3a-PAH1 domain was obtained from the NMR structure of the mSIN3b-PAH1 complex with the REST fragment by homology modeling using MOE software. Starting from the filtration of ZINC database (7 million molecules) by redundancy, drug likeness and diversity one million compounds were obtained, which further subjected to a consensus docking approach including two docking software programs, MOE and Autodock4. Finally, after force field refinement procedure, secondary docking with the AutoDock 4 package and redocking using both MOE and AutoDock 4, 94 compounds were selected to inhibit complex formation with a binding energy less than -6.5 kcal.mol-1, an arbitrarily chosen threshold. In the primary screening, 94 compounds selected by the virtual screening (VS) approach were examined in DiaNRSELuc8 cell line and quinolone-like compound 91 (C91) at a non-toxic nanomolar concentration was chosen and assessed in neural stem cell lines (NS) carrying the mutant huntingtin gene. At the non-toxic concentration of 250 nM, C91 had ability to reduce the silencing activity of RE1/NRSE in luciferase reporter assays by a RE1/NRSEBDNF-LUC (DiaNRSELuc8) construct. C91 was bound to the PAH1 hydrophobic cleft of mSIN3 through hydrophobic interactions between the 2-fluorobenzyl moiety and the Phe36 side chain (Fig. 1). In conclusion, authors demonstrated that, combining VS approaches to in vitro and in vivo experiments can lead to compounds inhibiting the PAH1-REST interaction, which might be helpful in HD and in other pathological conditions.
Fig. (1).

2D structure and predicted binding mode for C91 (highlighted in green).
Among glutamate-gated ion channels (iGluRs), N-methyl-D-aspartate receptors (NMDARs) are Ca2+ favoring glutamate-gated cation channels that their abnormal expression and deficiency have been associated with chronic neurodegenerative disorders like AD, PD, and HD. The allosteric modulation of N-terminal domain (NTD) of subunit NR2B of NMDARs by endogenous allosteric modulator like endogenous Zn2+ or by synthetic compounds like ifenprodil have a pivotal role in pathology by modulating pain processing. Marinelli et al. [22] presented a reliable 3D model of the NR2B–ifenprodil complex using homology modeling, which provided important clues for the development of NR2B selective antagonists. Docking calculations were used to define the ifenprodil binding mode at an atomic level and completely clarify all the accessible structure–activity relationships. Furthermore, MD simulations along with Molecular Mechanic/ Poisson-Boltzmann Surface Area (MM-PBSA) analysis were used to gain insight into the ifenprodil mechanism of action to find whether it binds and stabilizes an open or a closed conformation of the NR2B modulatory domain. The results revealed that the closed conformation of the R1-R2 domain instead of the open, forms the high affinity binding pocket for ifenprodil, so the closed conformation of the R1-R2 domain was considered for rational design and/ or for VS experiments (Fig. 2).
Fig. (2).

2D structure of ifenprodil and its binding modes in the a) open and b) closed conformation of R1-R2 modulatory domain.
3. Binding Pocket Identification
After determining the protein’s 3D structure by experimental or computational approaches, discovering binding pockets on that protein is an essential next stage in structure-based drug discovery which can be determined using a variety of computational binding pocket identifying algorithms. Binding pocket predicting algorithms can be classified into two main groups; geometry-based and energy-based methods [23]. One of the energy-based binding site identification servers is Q-SiteFinder [24]. As a geometry-based algorithm fpocket [25] program can be applied.
Pinhiero et al. [26] selected a set of 40 inhibitor molecules from the database BindingDB and executed a prediction of ligand binding site of beta-site amyloid precursor protein cleaving enzyme 1 (BACE-1) using the Q-SiteFinder webserver, which has a method based on purely energetic criterion: calculating van der Waals interaction energy of a methyl group with the submitted protein. Then, molecular docking using AutoDock Vina software and molecular interactions analyses were carried out to propose the binding mode of the inhibitors with the enzyme. 32 highly active compounds (with the lowest Ki values: 0.017 nM to 2.0 nM) were selected for pharmacophore perception calculation using the web server PharmaGist [17], which detects the pharmacophoric groups by multiple and flexible alignment of the ligands. New proposals based on molecular changes applied into the structure of the compound CID_46 888954 (binding affinity of -10.7 kcal.mol-1) together with a pharmacophore modeling as well as biological activity and synthetic accessibility predictions were made (Fig. 3).
Fig. (3).

Proposals of molecular changes to the inhibitor CID_46888954.
Pathak et al. [27] performed cheminformatics and molecular docking studies using Autodock Tools 4.2 on a series of 15 different cholinesterase inhibitors (ChEIs) to compare their inhibitory activity against acetylcholinesterase (AChE). This approach helped to determine the affinity of the interaction, mode of binding and to understand the selectivity of drug molecule for the treatment of AD. Q-SiteFinder was employed to evaluate the catalytic binding site of AChE, which uses the interaction energy between the protein and a simple imaginary van der Waals probe to retrieve energetically favourable binding sites. Docking results based on this kind of comparison revealed that huperzine A with inhibition constant of 0.009 μM is the best drug to treat AD patients among the 15 drugs available in market (Fig. 4).
Fig. (4).

2D structure of Huperzine A.
Glycogen synthase kinase 3 (GSK-3), a serine/threonine protein kinase, is one of the critical signaling molecules that regulates a number of prominent intracellular signaling pathways, which has a more instigative role in the etiology of AD as major signaling link between Aβ and tau pathology. Palomo et al. [28] performed a search for the druggable active sites on GSK-3 surface susceptible to small-molecule modulation using the free geometry-based algorithm fpocket and hpocket programs. Authors discovered four new potential allosteric sites on GSK-3 that could be utilized for future rational drug design and development of small molecule modulators as future therapies for NDDs where GSK-3 is up-regulated. Furthermore, they carried out the docking simulation taking into account the whole protein surface to find the preferential binding site of the quinoline derivative VP0.7 and to explore if it matches with any of fpocket’s results (Fig. 5). The predicted binding site matched with pocket no. 7 reported by hpocket with highly similar binding modes. Docking results proposed a change in the activation loop of the GSK-3 came about because of allosteric binding of VP0.7 (IC50 value on GSK-3 of 3.01 ± 0.14 μM) to the enzyme.
Fig. (5).

Chemical structure of the quinoline derivative VP0.7 and seven cavities found by hpocket.
The second most common chronic progressive neurological disorder, PD, is caused by death of dopaminergic neurons in the substantia nigra and other pigmented brainstem nuclei like the locus coeruleus. Dopamine receptor D3 (DRD3) serves as a therapeutic target for drugs used for the treatment of PD and schizophrenia because of less serious side effects and significant level of neuro-protection. According to the study performed by Usman Mirza and coworkers [29], 40 active phytochemicals against PD were retrieved from literature search and docked with DRD3 using AutoDock and AutoDockVina to find potent lead compounds. The binding residues of DRD3 were explored by using Computed Atlas of Surface Topography of Proteins (CASTp) server and Pocket Finder. CASTp gives an extensive and itemized quantitative identification and measurements of interior inaccessible cavities and surface accessible pockets of proteins, which are prominent concave regions on 3D structures of proteins and are frequently related with binding events. The docking results with phytochemicals showed that Thr369, Tyr373, Asp110, and Ile183 are likely target sites for designing drugs against PD. It is also concluded that glycyrrhetinic acid (binding energy of −12.7 kcal.mol-1) and E.resveratroloside (binding energy of −11.7 kcal.mol-1) as potential plant derived compounds can be evaluated as a template for future drug development against PD (Fig. 6).
Fig. (6).

2D structures of a) glycyrrhetinic acid and b) E.resveratroloside.
Dysfunction of Neuronal nicotinic receptors (nAChRs) has been implicated in a number of diseases and disorders such as mild cognitive impairment (MCI), attention deficit hyperactivity disorder (ADHD), AD, PD, schizophrenia, nicotine addiction, pain, and cancer. Mahasenan et al. [30] carried out a hierarchical structure-based VS protocol to discover novel chemotypes that can serve as new templates/scaffolds for the development of subtype selective nAChR antagonists. Human α4β2 nAChR extracellular domain homology models were built in an iterative fashion with MODELLER 9v1 based on PDB IDs: 1UW6, 2BYR, 2BJ0, and 2QC1. The VS template was prepared by docking the agonist epibatidine to 25 receptor conformation as extracted from a 5 ns molecular dynamics (MD) simulation. The putative ligand binding subpockets at the α4/β2 interface were screened using the SiteMap module of the Schrödinger. They identified compounds with similar scaffolds in the top hits; so, ligands with diverse chemotypes were chosen for in vitro test to obtain structurally diverse molecules for further examination. Eleven predicted active compounds and three negative control compounds were chosen for experimental assay based on structural diversity, binding pocket location, and standard error of the scoring results. Out of the eleven in silico hits tested for the activity in a preliminary single concentration assay, four compounds showed approximately 50% inhibition of hα4β2 nAChRs at concentrations of 50 μM (Fig. 7).
Fig. (7).

The hit molecules identified through structure-based VS.
Landon et al. [31] employed both the multiple solvent crystal structures (MSCS) method, an approach for detection of consensus solvent binding regions on protein surfaces, and the FTMap algorithm, a fragment-based method for the in silico identification of hot spots, in the identification of hot spots for DJ-1 and glucocerebrosidase (GCase), potential therapeutic targets for the treatment of Parkinson’s and Gaucher’s diseases, respectively. The FTMap algorithm consisted of five steps as follows: (1) rigid body docking of fragments; (2) minimization and re-scoring; (3) clustering and ranking; (4) identification of consensus sites; and (5) defining of the binding site. Authors identified non-catalytic binding regions that could serve as starting points for the discovery of pharmacological chaperones for DJ-1 and GCase. Comparison of data resulted from the MSCS experiments to hot spots derived from FTMap showed that FTMap is precise and robust alternative to the performance of costly and difficult MSCS methods. New hot spots were recognized on the surface of DJ-1 in two regions. The first region contained a residue whose oxidation may prevent PD and the second region was found in the dimer interface, where a pharmacological chaperone could be bound to enhance the stability of the dimeric structure. Moreover, three regions of interest were determined for GCase, with multiple hot spots emerging in the catalytic region.
The pathophysiology of the FRDA, an inherited neurodegenerative disease, is the consequence of frataxin deficiency in the mitochondria and cells. Rufinia et al. [32] identified a set of novel and more potent small molecules that more efficiently prevent frataxin ubiquitin-dependent degradation. These compounds which called ubiquitin-competing molecules (UCM) directly bind to frataxin protein and inhibit its ubiquitination. The NMR and x-ray structures of human frataxin were employed to identify the location of putative binding pockets on the solvent accessible area of the frataxin using MetaPocket. This method seeks consensus among eight different methods: ConCavity, Fpocket, GHECOM, LIGSITE, PASS, POCASA, Q-SiteFinder and SURFNET, by concentrating the analysis on the areas near to Lys147. VS experiment was conducted by AutoDock and AutoDock/Vina on in-house database of commercially available molecules including the sulfonyl-hydrazone scaffold with aromatic substituents utilizing the following principles: MW less than 500, logP less than 5, no atoms with undefined stereo, and no reactive groups. A total of 5000 molecules were recognized and docked on the X-ray structure of frataxin, which approximately 100 compounds were retrieved and purchased for biological evaluation using fluorescence spectroscopy via the investigation of the changes of the signal of the protein tryptophan residues in the presence of the different molecules. At the end, some of these compounds were predicted to bind with frataxin proximal to K147 (Table 1).
Table 1.
Chemical structure and activity of the compounds described in the Rufinia et al. study.

4. Molecular Docking Studies
Molecular docking is a standout amongst the most frequently utilized strategies in structure-based drug design which can be employed to model the interaction between a small molecule ligand and a biological target at the atomic level. This computational technique enables us to characterize the behavior of small molecules in a structurally defined site of the targeted proteins as well as to get information about essential biochemical mechanisms [33]. Furthermore, molecular docking algorithms execute quantitative predictions of the strength of association or binding, providing scoring function to rank docked ligands based on the binding energy of protein-ligand complexes [34]. The determination of the correct binding conformations requires two fundamental prerequisites: (i) searching of an extensive conformational space displaying different potential binding modes by incrementally modification of torsional (dihedral), translational and rotational orientations of the ligand relative to the protein by employing systematic and stochastic search techniques; and (ii) precise prediction of the binding energy related with each of the predicted binding poses [35]. Molecular docking programs play out these undertakings through a cyclical procedure, in which the ligand conformation is assessed by specific scoring functions. Ligand-protein docking explores conformations and orientations of small molecules (ligands) within the binding sites of macromolecular targets, and scoring functions are applied to rank different poses by a score, a quantity that ideally would correlate with the free energy of binding [36]. There are different docking programming packages accessible, in view of various search algorithms and scoring functions, such as AutoDock [37], Dock [38], FlexX [39], Glide [40], Gold [41], Surflex [42], ICM [43], Ligand- Fit [44], Drugster [45], and eHiTS [46]. An assortment of conformational search procedures have been created with a specific end goal to investigate the ligand conformational space; these are classified as following: (a) systematic search algorithms attempt to search different positions and orientations for the ligand in the binding site of receptor, (b) stochastic methods, such as random search about rotatable bonds that implement Monte Carlo and genetic algorithms to discover new low energy binding modes, and (c) Molecular Dynamics simulation techniques and energy minimization for probing the free-energy landscape of a molecule [47]. Docking programs utilize various methods of scoring functions which can be grouped into three main categories: (a) force field-based scoring functions take into account the sum of bonding interaction terms (bond stretching, angle bending, and dihedral variation) and non-bonding interactions terms (electrostatic and van der Waals interactions) between all atoms of the ligand and protein in the complex; (b) empirical scoring functions fit a set of different parameterized terms (polar–apolar interactions, loss of ligand flexibility, and desolvation effects), describing properties known to be decisive in molecular interaction, to formulate an equation for predicting experimental binding energies (such as LUDI, FlexX, F-Score, ChemScore and Fresno); and (c) knowledge-based functions are parameterized from statistical information of intermolecular contacts in a large set of known protein-ligand complexes (like Mean Force (PMF), DrugScore and SMoG (Small Molecule Growth)) [34]. In the present review, we give an overview of some successful examples of reported docking studies employed to design potential hits for NDDs which are essential from the polypharmacological point of view.
The endogenous cyclo-oxygenase (COX) enzyme catalyzes the generation of prostaglandin-H2 from arachidonic acid and is expressed in inflammatory reactions. COX has two isoforms, constitutive cyclooxygenase-1 (COX-1), which is expressed in almost all tissues and has an essential fuction in cell signaling and maintaining the homeostasis in normal cells, and inducible cyclooxygenase- 2 (COX-2), that is expressed and activated only in inflammatory situations. COX is competitively inhibited by a set of drugs named nonsteroidal anti-inflammatory drugs (NSAIDs) to overcome inflammation and for therapeutic purposes even in neuro inflammatory-based diseases like AD. Recently, Dadashpour et al. [48] designed and synthesized a series of novel COX-2 inhibitors for disrupting fibrillar Aβ aggregates. The accumulation of Aβ peptides as amyloid deposits within the brain results in mitochondrial impairment, oxidative damage, and finally causing neuronal damage. In this study, a series of newly designed derivatives based on the structure of a diaryltriazine lead was docked into the active site of both COX-1 (PDB 3N8Z, 2.90 Å) and COX-2 (PDB 3NT1, 1.73 Å) using AutoDock 4.2. To assess the reliability of the predicted enzyme-inhibitor complexes, authors estimated ligand-receptor binding free energy by evaluating the key intermolecular interactions, which showed higher activity for COX-2 rather than COX-1, in which Arg513 had a central role in selective COX-2 inhibition. Compound 6c, with methoxy group at the para position of phenyl ring of the ethyl 5-aryl-6-(4-methylsulfonyl)-1,2,4-triazine-3- thioacetate, with IC50 value of 10.1 μM was the most potent and selective COX-2 inhibitor (Fig. 8), which could remarkably destabilize the toxic Aβ plaques (94% inhibition for Aβ1–40 and 93% for Aβ1–42).
Fig. (8).

Chemical structure and binding mode of compound 6c in the COX-2 active site.
The pan neurotrophin receptor (p75NTR) is known for mediating neural loss and acts as a target for the treatment of neurodegenerative disease. It has been identified that the binding of Aβ to the ectodomain of p75NTR receptor induces apoptosis in nerve cells and activation of signalling cascade triggered by Aβ and gave the possibility that beta amyloid oligomer is a ligand for p75NTR. To study the atomic contact point responsible for molecular interactions and conformational changes of the p75NTR upon binding to Aβ42, Devarajan et al. [49] performed a molecular docking and simulation study to explore the binding behaviour of Aβ42 monomer with p75NTR ectodomain and represented a p75NTR- ectodomain-Aβ42 complex model. Cluspro 2.0 protein-protein docking algorithm was employed to study the molecular interactions and binding. The docking results indicated that, Aβ42 specifically recognizes cysteine rich domains (CRD1 and CRD2) and forms a “cap” like structure at the N-terminal of receptor which is stabilized by a network of hydrogen bond interactions. Molecular dynamics simulation was used to investigate the conformational stability of Aβ42 and p75NTR complex and demonstrated that Aβ42 shows distinct structural alterations at N- and C-terminal regions due to the influence of the receptor binding site. These findings provided an opportunity to analyze the intracellular signalling events mediated through various domains which lead to apoptosis.
To identify novel AChE inhibitors (AChEIs) based on their interaction with AChE, Rohit and coworkers [50] constructed a structure-based pharmacophore model from diverse series of compounds including flavonoids, cardenoloids, steroids, terpenes, vitamins, and phenols reported from leaves of Cassia tora, latex of Calotropis procera and seeds of Brassica campestris based on their interaction with AChE. The docking study revealed that calotropagenin (cardenolides) present in latex of C. procera, flavonoids and glucobrassicin (glucosinolate) showed better alignment at active site, by interacting with all major amino acid residues. The in silico method used in this study contributed to identification the lead compounds which further in vitro and in vivo researches could prove their therapeutic potential.
In AD, reduction in AChE activity, and an increase in butyrylcholinesterase (BChE) activity occur. González-Naranjo and collaborators [51] studied twenty five indazole compounds to perfect the dual activity of cannabinoid CB2 agonists and BChE inhibitors observed in indazole ethers. The binding affinity of the molecules was evaluated for cannabinoid receptors CB1 and CB2 and the AChE/BChE inhibitory activity was tested in vitro, and through flexible docking analysis. The best results were selected: compounds 3 and 24 were the most interesting, showing antioxidant properties as well (Table 2).
Table 2.
Structures for compounds 3 and 24 and their IC50 values.

Chen et al. [52] synthesized five tacrine-flurbiprofen hybrid compounds as multi-target-directed compounds for the treatment of AD, in which the tacrine-like heterocycle was connected to racemic flurbiprofen via alkylenediamine linkers, and evaluated them in vitro as inhibitors of the EeAChE (AChE from Electrophorus electricus) and BChE. All of the compounds displayed better or the same BChE inhibitory activity when compared to the reference drug tacrine and two of the them, (2-(2-fluorobiphenyl-4-yl)-N-[6-(1,2,3,4-tetrahydroacridin-9-ylamino)hexyl] propanamide (3d), and 2-(2-fluorobiphenyl-4-yl)-N-[8-(1,2,3,4- tetrahydroacridin-9-ylamino) octyl] propanamide (3e)) were more potent than tacrine (IC50 = 19.3 ± 3.4, and 34.5 ± 3.3 nM, respectively). Based on the crystal structure of AChE (PDB id: 2X8B), the inhibitory behavior of compound 3d was examined by molecular modeling study using CDOCKER module in Discovery Studion 3.0. (DS, Accelrys). The analysis of binding mode of 3d by docking simulation exhibited that 3d covered the binding gorge in a good position and mode, thus resulted in higher inhibitory affinity (IC50 of ACHE=19.3 nM, IC50 of BuCHE=3.7 nM). Tacrine fragment of 3d bound via strong parallel π–π stacking against the indole ring of Trp86, to near the bottom of the gorge (CAS), and at the mouth of the gorge, the benzene ring of flurbiprofen showed hydrophobic interactions with residue Try286, a key peripheral anionic site (PAS) residue. The results indicated the novel tacrine–flurbiprofen hybrids as multipotent anti-AD drug candidates, which can be used as lead compounds for the development of new potent anti-AD drugs.
Based on the multi-target-directed ligand (MTDL) concept, various authors have designed and synthesized compounds to treat AD. Azam and collaborators [53] studied twelve compounds from Ginger (Zingiber officinale) against Alzheimer drug targets: AChE, BChE, BACE, GSK-3, TNF-α converting enzyme (TACE), c-Jun N-terminal kinase (JNK), nitric oxide synthase (NOS), Human carboxylesterase, NMDA, COX1, COX2, Phosphodiesterase-5, and the angiotensin converting enzyme. The authors used a rigid protein and a flexible ligand whose torsion angles were identified (for ten independent runs per ligand). The calculations, reliability, and reproducibility of the molecular docking methodology were validated and docking parameters showed a correlation coefficient of R2 =0.931. In order to evaluate the pharmacokinetic profile of the compounds, molecular descriptors were calculated, including miLogP, the number of hydrogen bond donors, the number of hydrogen bond acceptors, the molecular mass of the compounds, the topological polar surface area (TPSA), the number of rotatable bonds, and violations of Lipinski’s rule of five. Based on the docking data, the authors built a “Ginger” model from structural requirements and interactions with the receptors.
Consensus scoring methods which comprised of an integration of several scoring functions have shown better performance and accuracy compared to one scoring function. In this regard, Cozza et al. [54] constructed a homology model for CK1d catalytic subunit as a target to combat various diseases such as neurodegenerative disorders, such as AD and PD. The generated homology model in this study was further used to find two new RSK2 NTD (N-terminal domain of p90 ribosomal S6 kinase 2) low micromolecular inhibitors from the National Cancer Institute (NCI) open repository. They have used an integration of four docking protocols (MOE-Dock, Glide, Gold and FlexX) and five different scoring functions (MOE-Score, GlideScore, Gold-Score, ChemScore and Xscore) to properly dock and rank all MMsINC (a large-scale chemoinformatics database) entries with a lead-likeness profile. A ‘FiTkconsensus scoring function’ was utilized to correctly rank the potential hit compounds. It has been observed that only few compounds (less than 150) have been scored with a full ‘FiTkconsen-sus’ from consensus structure-based VS protocol. Two anthraquinone derivatives were identified among them after visual inspection, which are amongst the most active and selective CK1d inhibitors known today (IC50= 0.3 and 0. 6 μM).
The diverse cerebral mechanisms involved in neurodegenerative disorders along with the heterogeneous and overlapping nature of phenotypes have shown that multitarget procedures might be proper for the enhanced treatment of complex brain diseases. Discovering dual-target-directed drugs that have dual functionality for both MAO-B and AA2AR represents a possible approach to prevent the progression of PD. C8-substituted caffeinyl derivatives are dual-target-directed drugs that inhibit MAO-B and AA2AR for the therapy of PD. Azam and coworkers [55] employed molecular docking technique to understand the dual mechanism of MAO-B inhibition as well as AA2AR antagonism at the molecular level by AA2AR antagonists with MAO-B inhibitory activities which were retrieved from the literature and subjected to in silico investigations. Molecular docking approach was established a good correlation (R2= 0.524 and 0.627 for MAO-B and AA2AR, respectively) between docking predicted and actual Ki values, which confirmed the reliability of molecular docking to understand the mechanism of dual interaction of caffeinyl analogs with MAO-B and AA2AR. Parameters for Lipinski's “Rule-of-Five” were also computed to predict the pharmacokinetic properties of caffeinyl derivatives. The docking studies reflected that (E)-styryl and 4-phenylbutadien-1-yl groups at C-8 position of the caffeinyl moiety utilize both cavities as potential binding targets making them potent MAO-B inhibitors (Ki= 31 μM- 1712 nM). These computational studies provided some advantageous hints in structural modification of C-8 substituted caffeinyl analogs for exploring new inhibitors as dual-target-directed drugs with favorable pharmacokinetic properties and also provided precious insight for comprehending the dual mechanism of MAO-B inhibition as well as AA2AR antagonism for the treatment of PD.
Signaling function of anandamide, a main actor of the endocannabinoid system, is terminated by fatty acid amide hydrolase (FAAH) via its hydrolysis in both the CNS and in peripheral tissues. So, inhibition of FAAH presents an interesting strategy to induce the cannabinoid receptor type 1 (CB1) stimulation and a valid pharmacological approach for the treatment of neurodegenerative and neuroinflammatory disorders like PD, AD, HD, and multiple sclerosis (MS). Poli et al. [56] employed VS study utilizing a mixed FLAP (fingerprints for ligands and proteins) consensus docking method to identify new noncovalent FAAH inhibitors. The main weakness point of the consensus docking approach was the needed computing time for subjecting a whole data set of molecules to all the docking procedures. Because of this, the application of a prefilter step capable to reduce the number of compounds to be analyzed was necessary, so in this study authors employed a FLAP prefilter for selecting potential noncovalent FAAH inhibitors. For FLAP analysis a database constituted by FAAH noncovalent inhibitors and decoys was generated and six different groups of compounds were obtained as the potent noncovalent FAAH inhibitors. A representative compound of each cluster was selected and inserted in the enriched database as active molecule. To evaluate the effectiveness of the FLAP software in discriminating FAAH inhibitors from decoys, the enriched database with six active molecules and 43629 decoys was employed. FLAP receptor–based approach was applied to prefilter a commercial database of approximately 1 million of compounds and selected set of molecules were introduced to the consensus docking analysis. This step was further followed by a total of 2ns MD simulation study to verify the stability of the docking pose for the best ranked compounds. Finally, ten most potent molecules were examined for their inhibitory activity against FAAH which two of them displayed low micromolar IC50 values (Fig. 9).
Fig. (9).

Structures of the compounds VS4 and VS7 with IC50 values of 34.6±2.0 μM and 62.3±5.3 μM, respectively.
γ-Aminobutyrate aminotransferase (GABA-AT), a pyridoxal phosphate dependent homodimeric enzyme of 50 kD subunits, is a target for neuroactive drugs containing drugs for Huntington’s disease (HD) because its inhibition alters the balance between its substrate 4-aminobutanoic acid (GABA) and the product L-glutamate. GABA-AT degrades the inhibitory GABA which is responsible for the regulation of muscle tone. Pareek et al. [57] conducted de novo drug designing approach and docking analysis to identify the potent inhibitors of GABA-AT which could be a promising drug to cure HD. Acetic acid and its derivatives were chosen as parent molecules and rigid docking simulation based on the potential distributions of the Tyr97 was performed between parent compounds and GABA-AT protein. The prediction of active inhibitory site for compounds were performed with the help of Ligsite. Seed molecules were matured in the identified inhibitory site after setting up the position of Tyr97 amino acid. The results revealed that the ligand 5 ((2 S)-3-[(3aR, 4S, 6R, 7aS)-6-methyloctahydro-1H-inden-4-yl]-2-(propanoylamino)propanoic acid) was the best suited to be a drug for HD base on Lipinski’s rule of five and ADME profiling. The ligand molecule 5 exhibited close interactions with the residues present in its binding site, including His72, Tyr97 and Ile100 and formed strong hydrogen bond with His72 and Tyr97 (Fig. 10).
Fig. (10).

3D representation of interaction between residues docking complex between GABA-AT and Ligand 5.
G protein-coupled receptor 17 (GPR17), a orphan receptor responding to both uracil nucleotides and cysteinyl-leukotrienes, has been suggested as a new therapeutic target for human neurodegenerative disorders. Eberini et al. [58] carried out comparative modelling to build 3D structure of GPCR by using four crystallized GPCRs as template and identified its binding site through the MOE Site. The in silico screening of 130,000 lead-like and non-targeted structural library was performed with the Dock program to identify putative GPR17-targeting ligands with diverse chemical scaffolds. The five top scoring compounds was kept and submitted to in vitro molecular pharmacology experiments, which suggested that thirty one amino acids were associated with the interaction between GPR17 and selected compounds as previously identified by the MOE Site Finder modul. Finally, to assess the functional activity of the 5 candidate molecules on GPR17, the selected compunds were examined in a well established cell-based GPCR assay (pKi= 10.5 – 16.6).
Familial ALS (FALS) appeares to result from a gain of toxic function or loss-of-nuclear function due to mutations in superoxide dismutase-1 (SOD1) which facilitate protein aggregation. In silico restricted docking calculations of a set of drug-like molecules that bind the SOD-1 dimer interface as pharmacological chaperones and disrupt accumulation of the mutant form of the protein were performed by Nowak et al. [59] to understand structural information and improve the binding specificity to identify new sites for modification on the parent molecules. The hydrophobic interior of the SOD monomer is comprised of Val148 and Val7 residues with a small number of charged or polar residues such as Lys9 and Asn53. The docked structures of aggregation inhibitors at the dimer interface binding pocket revealed that the azauracil/uracil imino groups form electrostatic and hydrogen bonds interactions with the backbone carbonyl of Val7 and side chain carbonyl of Arg53. According to this, they peformed docking simulations and VS on a library of about 2.2 million compounds with four hydrogen bonding constraints, which at last 20 new compounds were obtained. These novel compounds were analyzed for their ability to inhibit SOD1 A4V aggregation and bind to SOD1, which eventually six of these compounds indicated effective inhibitory activity (ΔG= 8.56 – 11.77 kcal.mol-1) and could be great starting points for drug development for ALS.
The FUS protein interacts with karyopherine β2 (Kapβ2) via its proline/tyrosine nuclear localization signal (PY-NLS) that allows for FUS protein nuclear localization. ALS with or without frontotemporal dementia arises from mutation in arginine residue in 521 position (R521) of PY-NLS of FUS protein and leads to incytoplasmic delocalization of mutant FUS. Swetha and coworkers [60] conducted protein-protein docking using HADDOCK and MD studies to examine the interaction behaviour of the mutants FUS (R521C) and FUS (R521H) with Kapβ2. In wild-type FUS structure, the contribution from cationic residues, lysine and arginine was more in binding with Kapβ2 and five H-bonds were observed between R521 and Kapβ2. The docking results indicated that the mutants had slightly low binding activity with Kapβ2 in comparison with wild FUS–Kapβ2 as proved by the lesser number of interactions found between the mutant FUS and Kapβ2. Subsequently, the wild and mutant complexes of FUS–Kapβ2 were subjected to MD simulation to evaluate the effects of mutation on the molecular and structural properties and binding with Kapβ2. They observed that wild FUS–Kapβ2 structure was the most stable structure among the investigated structures. Principle component analysis on the MD trajectories indicated that the concerted motions were increased in the mutant FUS (R521C)–Kapβ2 and mutant FUS (R521H)–Kapβ2 structures; therefore, the mutations in FUS reduced the stability of the protein relative to the wild FUS–Kapβ2 complex. These results provided better understanding of binding behavior of mutants FUS with Kapβ2 and could be a new channel for further experimental inspections on adult-onset motor neuron disease.
Rezaei Makhuri et al. [61] performed 3D-QSAR and molecular docking studies on a series of 47 CK1d inhibitors to identify the most important structural features required for designing of next generation compounds with increased bioactivity. Molecular docking simulation using GOLD protocol identified two different binding orientations: orientation 1, in which the benzothiazole ring of the inhibitors was located close to the hydrophobic center formed by Ile23 and Ile37, Ala36, Lys38, Met80, Met82 and Val81, and orientation 2, in which the benzene ring of the compounds was directed toward the hydrophobic area (Fig. 11). Finally, a two-stage VS approach was performed to find similar analogs using pharmacophore-based screening as ligand-based VS followed by structure-based VS using molecular docking.
Fig. (11).

The docking results of the a) compound 16 with orientation 2 and b) compound 24 with orientation 1.
The potent C5-substituted quinazolines which enhance survival motor neuron 2 (SMN2) gene expression were used to investigate other molecular targets for transcriptional activation of the SMN2 promoter. Therefore, by screening approximately 5,000 proteins on the protein microarray with a radiolabeled C5-substituted quinazoline tracer, scavenger decapping enzyme (DcpS) was identified as a molecular target [62]. DcpS is a nucleocytoplasmic shuttling protein that regulates RNA metabolism and hydrolyzes potentially toxic aggregation of capped mRNA structures. Singh and coworkers [62] performed molecular docking studies and cocrystal structure identification to determine the binding mode of the C5-substituted quinazolines to human DcpS as a potential therapeutic targe for the treatment of SMA. The molecular docking study of the potent C5-substituted quinazoline (D156844, Fig. 12) in the closed active site conformation of DcpS indicated that the 2,4-diaminoquinazoline moiety of the C5-substituted quinazolines nicely occupied the m7G binding pocket with excellent shape, hydrophilic, and hydrophobic complementarity. In the docked pose, the quinazoline ring displayed a potential for π-π stacking interaction with Trp175 and hydrophobic interaction with Leu206 side chain. This study represented DcpS as a new therapeutic target for modulating gene expression by a small molecule.
Fig. (12).

2D structure of D156844.
SMA is one of the most frequent autosomal recessive motor neuron disorders which is induced by deletions or mutations of the SMN1 gene but all patients retain a centromeric SMN gene, SMN2. The current main therapeutic strategy for SMA is the stimulation of expression of SMN2 gene by histone deacetylase (HDAC) inhibitors. Dayangaç-Erden and coworkers [63] carried out molecular docking simulations to predict the free energy and binding mode of E-resveratrol and known inhibitors trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA) and valproic acid against HDAC8 (Fig. 13). It was shown that E-resveratrol, which belongs to family of polyphenolic compounds, had more negative free energy of binding (-9.09 kcal.mol-1, with IC50 value of 0.219 μM) than known inhibitors. The binding mode of E-resveratrol in the HDAC8 binding cavity exhibited that the phenolic ring group of the ligand was fitted into the zinc binding cage encompassed by Asp178, Trp141, Gln263. E-resveratrol fitted the active site cavity by making diverse interactions between Gln263 and one of the hydroxyl group of the E-resveratrol (2.93 Å), Tyr306 side-chain hydroxyl group and one of the trans double bond carbon of the E-resveratrol (2.73 Å) and Phe208 and the phenolic group of the E-resveratrol (3.20 Å). These findings revealed that E-resveratrol has the highest binding capacity toward HDAC8 enzyme than known HDAC inhibitors and modifications of E-resveratrol could increase the possibility of discovering more active candidates to cure SMA.
Fig. (13).

Chemical structures of a) (E)-resveratrol with ΔG= -9.09 kcal.mol-1, b) TSA with ΔG= -8.59 kcal.mol-1, c) SAHA with ΔG= -7.48 kcal.mol-1 and d) valproic acid with ΔG = -4.41 kcal.mol-1.
Prion diseases, also named transmissible spongiform encephalopathies are fatal neurodegenerative conditions that are described by the formation and aggregation of an abnormal or scrapie form of the host-encoded prion protein (PrPSc) in the affected brains. In order to overcome the shortcomings of both NMR and X-ray crystallography techniques in the study of native PrP, Pagadala et al. [64] performed a robust in silico docking study using structural X- ray solvent to better comprehend the potential role of water in thiamine–PrP binding. They implemented structural solvent docking to identify the correct binding site and analyze water’s involvement in the binding and stabilization of thiamine (and its derivatives) to Syrian hamsterprion (ShPrP). Based on these studies, V conformation state was observed for thiamine (and its derivatives) upon the initial docking phase with the PrP, where the C4–NH2 of the pyrimidine ring contacted with the C2–H of the thiazolium, while the subsequent minimization with NMR-derived restraints allowed ligands to adopt an F conformation with the C2 carbon atom pointing over the pyrimidine ring (Fig. 14). In F conformation, the presence of 4-aminopyrimidine ring of thiamine allowed it to participate in π-stack interaction with Tyr150 and form hydrogen bonding between between the N4′ atom of thiamine and Asp147 residue. Furthermore, the terminal phosphate groups of thiamines interacted mainly through water-mediated hydrogen bonding with solvent molecules near the binding pocket.
Fig. (14).

Interaction maps for the V (a) and F (b) conformations.
Daisuke Ishibashi and coworkers [65] performed original docking simulations, termed Nagasaki University Docking Engine (NUDE), for PrPC conformation and small compounds in an original chemical compound library included approximately 210,000 compounds to identify several compounds with anti-prion effects. According this, they selected 96 compounds as candidates for anti-prion agents by analysing docking scores and similarities of chemical structures. The docking results revealed that the aromatic rings of these compounds were in contact with the amino acid residues, which were capable of generating a large attractive interaction with PrPC. Following virtual screening, the ability of candidate compounds, termed NPRs, to bind to PrPC using surface plasmon resonance (SPR) analysis, the thermal shift assay (TSA) were tested. Results from the in vitro and ex vivo drug screening showed that NPR-053 (IC50= 7.68 ± 2.64 μM) and NPR-056 (IC50= 3.72 ± 1.57 μM) significantly reduced PrPSc levels (Fig. 15). Molecular simulation and analysis of atomic level interactions between NPRs and PrPC indicated that the binding sites of NPR-053 and NPR-056 were located around the four amino acid residues — Asn159, Gln160, Lys194 and Glu196 — which was considered to be a “hot spot” for the pathogenic conversion of prion diseases. Binding conformations obtained from the docking simulation were analysed using the fragment molecular orbital (FMO) method to identify novel anti-prion drugs. FMO calculations clearly revealed that the detailed interaction mechanisms of NPR-053 and NPR-056 were different than GN8, as the positive anti-prion control drug. In the case of GN8, polar interactions, including hydrogen-bonding interactions with N159, Q160, K194 and E196 were critical while HF level calculations revealed that polar interactions were not important for the binding of NPR-053 and NPR-056. Instead, MP2 energies of several residues were negatively large, indicated that van der Waals interactions play an important role in complex stability.
Fig. (15).

Chemical structures of NPR-053 and NPR-056.
To incorporate receptor flexibility in docking methodologies, as it gives a more realistic depiction of the modeled system, Ensemble Docking (ED) can be used. ED includes docking a small chemical library against multiple rigid receptor conformations, in spite of the standard single rigid receptor docking methods. The ensembles for the execution of ED can be retrieved through NMR spectroscopy, X-ray crystallography or an integration of both and through computational approaches (MD simulations, homology modeling). According this, Tarcsay et al. [66] carried out explicit solvent membrane dynamics simulations of four protein ligand complexes from the X-ray structures and homology models of the GPCR family (CXCR4, D3, H4 and 5HT6) to create discrete protein conformations representing the intrinsic flexibility of the binding site. They investigated the capability of utilizing multiple receptor conformations for structure based drug design purposes. So, receptor conformations retrieved form molecular dynamics trajectories were used for docking. According to the results, retrieved frames from the MD trajectory outperformed X-ray structures and homology models in terms of enrichment factor. These conformations were beneficial for the determination of a “consensus” binding site with enhanced applicability in VS.
Synaptic vesicle protein 2A (SV2A) is an integral membrane protein necessary for the proper function of the CNS and is the molecular target of the anti-epileptic drug levetiracetam and its racetam analogs. Correa-Basurto et al. [67] performed an in silico study to explore the racetam binding site in SV2A and the ligand-receptor interactions between racetams and SV2A. A 3D model was built and refined by performing a MD simulation. Moreover, the interactions of SV2A with the racetams were determined by docking studies. Docking studies employing different SV2A snapshots suggested a consensus binding site for racetam ligands within SV2A constituted by five residues: Thr456, Ser665, Trp666, Asp670 and Leu689. Additionally, knowing the racetam binding site within SV2A could facilitate the synthesis of suitable radio-ligands to study treatment response and possibly epilepsy progression.
Deb et al. [68] constructed new receptor-specific prediction models using the stepwise-multiple linear regression (SW-MLR) based on consensus of various docking and their scoring functions (GOLD, Ligand Fit and GLIDE). A dataset of 91 compounds composed of 9 diverse groups of AChEIs, like tacrine-8-hydroxyquinoline hybrids, tacripyrines, donepezil-tacrine hybrids derivatives, tetrahydroacridine, benzofuran-based hybrids, rivastigmine analogs, tacrine-melatonin hybrids, carbamates of tetrahydro-furobenzofuran and methanobenzodioxepine, and diamine diamides, with an activity range of 0.008 - 281,000 nM were considered for receptor-specific 3D-QSAR models development. To construct the consensus models and perform the QSAR studies, 11 scoring functions and 24 docking descriptors were investigated. Based on statistical results, the model developed using consensus of docking scores of scoring functions, namely, Glide score, Gold score, Chem score, ASP score, PMF score, and DOCK score performed well in terms of accuracy. Summary of the other docking studies used in NDDs drug discovery was reported in Table 3.
Table 3.
Docking studies employed to identify potential inhibitors for neurodegenerative diseases.
| Author (Year of Publication) | Neurodegenerative Diseases | Method | Chemical Scaffold Under Study | Target | Significance of Study |
|---|---|---|---|---|---|
| Azam et al. (2011) [69] | PD, antiparkinsonism | Docking studies by using AutoDock | a set of 30 1-(substituted phenyl)-3-(naphtha[1,2-d]thiazol-2-yl) urea/thiourea | Adenosine A2A receptors (AA2AR) | The docking results signified that, molecules with methoxy group in the phenyl ring increased antiparkinsonian activity through H-bond interaction with Phe-168, Glu-169 and His-278 residues and hydrophilic and lipophilic interactions with AA2AR. |
| Sivaraman et al. (2016) [70] | AD | In silico docking screening using docking server | herbal leads such as arecoline, apigenin, chlorogenic acid, curcumin, kaempferol, luteolin, quercetin along with standard drug rasagiline and selegiline | MAO-B | Results revealed that all the seven compounds bound to the active site of enzyme with lower docking (D energy) when compared with standard drug rasagiline and selegiline. Compound luteolin exhibited quite tight binding against MAO-B enzyme with binding energy of -7.12 kcal.mol-1 and ranked first in the compound series. |
| Jayaraj et al. (2014) [71] | PD | Molecular docking simulations using FlexX docking approach | 5 different compounds namely (a) stimovul, (b) 7,8-dihydroxycoumarin, (c) etorphine, (d) propoxyphene and (e) pentazocine | α-synuclein (α-syn) |
Results indicated that stimovul had the higher binding capacity against the active site of α-syn with a docking score of -4.5122 and formed hydrogen bonds with Ser87 and Val95 amino acids of the active site. |
| Sehga et al. (2016) [72] | Depression, neurodegenerative disorder, and Charcot–Marie–Tooth (CMT) | Homology modeling molecular docking studies using AutoDock and AutoDock Vina, and pharmacophore-based virtual screening | fluoxetine, paroxetine, fluvoxamine, and ethacrynic acid | Heat Shock Protein Family B (HSPB8) | Docking analysis elucidated that Met37, Ser57, Ser58, Trp60, Thr63, Thr114, Lys115, Asp116, Gly117, Val152, Val154, Leu186, Asp189, Ser190, Gln191, and Glu192 are critical residues for ligand–receptor interactions. |
| Ray et al. (2005) [73] | FALS | Docking using glide v2.5 and in vitro screening | a library of about 1.5 million drug-like compounds from commercial data-bases | SOD1 | Docking study revealed that the aromatic group occupied the space between the two Val148 residues has favorable effect on stabilizing the dimer of A4V against aggregation. |
| Nagappan et al. (2015) [74] | PD | Docking studies using AutoDock 4.2 | hesperidin, bioflavonoid, and dopamine precursor levodopa (L-Dopa) | α- syn, MAO-B, COMT and UCHL-1 | The in silico results clearly demonstrated that the flavonoid hesperidin has similar binding sites and interactions with α- syn, MAO-B, COMT, UCHL-1 as that of the L-Dopa the standard drug. |
| Markandeyan et al. (2015) [75] | autoimmune diseases, heart failure, AD, and PD | Molegro virtual docker software | 22 phytochemicals extracted from Morinda citrifolia fruit including isoprincepin and balanophonin | p38α MAPK | The comparison between the docking scores of phytochemicals with the scores of native reference ligands, MW181 and GG5, indicated that isoprincepin and balanophonin (phytochemicals) display better docking scores. |
| Klein-Júnior et al. (2014) [76] | AD | Dock software | synthetic indole derivatives and indole alkaloids from the genus Psychotria (italic) L. Genus | AChE, butyrylcholinesterase (BChE), MAO-A and MAO-B | The findings indicated that the indolyl-hydantoin and indolylmethyl-thiohydantoin rings might consists of good scaffolds for the development of new MAO-A inhibitors possessing neuroprotective properties. |
5. Virtual Screening
VS is commonly employed to predict the binding of large libraries of drug-like compounds that are commercially available, to a particular target with the aim of identifying the most promising compounds from the database for further study. There are two VS methodology widely employed in CADD which are categorized as Ligand-Based Virtual Screening (LBVS) and SBVS. LBVS techniques utilize just ligand data for anticipating activity based on its similarity or dissimilarity to previous known active ligands. LBVS depends on the exploration of molecular descriptors collected from a set of known active compounds to describe the similarity between molecules. These 2D or 3D-similarity search are employed to choose compounds for experimental assessment and decrease the chemical space to be investigated in further screening steps [77, 78]. Another LBVS method is the use of structural features gathered from 3D structures of a set of known ligands to develop pharmacophore models which is normally utilized when some active compounds have been recognised however the 3D structure of the target protein is obscure. These ligand-based 3D pharmacophore models are the 3D-arrangement of main chemical functionalities that are identified by a receptor and are thus responsible for ligand-receptor interaction [78]. Producing a 3D pharmacophore model includs the following typical steps: (i) exploring the conformational space of a series of compounds with known activity; (ii) identifying reciprocal features; (iii) aligning the molecules according to the calculated features; and (iv) creating the pharmacophore model. On the contrary, the SBVS method utilizes different modeling approaches, often using a docking screening, to simulate the binding interaction of ligands to a biomolecular target. As a whole, SBVS involves the following steps: (i) receptor preparation; (ii) compound database selection; (iii) molecular docking of a small-database of known actives; and (iv) post-docking analysis. Rather than the individual utilization of ligand- or structure-based strategies, integrated techniques have also been suggested. It has been hypothesized that utilizing both ligand- and structure-based techniques against the same biological target can improve the strengths and diminish the disadvantages of every individual technique, in this manner bringing about more effective CADD. The integration of structure- and ligand-based techniques either in a consecutive, parallel or hybrid manner considers all available chemical and biological data. In the sequential method, ligand- and structure-based strategies are employed in the VS experiment to gradually screen the large databases until the number of retrieved hits is small enough for extensive biological evaluation. In the parallel approach, top-ranked hits retrieved with each strategy are chosen for biological testing. Hybrid methods include the combination of structural and ligand information into an independent strategy [79]. A few effective instances of reported VS studies used to recognize potential hits for NDDs have been reported in this section.
GSK-3 is a regulatory serine/threonine kinase which has been implicated in the pathogenesis of several diseases such as type-2 diabetics, AD, cancer, and chronic inflammation. With the aim of elucidating new biologically-active molecules with potent GSK-3β inhibitory profiles, Kim and co-workers [80] applied ligand-based sequential virtual screening, in which the first step of VS was the Catalyst/HipHop pharmacophore based VS and was followed by filtration by recursive partitioning model, docking pose and synthetic accessibility. HipHop hypothesis finds essential 3D common chemical feature present among a set of compounds for interacting with a specific biological target to generate a qualitative model without the use of activity data. For this study, all known inhibitor antagonists were collected and Hip-Hop pharmacophore model was developed from small sets of known inhibitors with considering the activity, structural rigidity, and diversity. The best model comprised of five pharmacophore features namely, three hydrogen bond acceptors, one hydrogen bond donor, and one hydrophobic feature. Then, the recursive partitioning (RP) model was built using the two-dimensional (2D) topological descriptors implying molecular shape information by comparing the known inhibitors against a set of decoys. In the next step, to identify potential new GSK-3β inhibitors, the authors applied VS with the models and an external library: ChemDiv library (600,970 compounds). The drug-likeness and ADME filtering were performed to reduce the time-consuming conformation generation step of pharmacophore-based virtual screening. Fifty six hit compounds were finally selected on the basis of predicted docking mode, structural diversity, and synthetic accessibility. The ultimate hit compounds were proposed for biological testing out of which a total of three compounds exhibited micromolar inhibitory activity. The best hit compound 7 (IC50=1.56 μM) fulfilled the best pharmacophore, by matching perfectly three hydrogen bond acceptors and one donor. The docking pose (Fig. 16) and conformation of the compound showed good alignment with the results from the ligand-based approach.
Fig. (16).

Chemical structure of compound 7 and docking pose.
Natarajan and co-workers [81] applied pharmacophore-based VS in combination with molecular docking and MD simulations to identify novel scaffolds which may bind to GSK-3β and thus play a role in the treatment of AD. The energy-based pharmacophore models were validated using enrichment analysis, and the four common e-pharmacophore models thus developed which had four features such as hydrogen bond acceptors (A), hydrogen bond donors (D), aromatic ring (R) and hydrophobic group (H). The four common e-pharmacophore models were employed for high-throughput VS against nine established small molecule databases using Phase v3 which had resulted in 1800 compounds. Rigid receptor docking (RRD) was carried out for 1800 molecules and the obtained leads were compared to 20 co-crystal ligands resulting in 18 leads among them, lead1 (2-amino-4(1-(carboxymethylcarbamoyl)-2-(9-hydroxy-7,8-dioxo-7,8,9,10-tetrahydro-benzo (cherysen-10-yl-sulfonyl)-ethyl carbamoyl-butyric acid)) had the lowest docking score, highest binding affinity (ΔG value of -91.398 kcal.mol-1) and better binding orientation toward GSK-3β. Further ligands obtained from RRD approach were taken for quantum polarized ligand docking (QPLD), where quantum mechanical (QM) and molecular mechanical calculations were calculated. Then, the flexibility of GSK-3β binding site considered for induced fit docking (IFD) protocol in Schrödinger was employed to the best docking complex obtained from QPLD. According to the docking results, Lead1 formed seven hydrogen bonds with ATP binding site residues such as Asn64, Ser66, Phe67 and Lys85 as well as with allosteric residues like Arg141, Asp181 and Asp200 of GSK-3β (Fig. 17). The 50 ns MD simulations run was used to assess the stability of GSK-3β-lead1 docking complex. The results from RRD, QPLD, IFD and MD simulations showed similar bonding pattern with better binding affinity in a stable orientation. The findings emphasized that the lead1 would have potential for treatment of GSK-3β -mediated AD.
Fig. (17).

Quantum polarized ligand docking interactions of lead1 with GSK-3β.
Reports from the literature provided evidences that targeting Aβ clearance by stimulating P-glycoprotein (Pgp) could be a useful strategy to prevent Alzheimer’s advancement. Shinde et al. [82] applied pharmacophore based virtual screening, molecular docking and MD studies to identify natural product based Pgp activators which can act as leads for developing drugs against AD. 37 molecules belonging to oleocanthal, benzopyrane, imidazobenzothiazole and tetrahydroisoquinoline class which were reported in the literature for their Pgp activating or inducing property were used to generate the common pharmacophore using Phase (Phase, version 4.3). The best pharmacophore was subjected as a 3D query for VS of around 103 bioactives which were mainly the phytoconstituents from 500 Indian Medicinal plants and are traditionally known for their ethanopharmacological biological activities. One hydrogen bond acceptor (A), one lipophilic/hydrophobic group (H) and two aromatic rings (R) were found as pharmacophoric features. Moreover, the Interbioscreen (IBS) library of natural products comprising of approximately 51,644 natural compounds was also used for VS using the generated best pharmacophore that resulted into 1,312 ligands based on the fitness score. Then, the docking study was carried out using Glide to find out which of the virtually screened hits interact with the protein (PDB ID: 3G60). The top 20 hits showed docking scores which were comparable to that of the reference ligand i.e. rifampicin. Amongst the hits obtained, the ligand BA_3691 showed strong interactions with all the sites, while STOCK1N-61426 and STOCK1N-59071 showed strong interactions with Msite only. The hits were further subjected to molecular dynamics simulation studies to understand the binding mechanism. The MD simulation results revealed that the ligands BA_3691, STOCK1N-61426 and STOCK1N-59071 are the most promising leads which can be evaluated further experimentally. The ligand BA_3691 showed three prominent H-bond interactions with residues Gln191, Glu871 and Ser948 and weak hydrophobic interactions with Met945. STOCK1N_59071 revealed H-bond interaction with Gln191 and Gln343 and strong hydrophobic interactions with Phe339. The ligand STOCK1N-61426 exhibited H-bond interaction with Gln191, strong hydrophobic interactions with Tyr303, Phe339 and weak hydrophobic interactions with Phe299 and Phe979 (Fig. 18). The scaffolds of the obtained leads can be further utilized to design around new synthetic derivatives which can effectively bind to Pgp and hence prevent the accumulation of the amyloid.
Fig. (18).

Docking interactions for, Rifampicin, BA_3691, STOCK1N-59071 and STOCK1N-61426 with Pgp M binding site.
BACE1 is the aspartic protease that initiates the generation of the neurotoxic Aβ peptide and is widely considered as a drug therapy target for AD. The active site of BACE1 contains two conserved aspartate residues that form the catalytic dyad. An N-terminal 11 residue long fragment antiparallel hairpin-loop known as flap is sheiding the catalytic Asp dyad at the active site. To identify the potent BACE1 inhibitors interacting with both Asp dyad and flap residues, Kumar and coworkers [83] applied SBVS of natural molecules from IBS database, followed by using a series of in silico methods such as 3D QSAR pharmacophore modeling using known 29 pyridinium inhibitors, ADME profiling, and MD simulation. Firstly, molecular docking was utilized for the docking of twenty six known inhibitors of BACE1 retrieved from the drug bank which DB02378 (-232.24) with the highest ranked MolDock score between them was selected out as control. Then, SBVS of IBS (Koelsch, 2008) database which composed of 50,536 natural compounds was performed against BACE1 (PDB ID 2VKM) active site and twenty six compounds showed promising docking scores lower than -250 (as control). The Hypo 1 including two hydrogen bond donor and three hydrophobic features maped on top hit compounds retrieved from VS IBS database to predict the activity and essential pharmacophoric region. The mapping identified 7 candidate compounds out of 26 which fitted well with the 3D spatial arrangement of 3D QSAR pharmacophore model and predicted IC50 value. According to docking study, 3D QSAR pharmacophore and ADME, ligands 2 (pIC50= 5.83) and 3 (pIC50= 5.64) were chosen as the potential leads to design novel BACE1 inhibitors. Furthermore, binding and interaction of ligand 2 with the BACE1 was also investigated using molecular docking and MD which revealed hydrophobic interaction as well as hydrogen bond interaction with flap region amino acid Pro70, Thr72, Gln73 and Asp dyad (Asp32 & Asp228) (Fig. 19).
Fig. (19).

a) Hydrophobic residues interacting with the ligand 2 and b) 2D view of ligand 3 and resi dues involved in hydrogen bond.
Sphingosine-1-phosphate (S1P) is a bioactive plasma-membrane lysophospholipid that decrease in its concentration stimulates T cells migration from the peripheral lymphoid organs to the blood circulation which disrupts the CNS and leads to diseases, including MS, brain ischemic stroke, schizophrenia and AD. The microsomal enzyme sphingosine-1-phosphate lyase (S1PL) degrades intracellular S1P and therefore, inhibition of S1PL activity is a promising therapeutic option against MS and AD. Deniz et al. [84] introduced an incorporated computational technique comprised of VS, molecular docking, substructure search and MD simulation in order to obtain more potent inhibitors against S1PL. Fourteen active ((4-benzylphthalazin-1-yl)-2-methylpiperazin-1-yl) nicotinonitrile derivatives with half maximal inhibitory concentration (IC50) in the range of 0.024 and 30 μM were employed to develope ligand-based pharmacophore models. Receptor-ligand pharmacophore model was generated based on unconstrained docking of 661 conformers of 441 unique fragments to the binding site of S1PL receptor (PDBcode:4Q6R) using the e-pharmacophores script. Ligand-based and structure-based pharmacophore models were used to screen 500,000 drug-like compounds from the ZINC databas and after filtering the database with the pharmacophore models, 10,000 ligands were subjected to molecular docking using Glide. The high-ranking poses obtained from docking were assessed in light of ligand-protein interactions, molecular weight, fitness values to the hypothesis, ADME properties and ligand strain energy. By using the hierarchical clustering tool and by performing scaffold decomposition, the similarity between compounds with GlideGscores higher than -7.0 kcal.mol-1 was resolved to obtain a diverse chemical scaffolds. The binding conformations of the 33 selected ligands were evaluated by treating the flexibility of protein side chain as well as ligand flexibility upon docking by induced filt docking algorithm. Fifteen compounds from different chemotypes with high IFD scores and favorable brain/blood partition coefficient (QPlogBB) values were chosen in the final hits set. MD simulations were performed on the ligand-protein complex using Desmond module, which showed that the ligand binds to S1PL around the intertwined dimer in a stable mode and the hydrogen bonding interactions, pi-stacking interactions and favorable contacts occur mostly with Asn473, Gln476, Tyr526, Gly384 and Ile386 residues. Furthermore, positive relationship between binding free energies of the proposed compounds which assessed through the Prime MM-GBSA panel and IFD scores was obtained. The best candidates for potential S1PL inhibitors had scaffolds including benzimidazole, tetrahydrofuran, and dihydropyrimidine, which have significant biological functions.
Kumalo and coworkers [85] introduced an enhanced pharmacophore model in which per-residue interaction energy decomposition footprints derived from MD simulations were used to account for receptor flexibility to determine new BACE1 inhibitors. The reliability of computational approach was assessed through a set of compounds with experimentally determined inhibitory activity towards BACE1. First, the per-residue free energy decomposition analysis was applied to highlight the most important protein residues involved in inhibitor binding. Then, all the pharmacophoric moieties were chosen based on the extent of energy contribution from interaction between the functional active residues of the BACE1 and the ligands to construct per-residue energy decomposition based pharmacophore model. The model was further utilized as a query to search 3D chemical databases like ZincPharmer to retrieve novel classes of lead compounds. Lipinski’s rule of five and ADME properties were applied as primary and secondary filters to eliminate undesirable molecules, which yielded a total of 530 hits. In order to reduce the number of false positives and to further refine the hit compounds, a molecular docking study was performed using AutoDock Vina with the crystal structure (PDB: 2VKM). Then, the top two protein–ligand docked complexes were introduced to molecular dynamics studies to assess the plausibility of the binding mode and determine the ligand–receptor interactions. In conclusion, the post MD analysis has shown that ZINC30028065 (ΔG= -10.3 kcal.mol-1) and ZINC29797869 (ΔG= -10.3 kcal.mol-1) (Fig. 20) are suitable inhibitors for further computational and experimental research to identify hits with higher anti-AD activity.
Fig. (20).

3D structures for a) ZINC30028065 and b) ZINC29797869.
Caulerpin is a bisindole alkaloid extracted from the Seaweeds of the genus Caulerpa, possesses potential inhibitory activity against monoamine oxidaseB, and has displayed considerable antinociceptive and anti- inflammatory activities. Lorenzo et al. [86] generated a database of 108 caulerpin and its analogs and assessed it with a ligand-based model that included Volsurf and Molegro descriptors and the machine learning Random Forest algorithm, as well as with a structure-based VS that included docking studies of MAO-B inhibitors. Molecular interaction fields (MIF) and non-MIF-derived descriptors were calculated totalizing 128 descriptors, which along with the class variables that classified the compound as active or inactive were employed to generate the random forest (RF) model. A structure-based methodology using molecular docking were also performed which the analogs that had energy lower than a −151.87 Moldock score were selected as active and a RF model was generated. Using the integrated ligand-based and structure-based VS, authors observed a consensus of 56.9% between the methodologies, and selected nine active analogs of caulerpin. The results indicated that the presence of an amide or acyl halide polar group in R and a short nonpolar allyl or butyl chain in R1 generate lower docking energies and higher predicted probability (Fig. 21).
Zhang et al. [87] carried out in silico study including pharmacophore modeling, VS, molecular docking simulation and in vitro bioassays to discover a promising class of AChEI for the treatment of AD. Six types of AChEI with IC50 values in the range of 1.1 – 10 nm were selected to generate common features in pharmacophore models within the HipHop module in Discovery Studio 4.0. Using the HipHop module, 10 hypotheses (Hypo) were generated with different combination of features and scores and Hypo 10, which was the best model, was then used to analyze the activity of the active compounds. Crystal complex of AChE (PDB ID: 4M0E) and ligand (dihydrotanshinone I, DHI) were respectively chosen as the receptor and ligand to generate the complex based pharmacophores. Two Hypo were generated, and Hypo 1 with desired Quality (0.712, Fair) was used to screening the library subsequently. Two hydrogen bond acceptor (HBA), one aromatic ring (RA) and one hydrophobic (HY) were figured out to be the critical features of the Hypo 1 model. About 220,000 small molecules were downloaded from SPECS database (www.specs.net), optimized and were then used as VS library. All the compounds were screened by the Lipinski’s “Rule of five” and Veber rules to build a drug-like library and 75,671 compounds were used in the subsequent VS and docking simulation. Hypo 1 of complex based pharmacophore was used to screen the drug-like database. Thus, top 500 (pharmacophore) molecules were used to compare with docking simulation using GOLD (Genetic Optimization of Ligand Docking) version 5.2.2. Clustering and comprehensive analysis of the top 500 (pharmacophore) molecules and top 500 (docking) molecules were carried out and 15 molecules with different skeletons were manually chosen, and purchased from SPECS to detect their bioactivities. SARs analyses showed that ZYQ1 (2-((3-cyano-4-(4-methoxyphenyl)-7,7- dimethyl-5-oxo-1,4,5,6,7,8-hexahydro-quinolin-2-yl)thio)-N-(4-isopropylphenyl)acetamide)) can bind with the structure of AChE stably through five hydrogen bonds, seven π bonds and multiple non-bonding interactions (IC50= 33.620 ± 1.862 μM). Four residues (Tyr133, Tyr124, Ser203 and Trp86) were suggested to be crucial for they can form hydrogen bonds with the ligand. Finally, ZYQ1 (Fig. 22) and its derivatives represented a promising starting point towards high-potent lead compounds in the treatment of AD.
Dighe and coworkers [88] applied in silico docking-based VS on 567,981 compounds, obtained from an integration of CoCoCo-SC Asinex database and ChemBridge CNS-Set™ library, using the Glide module of the Schrödinger software suite to find more potent and highly selective BChE inhibitors. Giving preference to compounds with significant binding interactions with the human BChE binding site residues, a set of 66 molecules were chosen for cross-docking studies with human AChE (hAChE) to anticipate selectivity towards human BChE (hBChE). Then, a set of 13 molecules retrived from in silico studies was evaluated in vitro for their inhibitory potentials against equine BChE (eqBChE) and EeAChE using Ellman’s assay. Four compounds (2, 5, 7, and 8) displayed significant micromolar inhibition of eqBChE, which compound 7 was the most potent hit among them with an IC50 value of 9.72 μM. Docking model of compound 7 into the binding site of hBChE proposed that the carbazole ring of molecule 7 has π-π interactions with the aromatic rings of Trp231 and Phe329 and the NH group of the hexahydroquinoline ring of 7 is H-bonded with His438. With the purpose of acquiring highly selective submicromolar inhibitor of BChE, substructure similarity search was conducted on compound 7 and seven analogs of 7 (compounds 14-20) were obtained (Fig. 23), which compound 16 was the most potent analog among them with IC50 value of 0.763 μM. In fact, cyclization of the methoxy ester of compound 7 to a tetrahydrofuran ring enhanced the activity by 13-fold and converted a micromolar inhibitor (7) into a submicromolar inhibitor (16). In vitro assay indicated that compound 16 was two-fold more potent in inhibiting hBChE, and it did not inhibit hAChE even at 10 μM, so, compound 16 was emerged as a promising submicromolar inhibitor of hBChE.
The major hallmark pathology needed for a diagnosis of AD is the extracellular deposits composed of Aβ, result from sequential cleavage of the AβPP by BACE-1 during the amyloidogenic pathway, which its inhibition is considered one of the most promising therapeutic methods in patients with AD. Semighini [89] applied a set of computational methods including LBVS, SBVS and combination of ligand-based and structure-based VS to find new inhibitors of BACE-1. First, X-ray crystallographic data of BACE-1 in complex with nine inhibitors were used to generate the pharmacophore model, which divided in three subpharmacophores. The subpharmacophore 1 had four chemical features, consisting of one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic aromatic, and one hydrophobic. The subpharmacophore 2 was comprised of five features including one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic aromatic, one hydrophobic, and one aromatic ring. The subpharmacophore 3 was composed of four features comprising of one hydrogen bond acceptor, one hydrogen bond donor, one hydrophobic, and one aromatic ring feature. Three subpharmacophores were employed in two different methodologies. The first one was a pure pharmacophore-based VS using the compound libraries CNS ZINC, CNS Chembridge, Diverset Chembridge, and Maybridge. The second approach mixed the SBVS and pharmacophore-based virtual screening, with a first run made with GLIDE5.5c on the CNS ZINC database and selected the best 10,000 results, and then, screened these 10,000 molecules by DISCOVERY STUDIO 2.5 and the three subpharmacophores without ligand flexibility. The best scored molecules obtained from two different VS methodologies were then evaluated due to its molecular interactions. Interaction with at least one of the catalytic aspartates (93 and 289) and one residue from the flap loop (Tyr132, Thr133 and Glu134), ensured the proper blockage of the catalytic site. The remaining molecules were then analyzed by MIF and ADME predictions. Finally, similarity search by PharmMappe was utilized to identify new inhibitors of BACE-1, which resulted in 10 promising structures. The 10 best structures were further evaluated by enzymatic assays, and, three of them exhibited inhibitory activity of BACE-1 at the submicromolar range.
The proteasome is a large multicatalytic protease that degrades polyubiquitinated proteins to small peptides and has been involved in many diseases including AD, HD, inflammatory bowel diseases, autoimmune diseases, multiple myeloma (MM) and other cancers. It is composed of two subtypes: the constitutive proteasome which is expressed in all eukaryotic cells and the immunoproteasome which is expressed in immune cells and can be stimulated in other cell types. Kasam and coworkers [90] used a VS approach combined with experimental assay to design and discovery of new, potent and selective inhibitors with diverse non-peptide scaffolds against the catalytic subunit β5i of immunoproteasome. First of all, the VS procedure was utilized to screen the UC/GRI compound library comprised of structural information for approximately 300,000 molecules by employing RRD using FRED, resulted in identification of top-25,000 compounds. Then, the subsequent binding free energy calculations between the protein and the compounds using Molecular Mechanics/Generalized Born SurfaceArea (MM/GBSA) method implemented in Amber9 software along with inspection of interactions with key amino acid residues (including Thr1, Ser21, Ser27, Gly47, Ala49, and Asp324) led to identification of top- 90 compounds. The obtained 90 compounds by in silico studies were further evaluated as inhibitors of the CT-like active site of the immunoproteasome, and nine of the tested compounds (Fig. 24) showed efficient inhibitory activity against the immunoproteasome. Compounds 1 and 2 at 5 μM inhibited the immunoproteasome by 85-62%, respectively. Furthermore, to examine the stability and conformational flexibility of compound 1 and 2, 1 ns molecular dynamics (MD) simulation was performed, and the results indicated that, residues Ser21 and Ser27 were common amino acids of the immunoproteasome that had favorable interactions with both compounds 1 and 2. These new inhibitors had significant selectivity for the immunoproteasome over the constitutive proteasome which was likely related with the favorable interaction between the inhibitors and the hydroxyl group of Ser27 side chain in the immunoproteasome. So, enhancement of the favorable interaction with Ser27 of the immunoproteasome was expected to enhance both the potency and selectivity of the immunoproteasome inhibitors.
Hajjo and coworkers [91] developed binary QSAR models of 194 compounds (102 actives and 92 nonactives) that were predicted to be active at serotonin-6 receptor (5-HT6R), a known target for the treatment of neurocognitive dysfunction in AD, and subsequently used these models for VS of the World Drug Index database (WDI) and Drug Bank to find putative 5-HT6R ligands among known drugs. Furthermore, they were interested in compounds whose chemogenomics data from the connectivity map were negatively correlate with the gene expression profile signatures of AD patients. A total of 13 common hits with the highest confidence level were examined in 5-HT6R radioligand binding characterization and ten of these hit compounds were experimentally confirmed as 5-HT6R ligands.
A rational computational-based strategy included MD stimulation, structure-based pharmacophore modeling, VS, molecular docking, and prediction of physicochemical and ADME properties were applied by Chen et al. [92] to identify new potent AChEI scaffolds. A structure-based pharmacophore was generated from the binding models obtained from the molecular docking and dynamics stimulation of AChE with the most prominent compounds by Discovery Studio platform. The structure-based pharmacophore model was then used to virtually screen a compounds library retrieved from ZINC commercial database with shape constraints. The hit compounds were scored through their molecular binding energies calculated by means of MM-PBSA. Fifteen compounds were selected and purchased in order to test their anti-AChE effects and seven of them showed inhibitory effects with IC50 values ranging from 1.5 to 9.8 μM.
Santos and coworkers [93] carried out a VS study for discovery of new AChEI with ability to interact with both the ‘catalytic’ and ‘peripherical’ anionic sites from the CERMN (Centre d’Etudes et de Recherche sur le Médicament de Normandie) Chemical Library. Two complementary screening approaches were conducted: first, using a 3D pharmacophore as LBVS, and second, based on the active-site topology as SBVS. The overlap of the resulting compounds of both screenings revealed that compounds 3 and 5 are in common in the two screening sets (Fig. 25). In vitro analysis on AChE indicated that compound 3 presented a very good inhibition activity (IC50 of 45 ± 10 nM), of the same order as donepezil. These findings showed the real complementary of both methods for the development of novel inhibitors.
Patil et al. [94] performed a combination of computational and bioassay methodologies to identify potent and selective AChEI. Dual binding site 3D-HipHop pharmacophore models were developed using six crystal structures of AChE bound with different inhibitors at its dual binding sites to understand the structural requirement for the design of improved anti-cholinesterase activity. The best HipHop AChEI pharmacophore (Hypo-1) model consisted of two ring aromatic features at 15 Å apart (RA), hydrophobe (HP), and aliphatic hydrophobe (AH) features. Then, sequential VS strategy was done using this model from the small molecule databases, which selected 61,208 molecules from 4,500,000 compounds. The filtered 125 molecules after ADME profiling of the molecules retrieved from pharmacophore based VS were subjected to molecular docking in order to understand their binding mode as well as the mode of interactions at the dual binding sites of the enzyme. At the result, five lead molecules were proposed showing good Glide docking scores at the AChE dual binding sites. These potential hits obtained from the final stages of VS were further evaluated by in vitro analysis by Ellman’s assay which indicated less AChE inhibitory activity compared to that of donepezil (a FDA approved drug).
Bottegoni et al. [95] applied a hit discovery strategy performed by a virtual ligand screening protocol to detect dual-acting fragments that possess simultaneous activities in the low- micromolar range at BACE-1 and GSK-3β, which is linked through different pathways to the pathogenesis of AD. Docking simulations and Tanimoto similarity assessment were conducted for VS in ZINC databases which topmost-ranking compounds determined by VS were submitted to in vitro study and one with reasonably balanced activities in the low-micromolar range at two structurally unrelated enzymes was identified as a hit.
Prolyl oligopeptidase (POP) is an endopeptidase with serine protease activity that cleaves peptides at the C-terminal side of L-proline residues, which has been found to be associated with several NDDs, including PD, AD, and MS. Literature reports claimed that inhibitors bearing a reactive functional group such as aldehyde, hydroxyacetyl or nitrile are predicted to form a covalent bond with the catalytic serine of the active site. For the discovery of covalent POP inhibitors, Cesco et al. [96] performed automated VS in lead-like compounds present in ZINC databases, containing either an aldehyde or a nitrile group with the software FITTED. This program was appropriately adjusted to automatically distinguish the presence of aldehydes, ketones, boronates, and nitriles reactive functional groups for covalent inhibition and then, the inhibitor will most likely covalently bind when the catalytic serine and reactive groups were properly positioned. To predict whether a compound is active or inactive, the RankScore scoring function implemented in Fitted was then utilized to assess the strength of enzyme/inhibitor binding in the covalent complex, revealing that nine known inhibitors were ranked highest in the list (within the top 1.5%), validating the VS protocol. A virtual hit molecule together with four analogues were selected for synthesis after visual inspection. The most active compounds exhibited high nanomolar inhibitory activities with IC50 = 200-890 nM in intact living human cells and acceptable metabolic stability. The success of the work can be ascribed to an “ad hoc” modification of the scoring function used to screen the compounds library, demonstrating that covalent binding and/or geometrical constraints to the ligand/protein complex may lead to an increase in bioactivity.
Chen et al. [97] conducted fragment-based ligand discovery by performing two prospective docking screens against a crystal structure of the AA2AR (PDB accession: 3EML), a drug target for inflammation and PD. Firstly, parallel NMR-based biophysical screening and molecular docking screening of an in-house library of 500 chemically diverse fragments against the AA2AR were carried out. The fragment library was computationally docked to the orthosteric site of an antagonist-bound crystal structure of the AA2AR7 using the program DOCK3.6. Then, every library molecule binding to the AA2AR was used in the NMR-based screen to assess the ability of molecular docking to discriminate between fragment ligands and experimentally verified nonbinders. The target immobilized NMR screening (TINS) was used to screen five hundred fragment that resulted in 94 primary hits, which were further pharmacologically characterized in radioligand displacement assay. Five fragments showed more than 30% displacement of the radioligand at 500 μM, which four of those fragments were among the top 50 fragments predicted in silico. While the computational method found most of the pharmacologically relevant fragments, it also predicted similar or better binding energies for 46 top-ranked inactive compounds. Twenty two top-ranked compounds were tested in radioligand binding assay which 14 molecules were subsequently shown to be AA2AR ligands with Ki values ranging from 2 to 240 μM. The structure–activity relationship (SAR) was explored to refine the lead fragments, guided by molecular docking and MD simulations.
Artificial neural networks (ANNs) and integration of virtual and high throughput screening were employed by Mueller et al. [98] to identify potent mGlu5 negative allosteric modulators (NAMs) which are used for the treatment of various neurological disorders such as anxiety, PD, and schizophrenia. In this study, for establishing QSAR models data obtained from an high-throughput screening (HTS) experiments including 345 confirmed NAMs and 155,774 inactive compounds were used to train ANNs through supervised back-propagation of errors. Subsequently, the ANN model with the significant theoretical enrichment value for mGlu5 NAMs (root mean square= 0.209) was utilized for VS of the ChemDiv discovery chemistry database of 708,416 molecules in order to further preselect 749 best candidates. The best candidates were submitted for HTS which further selected several new drug candidates of IC50 in the range of 75 to 124 nM.
Lavecchia et al. [99] applied multi-step SBVS, Calu66 cells-based assays and UF6-LC/MS based ligand binding assay for discovering new small molecule inhibitors targeting the frataxin/ubiquitin interaction for the treatment of FRDA. FRDA is an inherited recessive neurodegenerative disorder that causes progressive damage to the nervous system. Decreased expression or deficiency of the mitochondrial protein frataxin leads to FRDA, for which there is currently no effective treatment available especially for neurological deficits. In this work, the authors first reported findings that frataxin is degraded by means of the ubiquitin-dependent mechanism and residue K147 within frataxin is responsible for its ubiquitination. Then, a theoretical model of the frataxin-K147/ubiquitin complex was built considering the formation of a covalent isopeptide bond between the carboxyl group of the C terminal G76 of ubiquitin and the -NH2 group of frataxin K147 using the HADDOCK algorithm. Two structure based prediction program, WHISCY and ProMate, were used to identify the amino acid residues interacting across the frataxin-K147/ubiquitin complex interface for in silico targeting in VS. Afterwards, the authors went on to discover for small drug candidates with the ability to directly target the frataxin region that binds ubiquitin in order to prevent frataxin/ubiquitin association. So, a multi- step SBVS approach of a subset of approximately 65,000 lead-like molecules retrieved from the NCI Diversity Set was performed by using two different methods: AutoDock and Glide. Thirteen consensus hits with favorable docking scores for frataxin Ub-binding site were chosen and tested experimentally for their ability to prevent the frataxin ubiquitination. The most interesting molecule, compound (±)-11 (Fig. 26), was found to be the most potent in blocking the frataxin ubiquitination. Because compound 11 was tested as a mixture of equal amounts of both enantiomers, it was synthesized and resolved in its optical isomers (+)-11 and (-)-11, which were assayed individually and compared to the racemate. Compared to the racemate, (+)-11 resulted the active isomer capable to block the frataxin ubiquitination and indirectly incited a larger increment in the cellular concentration of mature frataxin by significantly restoring the endogenous level of frataxin precursor. Conversely, (-)-11 compound displayed no increase in the cellular concentration of frataxin, exhibiting a fine level of selectivity in the active site because of chiral geometry. The IC50 of 11 for prevention of frataxin ubiquitination was resolved to be 45 μM. Additionally, compound 11 revealed interesting ADME properties (predicted log octanol-water partition coefficient (QPlogPo/w) = 2.4; predicted log aqueous solubility coefficient (QPlogS) = -1.5; predicted apparent Caco-2 permeability in nm.sec-1 (QPPCaco-2) = 1,322; number of primary metabolites = 7). Similarity search on the basis of substructure on the most potent hit compound 11 resulted in a series of morpholino analogues with a key meta- and para-methoxy substituted phenyl ring that had activity in the micromolar range.
SOD1 mutations, which are associated to ALS disease, decrease protein stability and promote aggregation. To investigate the novel inhibitors of mutant SOD1 for inhibiting aggregation, Huang et al. [100] applied molecular docking to screen Chinese medicine (TCM) database against mutant SOD1 active site (PDB code: 4A7V). From scoring analysis, dopamine was regarded as control for comparing with TCM compounds. All docked ligands were ranked by Dock Score, and the results showed that hesperidin and 2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside (THSG) have high affinity to mutant SOD1. Docking poses of hesperidin displayed H bond with Glu21 and Glu100 which the surrounding residues included Trp32, Pro28, and Lys30. For THSG, there were three amino acids (Glu21, Lys30 and Glu100) generated H-bond interaction, and the surrounding residues were Lys23, Pro28, and Trp32. It is worth to know that Glu100 was the common residue for each ligand binding, and the Lys30 could be found in all binding residues of mutant SOD1. Authors further utilized MD simulation to verify the stability between protein and ligands for binding assay. For MD simulation analysis, hesperidin and THSG displayed similar value of RMSD (Root Mean Square Deviation) with dopamine, and the migration analysis revealed stable fluctuation at the end of MD simulation time. Moreover, RMSF (Root Mean Square Fluctuation) and DSSP (Define Secondary Structure of Proteins) assay indicated that the secondary structure of mutant SOD1 did not change significantly during MD simulation, suggesting that the docked ligands were not affected by protein structure. The result indicated that hesperidin and THSG might be potential lead compound to design inhibitors of mutant SOD1 for ALS therapy.
A large body of studies have shown that oxidative stress is involved in the pathophysiology of different neuropsychiatric disorders and conditions, including stroke, AD, PD, HD, progressive multiple sclerosis (PMS), and ALS. In this regard, Kanno et al. [101] used a new LBVS method, an integration of in silico molecular filtering and QSAR, together with NAIP-based in vitro drug screening and discovered a new small ligand, termed CPN-9 (Fig. 27), which selectively suppressed cells death from oxidative stress-induced damage through the upregulation of Nrf2– ARE (NF-E2-related factor 2- antioxidant responsive element) transcriptional pathway. CPN-9 was primarily examined for protection against pharmacologically induced oxidative stress and most strongly protected HeLa (human epithelial cells) cells from oxidative stress-induced damage. When tested against a number of cell-stress inducers, CPN-9 only protected against cellular death induced by oxidative stress pathways. To define the molecular basis for CPN-9-mediated antioxidative activities, the expression of stress-activated proteins HO-1 (Heme Oxygenase-1) and p21/CDKN1A (cyclin dependent kinase inhibitor 1A) was tested, which both stress-induced proteins showed increased expression, and activation of the Nrf2 transcription factor also increased. To analyze whether compound CPN-9 treatment induces ARE promoter activity in SH-SY5Y (Human SH-SY5Y cell line) cells, a luciferase reporter assay was performed. These data demonstrated that compound CPN-9 confers resistance to oxidative stress by upregulation of the Nrf2/ARE transcriptional pathway and represent a potential drug candidate for the treatment of ALS and other neurodegenerative disorders.
Rao et al. [102] performed various computer based premises including ligand based pharmacophore, active site prediction and VS to identify novel drugs to inhibit the abnormal PrP which causes wide array of degenerative neurological disorders that include Bovine Spongiform Encephalopathy (mad cow) and CJD. 3D structures of prion having mutations at E196K, V203I, and E211Q were designed by homology modelling using modeller and validated by Ramachandran plot. Amantidine, amphotericin, curcumin, ceparitin, pentosan sulphate, quinacrine, quinapyramine, tetracycline and thioflavine drugs which have been identified to work against the symptoms of the disease were used to generate pharmacophore model. Pharmacophore model was further proceeded for zinc pharmer and following VS and a collection of 27 drugs were obtained. Based on binding energy, two drugs were selected and subjected to toxicity prediction, which ZINC3830922 popularly known as idarubicin (-7.3 kcal.mol-1) was determined as a potent inhibitor for mutant PrP. They concluded that ZINC03830922 shows good binding interactions and a very good bioavailability and there will be no signs of carcinogenic and irritation.
Hyeon and coworkers [103] developed an integrated structure- and ligand-based VS strategy to find new anti-prion compounds which block the conversion of the physiological form of cellular prion protein (PrPC) to the pathogenic form PrPSc. Using the structure of PrPC-GN8 (a known anti-prion compound), a 3D pharmacophore model was generated and compounds were docked into the prion hotspot to determine their potential binding mode, which enabled the selection of a small number of molecules for in vitro testing. VS of in-house chemical database with the selected pharmacophore model yielded 1110 compounds, followed by cluster analysis, identified 37 compounds. All compounds were docked into the PrPC hotspot residues identified in a previous study of PrPC interaction with a known anti-prion compound (GN8). The compounds showed strong hydrogen bonds at Asn159 (stand S1) and Glu196 (helix B) within PrPC. These compounds were tested in vitro using a multimer detection system, cell-based assays, and SPR. The BMD42-29 (benoxazole compound) was most active (−7.87 kcal.mol-1) in the cell-based assay, interacted with conserved PrPC hotspot residues, indicating the importance of the two hydrogen bonds and the hydrophobic environment in prevent of the conversion of PrPC to PrPSc.
One ligand-based approach is the selection of new compounds based on chemical similarity of known active ones. This can be done using several fingerprint methods, which allow the representation of a molecule in a way that can be effectively compared against other molecules. These methods rely on the chemical information of compounds, giving a highly qualitative approach in the search of new more potent ligands. Ijjaali and co-workers [104] applied LBVS using bidimensional fingerprints to determine a novel series of T-type calcium channel blockers (TCCBs), which are implicated in epilepsy and neuropathic pain. A LBVS incorporating different bidimensional chemical and pharmacophoric fingerprints was made on a two million compound database using ChemAxon's PF (Pharmacophoric Fingerprints) and CCG's GpiDAPH3 fingerprints to test 38 molecules for their ability to affect the functional activity of recombinant human CaV3.2 (T-type calcium channels). Sixteen out of the 38 molecules were active hits as they displayed more than 50% blockade of the CaV3.2 mediated T-type current. Summary of the other reported virtual screening studies used to identify promising hits for NDDs has been listed in Table 4.
Table 4.
Virtual screening studies employed to identify potential inhibitors for neurodegenerative diseases.
| Author (Year of Publication) | Neurodegenerative Diseases | Method | Chemical Scaffold Under Study | Target | Significance of Study |
|---|---|---|---|---|---|
| Lin et al. (2016) [105] | Sporadic and familial forms of AD | VS by utilizing docking studies with the GOLD docking program | 1.1 million compounds in the ZINC and in-house databases | GSK-3β | The results revealed that among the tested compounds, VB-008 (anandamide transport inhibitor) with a polar head and a long nonpolar tail and one h-bond in the hinge region with Asp133, exerted the strongest overall effect on the examined GSK-3β activity, tau aggregation, and neuroprotection. |
| Noeske et al. (2007) [106] | PD and multiple sclerosis | 2D-pharmacophore-based VS | Six known mGluR1 antagonists | Metabotropic glutamate receptor 1 (mGluR1) | The most potent compound yielded an IC50 of 362 nM based on a coumarine scaffold which was further subjected to a hit optimization program, and a compound with an IC50 of 123 nM was obtained. |
| Daidone et al. (2012) [107] | PD and hypertension | VS of the ZINC database, docking-based screening and in vitro assay | In-house built database of known active and inactive DDC inhibitors | DOPA decarboxylase (DDC) | Authors found several competitive inhibitors of human DDC with Ki values in the low micromolar range. The most potent inhibitor with a Ki value of 500 nM emerged as a promising candidate for further lead optimization. |
| Lepailleur et al. (2014) [108] | AD | Common feature-based pharmacophore model to VS in combination with similarity based clustering method and molecular docking | 17,194 compounds of the CERMN chemical library | Histamine H3-receptor (H3R) and serotonin 4-receptor (5HT4R) | Results from the binding experiments confirmed that benzo[h]-[1,6]naphthyridine derivative retrieved by this VS method exerts high affinity for both H3R and 5HT4R. |
| Ferreira et al. (2011) [109] | AD, PD | Four-point pharmacophore | macrocycle diterpene derivatives with the greater number of compounds known to have multidrug resistance (MDR) -reversing activity | Pgp | The final 4-point pharmacophore model demonstrated the essential role of hydrophobic interactions and the presence of electron acceptor features for Pgp modulators which could be utilized for the development of novel multi-resistance modulators. |
| Lu et al. (2011) [110] | AD | Pharmacophore-based virtual screening approach of NCI chemical databases followed by molecular docking | known AChEI | AChE | The identified hits by VS were subjected to molecular docking study using the LibDock program which obtained 9 hits with the highest scoring structures that have ability to block simultaneously the catalytic and peripheral anionic sites of the enzyme. |
6. QSAR Study
The core objective of any QSAR modeling is to develop a relationship between an observed activity/property and structural features of a molecule. The approach depends on being able to represent the structure of a molecule in quantitative terms (descriptors) and then to develop a relationship between the quantitative values representing each structure and experimental activity/property value [111]. QSAR methods can be categorized into following groups, based on the way by which the descriptor values are computed. Zero-dimensional (0D)-QSAR model is developed based on descriptors extracted from molecular formula information including molecular weight, atom counts, number of individual types, sum of atomic properties; 1D-QSAR model correlates the activity/property with global molecular properties such as pKa, solubility, logP, functional groups; 2D-QSAR correlates the activity with structural patterns such as connectivity indices, Wiener index; 3D-QSAR considers the position and orientation of the molecule relative to the other molecules in 3D space, and correlates activity/property with non-covalent interaction fields (steric and electrostatic field) surrounding the molecules; four-dimensional (4D)-QSAR additionally incorporates the ligand conformational flexibility in 3D-QSAR, by representing each molecule in different conformations, stereoisomers, orientations, tautomers or protonation states; the fifth dimension (5D) in QSAR explicitly represents different induced-fit protocols in 4D-QSAR; sixth dimension (6D)-QSAR takes into consideration the simultaneous evaluation of various solvation models in 5D-QSAR; hierarchical technology for quantitative structure-activity relationship (HiT QSAR) depends on the simplex representation of molecular structure and its application allows one to derive a set of distinct QSAR models which complement each other. The spirit of HiT QSAR technology is a sequential solution to the QSAR problem by the series of enhanced models of molecular structure description from 1D to 4D [112]. Regression and classification are frequently applied in cheminformatics as pattern recognition algorithms which involve finding similarities and differences between chemical samples. Usually, regression analysis is used with naturally-occurring and continuous variables; however, many structural descriptors are scattered or Boolean variables, which have to be considered by classification approaches, such as supervised or non-supervised learning algorithms. In light of the sort of chemometric methods utilized, QSAR methods are classified as linear and non-linear. Linear methods include linear regression (LR), multiple LR (MLR), SW-MLR, principal component analysis, partial least-squares (PLS) and genetic function approximation (GFA) [113]. However, newer developments in the chemometric field have also created several new methods of building predictive models, which include non-LR and algorithmic techniques like support vector machine (SVM), ANN, k-nearest neighbors (kNN) and Bayesian neural nets [114,115].
Luan et al. [116] developed multiplexing quantitative structure–property relationship (mx-QSAR) model for multiplexing assays outcomes reported in chemical database of bioactive molecules (CHEMBL) for neurotoxicity/neuroprotective effects of drugs (Fig. 28). The data was extracted from public databases like CHEMBL. Authors reported the first mx-QSAR model capable of predicting whether a drug with a determined molecular structure could possibly give a positive result in various multiplexing assay conditions. The best reported model was found to correctly classify 4,393 out of 4,915 compounds comprising of both training and validation set compounds. The overall parameters accuracy, sensitivity and specificity were found to be 90, 98 and 80%, respectively.
With the aim of looking for improved multi-target designed ligands, Bautista-Aguilera et al. [117] applied pharmacophore and 3D-QSAR studies to design a series of new structurally derived compounds from ASS234 as donepezil-indolyl hybrids which are able to inhibit both MAO A/B, AChE and BChE enzymes to provide additional benefits in AD therapy (Fig. 29). Authors have identified that N-((5-(3-(1-benzylpiperidin-4-yl)propoxy)-1-methyl-1H-indol-2-yl)methyl)prop-2-yn-1-amine (donepezile-indolyl hybrid 15, Fig. 30) as a potent, in the nanomolar range, exhibited the most interesting profile as a potent MAO A inhibitor (IC50 = 5.5 nM) moderately able to inhibit MAO B (IC50 = 150 nM), AChE (IC50 = 190 nM), and BChE (IC50 = 830 nM). Molecular modeling analysis suggested that donepezile-indolyl hybrid 15 is a mixed-type eel AChE (EeAChE) inhibitor which its linear conformation allows to span both the catalytic and peripheral sites (CAS and PAS) of this enzyme. The results indicated that propargylamine group, that bears a N-benzylpiperidine moiety from donepezil and a 8-hydroxyquinoline group, is probably going to be a critical feature for these derivatives to display both AChE and BChE inhibitory activities. Based on the best observed drug-like characteristics and ADME properties, donepezile-indolyl hybrid 15 exhibited proper drug-likeness properties and good brain penetration that deserves further analysis as a potential drug for the prevention and treatment of AD.
For the designing of multi-target inhibitors as versatile inhibitors against five proteins targets associated with AD, Speck-Planche et al. [118] developed the fragment-based approach by exploring QSAR. Their approach was centered around the development of a multi-target QSAR (mt-QSAR) discriminant model, which allowed the simultaneous classification and prediction of inhibitors against the targets. Further, the mt-QSAR discriminant model was utilized for the automatic and quick extraction of fragments in charge of the inhibitory action against the five targets, and new molecular entities were proposed.
Prado-Prado et al. [119] developed 3D-mt-QSAR models for the prediction of AChE and its inhibitors using the 3D MI-DRAGON technique based on two different software, namely MARCH-INSIDE (MI) and DRAGON to carry out a rational Drug-Protein Interactions (DPIs) prediction. MI and DRAGON softwares were used to compute 3D structural descriptor for targets and 3D parameters of all DPIs present in the DrugBank database (US FDA benchmark dataset), respectively. Both groups of descriptors were utilized as input of different ANN algorithms using as benchmark dataset to identify a better model using a non-linear approach. The developed mt-QSAR classifier predicted the binding of ligands to more than 500 diverse biological targets covered by the FDA approved drugs with accuracy 85%, and can be used for the prediction of the affinity of new compounds against different targets.
Makhaeva et al. [120] applied different QSAR approaches to study bioactivity of 58 O- phosphorylated oximes against four serine esterases, namely, AChE, BChE, carboxylesterase and neuropathy target esterase. QSAR techniques were performed using linear regression and backpropagation neural networks in combination with fragmental descriptors containing labeled atoms, molecular field topology analysis (2D QSAR), and comparative molecular similarity indices analysis (CoMSIA) (3D QSAR). All methods provided mutually consistent and complementary insights into structural features controlling the anti-EOH (Oxime reactivation of serine esterases) activity and selectivity of POXs (O-phosphorylated oximes). In conclusion, QSAR models were utilized to design a library of compounds having a cognition-enhancement esterase profile suitable for potential application to the treatment of AD.
Ambure and Roy [121] developed 2D-QSAR, group-based QSAR (G-QSAR) and quantitative activity-activity relationship (QAAR) models based on a congeneric series of 224 cyclin-dependant kinase 5/p25 (CDK5/p25) inhibitors (Fig. 31) to explore structural features needed for CDK5/p25 inhibition considering activity against CDK5/p25 and selectivity over CDK2. The structural features needed for improving activity was studied using 2D-QSAR and G-QSAR models, while the QAAR model facilitated the better understanding of features needed for selectivity of the inhibitors. Furthermore, docking analysis was carried out using GLIDE to predict the main active site residues and structural features essential for correct binding in the active site of the CDK5/p25 complex (PDB id: 4AU8). The results suggested that: i) presence of branching or ring structure at R2 position affects the activity as well as selectivity; ii) presence of –NH2 group at R2 position is important for the activity; iii) a chlorine atom at R3 is found to be essential for the activity as well as selectivity; iv) presence of a ring structure like 4-chloro-benzyl group at R1 position is required for the activity; v) presence of a –NH– fragment is found to be essential, since it interacts with an active site residue (Ile10) which was identified to be an important residue responsible for biological activity.
Araújo et al. [122] constructed receptor-dependent 3D-QSAR (RD-3D-QSAR) models based on a series of 60 benzylpiperidine inhibitors of hAChE to identify the relationship between the chemical structure and the biological activity of compounds that inhibit the hAChE. These models were retrieved from 12 databases (DBs) derived from three main groups, which were submitted to a combined Genetic Function Approximation (GFA) and partial least square techniques to construct the QSAR equations. The best two models suggested that the hydrophobic residues of the active site of hAChE were more important in the interaction with this series of inhibitors when compared to polar residues. Residues of the aromatic gorge (Tyr341 and Trp439) and catalytic triad (His447) in AChE were related to both equations displaying the consistency of these models with the SAR. Based on those models, four new benzylpiperidine derivatives were proposed and then, the inhibitory concentration at 50% predicted for each molecule. The good predicted potency of the benzylpiperidine derivative, IIa (IC50 around sub-picomolar order), indicated that it could be a potential candidate as a new AChEI (Fig. 32).
Saracoglu and Kandemirli [123] have carried out QSAR analyses of AChEIs by means of the Electron-Topological Method (ETM) related to tacrine and 11 H-indeno-[1,2-b]-quinolin-10-yl-amine tetracyclic tacrine analogues, a drug currently in use for the treatment of the AD. The ETM was implemented with the ETM software on a training set of 44 molecules, which considered both structural and electronic characteristics of compounds. All conformational and quantum-chemical data were retrieved by means of the MMP2 method of the molecular mechanics (MM) and a semi-empirical quantum-chemistry method known as AM1. Structural fragments being specific for active and inactive compounds were revealed by using ETM. Based on pharmacophores and anti-pharmacophores calculated as sub-matrices including spatial and quantum chemistry characteristics, a system for the activity prognostication was developed.
It has been suggested that the histamine H3-receptor (H3R) antagonists may play a role in the treatment of several neurological diseases such as epilepsy, obesity, arousal, attention-deficit hyperactivity disorder (ADHD), schizophrenia, AD and PD. Dastmalchi and coworkers [124] conducted the QSAR studies on a set of arylbenzofuran H3R antagonists using both 2D (MLR and ANN) and 3D (hypothetical active site lattice (HASL) QSAR methods to compare the predictive powers of three different QSAR methods. GA coupled partial least square in addition to stepwise multiple regression methods were employed to choose the minimum number of molecular descriptors to be used in MLR and ANN-based QSAR studies. The results demonstrated that the both MLR and ANN methods performed equally well in predicting the receptor binding affinities of the arylbenzofuran derived histamine H3R antagonists. Both of these 2D-QSAR methods were superior to HASL, a 3D-QSAR method, in predicting the activity of the arylbenzofuran H3R antagonists.
Gupta et al. [125] identified the dual binding AChEIs via in vitro HTS of library consisting of 56,000 compounds and then their comparative 2D-QSAR models were developed using linear (GFA and G/PLS), and non-linear (SVM and ANN) techniques. SVM, a non-linear model, was found to be superior to the corresponding linear models. Among the eight descriptors that were utilized in QSAR models development, four descriptors, namely, electrotopological descriptor (S_ssS), two thermodynamic descriptors (Atype_H_46, Atype_C_3) and an electronic descriptor (LUMO - lowest unoccupied molecular orbital), were found common for both the models. LUMO and electrotopological index descriptors were found to be decisive in determining the AChE inhibitory activity. It was suggested that the energy contribution must be taken into consideration while designing new dual binding site AChEIs.
Ajmani et al. [126] reported QSAR studies on diverse chemical classes as Gamma secretase (GS) inhibitors comprising 233 compounds (arylsulfones, aryl sulfonamide, benzobicyclononane, cyclosulfamide, cyclohexenone, cyclohexyl sulfones, fused cyclohexylsulfones), which were collected from the ChEMBL database. Continuous (PLS regression and neural network (NN)) and categorical QSAR models (NN and linear discriminant analysis (LDA)) were built to find the most important descriptors. This study indicated the importance of electronegative substitutions on aryl rings (Partial equalization of orbital electronegativity (PEOE3)) in determining variation of GS inhibitor potency. Moreover, substitution of acyclic amines with N-substituted cyclic amines contributed to inhibitor potency by increasing the values of sssN_Cnt (count of atom-type) and number of aliphatic rings.
H3R are expressed in the CNS and to a lesser extent the peripheral nervous system (PNS) of many species and suggest a potential therapeutic role for their inhibitors in treatment of several neurological disorders such as AD, epilepsy, schizophrenia and PD. Dastmalchi and coworkers [127] identified the structural requirements for H3R antagonistic activity via QSAR studies and docking techniques on a series of 58 arylbenzofuran derivatives as H3R antagonists. A combination of PLS and GA was used in the QSAR approach to select the structural descriptors. The 3D model of human H3R was built based on bovine rhodopsin structure and evaluated by MD to investigate the stability of the model. QSAR models suggested the role of charge transfer interactions in the ligand-receptor interaction, which was verified using the molecular docking analysis.
Nicolotti et al. [128] explored SAR of a wide series of 270 nicotinic agonists from diverse chemical classes. Within each congeneric series, 2D-QSAR equations indicated detrimental steric effects for the response as modeled by molar refractivity (MR), whereas comparative molecular field analysis (CoMFA) allowed authors to merge progressive models obtained for each congeneric class into a general model. In 2004, Nicolotti et al. [129] again explored about 300 nicotinic agonists via 2D-QSAR, CoMFA and Multi-objective Genetic QSAR (MoQSAR) analysis with similar conclusion. MoQSAR was used to analyze a dataset of 58 highly active nicotinoids characterized by the descriptors, namely, logP, MR and low inter-correlated weighted holistic invariant molecular indices.
Asadabadi et al. [130] developed quantitative models to describe the SAR in dual binding site inhibitors of AChE, as well as to introduce the structural determinants of their bioactivity as the future potent drugs of AD. The study was devoted to extract the most significant descriptors of these inhibitors from among a large number of quantitative descriptors. An efficient feature selection method was emphasized to find out what structural properties of dual binding site inhibitors determine their inhibition potency against AChE, utilizing the results of different routine and novel feature selection methods, for example, using LDA, binary logistic regression (BLR), genetic algorithm-based neural networks, ANN. The selected descriptors were reported and discussed accordingly.
In the multivariate image analysis-quantitative structure–activity relationship (MIA-QSAR) methodology, the descriptors are pixels of 2D images corresponding to chemical structures with biological activities and it can easily handle a large amount of information, being effective for the prediction of new molecules that might present biological activity. Bitencourt et al. [131] performed MIA-QSAR on a set of 34 compounds, including quaternary amines and carbamates with known anti-AChE activity. Structural analysis suggested the importance of a phenol group together with a carbamate scaffold in meta position of the benzene ring, to improve the AChE inhibitory activity.
To explore the mechanism of inhibition of BACE1 inhibitors, Liu et al. [132] employed 46 X-ray crystallographic BACE1/inhibitor complexes to derive QSAR models. The COMparative BINding Energy (COMBINE) software was utilized to perform COMBINE analysis on these 46 complexes. The major benefit of the COMBINE analysis is that it can quantitatively extract key residues involved in ligand binding and also identify the nature of the interactions between the ligand and receptor. The QSAR models provided some vital insights into the design of novel inhibitors via the optimization of the interactions between ligands and key residues of BACE1.
To extend the boundaries of the QSAR paradigm, and to rationalize fragment-based drug design using in silico approach, Manoharan et al. [133] proposed a fragment-based QSAR (FB-QSAR) methodology. The FB-QSAR methodology was carried on a dataset consisting of 52 hydroxyethylamine derivatives as BACE1 inhibitors, disclosed by GSK as potential anti-AD agents. A heat map constructed (based on the activity and selectivity profile of the individual R- group fragments) was used to identify superior R-group fragments. Further, they also performed multi-objective QSPR (Quantitative Structure-Property Relationship) using Derringer and Suich desirability algorithm to identify the best descriptors that can confer a trade-off between selectivity and activity.
The low BBB permeability of most of the tau aggregation inhibitors is the key issue that needs to be resolved for future development of new generation of aggregation inhibitors. In this regard, as most potent inhibitors including methylthionium chloride are negatively or positively charged, the neutral phenylthiazolylhydrazides (PTH) were used by Park et al. [134] to develop a 3D- QSAR model to provide insights into designing of novel tau aggregation inhibitors with improved activity and reduced cytotoxicity. The CoMFA 3D-QSAR model which was constructed by using 22 PTH derivatives along with the information gathered from the 3D contour maps revealed that the relative orientation of the two aromatic rings attached to both ends of the tweezer-like structure of the PTH plays an essential role in its biological activity.
Fang et al. [135] proposed a new lead identification protocol that combines ligand and structure-based approaches to predict new GSK-3b ATP (adenosine triphosphate) competitive inhibitors with topologically diverse scaffolds. According to this protocol, first the 3D-QSAR models were built and validated utilizing benzofuran-3-yl-(indol-3-yl) maleimides derivatives as GSK-3β inhibitors. Second, maleimide derivatives from thePubChem database were filtrated via Lipinski’s rule. Third, the FlexX-dock program was employed to virtually screen the remaining compounds against GSK-3β. Fourth, the 3D-QSAR models were used to predict the GSK-3β inhibition values. Finally, from the 93 predicted active hits, 23compounds were confirmed as GSK-3β inhibitors from the literatures and their GSK-3β inhibition ranged from 1.3 to 480 nM.
Zheng et al. [136] contructed ANN, MLR, and docking-QSAR models using a set of 91 BChE inhibitors divided into training (62 compounds) and test sets (31compounds). An ANN model of 10-2-1 architecture was optimized using 10 molecular descriptors describing topological and topographical features of BChE inhibitors. The same descriptors were used to develop the MLR model. Further, these BChE inhibitors were docked at the catalytic site of BChE, and their binding free energy and torsional energy were calculated. The docking-QSAR model was developed by using binding free energy and torsional energy as independent variables and BChE inhibitory activity as the dependent variable. The ANN model showed the highest correlation coefficient and the cross-validated coefficient along with lower root-mean-square deviation (RMSD) and leave-one-out root mean square deviation (LOORMSD) than MLR and docking-QSAR models. The study clearly demonstrated that the ANN-QSAR model is much robust than MLR and docking-QSAR models.
Fernández et al. [137] have modeled the AChE inhibitory activity of a set of tacrine analogues includes 136 compounds with the biological activity reported as IC50 values by using Bayesian-Regularized Genetic Neural Networks (BRGNNs). The Bayesian-regularization avoided overtraining, while the GA approach allowed exploring an ample pool of 3D-descriptors generated by the Dragon software. In addition, the capacity of the selected variables for discriminating the data was assessed by means of the unsupervised training of Kohonen Self-Organizing Maps (SOMs). The resulted model was evaluated by averaging multiple validation sets generated as members of diverse-training set neural network ensembles (NNEs). When considering 40 assembled members, the NNE provided reliable statistics.
Simeon et al. [138] investigated QSAR studies of a large deta set of 2,570 compounds with inhibitory activity towards AChE using 12 sets of molecular fingerprints and RF learning method with 100 different data splits. Furthermore, in order to identify the structural requirement for AChE inhibition molecular docking as structure-based approach was carried out on a chemically diverse set of compounds selected from active AChEIs using the Kennard–Stone algorithm. Y-scrambling was also applied to further verify the predictive performance of the selected model and assess the possibility of chance correlation. QSAR models identified “C ONS bond,” “secondary mixed amine,” “heterocyclic” and “hetero N non-basic” as the important features for AChE inhibition. According to molecular docking and QSAR models, authors concluded that the aromatic, heteroaromatic and heterocyclic rings are preferable moieties for interacting with the hydrophobic pocket of AChE and this information could be employed as guidelines for the development of new and robust AChEIs.
Subramanian et al. [139] applied multiple in silico ligand based modeling approaches starting from simple Bayesian methods to sophisticated machine-learning methods to model the binding affinities (IC50) reported for diverse structural and chemical classes of human BACE-1 inhibitors in literature. The affinities were modeled using qualitative classification or quantitative regression schemes involving linear, non-linear, and deep neural network (DNN) machine-learning methods which linear, radial, dendritic, and MolPrint2D fingerprints were used to develop qualitative classification models while the constitutional, physicochemical and topological descriptors computed using Canvas were used to build quantitative regression models. The results indicated that DNN and RF machine learning methods with Canvas descriptors resulted in robust classification accuracy and are shown to exhibit superior performance compared to traditional Bayesian techniques. Qualitative classification models identified ECFP6 and MolPrint2D to be better fingerprint schemes among the ones considered. The success of the 2D descriptor based machine learning approach when compared against the 3D field based techniques (CoMFA,CoMSIA, atom based QSAR modeling (ABM), FQSAR_gau (Field QSAR using gaussian approximation), and QSAR_ff (Field QSAR using forcefields) suggested that 2D descriptor based statistical techniques such as DNNs or RFs using Canvas descriptors can achieve statistical accuracy similar to 3D field based techniques that often require molecular alignment of diverse chemical scaffolds. This study provided a strong impetus for systematically applying such methods during the lead identification and optimization efforts for other protein families as well.
Salum and Andricopulo [140] presented a methodology that incorporates ligand-based method with structural information derived from the receptor to derive consistent 3D-QSAR models. They selected about 128 hydroxyethylamines derivatives as BACE1 inhibitors recently disclosed by GlaxoSmithKline R&D. A new fragment guided approach including HQSAR was designed to integrate the structural information from multiple crystal structures into a CoMFA study. The methodology was systematically compared to other popular approaches, such as docking, for performing molecular alignment. The models derived from the fragment-guided approach indicated substantially better external validation power (R2 pred= 0.72) than best docking derived models. Finally, the contour maps further provided required information for the development of analogs with improved potency.
Dual specificity tyrosine-phosphorylation-regulated kinase-1A (DYRK1A) is a proline- and arginine-directed Ser/Thr kinase which is associated with AD, since increased expression of this enzyme leads to hyper-phosphorylation of Tau protein and amyloid precursor protein, which results in β-amyloidosis and microtubule instability and subsequent NFT formation. Leal et al. [141] developed local HQSAR models of a series of 46 6-arylquinazolin-4-amine DYRK1A inhibitors using the commercial SYBYL software to find structural fragments with favorable contribution to the inhibitory activity. The color of each molecular fragment provided information about contribution of each molecular fragments to the inhibitory activity of this series where the yellow-to-green color exhibited the positive contribution and orange-to-red colors indicated negative contributions of the fragments of the molecules under study. The developed HQSAR model recommended that the DYRK1A inhibitors should possess the following features to increase the activity (Fig. 33): i) a phenyl ring substituted with a hydrophilic and electron-withdrawing group in R1 position; ii) heterocyclic ring substituted with a hydrophobic group in R3 position; and iii) the nitrogen atom of the amine group is substituted with a bulky hydrophobic group.
Kumar et al. [142] performed molecular docking, HQSAR and lead optimization studies to design of novel Choline acetyltransferase (ChAT) ligands. Robust statistical fragment HQSAR models were developed based on 26 known potent ChAT ligands. The models revealed the fragments essential for the activity of these ChAT ligands (Fig. 34) and indicated that a pyridine ring, which is one of the most common functional group among the parent compounds, had major contribution towards the affinity for ChAT. Molecular docking was also performed to probe the mechanism of their interaction with the active site of ChAT using Surflex-Dock GeomX (SFXC) module of SYBYL-X2.1.1 suit. The results indicated that the Tyr552 and His324 amino acid residues were of outmost importance for stabilization of an active conformation of ligands of ChAT by forming π-π and/or π-cation interactions with certain functional moiety of the ChAT ligands. The results from the HQSAR and consequential molecular docking allowed authors to select sixteen most potent compounds for use as reference and seed structures to generate novel ligands based on the pharmacophoric and shape similarity scoring function.
Dual-targeting MAO-B and the adenosine A2A receptors (AA2AR) by multipotent ligands gives a promising strategy for the treatment of NDDs, such as AD and PD. Bhayye et al. [143] performed ligand-based and structure-based modelling using deazaxanthine and benzothiazine derivatives to describe the major pharmacophoric properties responsible for inhibitory activity against MAO-B and AA2AR. To generate robust QSAR models, pharmacophore-based alignment was employed to generate a 3D pharmacophore hypothesis, based on the atom-based QSAR and HQSAR models. The developed 3D-QSAR and HQSAR models and activity cliff studies for dual antagonists of AA2AR and MAO-B provided structural requirement that serve as building blocks in designing drug-like molecules that could eventually help for treating NDDs. QSAR studies indicated that an electron withdrawing group on the heterocyclic ring of benzothiazine was essential to increase activity against both AA2AR and MAO-B and showed H-bond interaction with Tyr435 of MAO-B and Asn253 of AA2AR, which confirmed by docking and MD simulation studies. Authors designed new compounds as dual inhibitors through modifying chemical structures from the dataset and evaluated their inhibitory activity via developed QSAR models and molecular docking.
NMDARs are a subclass of glutamate receptors that are associated with many of the primary functions and developmental mechanisms of the nervous system, and implicated in various neurological conditions, AD, HD, and PD. NMDARs are heterotetramers composed of four subunits which the NR2 subunit of NMDARs plays an important role in the receptor regulation and can be further classified into four types, namely, NR2A, NR2B, NR2C and NR2D. NR2B subunit-selective antagonists are considered to have potential therapeutic benefit for the treatment of schizophrenia, PD, AD, cognitive disorders, depression, neuropathic pain, stroke, traumatic brain injury, and epilepsy. Zambre and coworkers [144] built 2D- and 3D-QSAR models to explore the structural features needed for pyrazine and related analogs as NR2B site antagonists of the NMDA. 2D-QSAR analyses using MLR and PLS revealed the major importance of Baumann's alignment independent topological descriptors, such as T_2_F_7,T_C_O_7 and T_T_T_6, in predicting pyrazine and related derivatives antagonistic activity against the NMDAR. Moreover, 3D-QSAR analyses using k-Nearest neighbor molecular field analysis, stepwise forward-backward and simulated annealing methods have shown that steric and electrostatic features are playing essential roles in order to optimize the activity and selectivity of pyrazine and related lead compounds. According to 3D-QSAR study, the general structure of the pyrazine derivatives was classified into three regions (Fig. 35) including central scaffold, right hand side of the central scaffold, and left hand side of the central scaffold. More fused aromatic/hetero-aromatic rings at central scaffold were required for optimization of the NMDAR binding activity of the lead compounds, and the right hand side accommodated substitutions that hold electronegative atoms with less steric electropositive groups. The positive range for electrostatic interaction for left hand side substitutions indicated that the presence of electropositive groups on the phenyl ring is detrimental for the antagonistic activity. Furthermore, a 3D chemical feature based pharmacophore model was built using docking study, which consisted of two aromatic rings, one hydrogen bond donor and one hydrogen bond acceptor. Authors concluded that the developed pharmacophore model could be used as a 3D structural query for VS of commercially available chemical databases of diverse chemical compounds to retrieve new potential drugs, and 2D- and 3D-QSAR models could be used to predict the activities of identified hits obtained from the VS.
Computational target finding approaches can predict protein targets and therapeutic activities of small molecules against the whole set of targets to be assessed. Application of these cheminformatic and 3D-QSAR approaches is needed to significantly decrease the animal or human experiments in the process of drug discovery. Nikolic et al. [145] studied multi-potent ligands targeting MAO-A and B; AChE and BChE; and also histamine N-methyltransferase (HMT) and Histamine H3 receptor (H3R) which are promising novel drug candidates for treatment of neurological conditions, such as depression, AD, obsessive disorders, and PD. In this study, authors used cheminformatic approach to predict the pharmaceutical targets associated with the 134 novel multi-target ligands able to interact with MAO-A and B; AChE and BChE; or with HMT and H3R. Data collected from three classes of novel multi-target ligands was used to build 3D-QSAR models for activity evaluation at the selected targets and identify the main pharmacological groups of the compounds. The first group of ligands contained of novel carbonitrile–aminoheterocyclic compounds, determined to have activity on both MAO-A and B. Amongst these, dicarbonitrile aminofuran derivatives were found to be potent and selective MAO-A inhibitors. The second set consisted acetylene, indol, piperidine and pyridine derivatives that simultaneously exhibited strong inhibitory activity towards MAO-A, MAO-B, AChE, and BChE which are putative multi-target inhibitors for therapy of neurodegenerative AD. The third group of compounds included pyridine, quinoline and piperidine derivatives that possess possible inhibitory effects against H3R, HMT and AChE/BChE. The cheminformatics-based target predictions were in good accordance with four 3D-QSAR (H3R/D1R (dopamine receptor)/D2R/5-HT2aR (5-hydroxytryptamine 2A receptor) models for the various receptors. As a result of this work, multi-target AChE/BChE /MAO-A/MAO-B, and also D1/D2/ 5-HT2a/H3R inhibitors, such as 63/Donz-D9 and 71/MBA-VEG8 (Ki for 5-HT1aR = 108 nM and the Ki for 5-HT2aR = 14.2 nM) (Fig. 36), were identified as new drug candidates which could be selected for further investigation to facilitate the development of other new agents with enhanced activity.
For development of dual inhibitors which are able to cleavage the interactions between AβPP and BACE-1 and simultaneously can interact at the peripheral anionic site of AChE, Goyal and coworkers [146] peformed Group-based QSAR analysis on a series of 20 1,4-dihydropyridine (DHP) derivatives inhibiting BACE-1 to find out the essential structural requirements of the molecular fragments of these molecules that are crucial for their biological activity. To perform G-QSAR analysis, each selected compounds was divided into 4 fragments based on four different R-groups namely, R1, R2, R3, and R4 on a common DHP moiety (Fig. 37). A total of 705 physicochemical descriptors including electro-topological state index, element count, Merck molecular force field atom type count, and molecular topological index and alignment independent descriptors were generated using Vlife MDS. Then, simulated annealing as variable selection method along with PLS regression as model building method was used to build G-QSAR model. A combinatorial library of 86,400 compounds was generated and subjected to generated G-QSAR model to biological activity prediction. The 3,405 molecules with highest activity values were introduced to docking studies and two top scoring compounds (XP score of −15.20 kcal.mol-1 for BACE-1 and −11.92 kcal.mol-1 for ACHE) were selected to investigate of their molecular interaction with the target proteins, which indicated significant binding affinity towards BACE-1 and AChE.
Polychronopoulos et al. [147] constructed a quantitative model correlated the IC50 activities of indirubins, a family of bis-indoles isolated from various natural sources, with calculated interaction energies extracted from molecular mechanics docking-scoring calculations, utilizing recent co-crystal structures of various indirubins with GSK-3β, CDK2 and CDK5/p25. This modeling approach allowed them to understand the molecular basis of indirubins' action and selectivity and to predict improvements of this family of bis-indoles as kinase inhibitors. The method contained two main steps: (a) correlation-coupled receptor minimization and (b) unconstrained ligand relaxation/Monte Carlo search. Based on the results for the main model, molecules including 6-substituted and 5,6-disubstituted indirubins were predicted and experimentally evaluated as inhibitors. The authors suggested that the affinity of indirubins for GSK-3α/β depends principally on the hydrophobic van der Waals energy term, which considers 66–92% of the sum of the three energy terms (VDW, electrostatics, and H-bonding).
The advantage of PCM over QSAR is the addition of 3D protein target information in which non-covalent interactions between protein and ligands are numerically encoded by the concatenation of ligand and target descriptors. As an example, Ain et al. [148] used 12,625 distinct protease inhibitors and their bioactivity against 67 targets of the serine protease family (20 213 data point) to model the multi-target inhibitory profiles of serine proteinase inhibitors. In order to evaluate whether the inclusion of explicit protein information enhances model performance, authors built separate sets of RF models, some using only ligand descriptors, which they called “individual and global QSAR models”. The “PCM models” were also built using the inclusion of explicit target information. Authors found that the models including protein descriptors (PCM) performed substantially better than ligand-only ones, in terms of both R2 (0.64 v 0.35) and root mean squared error (0.66 v 1.05 log units). They anticipated that using the binding site amino acids and the protein sequence descriptors in their models contribute to increase model performance.
Caspase-3 is a key executioner member of the caspase family which inappropriate control of it has been implicated in many diseases, including neurodegenerative disorders, cancer, and autoimmune diseases. Firoozpour et al. [149] used linear (MLR), non-linear (ANN) methods as global models and an approach based on ‘Extended Classifier System in Function approximation (XCSF)’ as a local model to model the bioactivity of 658 caspase-3 inhibitors. In total 1,481 descriptors were calculated which after feature selection 24 descriptors remained for the linear model and seven variables for the non-linear models. The results showed that the XCSF as a local modeling strategy estimate caspase-3 inhibition activity better than the global models such as linear regression and ANN. The atom-centered fragments type CR2X2, electronegativity, polarizability, atomic radius and also the lipophilicity of the molecule were found to be the key features contributing to the caspase-3 inhibition activity, which can be exploited for further development of new caspase-3 inhibitors.
The dopamine receptors have been implicated in PD and schizophrenia. Unfortunately, no crystal structure is currently available and thus the search for new antagonists has used QSAR models. Oloff et al. [150] employed four different QSAR methods (CoMFA, simulated annealing-partial least squares (SA-PLS), kNN, and SVM) on a set of 48 compounds, and training as well as testing statistics were generated. With the exception of CoMFA, these approaches employed 2D topological descriptors generated with the MolConnZ software package. Each of the validated KNN and SVM models were also used to mine compound databases of over 750 000 molecules that resulted in 54 consensus hits with moderate to high predicted affinities. Five of these hits had experimentally confirmed binding to the dopamine D1 receptor (pKi= 5.6 - 8) and were not present in the training set, while other suggested hits did not contain the catechol group normally seen in most dopamine inhibitors.
Bolisetty et al. [151] performed QSAR and docking studies in order to find out the structural relationship with the activity and the interaction between aphorphine inhibitor (C17H17N), a heterocyclic quinoline compound, and Dopamine Receptors (D2) which are the main drugs used in the treatment of PD. QSAR technique was applied to the twenty five aphorphine analogues that were varied at the positions of different substituent’s. Quantum chemical calculations at the DFT/RB3LYP/631G* (restricted B3LYP), RHF/6-31G* (restricted Hartree-Fock), AM1 (Austin Model 1) and PM3 (Parameterized Model number 3) semi empirical theory levels, were employed for full optimization of the selected neutral compounds. The biological activity data and the physicochemical properties IPV (vertical ionization potentials), IP (ionization potential), EA (electron affinity), EI (electrophilic index), EN (electro negativity), Hardness, Softness, LogP (partition coefficient), HE (hydration energy) and POL (polarisability) of the aphorphine derivatives were subjected to regression analysis. The AM1 and PM3 semi-empirical methods have been used to estimate the predictive power of final QSAR equations. The results revealed that higher values of electron affinity (EA), Hardness and Softness were responsible for higher inhibitory activity nature for D2 enzyme. Moreover, QSAR coupled with molecular docking studies indicated that, [6aR]-6,10-dimethyl-5,6,6a,7-tetrahydro-4H-dibenzo [de, g] quin derivative of aphorphine with the highest percentage of concentration can become a potential lead for treating PD.
A perfect example for the excellence of the ANNs and combination of VS and HTS was demonstrated by Mueller [152] to identify novel positive and negative allosteric modulators of mGlu5 (Metabotropic Glutamate Receptor 5) used in the treatment of several CNS diseases as anxiety, PD, schizophrenia. Authors first performed a traditional HTS of approximately 144,000 compounds for the identification of positive allosteric modulators (PAMs). This screen yielded a total of 1,356 hits which was then used to develop a QSAR model that could be applied to a VS. To generate the QSAR model, a set of 1,252 different descriptors across 35 categories were calculated using the ADRIANA software package which comprised scalar, 2D and 3D descriptor categories. A statistical model was created with an ANN and the authors iteratively removed the least sensitive descriptors through several rounds in order to create the optimal set. This final set included 276 different descriptors, including scalar descriptors such as molecular weight up to 3D descriptors including the radial distribution function weighted by lone-pair electronegativity and pielectronegativity. A VS was performed against approximately 450,000 commercially available compounds in the ChemBridge database. 824 compounds were tested experimentally for the potentiation of mGlu5 signaling. Of these compounds, 232 were confirmed as potentiators or partial agonists. This hit rate of 28.2% was approximately thirty times greater than that of the traditional HTS and the VS took approximately one hour to complete once the model had been optimized.
NMDA receptor (NMDAR) belongs to the family of ionotropic glutamate receptors that requires both binding of glutamate and partial membrane depolarization for its activation. It has become increasingly clear that CNS disease can arise from both NMDAR hypofunction and NMDAR hyperactivity in different pathways, which hypofunction of NMDAR occurs in schizophrenia and hyperactivity leads to neuronal death as in HD. Chtitaa et al. [153] carried out multiple linear and non-linear regression and an ANN to construct a QSAR for non-competitive antagonists of NMDAR by studying a series of 48 substituted dibenzo[a,d]cycloalkenimine derivatives. Geometrical optimization of 48 antagonists of NMDARs was performed by Lee–Yang– Parr exchange correlation functional with the 6-31G (d) basic set. The quantum chemistry descriptors were obtained for the model from the density functional theory calculations as follows: total energy (E), EHOMO, ELUMO, HOMO-LUMO gap, μ, hardness, χ and the reactivity index. The results revealed that the ANN had substantially better predictive capability than the other two models and showed good stability to data variation in leave-one-out cross-validation with greater predictive power.
The cyclic nucleotide phosphodiesterases (or PDE’s) are enzymes that control the cellular levels of the second messengers, cAMP (Cyclic adenosine monophosphate) and cGMP (Cyclic guanosine monophosphate), by regulating their rates of degradation. Inhibitors of other PDE enzymes are being explored for coronary heart disease, dementia, depression, and schizophrenia. Dong and Zheng [154] utilized a novel structure-based QSAR strategy to investigate the SAR of 35 indole derivative-based PDE-4 inhibitors. This new formalism described molecular descriptors based on the matching of their pharmacophore feature pairs with those (the reference) of the target binding pocket. The reference was derived from the X-ray crystal structures of the target under study (1xon.pdb). The general process of Structure-based Pharmacophore Key (SB-PPK) was as follows: first, the LigandScout program was employed to derive structure-based pharmacophore centers from the target binding site, then, the LigandScout program was used to perceive the pharmacophoric groups on small organic molecules, finally, once the pharmacophore feature pairs for both the receptor and ligand molecules were generated, authors compared them to determine pattern matches. The descriptors of the PDE-4 inhibitors were then determined based on receptor’s pharmacophore feature pairs matching with small molecule’s pharmacophore pairs. PLS method with different number of principle components used in the regression model was employed to establish predictive QSAR models. The new structure-based descriptors could offer structural insights into the critical features responsible for the potency of the inhibitors and overcomed the drawbacks of traditional descriptors that ignore the binding pocket information.
Sinha et al. [155] carried out fragment based G-QSAR, molecular docking and MD simulations studies on HDAC inhibitors having hydroxamic moiety for elucidating its role towards ataxia. G-QSAR model was generated based on a set of 44 hydroxamate class of compounds that have pivotal role in inhibiting HDAC enzymes and was used to determine the structural modifications needed for hydroxamic derivatives as anti-ataxia compounds. In G-QSAR a variety of 2D descriptors were employed in the QSAR model generation, which after removing of the invariable molecular descriptors 288 descriptors were used. Two combinatorial libraries constituted of 3,180 compounds with hydroxamate moiety as the template with the substitution site R1 were generated using the Leadgrow module in VLife MDS (Molecular Design Suite) and biological activity of these compounds were predicted using G-QSAR model. G-QSAR model prediction yielded 53 molecules with highest predicted activity ranging from 6.14 to 7.28 M, which were selected and docked at the His-Asp dyad active site of HDAC4. In order to gain insights into inhibitory roles of hydroxamic acid derivatives, molecular docking and 20 ns long MD simulations were performed to understand the mode of interaction for these inhibitors with regard to hydrogen bond and hydrophobic interaction with His802, Asp840, Pro942 and Gly975 residues of HDAC4. Furthermore, to capture the dominant modes of the protein motions dominant and collective modes of the protein, principal component analysis was performed using trajectory data obtained from MD simulation. Overally, compounds HIC (N-hydroxy-5-(1H- imidazole-4yl) thiopene-2-carboxamide) and DHC ([(1r,4as,8aR) decahydronapthelene-1yl-3'-N-hydroxy-thiopene-2-carboxamide]) with pIC50 (activity) of 7.28 and 7.04 M were displayed good binding activity which could be introduced as potent therapeutic leads against ataxia (Fig. 38).
The cytochrome P450 isoenzyme 2D6 (CYP2D6) involved in the metabolism and elimination of about 20–25% of clinically used drugs. Ringsted et al. [156] developed QSAR models using a training set of 747 chemicals for the CYP2D6 utilizing the three modeling systems included MultiCASE, Leadscope Predictive Data Miner and MDL QSAR. Moreover, the constructed models were employed to screen a structure set of 57,014 discrete organic chemicals, including all discrete organic European Inventory of Existing Commercial Chemical Substances (EINECS) chemicals for CYP2D6 substrates and non-substrates. The differences in models and applicability domains of models in MultiCASE, Leadscope and MDL QSAR were observed, which MDL QSAR model had the highest domain (90% of the screened EINECS), next was MultiCASE (47% of the screened EINECS) and last was Leadscope (22% of the screened EINECS). The percentages of the screened EINECS chemicals that were identified positive as CYP2D6 substrate (among the EINECS chemicals set) varied from 8% for the MultiCASE model to 18% for the MDL QSAR model. The generated QSAR models for the CYP2D6 can be employed to screen CYP2D6 substrates in untested chemicals and recognize the potential risk associated with exposure to environmental chemicals.
Gharaghani and coworkers [157] performed molecular docking, MD simulation and structure-based QSAR studies to explore structural features and binding mechanism of a series of naphthalene and non-naphthalene derivatives (n=38) as potent inhibitors of CYP2A6. A 6000 ps MD simulation was performed to generate the 3D structure of CYP2A6 in a water environment and 2-bromonaphthalene (most potent inhibitor with pIC50= 6.26) was docked into the final structure derived from the MD trajectories of enzyme to obtain binding site of CYP2A6. Then, the docked configurations of the inhibitors with the lowest free energy were used to calculate the most feasible descriptors to build a QSAR model using MLR and least squares support vector regression (LS-SVR). The results revealed that the performance of LS-SVR model in terms of root-mean-square error (RMSE), correlation coefficient, and predictive squared correlation coefficient (Q2) for the test set was better than the MLR model. The docking analysis showed that π–π interaction of the inhibitors with four phenylalanine residues at positions of 107,111, 118, and 480 plays an important role in the activities of the inhibitors. Moreover, the complex of enzyme with 2-bromonaphthalene (the most potent inhibitor) was used in 6000 ps MD simulation to explore the conformation changes of the complex, which indicated that the structure of the CYP2A6 in the presence of 2- bromonaphthalene has not altered.
Mandi and coworkers [158] developed QSAR models of forty seven 2-aminothiazole derivatives with inhibitory activity against PrP using machine learning approaches, which consisted of MLR, ANN and SVM. Molecular graphics were drawn using ChemAxon Marvin and the geometries were optimized at the density functional theory (DFT) level using hybrid functional (B3LYP) in combination with the 6-31G(d) basis set. Then, a set of quantum chemical descriptors was acquired from the low energy conformer of the structures which included the total energy of the molecule (Etotal), highest occupied molecular orbital energy (EHOMO), lowest unoccupied molecular orbital energy (ELUMO), dipole moment (μ) of the molecule, electron affinity (EA), ionization potential (IP), energy difference of HOMO and LUMO (HOMO-LUMOgap), Mulliken electronegativity (χ), Hardness, Softness, Electrophilicity (ω), EI, most negative atom in the molecule (Qneg), most positive atom in the molecule (Qpos) and the mean absolute atomic charge. Descriptors having invariable value and pairs of variables with correlation coefficient greater than 0.9 were removed using the Unsupervised Forward Selection algorithm. The independent variables (e.g. quantum chemical and molecular descriptors) and the dependent variable (e.g. pEC50) were subjected to multivariate analysis using MLR and machine learning techniques, particularly SVM and ANN. Of the tested learning methods, SVM was demonstrated to be the best learning approach for predicting the anti-prion activity while ANN and MLR gave similar level of performance. This study provided guideline for future structural modifications of 2-aminothiazoles as therapeutic agents against prion with potentially higher potency and less toxicity.
Tetracyclines are potentially effective drugs in the treatment of human prion disease, a group of infectious fatal NDDs in which PrPC change in conformation to become an abnormal misfolded isoform (PrPSc). Base on antifibrillogenic tests on aggregates formed by PrP106–126 with tetracycline and 14 derivatives, Cosentino et al. [159] performed a 3D-QSAR study to investigate the stereoelectronic features affecting the anti-fibrillogenic activity. Authors carried out a molecular descriptors selection step using statistical extrema technique (SESAME) to obtain a predictive QSAR models and then the best QSAR models was searched using the genetic algorithm-variable subset selection (GA-VSS) method. Finally, a 6-variable model exhibited good statistical quality and the best predictability of the anti-fibrillogenic activity. The 3D-QSAR investigation highlighted that hydroxyl group introduction in positions 5 and 6, electrodonor substituents on the aromatic D-ring, alkylamine substituent at the amidic group in position 2 and non-epi configuration of the NMe2 group were the best tetracycline substitution patterns (Fig. 39).
Kynurenine monooxygenase (KMO) enzyme is a promising drug target to address the neurodegenerative disorders such as HD. Amin et al. [160] used regression and classification based multi-QSAR modeling (such as multiple linear regression, ANN, SVM, LDA, Bayesian classification), pharmacophore mapping and molecular docking approaches to identify important physicochemical and structural features of fifty six arylpyrimidine KMO inhibitors. Pharmacophore mapping showed the importance of two hydrogen bond acceptors (HBA), one hydrophobic (HY) and one ring aromatic (RA) feature. The pharmacophore mapping results were in agreement with the molecular docking study. As the MLR model was found to be the best over the ANN and SVM model, it was used for predicting new molecules that may have better inhibitory properties compared to the observed molecules. Ten new compounds (IC50= 0.00015 - 0.00163 μM) were proposed and validated through the pharmacophore mapping and docking analysis.
REST/NRSF, also known as Repressor Element 1/Neuron- restrictive silencer element (RE1/NRSE), is a multifunctional transcription factor that regulates gene expression by binding to a DNA regulatory motif. An increase in RE1/NRSE genomic binding is found in HD, causing to the RE1/NRSE sites activation and represses transcription of several important neuronal genes. Leonea et al. [161] carried out SAR and 3D-QSAR pharmacophore studies on a library of commercially available 2-aminoisothiazoles, which variously substituted at the amino group or at position 4, as most active modulators of the RE1/NRSE silencing activity. To build a library of 2-aminothiazoles with differences in regions 1 (in the amino group) and 2 (position 4) (Fig. 40), a similarity search was done using the Ligand-Info system, which searched similar compunds in a database and selected molecules with modified Tanimoto coefficient of > 0.72. The SAR analysis revealed that the para-position of aromatic ring in region 1 should be substituted by a lyophilicity and moderate steric encumbrance, but H-bond acceptor capabilities were also favorable and region 2 should be characterised by an aromatic bicycle containing heteroatoms with H-bond acceptor capabilities. A 5-feature pharmacophore model were developed using Phase methodology based on a set of eight diversely substituted and active 2-aminothiazoles which additionally supported the observations based on the SAR analysis. This study could be useful for identifying novel closely related derivatives and the generated pharmacophore model could be also utilized as 3D query for VS of large chemical databases with the aim of discovering new classes of RE1/NRSE silencer modulators.
Ensemble methods (EMs) are computational algorithms that construct numerous base models using multiple machine learning algorithms and combine them into a generalizable model that predicts the properties of new compounds based on the combination of single classifiers predictions to obtain reliable and more accurate predictions. Helguera et al. [162] proposed a new EM, which uses a GA-based search to find the best ensemble combination providing the best VS performance in the identification of dual target ligands for AA2AR binding and MAO-B inhibition, as potential therapeutics for PD. In this regard, two sets of QSAR models were independently developed for each target, in which three classes of base classifiers and three feature selection methods were utilized to generate 9 different classifier pairs. To train and validate the nine diverse classifier types, three modeling set partitions namely, 1M, 2M and 3M, obtained after applying the sphere exclusion algorithms were used . In outcome, 27 distinctive classification experiments per target (hMAO-B inhibition and antagonism of the AA2AR) were carried out. The so-called external evaluation set is employed to verify the real predictive power of 27 best performing models per endpoint. The whole pool of classification models for AA2AR binding and MAO-B inhibition were first filtered to obtain the accurate, robust and predictive models as ensemble members. For retrieving of dual-target ligands, authors selected a set of representative models for each target using EF and BEDROC to guarantee the diversity in the pool of base models for further ensemble generation. Subsequently, authors took into account the applicability domain for the final pool of models forming the ensemble, and then, the scores computed by each model for the compounds within model’s applicability domain were utilized to get a relative ranking of each sample. Immediately after the relative rank of every sample in each model was computed, the final aggregated score was obtained by averaging these relative rank values over the models. At last, the final ensemble ranking was obtained by sorting the compounds in ascending order based on this aggregated score. The fundamental preferred standpoint of the proposed algorithm was that it was capable of finding the combination of models giving better performance for ligand-based drug discovery problem. Results indicated that the obtained ensembles exhibit an improved predictive performance as compared to the best individual model based on the evaluated enrichment metrics.
MAO isoenzymes A and B present a significant pharmacological interest due to their role in the breakdorn of neurotransmitters like norepinephrine, epinephrine and dopamine, which have a key role in neurodegenerative and stress-related disorders. Helguera et al. [163] presented ensemble methodology that successfully integrates a set of accurate and representative QSAR models to the classification of hMAO inhibitors. They first selected a large and diverse dataset of heterocyclic compounds for making QSAR models with a broad applicability, which included chromones, homoisoflavonoids, coumarins, chalcones, thiazolylhydrazones and pyrazole. The diverse base classifiers were generated by applying LDA to data extracted from different subsets of molecular descriptors obtained from DRAGON, MOE and MODESLAB structural representation and different feature selection algorithms. The final models predicted the inhibitory activity and selectivity toward hMAO with acceptable accuracy using a external set compounds, comprising 10 new chromones derivative and 15 coumarins with unknown activity. By considering different combination schemes, authors proved that the ensemble model can improve performance over the base classifiers. Summary of the other reported QSAR studies employed to identify potential inhibitors for neurological targets has been listed in Table 5.
Table 5.
Reported QSAR studies used in neurodegenerative diseases drug discovery.
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rezaei Makhuri et al. (2015) [61] | ALS | 3D-QSAR using CoMFA, CoMSIA and Auto GPA | A series of 47 N-(benzothiazolyl)-2-phenyl-acetamides | CK-1δ | The results of 3D-QSAR analyses revealed that hydrophobic and negatively charged groups at 6th position of benzothiazole ring and positively charged and bulky groups at ortho position of phenyl ring in N-(benzothiazolyl)-2-phenyl-acetamides were favorable for high bioactivity. | |||||
| Zhu et al. (2006) [164] |
PD, AD | 3D-QSAR using CoMFA and CoMSIA techniques | A set of 55 tripeptide aldehyde inhibitors | 20S proteasome | The contour maps corroborated with the structural features of the binding pocket of β5 subunit of 20S proteasome, which proposed that the built models could be applied to pre-screen compounds to expedite the development of lead-compounds with optimized pharmacokinetic properties. | |||||
| Fresqui et al. (2013) [165] |
anti-AD, antidepressant and anti-PD | 3D-QSAR | A set of 34 amphetamine derivatives (the R and S configurations of a series of MAO A inhibitors) | MAO A | Six descriptors, namely, CHELPG atomic charges C3, C4 and C5, electrophilicity, molecular surface area and logP were found to be significant, considering both the configurations. | |||||
| Bharate et al. (2013) [166] |
AD | Descriptor based QSAR and pharmacophore based QSAR studies | A series of meridianin analogs | Dyrk1A | This study revealed that Kier Chi4 path/cluster (molecular connectivity index), total lipole (measure of the lipophilic distribution in a 3D space), VAMP polarization (polarizability coordinate), Dp and logP play vital role in Dyrk1A inhibition. | |||||
| Tong et al. (1996) [167] |
AD | 3D-QSAR (CoMFA) study | A series of 1-benzyl-4-[2-(N- benzoylamino)ethyl] piperidine derivatives and of N-benzylpiperidine benzisoxazoles | AChE | i) Substitutions with bulky and/or lipophilic groups at the benzisoxazole and benzoyl moieties are important for the activity; ii) The oxygen in isoxazole ring, if replaced with less electronegative atom like nitrogen or sulfur, is found to diminish the potency; iii) The basicity of the nitrogen atom in N-piperidine ring is important in contributing to the activity; iv) Occupying the ortho position of the benzoyl moiety with steric bulk negatively affects the activity. |
|||||
| Ponmary et al. (2010) [168] |
PD | SW-MLR method | Compounds structurally similar to glycerol | Parkinson’s disease causing targets | The results demonstrated the high robustness and real predictive power of IC50 model. | |||||
| Jung et al.(2007) [169] | AD | (GA)-MLR and (SA)-MLR |
Tacrine derivatives (a set of 80 structurally heterogeneous compounds composed of 11H-indeno-[1,2-b]-quinolin-10-ylamine derivatives, thiopyranoquinolines, pyranoquinolines and benzonaphthyridines, tacrine-E2020 hybrids, bis-tacrine congeners, and tacrine-hurprine heterodimers) | AChE | The best equation was obtained from SA MLR with greater explanatory and prediction capability. The results suggested the important roles of hydrophobic and electrostatic interactions on increasing the structure’s AChE activity. | |||||
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
| Chen et al. (2012) [170] |
Neuroprotect-ive profile and a moderate Ca2+ channel blockade effect | 3D-QSAR models based on the flexible docking alignment using CoMFA and CoMSIA | multi-target-directed AChEIs of tacrine-nimodipine dihydropyridine | AChE | The results indicated that the IC50 can be improved by means of increasing the electronegativity and introducing small volume substituent at 3-position of the DHP (1,4-dihydropyridines) and hydrophobic like methoxy group was favorable to the 4-position of the benzene ring of DHP. | |||||
| Hoeglund et al. (2010) [171] | Cancer progression, MS, obesity, diabetes, AD, and chronic pain | Integrated ligand-based computational strategies (binary QSAR), medicinal chemistry, and experimental enzymatic assays | Analogues of the most potent hit (H2L 7905958, IC50 of 1.6 ± 0.4 μM) | Autotaxin (ATX) | Analogues of the lead compound were examined and four of the 30 indicated IC50 less than or equal to the lead. The most potent analog indicated an IC50 of 900 nM with respect to ATX-mediated FS-3 hydrolysis with a Ki of 700 nM, making this compound approximately 3-fold more potent than the lead. | |||||
| Recanatini et al. (1997) [172] | AD | Comparative 2D-QSAR studies | Three classes of AChEIs, for example, physostigmine analogs, 1,2,3,4-tetrahydroacridines (tacrine analogs) and benzylamines | AChE | i) Hydrophobicity plays a crucial role in both the physostigmine and the benzylamine-derived classes; ii) electronic effects are vital for the interactions shown by the variable portion of benzylamine derivatives; and iii) steric factors are also important. | |||||
| Jain and Jadhav (2013) [173] | AD | 2D-QSAR using MLR | Aminoimidazoles dataset | β-Secretase (BACE-1) | The study revealed that thermodynamic descriptors (MR, logP, van der Waals energy, polar surfacearea) and steric descriptors (Harary index, Randic index) play important role in β-secretase inhibition. | |||||
| Debord et al. (1997) [174] | AD | 2D-QSAR | Derivatives of 2-amino-4,6-dimethylpyridine, aryl (alkyl) carboxamides, thiocarbamides and amidrazones | AChE and BChE | The binding affinity was improved by the structural changes like: i) increase in molecular volume; ii) decrease in the energy of the LUMO; iii) insertion of a methylene group between the amide carbonyl and the aromatic ring; and iv) replacement of the amide oxygen by sulfur. | |||||
| Huang et al. (2013) [175] | AD | 3D-QSAR using Topomer CoMFA | 125 BACE-1 inhibitors |
BACE1 | Topomer search was used for VS in lead-like compounds present in ZINC databases and as a result, they successfully designed 30 new molecules with better activity than those present in the dataset. | |||||
| Hossain et al. (2013) [176] | AD | QSAR (3D-QSAR, HQSAR) and pharmacophore mapping studies | Structurally diverse BACE inhibitors | BACE | Both types of studies confirmed the importance of terminal meta-tolyl sulfonamide piperazine core and heterocyclic ring along with adjacent nucleophilic hydroxyl group and amide linkage. Finally, it was concluded from the findings that hydrogen bond donor and acceptor, hydrophobicity, electrostatic and steric properties of ligand are the important features for interaction with receptor cavity. | |||||
| Dessalew et al. (2007) [177] | cancer, chronic inflammation, bipolar disorders and AD | 3D-QSAR studies using CoMFA and CoMSIA | Novel class of pyrazolopyrimidine derivatives | GSK-3 | Based on the contour analysis, authors deduced that improvement in GSK-3β binding affinity can be achieved through conformationally restricted substitution at N1 position near region A and keeping the electronegative group to the central core in region B.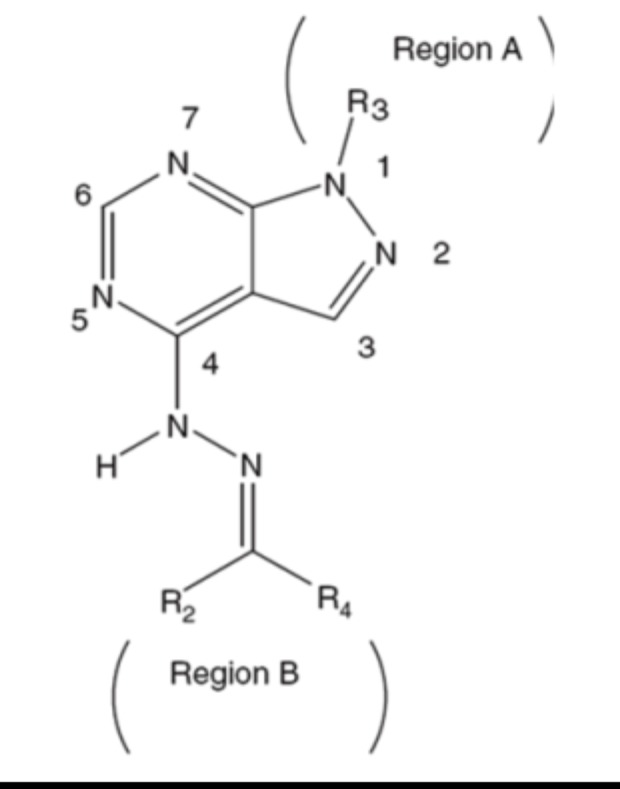
|
|||||
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
| Garcı´a et al. (2010) [178] | anti-AD, anti-parasitic, anti-fungi and antibacterial | Multi-target LDA | Heterogeneous structural GSK-3 inhibitors | GSK-3 | The reported LDA model was significant, since one can use a single equation to predict the results of heterogeneous series of organic compounds in 42 different experimental tests instead of developing and using 42 different QSAR models. | |||||
| Bhadoriya et al. (2014) [179] | AD | 3D-QSAR using kNN-MFA | A series of 34 fused 5,6-bicyclic heterocycles | g-secretase | The developed kNN-MFA model highlighted the importance of shape of the molecules, that is, hydrophobic and steric descriptors at the grid points His83 and Ser183, Ser227 for γ-secretase binding interaction. | |||||
| Barreca et al. (2003) [180] | Anticonvulsa-nts and neuroprotectants | A four-point Catalyst HIPHOP pharmacophore | 14 noncompetitive AMPA receptor antagonists | AMPA | This hypothesis, which consisted of two hydrophobic regions, one hydrogen bond acceptor and one aromatic region was employed to screen the Maybridge database and select eight compounds for testing of which six of these were found to be active in vivo as anticonvulsants. | |||||
| Valasani et al. (2013) [181] | AD | 2D-QSAR | Frentizole, benzothiazole-urea derivatives | ABAD | Based on QSAR studies of frentizole and benzothiazole-urea derivatives, authors designed and synthesized novel small drug molecules as benzothiazole-urea and frentizole phosphonate derivatives, which might have the capacity to cross the BBB and inhibit ABAD interaction. | |||||
| Kaur et al. (2000) [182] | AD | 2D-QSAR | Derivatives of physostigmine, tacrine, donepezil, huperzine A | AChE | It was concluded that all inhibitors were of hydrophobic nature as suggested by the presence of logP in the majority of QSAR models. Additionally, it was observed that all classes of inhibitors contained ionizable nitrogen. | |||||
| Zhou et al. (2015) [183] | AD | 3D-QSAR (CoMFA and CoMSIA), molecular docking, and MD | 60 tacrine derivatives | AChE | The contour maps for five fields obtained from the optimal 3D-QSAR models revealed that the steric and H-bond fields of these compounds were essential for their activities. Some key residues such as Tyr70, Trp84, Tyr121, Trp279, and Phe330 at the binding site of AChE were identified from molecular docking. | |||||
| Pourbasheer et al. (2015) [184] | PD | 3D-QSAR using CoMFA | A series of pyrimidines such as AA2AR antagonists | AA2AR | Based on the derived results some novel potent AA2AR antagonists have been designed and the proposed models were used to predict the AA2AR antagonist activity of newly designed compounds. | |||||
| Dinata et al. (2013) [185] | CJD | QSAR using LR, a statistical model of parabolic regression and multiple regression. | 2-aminothiazole derivatives | PrPSc | The results indicated that steric and lipophilic were the parameter most closely related to improve the biological activity of the compound 2-aminothiazole derivatives. | |||||
| Hajimahdi et al. (2016) [186] | AD, HD and PD | SW-MLR | A series of 53 potent 1,2-benzisothiazol-3-one derivatives | Caspase-3 (cysteine-dependent aspartyl-specific protease) | The results indicated that atomic masses, atomic Sanderson electronegativities, atomic van der Waals volumes and atom-centered fragments had a key role in regulating the caspase-3 inhibitory activity. | |||||
7. Quantum Mechanics Study
QM methods add an extra layer of detail to the classical potential descriptors, which account for the electronic changes that occur upon ligand binding to a receptor. These descriptors are useful in giving qualitative information regarding protein–ligand interactions, which can then be used to guide medicinal chemistry in ligand design [187]. QM methods have been applied for many CADD problems, such as: describing molecular interactions, providing estimates of binding affinities, determining ligand energies, refining molecular geometries, scoring of docked protein–ligand poses, describing molecular similarity, and deriving descriptors for QSAR. Divided into two sections, the first will examine QM methods in structure-based drug discovery (SBDD), particularly in optimizing structures and calculating interaction energies. The second section will focus on ligand-based applications, particularly in the exploration of conformational space, similarity measurements and in the era of QM-based QSAR descriptors [188].
Rahman et al. [189] employed density functional theory to design a series of halogen-directed donepezil drugs to repress AChE activity. Nonplanar piperidine rings can adopt either the chair or boat-like conformation, so according this, authors considered the chair as well as the boat conformation prior to halogen-directed modifications on donepezil. DFT employing B3LYP/MidiX and B3LYP/6-311G+(d,p) level of theory had been applied to optimize both the chair and boat conformers of the donepezil and halogenated ligands. Gibbs free energy, enthalpy, partial charge distribution, molecular μ and molecular orbitals calculations of these ligands were also explored to comprehend how halogenation affect the ligand structure and control the non-bonding interactions with the acetyl cholinesterase. The HOMO-LUMO gap of these modified ligands were reasonably lower compared to that of donepezil, which revealed that these compounds were more chemically reactive. Moreover, with the aid of molecular docking calculation, they reported halogenated drugs interaction with different binding sites of AChE and indicated that donepezil and halogenated chair-formed ligands have non-covalent interactions mostly hydrophobic and π-stacking type with hydrophobic gorges and anionic subsites of AChE. Generally, modification with halogens remarkably enhanced the μ and polar nature of the modified ligands which made them thermodynamically more stable as apparent from enthalpy and Gibbs free energies.
Silva et al. [190] proposed two novel drug candidates with higher and selective inhibitory activity towards AChE using CADD techniques including MD simulations, molecular interaction field, density functional calculation, docking single calculations, VS along with ADME screening. In this study, the Density Functional method employing B3LYP/6-31G* level of theory was utilized to obtain reliable initial 3D geometry of ligands. The Proposal 1 was gotten by altering donepezil through introduction of an amino group on the phenyl ring at the para position in regard to the piperidine ring, prompting to new extra hydrogen bond with AChE. Proposal 2 did not display good ADME parameters. The second potential AChEI presented in this study was the Proposal 3, which had a familiar benzodiazepenic framework and was chosen by VS in a drug-like collection (Fig. 41). Through the interaction analysis of the AChE complexes of these new two proposed potential pharmaceuticals by docking studies using GOLD 3.1.1, authors acquired a GolsScore of 56.2, 63.2, and 62.0 for donepezil, Proposal 1, and the Proposal 3, respectively, suggested hypothetically that the proposed new potential pharmaceuticals could be promising AChEIs. Moreover, the potential of the proposals to cross the BBB was evaluated by polar surface area (PSA) and log P values proposed that the two new proposed molecules, Proposal 1 and 3, presented good ADME characteristics.
Small molecule inhibitors that bind tau protein-bearing NFT present the promising strategy for optimization of binding properties for the premortem differential diagnosis and staging of AD and other tauopathic NDDs. To investigate the correlation between ligand polarizability and site occupancy, Cisek et al. [191] examined the ability of two closely related benzothiazole derivatives ((E)-2-[[4-(dimethylamino)phenyl]azo]-6- methoxybenzothiazole) and ((E)-2-[2-[4(dimethylamino)phenyl]ethenyl]-6- methoxybenzothiazole)) in a different polarizability state to displace probes of high (thioflavin S) and low (radiolabeled (E,E)-1-iodo-2,5-bis(3-hydroxycarbonyl-4-methoxy) styrylbenzene; IMSB) density sites. Quantum property calculations using hybrid density functional B3LYP and the 6-311++G(d,p) basis set were conducted to obtain electronic properties, as implemented in Gaussian 09 (G09) software package. QM computations indicated that highly delocalized electronic structure exhibited considerable π-electron delocalization improved by the introduction of electron donating and accepting moieties. The findings suggested that electron delocalization stimulated the displacement of Thioflavin S and IMSB probes from tau lesions that exist at high density and that enhancing this property of ligand structure presented a strategy for optimization of binding properties for superior diagnostic and therapeutic performance (Table 6).
Table 6.
Compound structures and characteristics.

8. QM/MM Study
The hybrid QM/MM computation is a compromise between the speed and accuracy, which allows for detailed analysis of ligand association for understanding or predicting their interactions. The QM/MM methods have been widely employed during the last decades to study chemical processes such as enzyme-inhibitor interactions [192].
Da Cunha et al. [193] applied docking technique to study some piperidine derivative inhibitors of AChE and further proposed structures of six new AChEIs as potential new drugs against neurodegenerative disorders. Initially, the 3D structure of each piperidine derivatives was geometry-optimized with no restriction in vacuum, and subsequently using the AM1 semiempirical method, in order to assign the partial atomic charges. Then, the compounds were docked into the AChE binding site using the Molegro Virtual Docker (MVD), and some relevant residues of the protein were considered flexible during the docking simulation. The relative binding energy was computed at the QM/MM level from the selected orientations obtained from docking calculations to investigate the influence of electronic effects on the relative binding energy. In this hybrid QM/MM strategy, a specified region around the active center was treated at an appropriate level of quantum chemistry theory, while the rest of the protein was described by a MM force field. The QM calculations were carried out with the B3LYP hybrid functional, which consists of HF exchange, Slater exchange, Becke exchange, Vosko–Wilk–Nusair correlation and the correlation functional of Lee, Yang, and Parr (LYP). For the MM part of the QM/MM calculations, Amber force field was used. The relative binding energies at the QM/MM level showed a very good agreement with the docking energy calculation. The best inhibitor proposed was submitted to additional MD simulations steps using the GROMACS 4.5.4 package. The results indicated that compound G had presented better values than donepezil. Compounds D, E, J, K, and N showed theoretical inhibitory potencies better than experimentally tested drugs (Table 7).
Table 7.
Proposed structure of new potential inhibitors.

HDACs are Zn2+-dependent metalloenzymes that catalyze the hydrolysis of acetyl functional groups from the ε-N-acetyl lysine amino acids of both histone and nonhistone proteins. It is much more challenging to simulate metalloproteins based on the empirical MM techniques, so, higher level theoretical models to investigate of protein–ligand binding, and also reactivity, are needed. Gleeson and Gleeson [194] employed hybrid QM/MM methods to study substrate binding and reactivity at HDAC8, which has a putative pathogenic role in neurological conditions including AD and FRDA. The aim of this study was to develop and validate QM and QM/MM HDAC8 models for predicting new Zn binding moieties, and especially analyzing the interactions between new inhibitors and HDAC proteins that are suboptimally described by traditional molecular mechanic methods. QM and QM/MM simulations were carried out using the Gaussian 09 program package, which the former used a polarizable continuum solvent model consisting of water while the latter employed the n-layered integrated molecular orbital molecular mechanics (ONIOM) method with the electronic embedding scheme. The calculations showed that the results obtained from relatively modest QM/MM methods for HDAC8 deacetylation are qualitatively similar to those obtained from higher basis set calculations or results where free energy effects have been incorporated.
Lucas et al. [195] used the combined QM/MM-QTAIM (Quantum Theory of Atoms in Molecules) analysis to describe the interactions between bapineuzumab and different forms Aβ peptide ((AβWT and AβN3 (pE)) which allowed to understand why the missing of two first residues (Asp1 and Ala2) from the isoforms decreased the affinity between the antibody and peptides which begining with a pyroglutamate residue. The ONIOM scheme permits a partition of the molecular system into high and low level layers, which in this study the paratope residues obtained from per residue energy decomposition, the Aβ peptide and the water molecules were considered at he QM level, and rest of the system was in the low-level MM layer, using the AMBER force field. The binding energy of each Aβ–antibody complexes was computed as single-point energy calculations at B3LYP/6-31G (d) level of theory, with basis set superposition error (BSSE) corrections, using the geometries obtained by QM/MM calculations.
9. Monte Carlo Study
Search algorithms like quick explore (QXP) are based on Monte Carlo searching gotten from the strategy for Monte Carlo perturbation followed by energy minimization in Cartesian space which has been employed to design potent and selective inhibitors of hBACE1 for preventing AD by lessening the formation of neurotoxic Aβ aggregates [196]. In this study, flexible ligand docking calculations was carried out using the QXP Monte Carlo docking algorithm mcdock along with CombiDOCK. QXP optimized grid map energy and internal ligand energy to identify the structures of the receptor-ligand complex with minimum free energy. The search algorithm executed a rigid body alignment of ligand-receptor complex with Monte Carlo minimization translation and rotation of ligand, which was coupled with another rigid body alignment and scoring using energy grid map.
10. MD Simulation Study
In some cases, MD simulations of the protein target have been carried out before docking to explore the conformational space of the protein receptor, which differs from the available crystal structure(s). Moreover, MD simulations after docking could be performed to optimize the final structures, analyze the stability of different complexes, and account for solvent effects as final filter in silico or to guide chemical synthesis for hit optimization [197]. Identification of cryptic or allosteric binding sites, thermodynamics and kinetics of small molecules binding to proteins, generation of protein conformation ensembles for in silico drug design, validation of binding modes predicted by docking, elucidation of mechanism of action of drugs and natural ligands are important applications of MD in drug discovery [198]. Enhanced sampling methods are often coupled to MD to accelerate this process and to retrieve useful thermodynamic and kinetic data. Of the enhanced sampling techniques developed in recent years, we focus here on free-energy perturbation (FEP), steered MD, and metadynamics [199]. FEP calculations are based on MD simulations that explicitly consider conformational flexibility and entropy effects through the use of a physics-based force field to describe molecular interactions and explicit solvent to model the real environment of the protein binding site. FEP calculations have demonstrated astonishing potential for rational lead optimization [200]. Steered MD is a time-dependent external potential which is applied to the ligand to facilitate its unbinding from the target protein. Ligand undocking is therefore accelerated by acting on a descriptor (or CV), which is usually the protein−ligand distance or a vector describing the ligand exit pathway [201]. Metadynamics allows fast exploration of the underlying free-energy landscape of rare events using a set of order parameters, usually referred to as collective variables (CVs), that approximate the true reaction coordinate of the process [202]. Here, we provided an overview of recent applications of MD in drug design for NDDs.
As a valuable example describing FEP as a tool for drug design, Ciordia and coworkers [203] applied FEP/MD-guided lead optimization to design of novel series of BACE1 inhibitors. The authors explored FEP to design and prioritize molecules with substituents to fill the P1−P3 pockets. They first reported a retrospective application studied 32 molecules and showed good correlation between predicted and experimental binding energies. Then they performed prospective FEP binding energy predictions on a set of 18 molecules from spiroaminodihydropyrrole scaffold and subsequently 9 examples were synthesized based on the results. The results indicated good correlation between predicted and experimental binding energies, providing further evidence that FEP can be used as a tool to assist lead optimization, even for BACE1. The FEP approach outperformed docking and MM-GBSA methods.
MAO is a flavoenzyme bound to the outer mitochondrial membrane in most cell types in the body, which exists in two subtypes, designated MAO-A and MAO-B, each having different substrate preference and inhibitor specificity. Deficiency in the catabolism of monoaminergic neurotransmitters and oxidative damage by MAO play an important role in the physiology of NDDs including AD, anti-depressant and PD. So, Braun et al. [204] employed a wide range of computational approaches including VS simulations, flexible docking using GOLD 3.1.1 and MD to identify new inhibitors with higher selectivity against MAO-B enzyme. Selective and potent inhibitors of MAO-B, like rasagiline analogues as irreversible inhibitors and lazabemide as reversible one, with different structural features were taken from the published literature and four new therapeutic derivatives with higher activity than orginal ones were designed by molecular modifications based on molecular hybridization of the pharmaceuticals rasagiline and lazabemide. Furthermore, two proposals were derived from VS simulations using the ChemBridge EXPRESS-Pick compound collection containing 448,532 compounds. The proposed compounds (Fig. 42) were optimized at B3LYP/6-31G* level of theory using Gaussian 03 series of programs. The four proposals derived from molecular hybridization interacted with the MAO-B binding site through Tyr326, Tyr435, and Tyr398. Proposals 1 and 2 made π-stacking interactions with MAO-B, while proposals 3 and 4 made both hydrophobic and hydrophilic interactions with the MAO-B active site. The main interactions for two proposals derived from VS observed with the conserved tyrosine residues and Phe168. Computation of MIFs for Proposals 2 and 4 using water probe exhibited a good correlation with the water MIF, represented a favorable hydrogen bond interaction between active site of MAO-B and respective moieties of the proposed compounds. The results of MIF studies for the molecules (compounds 1 and 2) retrieved from VS suggested a favorable hydrogen bonded interaction between the molecules of MAO-B and one of the aromatic rings of the compound 1 and also with the two side chains of the compound 2. Based on the docking studies and Lipinski's RO5 criteria, Proposal 3 was suggested as the most promising inhibitor and a 1500 ps MD simulation of Proposal 3 inside the MAO-B active site revealed that Proposal 3 was stable after a long trajectory. Finally, the potential toxicity we also have investigated for the best molecule, Proposal 3.
Integrating an IFD method along with MD and/or QM/MM simulations can be useful for the efficient description of induced molecular flexibilities within the protein–ligand complexes and also for accurate binding-mode analysis of ligands. In this regard, Distinto and coworkers [205] used IFD and MD simulations to unravel the putative binding modes and activities of 1-aryliden-2-[4-(4-chlorophenyl)thiazol-2-yl]hydrazines against the MAO-B enzyme, a therapeutic target for the treating of neurodegenerative disorders. By structural alignment of twenty X-ray structures of MAO-B co-crystallised with different inhibitors, it was found that the enzyme adopted induced-fit changes with respect to the bound ligands. Hence, the authors initially performed IFD using the Schrodinger drug discovery suite, during which the side chains near the inhibitor were kept flexible. The results from the IFD explained how ligand binding tended to induce structural changed in the protein. However, many of the compounds showing two binding modes were ranked high in IFD. To determine the best binding mode of the inhibitors, the authors performed 3–5 ns long MD simulations for both the binding modes of two of the top-ranking compounds from IFD. The MD results followed by the free energy calculations highlighted the significance of the fluorine atom interacting with water near the cofactor and the influence of the steric bulkiness of substituents in the arylidene moiety. The authors proposed that the pharmacophore features of these experimentally synthesised compounds, developed using combined IFD, MD and free energy calculations, should be useful for achieving novel high-affinity MAO-B inhibitors for the treatment of NDDs.
Recent experimental reports have shown Abelson tyrosine kinase (c-Abl), a non-receptor kinase, as a new target for NDDs including AD and PD. Palakurti and Vadrevu [206] applied energy optimized multiple pharmacophore modeling, VS and docking coupled with MD simulations to identify highly potent inhibitors against c-Abl. The e-pharmacophore models generated from four c-Abl crystal structures bound with ligands in the inactive ATP binding conformation (DFG-out). The energy-based pharmacophore models were validated using the percentage of yields, enrichment factor, false positives, false negatives and goodness of hit score. The developed models were then employed for high-throughput VS using the ChemBridge_CNS database and docking to identify leads which led to the identification of 10 top tanked molecules. MD simulations of the best three complexes indicated that compounds A and C (Fig. 43) were the most stable systems with persistent interactions with the active site residues, which were in very good agreement with the computed binding affinities for the top three hit compounds.
In addition to using experimental techniques to structurally characterize the receptor in ensemble docking, MD simulations are an obvious way to obtain multiple conformations of macromolecular targets. An example of using MD to incorporate target flexibility into standard docking calculations is the work by Pang and Kozikowski [207] which extracted multiple conformations of the AChE enzyme from a 40 ps trajectory and used these to successfully predict, through rigid docking, the bound pose of huperzine A. Based on docking results, the authors predicted that huperzine A binds to the bottom of the binding cavity of AChE (the gorge) with its ammonium group interacting with Trp84, Phe330, Glu199 and Asp72 (catalytic site).
Cavalli et al. [208] performed combined docking and MD to provide an explanation of the molecular mechanism of hAChE inhibition carried out by the small-molecule peripheral site ligand propidium. Initially, two different docking protocols followed by cluster analyses were carried out using the DOCK 4.0.1 package to identify several orientations of propidium within the PAS of hAChE and then these initial poses investigated using MD which eventually two alternative binding modes were identified. Both binding modes were compatible with the electron density map, with one resembling the crystallographic pose and the other being flipped by 180°. This study showed that even a few ns of MD simulations could discriminate good docking poses from bad, when surface solvent-exposed binding sites were considered.
Because of the high plasticity of the flap and loop regions of BACE1, the binding site of this enzyme exhibits remarkable conformational flexibility, which explains the importance of considering multiple pocket rearrangements in structure-based hit discovery. Multiple receptor conformations (MRC) virtual ligand screening was indeed adopted by several groups as a straightforward approach to consider the receptor flexibility, and it contributed to identify new hits in BACE-1 inhibitors drug discovery campaigns. To tackle multiple protonation states of the BACE-1 catalytic dyad and the conformational flexibility of the active site at the same time, Kacker et al. [209] used an integration of QM calculations, docking, MD, and conformational ensemble virtual ligand screening to define the protonation state of BACE-1’s catalytic machinery in complex with different ligands. The DFT study was performed on a representative structure from seven clusters, obtained based on the functional groups of the 47 inhibitors interacting with the BACE-1 catalytic dyad, to find the most energetically favorable dyad protonation state in presence of a certain interacting group. The MD simulation was used to remove the protonation states in which strong H-bond interactions with dyad and a stable binding of the substrate could not be kept up throughout the trajectories. Self-docking simulations were done on each receptor conformation considering independently the all five protonation states to evaluate the specific protonation states in ligand binding. After highlighting the contribution of dyad titration in complex formation by means of self-docking, cross-docking simulations were permored to explore the impact of receptor plasticity upon inhibitor binding. Based on the information obtained from QM and MD simulations, two sets of BACE-1 variants were compiled and tested in an virtual ligand screening protocol for hit discovery campaign.
HD is caused by an expanded CAG repeat in the 5’-end of the HTT mRNA. Discovering ligands with high affinity for mRNA transcripts of pathogenic length constitutes a potential strategy against HD. Bochicchio and coworkers [210] used well-tempered metadynamics (WT-meta) to evaluate binding pose and free energies of two ligands, 4-((diaminomethylene)amino)phenyl-4-((diaminomethylene)amino)benzoate (ligand1) with Kd=60(30) nM and 6-(4,5-dihydro-1H-imidazol-2-yl)-2-(4-(4,5-dihydro-1H-imidazol-2-yl)phenyl)-1H-indol-3-amine (ligand 2) with Kd=700(80) nM, for which the experimental affinities have been previously determined. The results reproduced the experimental affinities and uncovered the recognition pattern between ligands’ and their RNA target.
Biarne´s and coworkers [211] reported a new approach named enhanced molecular docking (EMD), which integrates molecular docking calculations with metadynamics-based free energy simulations, to investigate the association between PrPC and ligand GN8 (2-pyrrolidin-1-yl-N-[4-[4-(2-pyrrolidin-1-yl-acetylamino)-benzyl]-phenyl]-acetamide), the main agent involved in prion diseases (Kd= 3.9 μM). To find multiple binding sites pattern of GN8 onto PrPC surface, ligand GN8 with three protonation states (0, +1, +2) was docked against PrPC. MD simulations were used initially to relax the structures and to explore the role of hydration. Secondly, representative PrPC structures were chosen by statistical analysis of the MD simulation. Standard docking calculations were then used to predict the formation of ligand-target complexes between the ligand GN8 and these protein structures, provided a first guess of putative binding regions. The CVs were selected from analysis of the standard molecular docking protocols outcomes. The results indicated that GN8 may bind in opposite parts of the protein surface. The authors concluded that, this new protocol in which standard molecular docking protocols are extended with enhanced sampling simulations can be useful in the identification of small organic molecules that interfere with cavity-less proteins such as PrPC.
Purine nucleoside phosphorylase (PNP) impairment raises d-guanosine concentrations in the blood, leading to apoptosis in both dividing and nondividing lymphoid cells due to the aggregation of dGTP, which can inhibit ribonucleotide reductase activity. PNP inhibitors could potentially be employed to cure T-cell mediated diseases and autoimmune diseases such as transplant (allograft) rejection, ALS, rheumatoid arthritis, gout and MS. Decherchi et al. [212] ran extensive unbiased MD simulations (about 1μs each) with machine learning algorithms to capture the key structural and dynamical features of a transition state analog (DADMe-immucilin H) inhibitor (Fig. 44) binding to PNP. Authors implemented k-medoids algorithm to find a set of interpretable and meaningful mesostates that were generated from 13 μs of MD simulations. Consequently, three independent binding paths were recognized, prompting to various PNP–DADME binding configurations. They biased the simulations by carrying out scaled MD runs to provide an estimate of the relative residence time of DADME in the three ensembles (A, B and C) retrieved from the clustering protocol. Despite observing spontaneous binding through different routes, the authors used the pathways obtained by MD simulations to determine the free-energy profiles associated with the diverse binding mechanisms. To construct the free energy profile, authors employed Kernel-based Regularized Least Squares algorithm, a machine learning method, to nonlinearly interpolate the mean force and obtain smooth free energy curves. In general, by combining MD with machine learning and enhanced sampling methods, authors could observe the binding and estimate the associated kinetics (konandkoff) and thermodynamics which is in good agreement with available experimental data. In addition, they advanced a hypothesis for the slow-onset inhibition mechanism of DADMe–immucillin-H against PNP.
In a recent study, Srinivasan and Rajasekaran [213] proposed a molecular mechanism for the inhibition of aggregation and destabilization in the Ala4Val mutant SOD1, with the natural polyphenolcurcumin using docking, MM-PBSA and SMD for providing insight into the cures for the predominant neurodegenerative disorders. In view of that, curcumin was computationally docked with both the native and mutant SOD1, using Autodock and results elucidated that curcumin greatly binds to mutant SOD1 with increased hydrophobic interactions as compared to native SOD1. Further investigations were accomplished, using steered MD and conformational sampling on both the bound complexes of native and mutant SOD1 with curcumin to unravel the effect of disaggregation. Moreover, authors also elucidated the variations in the free energy landscape of native and mutant SOD1 in their unbound and bound states which indicated a reduction in the toxic aggregates formed in the mutant SOD1 upon binding with the curcumin.
Padhi and coworkers [214] employed extensive MD simulations to gain insights into the molecular mechanisms of all known ALS-related angiogenin mutants and hence diseases pathogenesis. They indicated that the loss of ribonucleolytic function is caused by conformational change of catalytic residue His114, while reduction in solvent-accessible surface area (SASA) of 31RRR33 (signal residues) due to local misfolding resulted in loss of nuclear translocation activity. The results agreed very well with the reported experimental findings.
Niemann Pick C2 (NPC2) is a sterol transfer protein in late endosomes and lysosome of mammalian cells with broad sterol ligand specificity. Absence or dysfunction of this protein causes NPC2 diseases, a neurodegenerative disorder with endosomal accumulation of cholesterol and other lipids. Poongavanam and coworkers [215] employed MD simulations based ligand binding free energy calculations (MM-PBSA calculations) to obtain insight into the broad sterol ligand specificity of NPC2 and investigate the structural dynamics of the sterol -NPC2 complexes of the wild type NPC2 protein and of various NPC2 mutations. Using MD simulations and MM-PBSA calculations, authors overall ranked the various sterol ligands correctly compared to the experimentally determined binding affinity relative to the fluorescent sterol DHE (dehydroergosterol). They showed that a structural requirement of high affinity sterol ligands to NPC2 is an aliphatic side chain buried inside the NPC2 binding pocket. All sterols were docked into the NPC2 binding pocket to obtain an initial binding orientation (pose) and this was followed by MD simulations based free energy binding calculations i.e. MM-PBSA. From MD simulations, they proposed a general mechanism for NPC2 mediated sterol transfer, in which Phe66, Val96 and Tyr100 act as reversible gate keepers. These residues stabilize the sterol in the binding pose via π-π stacking but move transiently apart during sterol release. Furthermore, they found that an aliphatic side chain in the sterol ligand results in strong binding to NPC2, while side chain oxidized sterols gave weaker binding.
CB enzyme is one of the well-characterized lysosomal cysteine proteinase of the papain family which is involved in the pathogenesis of NDDs through neuronal apoptosis and activation of caspase 3. CA-074Me inhibitor is a cell-permeable derivative of CA-074 selective for the CB as verified by in vivo investigations. Due to the lack of X- ray crystal structure of this inhibitor with the hCB protein, Mashamba-Thompson and Soliman [216] employed a variety of computational approaches including MD simulations and MM-GBSA binding free energy calculations to explore the binding mode of CA-074Me to hCB. Furthermore, this study was also identified new CB inhibitors based on the structural features of CA-074Me, the most known prototype, using fragment-based scaffold hoping; structure-based VS and validation of docking protocol by MD simulations and binding free energy calculations. Mcule scaffold hopping tool was employed to generate novel structural scaffolds based on CA-074Me structural features and structure-based VS using Autodock Vina screening software was then performed to rank the created compound library with respect to binding affinities towards CB protein. MD simulations and free energy calculations of binding were also carried out to assess the validity of docking simulations and ensure the binding stability of the compounds in the active site of the hCB. Per-residue energy decomposition revealed that amino acid residues Cys29, Gly196, His197, and Val174 contributed most to the total binding energy. Two new compounds, Hit1 (ΔG= -46.40 kcal.mol-1) and Hit2 (ΔG= -45.01 kcal.mol-1) (Fig. 45), with better binding activities relative to prototype inhibitor, CA-074Me (ΔG= -44.38 kcal.mol-1), were identified in which the insertion of heterocyclic rings attached to the epoxy ring enhaned ligand binding.
Proteins participate in biochemical interaction networks by switching among structural substates, which are induced by ligand binding and fine-tune their functions. Recently, Sattin and coworkers [217] designed a library of small molecules that activate 90 kDa Heat Shock Protein (Hsp90) ATPase by targeting an allosteric site, with the goal of exploring their biochemical and cellular effects. The Hsp90 molecular chaperone is hub protein that the disregulation of function is associated with its overexpression and has been shown to be at the basis of disease states, such as cancer and neurodegeneration. In the next study [218], through the use of docking calculations and comparative analyses of MD simulations of different complexes of Hsp90 with the allosteric ligand compounds, and known inhibitors as controls, they provided molecular level insights into how ligand binding at an allosteric site can affect protein structure, dynamics and, consequently, enzymatic activity. They developed structure-dynamics-activity relationship (SDAR) model that links the physico-chemical properties of the small molecules to experimentally measured activation effects. To achieve this model, they performed a Docking over Multiple Receptor Structures (DMRS) approach, originating from the ATP-only dynamics: the 10 representatives of the most populated conformational clusters obtained from the ATP-only simulation were extracted from the MD trajectory and compounds 1, 4, 10, 12, 16, 18 (Fig. 46) were re-docked into each of the 10 representative structures. This study provided the stage for the use of computational approaches to rationally design chaperone activators with considering the conformational dialogue between allosteric ligands and protein conformations.
Bromodomains are protein modules found in chromatin-associated proteins, that specifically read the acetylation marks of histones in the epigenetic regulation. Mounting evidences correlate the dysfunction of bromodomain contained proteins with human diseases, such as neurodegeneration, cardiovascular disease and cancers. Ran and coworkers [219] studied a series of 20 known bromodomain inhibitors (IC50=35-51200 nM) using an integration of computational methods, including molecular docking, interaction fingerprints by protein-ligand interaction fingerprint (PLIF), MD simulation and binding free energy calculation to understand the interaction mechanism of bromodomain inhibitors which may facilitate the rational design of novel small molecule bromodomain inhibitors. To obtain detailed information of protein-ligand interaction, interaction fingerprints by PLIF was employed on bromodomain-inhibitor complexes acquired
from crystallization and molecular docking, and five fingerprints were present in twenty bromodomain-inhibitor complexes, including surface contact interaction (C), backbone H:bond acceptor (a) and donor (d), and sidechain H:bond acceptor (A) and donor (D). MD simulations in aqueous solution were done on all twenty bromodomain-inhibitor complexes to investigate the residue flexibility and the hydrogen bonds in the bromodomain-inhibitor interaction. The binding free energy calculation using MM-PBSA and MM-GBSA was implemented on the ensemble conformation obtained by MD simulation, and then energy decomposition was performed to compute the interaction in terms of energy contribution from individual residues. These integrated results determined two hot spots in the active site of the bromodomain, where the hydrophobic hot spot formed by Trp81, Val87, Leu92 and Ile146 played a critical role in the interaction, and the hydrogen-bond hot spot mediated by Asn140 displayed moderate contribution to the binding affinity of the bromodomain inhibitors.
MD simulations can also be used to identify druggable allosteric sites. Activators can help identify and characterize allosteric sites and mechanisms for the discovery of novel drug candidates, overcoming the limits of classical active-site oriented drug design. Morra et al. [220] developed a new methodology for computational design of allosteric inhibitors of molecular chaperones, which has proven as a key target for cancer and neurodegeneration drug discovery. They integrated MD/dynamic pharmacophore approach for identifying and targeting allosteric hot spots on the NTD of the molecular chaperone Hsp90. For generating dynamic pharmacophore model, all protein conformations from the MD were overlaid, and local MIF minima were computed at the allosteric site with the GRID v22a with the probes DRY (hydrophobic), O (sp2 carbonyl oxygen) and N1 (neutral flat amide NH). The allosteric dynamic pharmacophore model composed of a 3D arrangement of six features (i.e., four hydrophobic regions and one each hydrogen-bond acceptor and donor) was used to screen NCI repository to obtain small molecules with the functional and conformational properties required to bind these “hot spot” allosteric sites. Autodock Tools v1.5 was employed to characterize the molecular interactions of the newly discovered compounds with the Hsp90 C-terminal domain. Experimental tests indicated that fourteen of the selected compounds obtained from the VS displayed antiproliferative effects in various tumor cell lines, while not affecting proliferation of normal human cells.
HtrA2 (High temperature requirement protease A2) is a mitochondrial serine protease protein with a single PDZ (post-synaptic density-95/discs large/zonula occludens-1) domain per monomer, which is involved in important biological functions and pathogenicity like cancer and neurodegenerative disorders. Bejugam et al. [221] applied an in silico and biochemical method to show that a new non-canonical selective binding pocket in HtrA2 take part in the allosteric activation of HtrA2. To understand the conformational dynamics and structural plasticity during HtrA2 allosteric activation, MD simulation with a bound peptide activator was employed. Authors modelled the entire mature protease by completing in the missing residues using Prime and identified the putative binding site(s) on HtrA2 using SiteMap 2.5. Site2 or selective binding pocket (SBP) that encompasses the groove created by SPD (serine protease domain)-PDZ linker, protease, and PDZ domains was selected based on optimum energy parameters among the five possible putative binding sites. Generated peptide library based on literature reports and structural complementarities were docked at SBP and MD simulation of the docked structures was employed by Desmond 2010, which yielded important information on loop and linker movements in HtrA2. These findings along with mutational and enzymology investigations suggested that during binding of the peptide activator at this alternative non-canonical PDZ binding site, the linker at the PDZ-protease interface and loops L1, LA and LD around the catalytic groove undergo conformational fluctuations in order to create an efficient active site pocket.
Concluding Remarks
Computational techniques, for example, data mining, homology modeling, MD simulation, cheminformatics, VS, molecular docking and QSAR modeling have provided a powerful toolbox for target identification, discovery and optimization of drug candidate molecules. This review concentrates on different computational approaches effectively applied in NDDs drug discovery processes (years between 1996 and 2016). Detailed analysis of recently published examples of case studies uncovered that the dominant part of them utilize a successive integration of ligand- and structure-based methods in VS, with specific concentrate on pharmacophore and docking modeling. From this review, it is observed that, most of the in silico studies performed have been for single biological targets, but as we have discussed the NDDs have a multifactorial pathoetiological origin, which involves concurrent malfunctioning of more than single target. Considering this fact, recently, scientists have become persuaded that a multi-target therapeutic strategy aimed at the simultaneous targeting of multiple proteins (and therefore etiologies) involved in the development of a disease are recommended in future. Thus, the rational design of new leads as versatile inhibitors for different targets associated with NDDs constitutes a major goal.
Fig. (21).

Chemical structure of analogs of caulerpin.
Fig. (22).

2D structure of ZYQ1.
Fig. (23).

Parent compound 7 and its analogs (14-20).
Fig. (24).

Molecular structures of the identified new inhibitors of the immunoproteasome.
Fig. (25).

2D strucrures of compounds 3 and 5.
Fig. (26).

Compound (±)-11.
Fig. (27).

Chemical structure of CPN-9 (N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide).
Fig. (28).

Workflow of the mx-QSAR study.
Fig. (29).

General structure of donepezil, ASS234, PF9601N, and the novel MAOI/ChEI hybrids I and II, described in this work.
Fig. (30).

Chemical structure and binding mode of inhibitor 15.
Fig. (31).
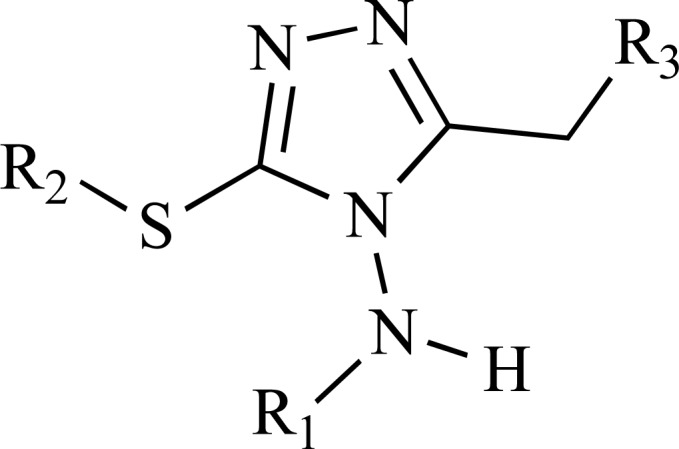
Chemical structure of congeneric series of 224 4-amino-5-methyl-4H-1,2,4-triazole-3-thiol derivatives.
Fig. (32).

2D representation of benzylpiperidine derivative IIa.
Fig. (33).

a) 2D structure of common moiety for a series of 6-arylquinazolin-4-amine derivatives and b) The HQSAR contribution maps of the most (24, left) and least (6, right) active compounds, according to the two best HQSAR models.
Fig. (34).

Fragment contribution map for the most potent and least potent compounds obtained using HQSAR.
Fig. (35).

Three regions of molecules considered in the study.
Fig. (36).

Chemical scaffolds of a) 63/Donz-D9 and b) 71/MBA-VEG8.
Fig. (37).

2D structure of common moiety of DHP derivatives.
Fig. (38).

Graphical representation of (A) HIC, and (B) DHC.
Fig. (39).

Chemical structure of tetracycline with atom numbering and ring labels of the hydronaphthacene moiety.
Fig. (40).

Selected regions of interest for SAR evaluation.
Fig. (41).

Molecular structures of AChE inhibitors: (A) new proposed inhibitor by modifying Donepezil; (B) Donepezil; (D) hybrid; (E) inhibitor obtained by virtual screening.
Fig. (42).

Structures of proposals 1 to 4 of molecular hybridization of rasagiline with lazabemide, and two compounds selected by virtual screening from the database.
Fig. (43).

2D structure of compounds A and C.
Fig. (44).

2D structure of DADMe-immucillin-H.
Fig. (45).

2D representation of a) CA-074Me, b) Hit 1 and c) Hit 2.
Fig. (46).

Chemical structures of compounds 1, 4, 10, 12, 16, and 18.
Acknowledgements
Declared none.
List of abbreviations
- 0D
Zero-dimensional
- 1D
One-dimensional
- 2D
Two-dimensional
- 3D
Three-dimensional
- 4D
Four-dimensional
- 5D
Fifth dimension
- 5-HT
Serotonin
- 5HT4R
Serotonin 4-receptor
- 5-HT6R
Serotonin-6 receptor
- 6D
Sixth dimension
- AA2AR
Adenosine A2A receptors
- ABAD
Amyloid-binding alcohol dehydrogenase
- AChE
Acetylcholinesterase
- AChEI
AChE inhibitors
- AD
Alzheimer's disease
- ADHD
Attention deficit hyperactivity disorder
- AGD
Argyrophilic grain disease
- ALS
Amyotrophic lateral sclerosis
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor
- ANN
Artificial neural networks
- Asp
Aspartate
- ATX
Autotaxin
- Aβ
Amyloid-β
- Aβ
Neurotoxic β-amyloid
- AβPP
Amyloid-β precursor protein
- BACE
β-Site amyloid precursor protein cleaving enzyme or amidine containing spirocyclic β-secretase 1
- BBB
Blood-brain barrier
- BChE
Butyrylcholinesterase
- BLAST
NCBI Basic Local Alignment Search Tool
- BLR
Binary logistic regression
- BRGNNs
Bayesian-Regularized Genetic Neural Networks
- BSSE
Basis set superposition error
- c-Abl
Abelson tyrosine kinase
- CADD
Computer-aided drug design
- CASTp
Computed Atlas of Surface Topography of Proteins
- CB
Cannabinoid receptors
- CB
Cathepsin B
- CBD
Corticobasal degeneration
- CDK5/p25
Cyclin-dependant kinase 5/p25
- CERMN
Centre d’Etudes et de Recherche sur le Médicament de Normandie
- ChE-I
Cholinesterase inhibitor
- CJD
Creutzfeldt-Jakob disease
- CK1d
Casein kinase CK1 delta
- COMBINE
COMparative BINding Energy
- CoMFA
Comparative molecular field analysis
- CoMSIA
Comparative molecular similarity indices analysis
- COMT
Catecholamine-O-methyltransferase
- COX
Cyclo-oxygenase
- CVs
Collective variables
- CYP
Cytochrome P450
- CYP2A6
Cytochrome P450 isoenzyme 2A6
- CYP2D6
Cytochrome P450 isoenzyme 2D6
- DB
Database
- DcpS
Scavenger decapping enzyme
- DDC
DOPA decarboxylase
- DFT
Density functional theory
- DHC
[(1r,4as,8aR) decahydronapthelene-1yl-3'-N-hydroxy-thiopene-2-carboxamide]
- DHI
Dihydrotanshinone I
- DHP
1,4-dihydropyridine
- DLB
Dementia with Lewy bodies
- DMRS
Multiple Receptor Structures
- DNN
Deep neural network
- DPI
Drug-Protein Interaction
- DRD3
Dopamine receptor D3
- DYRK
Dual-specificity protein kinases
- DYRK1A
Tyrosine-phosphorylation-regulated kinase-1ª
- EA
Electron affinity
- ED
Ensemble Docking
- EHOMO
Highest occupied molecular orbital energy
- EINECS
European Inventory of Existing Commercial Chemical Substances
- ELUMO
Lowest unoccupied molecular orbital energy
- EM
Ensemble methods
- EMD
Enhanced molecular docking
- eqBChE
Equine BChE
- ETM
Electron-Topological Method
- Etotal
Total energy of the molecule
- EWSR1
Ewing’s sarcoma RNA-binding protein 1
- FAAH
Fatty acid amide hydrolase
- FALS
Familial ALS
- FB-QSAR
Fragment-based QSAR
- FEP
Free-energy perturbation
- FFI
Fatal familial insomnia
- FLAP
Fingerprints for ligands and proteins
- FMO
Fragment molecular orbital
- FRDA
Friedreich ataxia
- FSI
Sporadic fatal insomnia
- FTLD
Frontotemporal lobar degenerations
- FUS
Fused-in-sarcoma
- GA
Genetic algorithms
- GABA-AT
γ-Aminobutyrate aminotransferase
- GA-VSS
Genetic algorithm-variable subset selection
- GCase
Glucocerebrosidase
- GFA
Genetic function approximation
- GGT
Globular glial tauopathies
- GOLD
Genetic optimization of ligand docking
- G-QSAR
Group-based QSAR
- GS
Gamma secretase
- GSK-3
Glycogen synthase kinase 3
- GSK-3β
Glycogen synthase kinase 3β
- GSS
Gerstmann-Sträussler-Scheinker syndrome
- H3R
Histamine H3-receptor
- hAChE
Human AChE
- HASL
Hypothetical active site lattice
- hBChE
Human BChE
- hCOMT
Human COMT
- HD
Huntington’s disease
- HDAC
Histone deacetylase
- HIC
N-hydroxy-5-(1H- imidazole-4yl) thiopene-2-carboxamide
- HiT QSAR
Hierarchical technology for quantitative structure-activity relationship
- HMN
Hereditary motor neuropathy
- HMT
Histamine N-methyltransferase
- HOMO-
Energy difference of HOMO and LUMO
- HOMO
Highest occupied molecular orbital
- HQSAR
Hologram quantitative structure activity relationship
- Hsp90
Heat Shock Protein
- HtrA2
High temperature requirement protease A2
- HTS
High-throughput screening
- HTT
Huntingtin
- Hypo
Hypotheses
- IBS
Interbioscreen
- IFD
Induced fit docking
- iGluRs
Glutamate-gated ion channels
- IMSB
Radiolabeled (E,E)-1-iodo-2,5-bis(3-hydroxycarbonyl-4-methoxy) styrylbenzene
- IP
Ionization potential
- Kapβ2
Karyopherine β2
- KMO
Kynurenine monooxygenase
- kNN
k-nearest neighbors
- LBVS
Ligand-Based Virtual Screening
- LDA
Linear discriminant analysis
- L-Dopa
Levodopa
- Lewy body
(LB)-associated disorders
- LNK
c-Jun N-terminal kinase
- LOORMSD
Leave-one-out root mean square deviation
- LPA
Lysophosphatidic acid
- LPC
Lysophosphatidyl choline
- LR
Linear regression
- LS-SVR
Least squares support vector regression
- LUMO
Lowest unoccupied molecular orbital LUMOgap
- LYP
Lee, Yang, and Parr
- MAO
Monoamine oxidase
- MAP
Mitogen activated protein
- MAPT
Microtubule-associated protein tau
- MCI
Mild cognitive impairment
- MD
Molecular dynamic
- MDR
Multidrug resistance
- MFTA
Molecular field topology analysis
- mGluR1
Metabotropic glutamate receptor 1
- MI
MARCH-INSIDE
- MIA-QSAR
Multivariate image analysis-quantitative structure-activity relationship
- MIF
Molecular interaction fields
- MLR
Multiple LR
- MM
Multiple myeloma
- MM/GBSA
Molecular Mechanics/Generalized Born Surface Area
- MoQSAR
Multi-objective Genetic QSAR
- MR
Molar refractivity
- MRC
Multiple receptor conformations
- MS
Multiple sclerosis
- MSA
Multiple system atrophy
- MSCS
Multiple solvent crystal structures
- MTDL
Multi-target-directed ligand
- mt-QSAR
Multi-target QSAR
- MVD
Molegro Virtual Docker
- mx-QSAR
Multiplexing quantitative structure–property relationship
- nAChRs
Nicotinic receptors
- NCI
National Cancer Institute
- NDDs
Neurodegenerative diseases
- NFT
Neurofibrillary tangle
- NMDA
N-methyl-D-aspartate
- NMDAR
N-methyl-D-aspartate receptor
- NMR
Nuclear magnetic resonance
- NNE
Neural network ensemble
- NOS
Nitric oxide synthase
- NPC2
Niemann Pick C2
- NS
Neural stem cell lines
- NSAIDs
Nonsteroidal anti-inflammatory drugs
- NTD
N-terminal domain
- NUDE
Nagasaki University Docking Engine
- p38α MAPK
p38α mitogen-activated protein kinase
- p75NTR
Neurotrophin receptor
- PAH1
Alpha-Helix 1
- PAM
Positive allosteric modulators
- PCM
Proteochemometrics modeling
- PD
Parkinson's disease
- PDE
Phosphodiesterase
- Pgp
P-glycoprotein
- PH
Pharmacophore hypothesis
- PiD
Pick disease
- PLIF
Protein-ligand interaction fingerprint
- PLS
Partial least-squares
- PMF
Mean Force
- PMS
Progressive multiple sclerosis
- PNP
Purine nucleoside phosphorylase
- PNS
Peripheral nervous system
- POP
Prolyl oligopeptidase
- PrP
Prion protein
- PrPC
Cellular prion protein
- PrPSc
Pathogenic prion protein
- PSA
Polar surface area
- PSP
Progressive supranuclear palsy
- PTH
Phenylthiazolylhydrazides
- PY-NLS
Proline/tyrosine nuclear localization signal
- Q2
Predictive squared correlation coefficient
- QAAR
Quantitative activity-activity relationship
- QM
Quantum mechanics
- QM/MM
Quantum mechanical and molecular mechanical
- Qneg
Most negative atom in the molecule
- QPLD
Quantum polarized ligand docking
- QPlogBB
Brain/blood partition coefficient
- QPlogPo/w
Log octanol-water partition coefficient
- QPlogS
Log aqueous solubility coefficient
- Qpos
Most positive atom in the molecule
- QPPCaco- 2
Apparent Caco-2 permeability in nm.sec-1 QSAR
- QSAR
Quantitative structure activity relationship
- QXP
Quick explore
- R
Correlation coefficient
- rCOMT
Rat COMT
- RD-3D-
Receptor-dependent 3D-QSAR
- RE1/NRSE
Repressor Element 1/Neuron- restrictive silencer elemento
- RF
Random forest
- RMSE
Root-mean-square error
- RP
Recursive partitioning
- RRD
Rigid receptor docking
- S
Softness
- S1P
Sphingosine-1-phosphate
- S1PL
Sphingosine-1-phosphate lyase
- SA
Simulated annealing
- SAM
S-adenosylmethionine
- SA-PLS
Simulated annealing-partial least squares
- SAR
Structure-activity relationship
- SASA
Solvent-accessible surface área
- SBDD
Structure-based drug discovery
- SBMA
Spinal and bulbar muscular atrophy - Kennedy´s disease
- SBP
Selective binding pocket
- SB-PPK
Structure-based Pharmacophore Key
- SBVS
Structure-Based Virtual Screening
- SDAR
Structure-dynamics-activity relationship
- SFXC
Surflex-Dock GeomX
- ShPrP
Syrian hamster prion
- S-MLR
Stepwise MLR
- SMN1
Survival motor neuron gene
- SMoG
Small MoleculeGrowth
- SOD1
Superoxide dismutase-1
- SOMs
Self-Organizing Maps
- SPR
Surface plasmon resonance
- SV2A
Synaptic vesicle protein 2A
- SVM
Support vector machine
- SW
Stepwise
- SW-MLR
Stepwise-multiple linear regression
- TACE
TNF-α converting enzyme
- TAF15
TATA-binding protein-associated factor 15
- TCCBs
T-type calcium channel blockers
- THSG
2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside
- TINS
Target immobilized NMR screening
- TPSA
Topological polar surface area
- TSA
Thermal shift assay
- UCM
Ubiquitin-competing molecules
- VS
Virtual screening (VS)
- WDI
World Drug Index database
- WT-meta
Well-tempered metadynamics
- α-syn
Protein α-synuclein
- η
Absolute hardness
- μ
Dipole moment (Debye)
- χ
Absolute electronegativity (Mulliken)
- ω
Electrophilicity
- ωi
Electrophilic index
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Carrell R.W., Lomas D.A. Conformational disease. Lancet. 1997;350(9071):134–138. doi: 10.1016/S0140-6736(97)02073-4. [http://dx.doi.org/10.1016/S0140-6736(97) 02073-4]. [PMID: 9228977]. [DOI] [PubMed] [Google Scholar]
- 2.Przedborski S., Vila M., Jackson-Lewis V. Neurodegeneration: what is it and where are we? J. Clin. Invest. 2003;111(1):3–10. doi: 10.1172/JCI17522. [http://dx.doi.org/10.1172/JCI200317522]. [PMID: 12511579]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng W., Wangc C. Classification of neurodegenerative diseases using gait dynamics via deterministic learning. Inf. Sci. 2015;317:246–258. [http://dx.doi.org/10.1016/j.ins.2015.04.047]. [Google Scholar]
- 4.Pendlebury W.W. The neuropathology of neurodegenerative diseases causing dementia. Diagn. Histopathol. 2016;22:424–430. [http://dx.doi.org/10.1016/j.mpdhp.2016.10.009]. [Google Scholar]
- 5.Kovacs G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016;17(2):189. doi: 10.3390/ijms17020189. [http://dx.doi.org/10.3390/ijms17020189]. [PMID: 26848654]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedersen J.T., Heegaard N.H.H. Analysis of protein aggregation in neurodegenerative disease. Anal. Chem. 2013;85(9):4215–4227. doi: 10.1021/ac400023c. [http://dx.doi.org/10.1021/ac400023c]. [PMID: 23472885]. [DOI] [PubMed] [Google Scholar]
- 7.Kovacs G.G., Rahimi J., Ströbel T., Lutz M.I., Regelsberger G., Nathalie Streichenberger N., Perret-Liaudet A., Höftberger R., Liberski P.P. Budka. H.; Sikorska, B. Tau Pathology in Creutzfeldt-Jakob Disease Revisited. Brain Pathol. 2016;2016:1–13. doi: 10.1111/bpa.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari R., Kapogiannis D., Huey E.D., Momeni P. FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 2011;8(3):273–294. doi: 10.2174/156720511795563700. [http://dx.doi.org/10.2174/156720511795563700]. [PMID: 21222600]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns N.J., Neumann M., Bigio E.H., Holm I.E., Troost D., Hatanpaa K.J., Foong C., White C.L., III, Schneider J.A., Kretzschmar H.A., Carter D., Taylor-Reinwald L., Paulsmeyer K., Strider J., Gitcho M., Goate A.M., Morris J.C., Mishra M., Kwong L.K., Stieber A., Xu Y., Forman M.S., Trojanowski J.Q., Lee V.M.Y., Mackenzie I.R.A. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007;171(1):227–240. doi: 10.2353/ajpath.2007.070182. [http://dx.doi.org/10.2353/ ajpath.2007.070182]. [PMID: 17591968]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Przedborski S., Vila M., Jackson-Lewis V. Neurodegeneration: what is it and where are we? J. Clin. Invest. 2003;111(1):3–10. doi: 10.1172/JCI17522. [http://dx.doi.org/10.1172/JCI200317522]. [PMID: 12511579]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekins S., Mestres J., Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007;152(1):9–20. doi: 10.1038/sj.bjp.0707305. [http://dx.doi.org/10.1038/sj.bjp. 0707305]. [PMID: 17549047]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsila T., Spyroulias G.A., Patrinos G.P., Matsoukas M.T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016;14:177–184. doi: 10.1016/j.csbj.2016.04.004. [http://dx. doi.org/10.1016/j.csbj.2016.04.004]. [PMID: 27293534]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapetanovic I.M. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem. Biol. Interact. 2008;171(2):165–176. doi: 10.1016/j.cbi.2006.12.006. [http://dx.doi.org/10.1016/ j.cbi.2006.12.006]. [PMID: 17229415]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sliwoski G., Kothiwale S., Meiler J., Lowe E.W., Jr Computational methods in drug discovery. Pharmacol. Rev. 2013;66(1):334–395. doi: 10.1124/pr.112.007336. [http://dx.doi.org/10.1124/pr.112.007336]. [PMID: 24381236]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson A.C. The process of structure-based drug design. Chem. Biol. 2003;10(9):787–797. doi: 10.1016/j.chembiol.2003.09.002. [http://dx.doi.org/10.1016/j.chembiol. 2003.09.002]. [PMID: 14522049]. [DOI] [PubMed] [Google Scholar]
- 16.Vyas V.K., Ukawala R.D., Ghate M., Chintha C. Homology modeling a fast tool for drug discovery: current perspectives. Indian J. Pharm. Sci. 2012;74(1):1–17. doi: 10.4103/0250-474X.102537. [http://dx.doi.org/10.4103/ 0250-474X.102537]. [PMID: 23204616]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinformatics; 2013. pp. 1–7. [PMID: 23749753] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leelananda S.P., Lindert S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016;12:2694–2718. doi: 10.3762/bjoc.12.267. [http://dx. doi.org/10.3762/bjoc.12.267]. [PMID: 28144341]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J.Y., Kim Y. Comparative homology modeling and ligand docking study of human catechol-O-methyltransferase for antiparkinson drug design. B. Kor. Chem. Soc. 2005;26:379–385. [Google Scholar]
- 20.Dhanavade M.J., Jalkute C.B., Barage S.H., Sonawane K.D. Homology modeling, molecular docking and MD simulation studies to investigate role of cysteine protease from Xanthomonas campestris in degradation of Aβ peptide. Comput. Biol. Med. 2013;43(12):2063–2070. doi: 10.1016/j.compbiomed.2013.09.021. [http://dx.doi.org/10.1016/j.compbiomed.2013. 09.021]. [PMID: 24290922]. [DOI] [PubMed] [Google Scholar]
- 21.Conforti P., Zuccato C., Gaudenzi G., Ieraci A., Camnasio S., Buckley N.J., Mutti C., Cotelli F., Contini A., Cattaneo E. Binding of the repressor complex REST-mSIN3b by small molecules restores neuronal gene transcription in Huntington’s disease models. J. Neurochem. 2013;127(1):22–35. doi: 10.1111/jnc.12348. [PMID: 23800350]. [DOI] [PubMed] [Google Scholar]
- 22.Marinelli L., Cosconati S., Steinbrecher T., Limongelli V., Bertamino A., Novellino E., Case D.A. Homology modeling of NR2B modulatory domain of NMDA receptor and analysis of ifenprodil binding. Chem.Med.Chem. 2007;2(10):1498–1510. doi: 10.1002/cmdc.200700091. [http://dx. doi.org/10.1002/cmdc.200700091]. [PMID: 17849398]. [DOI] [PubMed] [Google Scholar]
- 23.Dukka B.K.C. Structure-based methods for computational protein functional site prediction. Comput. Struct. Biotechnol. J. 2013;8:e201308005. doi: 10.5936/csbj.201308005. [http://dx.doi.org/10.5936/csbj.201308005]. [PMID: 24688745]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurie A.T.R., Jackson R.M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21(9):1908–1916. doi: 10.1093/bioinformatics/bti315. [http://dx.doi.org/10.1093/ bioinformatics/bti315]. [PMID: 15701681]. [DOI] [PubMed] [Google Scholar]
- 25.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [http://dx.doi.org/10.1186/1471-2105-10-168]. [PMID: 19486540]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinhiero A.A., Silva K.R.d., Silva A.E.S., Braga F.S., Silva C.H.T.P.d., Santos C.B.R., Hage-Melim L.I.S. In silico identification of novel potential BACE-1 inhibitors for alzheimer’s disease treatment: Molecular docking, pharmacophore modeling and activity and synthetic accessibility predictions. Br. J. Pharm. Res. 2015;7:217–229. [http://dx.doi.org/10.9734/BJPR/2015/18013]. [Google Scholar]
- 27.Pathak A., Madar I.H., Raithatha K., Gupta J.K. In-Silico Identification of Potential Inhibitors Against AChE Using Cheminformatics Approach. MOJ Proteomics Bioinform. 2014;1:1–5. [Google Scholar]
- 28.Palomo V., Soteras I., Perez D.I., Perez C., Gil C., Campillo N.E., Martinez A. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011;54(24):8461–8470. doi: 10.1021/jm200996g. [http://dx.doi.org/10.1021/jm200996g]. [PMID: 22050263]. [DOI] [PubMed] [Google Scholar]
- 29.Mirza M.U., Mirza A.H., Ghori N.U.H., Ferdous S. Glycyrrhetinic acid and E.resveratroloside act as potential plant derived compounds against dopamine receptor D3 for Parkinson’s disease: a pharmacoinformatics study. Drug Des. Devel. Ther. 2014;9:187–198. doi: 10.2147/DDDT.S72794. [http://dx.doi.org/10.2147/DDDT.S72794]. [PMID: 25565772]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahasenan K.V., Pavlovicz R.E., Henderson B.J., González-Cestari T.F., Yi B., McKay D.B., Li C. Discovery of novel α4β2 neuronal nicotinic receptor modulators through structure-based virtual screening. ACS Med. Chem. Lett. 2011;2(11):855–860. doi: 10.1021/ml2001714. [http://dx.doi.org/10.1021/ml2001714]. [PMID: 24936233]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landon M.R., Lieberman R.L., Hoang Q.Q.J.S., Ju S., Caaveiro J.M., Orwig S.D., Kozakov D., Brenke R., Chuang G.Y., Beglov D., Vajda S., Petsko G.A., Ringe D. Detection of ligand binding hot spots on protein surfaces via fragment-based methods: application to DJ-1 and glucocerebrosidase. J. Comput. Aided Mol. Des. 2009;23(8):491–500. doi: 10.1007/s10822-009-9283-2. [http://dx.doi.org/10.1007/s10822-009-9283-2]. [PMID: 19521672]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rufini A., Cavallo F., Condò I., Fortuni S., De Martino G., Incani O., Di Venere A., Benini M., Massaro D.S., Arcuri G., Serio D., Malisan F., Testi R. Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol. Dis. 2015;75:91–99. doi: 10.1016/j.nbd.2014.12.011. [http://dx.doi.org/10.1016/j.nbd. 2014.12.011]. [PMID: 25549872]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferreira L.G., Dos Santos R.N., Oliva G., Andricopulo A.D. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384–13421. doi: 10.3390/molecules200713384. [http://dx.doi.org/10.3390/ molecules200713384]. [PMID: 26205061]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng X.Y., Zhang H.X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [http://dx.doi.org/ 10.2174/157340911795677602]. [PMID: 21534921]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitchen D.B., Decornez H., Furr J.R., Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004;3(11):935–949. doi: 10.1038/nrd1549. [http://dx.doi.org/10.1038/nrd1549]. [PMID: 15520816]. [DOI] [PubMed] [Google Scholar]
- 36.Kroemer R.T. Structure-based drug design: docking and scoring. Curr. Protein Pept. Sci. 2007;8(4):312–328. doi: 10.2174/138920307781369382. [http://dx.doi.org/10. 2174/138920307781369382]. [PMID: 17696866]. [DOI] [PubMed] [Google Scholar]
- 37.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [http://dx.doi.org/10. 1002/jcc.21256]. [PMID: 19399780]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ewing T.J., Makino S., Skillman A.G., Kuntz I.D. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15(5):411–428. doi: 10.1023/a:1011115820450. [http://dx.doi.org/10.1023/A:1011115820450]. [PMID: 11394736]. [DOI] [PubMed] [Google Scholar]
- 39.Rarey M., Kramer B., Lengauer T., Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261(3):470–489. doi: 10.1006/jmbi.1996.0477. [http://dx.doi.org/10.1006/jmbi. 1996.0477]. [PMID: 8780787]. [DOI] [PubMed] [Google Scholar]
- 40.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., Shaw D.E., Francis P., Shenkin P.S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [http://dx.doi.org/10.1021/jm0306430]. [PMID: 15027865]. [DOI] [PubMed] [Google Scholar]
- 41.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [http://dx.doi.org/10.1006/ jmbi.1996.0897]. [PMID: 9126849]. [DOI] [PubMed] [Google Scholar]
- 42.Jain A.N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003;46(4):499–511. doi: 10.1021/jm020406h. [http://dx.doi.org/10.1021/jm020406h]. [PMID: 12570372]. [DOI] [PubMed] [Google Scholar]
- 43.Abagyan R., Totrov M., Kuznetsov D. ICM: a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994;15:488–506. [http://dx.doi.org/10.1002/jcc.540150503]. [Google Scholar]
- 44.Venkatachalam C.M., Jiang X., Oldfield T., Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003;21(4):289–307. doi: 10.1016/s1093-3263(02)00164-x. [http://dx.doi.org/10.1016/S1093-3263(02)00164-X]. [PMID: 12479928]. [DOI] [PubMed] [Google Scholar]
- 45.Vlachakis D., Tsagrasoulis D., Megalooikonomou V., Kossida S. Introducing Drugster: a comprehensive and fully integrated drug design, lead and structure optimization toolkit. Bioinformatics. 2013;29(1):126–128. doi: 10.1093/bioinformatics/bts637. [http://dx.doi.org/10.1093/bioinformatics/ bts637]. [PMID: 23104887]. [DOI] [PubMed] [Google Scholar]
- 46.Zsoldos Z., Szabo I., Szabo Z.A.P.J. Software tools for structure based rational drug design. J. Mol. Struct. 2003;666-667:659–665. [http://dx.doi.org/10.1016/j.theochem.2003.08.105]. [Google Scholar]
- 47.Lionta E., Spyrou G., Vassilatis D.K., Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 2014;14(16):1923–1938. doi: 10.2174/1568026614666140929124445. [http://dx.doi.org/10.2174/1568026614666140929124445]. [PMID: 25262799]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dadashpour S., Tuylu K.T., Unsal T.O., Ozadali K., Irannejad H., Emami S. Design, synthesis and in vitro study of 5,6-diaryl-1,2,4-triazine-3-ylthioacetate derivatives as COX-2 and β-amyloid aggregation inhibitors. Arch. Pharm. (Weinheim) 2015;348(3):179–187. doi: 10.1002/ardp.201400400. [http://dx.doi.org/10.1002/ardp.201400400]. [PMID: 25690564]. [DOI] [PubMed] [Google Scholar]
- 49.Devarajan S., Sharmila J.S. Computational Studies of Beta Amyloid (Aβ42) with p75NTR Receptor: A Novel Therapeutic Target in Alzheimer’s Disease. Adv. Bioinforma. 2014;2014:1–6. doi: 10.1155/2014/736378. [http://dx.doi.org/10.1155/2014/736378]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohit M., Ashok T., Vijaykumar R., Kashniyal K. Molecular docking study of cassia tora, brassica campestris and calotropis procera as acetylcholinesterase inhibitor. Indian J. Pharm. Educ. 2016;50:116–122. [Google Scholar]
- 51.González-Naranjo P., Pérez-Macias N., Campillo N.E., Pérez C., Arán V.J., Girón R., Sánchez-Robles E., Martín M.I., Gómez-Cañas M., García-Arencibia M., Fernández-Ruiz J., Páez J.A. Cannabinoid agonists showing BuChE inhibition as potential therapeutic agents for Alzheimer’s disease. Eur. J. Med. Chem. 2014;73:56–72. doi: 10.1016/j.ejmech.2013.11.026. [http://dx.doi.org/10.1016/j.ejmech.2013.11.026]. [PMID: 24378710]. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y., Sun J., Peng S., Liao H., Zhang Y., Lehmann J. Tacrine-flurbiprofen hybrids as multifunctional drug candidates for the treatment of Alzheimer’s disease. Arch. Pharm. (Weinheim) 2013;346(12):865–871. doi: 10.1002/ardp.201300074. [http://dx.doi.org/10.1002/ardp.201300074]. [PMID: 24203864]. [DOI] [PubMed] [Google Scholar]
- 53.Azam F., Amer A.M., Abulifa A.R., Elzwawi M.M. Ginger components as new leads for the design and development of novel multi-targeted anti-Alzheimer’s drugs: a computational investigation. Drug Des. Devel. Ther. 2014;8:2045–2059. doi: 10.2147/DDDT.S67778. [http://dx.doi. org/10.2147/DDDT.S67778]. [PMID: 25364231]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cozza G., Gianoncelli A., Montopoli M., Caparrotta L., Venerando A., Meggio F., Pinna L.A., Zagotto G., Moro S. Identification of novel protein kinase CK1 delta (CK1delta) inhibitors through structure-based virtual screening. Bioorg. Med. Chem. Lett. 2008;18(20):5672–5675. doi: 10.1016/j.bmcl.2008.08.072. [http://dx.doi.org/10.1016/j.bmcl. 2008.08.072]. [PMID: 18799313]. [DOI] [PubMed] [Google Scholar]
- 55.Azam F., Madi A.M., Ali H.I. Molecular docking and prediction of pharmacokinetic properties of dual mechanism drugs that block MAO-B and adenosine A(2A) receptors for the treatment of parkinson’s disease. J. Young Pharm. 2012;4(3):184–192. doi: 10.4103/0975-1483.100027. [http://dx. doi.org/10.4103/0975-1483.100027]. [PMID: 23112538]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poli G., Giuntini N., Martinelli A., Tuccinardi T. Application of a FLAP-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J. Chem. Inf. Model. 2015;55(3):667–675. doi: 10.1021/ci5006806. [http://dx.doi.org/10.1021/ci5006806]. [PMID: 25746133]. [DOI] [PubMed] [Google Scholar]
- 57.Pareek H., Thakur P., Ray D. Modeling and Docking Studies Of 4-Aminobutyrate Aminotransferase for Huntington’s Disease. Int. J. Pharma Bio Sci. 2011;2:539–549. [Google Scholar]
- 58.Eberini I., Daniele S., Parravicini C., Sensi C., Trincavelli M.L., Martini C., Abbracchio M.P. In silico identification of new ligands for GPR17: a promising therapeutic target for neurodegenerative diseases. J. Comput. Aided Mol. Des. 2011;25(8):743–752. doi: 10.1007/s10822-011-9455-8. [http://dx.doi.org/10.1007/s10822-011-9455-8]. [PMID: 21744154]. [DOI] [PubMed] [Google Scholar]
- 59.Nowak R.J., Cuny G.D., Choi S., Lansbury P.T., Ray S.S. Improving binding specificity of pharmacological chaperones that target mutant superoxide dismutase-1 linked to familial amyotrophic lateral sclerosis using computational methods. J. Med. Chem. 2010;53(7):2709–2718. doi: 10.1021/jm901062p. [http://dx.doi.org/10.1021/jm901062p]. [PMID: 20232802]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swetha R.G., Ramaiah S., Anbarasu A. R521C and R521H mutations in FUS result in weak binding with Karyopherinβ2 leading to Amyotrophic lateral sclerosis: a molecular docking and dynamics study. J. Biomol. Struct. Dyn. 2017;35:2169–2185. doi: 10.1080/07391102.2016.1209130. [PMID: 27381509]. [DOI] [PubMed] [Google Scholar]
- 61.Makhuri F.R., Ghasemi J.B. Computer-aided scaffold hopping to identify a novel series of casein kinase 1 delta (CK1d) inhibitors for amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 2015;78:151–162. doi: 10.1016/j.ejps.2015.07.011. [http://dx.doi.org/10.1016/j.ejps.2015.07.011]. [PMID: 26206296]. [DOI] [PubMed] [Google Scholar]
- 62.Singh J., Salcius M., Liu S.W., Staker B.L., Mishra R., Thurmond J., Michaud G., Mattoon D.R., Printen J., Christensen J., Bjornsson J.M., Pollok B.A., Kiledjian M., Stewart L., Jarecki J., Gurney M.E. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem. Biol. 2008;3(11):711–722. doi: 10.1021/cb800120t. [http://dx.doi.org/ 10.1021/cb800120t]. [PMID: 18839960]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dayangaç-Erden D., Bora G., Ayhan P., Kocaefe C., Dalkara S., Yelekçi K., Demir A.S., Erdem-Yurter H. Histone deacetylase inhibition activity and molecular docking of (e)-resveratrol: its therapeutic potential in spinal muscular atrophy. Chem. Biol. Drug Des. 2009;73(3):355–364. doi: 10.1111/j.1747-0285.2009.00781.x. [http://dx.doi.org/10.1111/j.1747-0285. 2009.00781.x]. [PMID: 19207472]. [DOI] [PubMed] [Google Scholar]
- 64.Pagadala N.S., Bjorndahl T.C., Blinov N., Kovalenko A., Wishart D.S. Molecular docking of thiamine reveals similarity in binding properties between the prion protein and other thiamine-binding proteins. J. Mol. Model. 2013;19(12):5225–5235. doi: 10.1007/s00894-013-1979-5. [http://dx.doi.org/10.1007/s00894-013-1979-5]. [PMID: 24126825]. [DOI] [PubMed] [Google Scholar]
- 65.Ishibashi D., Nakagaki T., Ishikawa T., Atarashi R., Watanabe K., Cruz F.A., Hamada T., Nishida N. Structure-based drug discovery for prion disease using a novel binding simulation. EBioMedicine. 2016;9:238–249. doi: 10.1016/j.ebiom.2016.06.010. [http://dx.doi.org/10.1016/j. ebiom.2016.06.010]. [PMID: 27333028]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tarcsay A., Paragi G., Vass M., Jójárt B., Bogár F., Keserű G.M. The impact of molecular dynamics sampling on the performance of virtual screening against GPCRs. J. Chem. Inf. Model. 2013;53(11):2990–2999. doi: 10.1021/ci400087b. [http://dx.doi.org/10.1021/ci400087b]. [PMID: 24116387]. [DOI] [PubMed] [Google Scholar]
- 67.Correa-Basurto J., Cuevas-Hernández R.I., Phillips-Farfán B.V., Martínez-Archundia M., Romo-Mancillas A., Ramírez-Salinas G.L., Pérez-González Ó.A., Trujillo-Ferrara J., Mendoza-Torreblanca J.G. Identification of the antiepileptic racetam binding site in the synaptic vesicle protein 2A by molecular dynamics and docking simulations. Front. Cell. Neurosci. 2015;9:125. doi: 10.3389/fncel.2015.00125. [http://dx.doi.org/10.3389/fncel.2015.00125]. [PMID: 25914622]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deb P.K., Sharma A., Piplani P., Akkinepally R.R. Molecular docking and receptor-specific 3D-QSAR studies of acetylcholinesterase inhibitors. Mol. Divers. 2012;16(4):803–823. doi: 10.1007/s11030-012-9394-x. [http:// dx.doi.org/10.1007/s11030-012-9394-x]. [PMID: 22996404]. [DOI] [PubMed] [Google Scholar]
- 69.Azam F., Prasad M.V.V., Thangavel N., Ali H.I. Molecular docking studies of 1-(substituted phenyl)-3-(naphtha [1, 2-d] thiazol-2-yl) urea/thiourea derivatives with human adenosine A(2A) receptor. Bioinformation. 2011;6(9):330–334. doi: 10.6026/97320630006330. [http://dx.doi. org/10.6026/97320630006330]. [PMID: 21814389]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sivaraman D., Srikanth J. Discovery of novel monoamine oxidase-B inhibitors by molecular docking approach for alzheimer’s and parkinson’s disease Treatment. Int. J. Pharm. Sci. Rev. Res. 2016;40:245–250. [Google Scholar]
- 71.Jayaraj R.L., Ranjani V., Manigandan K., Elangovan N. In silico docking studies to identify potent inhibitors of alpha-synuclein aggregation in parkinson disease. Asian J. Pharm. Clin. Res. 2014;6:127–131. [Google Scholar]
- 72.Sehgal S.A., Mannan S., Ali S. Pharmacoinformatic and molecular docking studies reveal potential novel antidepressants against neurodegenerative disorders by targeting HSPB8. Drug Des. Devel. Ther. 2016;10:1605–1618. doi: 10.2147/DDDT.S101929. [http://dx.doi.org/10.2147/DDDT. S101929]. [PMID: 27226709]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ray S.S., Nowak R.J., Brown R.H., Jr, Lansbury P.T., Jr Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc. Natl. Acad. Sci. USA. 2005;102(10):3639–3644. doi: 10.1073/pnas.0408277102. [http://dx.doi.org/10.1073/pnas.0408277102]. [PMID: 15738401]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagappan P., Krishnamurthy V. Structural prediction and comparative molecular docking studies of hesperidin and L-dopa on A-synuclein, MAO-B, COMT and UCHL-1 inhibitors. Int. J. Pharm. Clin. Res. 2015;7:221–225. [Google Scholar]
- 75.Markandeyan D. Santhalingam, k.; Kannaiyan, S.; Suresh, S.; Paul, B. Virtual screening of phytochemicals of morinda citrifolia as anti-inflammatory and anti-alzheimer agents using molegro virtual docker on P38-A mitogen activated protein kinase enzyme. Asian J. Pharm. Clin. Res. 2015;8:83–87. [Google Scholar]
- 76.Klein-Junior L.C., Passos C.S., Moraes A.P., Wakui V.G. ; Konrath E.L., Nurisso A., Carrupt P.A. Oliveira, Cecilia M. A.; Kato, L.; Henriques, A. T. Indole alkaloids and semisynthetic indole derivatives as multifunctional scaffolds aiming the inhibition of enzymes related to neurodegenerative diseases - a focus on psychotria L. genus. Curr. Top. Med. Chem. 2014;14:1056–1075. doi: 10.2174/1568026614666140324142409. [http://dx.doi.org/10.2174/1568026614666140324142409]. [PMID: 24660679]. [DOI] [PubMed] [Google Scholar]
- 77.Laeeq S., Sirbaiya A.K., Siddiqui H.H. Zaidi1, S. M. H. AN overview of the computer aided drug designing. Int. J. Pharm. Pharm. Sci. 2014;3:963–994. [Google Scholar]
- 78.Spitzer G.M., Heiss M., Mangold M., Markt P., Kirchmair J., Wolber G., Liedl K.R. One concept, three implementations of 3D pharmacophore-based virtual screening: distinct coverage of chemical search space. J. Chem. Inf. Model. 2010;50(7):1241–1247. doi: 10.1021/ci100136b. [http://dx.doi.org/10.1021/ci100136b]. [PMID: 20583761]. [DOI] [PubMed] [Google Scholar]
- 79.Katsila T., Spyroulias G.A., Patrinos G.P., Matsoukas M.T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016;14:177–184. doi: 10.1016/j.csbj.2016.04.004. [http://dx. doi.org/10.1016/j.csbj.2016.04.004]. [PMID: 27293534]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim H.J., Choo H., Cho Y.S., No K.T., Pae A.N. Novel GSK-3beta inhibitors from sequential virtual screening. Bioorg. Med. Chem. 2008;16(2):636–643. doi: 10.1016/j.bmc.2007.10.047. [http://dx.doi.org/10.1016/j.bmc. 2007.10.047]. [PMID: 18006321]. [DOI] [PubMed] [Google Scholar]
- 81.Natarajan P., Priyadarshini V., Pradhan D., Manne M., Swargam S., Kanipakam H., Bhuma V., Amineni U. E-pharmacophore-based virtual screening to identify GSK-3β inhibitors. J. Recept. Signal Transduct. Res. 2016;36(5):445–458. doi: 10.3109/10799893.2015.1122043. [http:// dx.doi.org/10.3109/10799893.2015.1122043]. [PMID: 27305963]. [DOI] [PubMed] [Google Scholar]
- 82.Shinde P., Vidyasagar N., Dhulap S., Dhulap A., Hirwani R. Natural products based P-glycoprotein activators for improved β-amyloid clearance in alzheimer’s disease: An in silico approach. Cent. Nerv. Syst. Agents Med. Chem. 2015;16(1):50–59. doi: 10.2174/1871524915666150826092152. [http:// dx.doi.org/10.2174/1871524915666150826092152]. [PMID: 26306632]. [DOI] [PubMed] [Google Scholar]
- 83.Kumar A., Roy S., Tripathi S., Sharma A. Molecular docking based virtual screening of natural compounds as potential BACE1 inhibitors: 3D QSAR pharmacophore mapping and molecular dynamics analysis. J. Biomol. Struct. Dyn. 2016;34(2):239–249. doi: 10.1080/07391102.2015.1022603. [http:// dx.doi.org/10.1080/07391102.2015.1022603]. [PMID: 25707809]. [DOI] [PubMed] [Google Scholar]
- 84.Deniz U., Ozkirimli E., Ulgen K.O. A systematic methodology for large scale compound screening: A case study on the discovery of novel S1PL inhibitors. J. Mol. Graph. Model. 2016;63:110–124. doi: 10.1016/j.jmgm.2015.11.004. [http://dx.doi.org/10.1016/j.jmgm.2015.11.004]. [PMID: 26724452]. [DOI] [PubMed] [Google Scholar]
- 85.H. M. SOLIMAN, M. Per-residue energy footprints-based pharmacophore modeling as an enhanced in silico approach in drug discovery: A case study on theIdentification of novelb-secretase1 (BACE1) inhibitors as anti-alzheimer agents. Cell. Mol. Bioeng. 2016;9:175–189. [http://dx.doi.org/10.1007/s12195-015-0421-8]. [Google Scholar]
- 86.Lorenzo V.P., Filhoa J.M.B., Scottia L. Scottia, Marcus, T. Combined structure-and ligand-based virtual screening to evaluate caulerpin analogs with potential inhibitory activity against monoamine oxidase B. Rev. Bras. Farmacogn. 2015;25:690–697. [http://dx. doi.org/10.1016/j.bjp.2015.08.005]. [Google Scholar]
- 87.Zhang Y., Zhang S., Xu G., Yan H., Pu Y., Zuo Z. The discovery of new acetylcholinesterase inhibitors derived from pharmacophore modeling, virtual screening, docking simulation and bioassays. Mol. Biosyst. 2016;12(12):3734–3742. doi: 10.1039/c6mb00661b. [http://dx.doi.org/ 10.1039/C6MB00661B]. [PMID: 27801451]. [DOI] [PubMed] [Google Scholar]
- 88.Dighe S.N., Deora G.S., De la Mora E., Nachon F., Chan S., Parat M.O., Brazzolotto X., Ross B.P. Discovery and structure-activity relationships of a highly selective butyrylcholinesterase inhibitor by structure-based virtual screening. J. Med. Chem. 2016;59(16):7683–7689. doi: 10.1021/acs.jmedchem.6b00356. [http://dx.doi.org/10.1021/acs.jmedchem. 6b00356]. [PMID: 27405689]. [DOI] [PubMed] [Google Scholar]
- 89.Semighini E.P. In Silico design of beta-secretase inhibitors in Alzheimer’s disease. Chem. Biol. Drug Des. 2015;86(3):284–290. doi: 10.1111/cbdd.12492. [http://dx.doi.org/10.1111/cbdd.12492]. [PMID: 25476252]. [DOI] [PubMed] [Google Scholar]
- 90.Kasam V., Lee N.R., Kim K.B., Zhan C.G. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg. Med. Chem. Lett. 2014;24(15):3614–3617. doi: 10.1016/j.bmcl.2014.05.025. [http://dx.doi.org/10.1016/j.bmcl.2014.05.025]. [PMID: 24913713]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hajjo R., Setola V., Roth B.L., Tropsha A. Chemocentric informatics approach to drug discovery: identification and experimental validation of selective estrogen receptor modulators as ligands of 5-hydroxytryptamine-6 receptors and as potential cognition enhancers. J. Med. Chem. 2012;55(12):5704–5719. doi: 10.1021/jm2011657. [http://dx.doi. org/10.1021/jm2011657]. [PMID: 22537153]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen Y., Fang L., Peng S., Liao H., Lehmann J., Zhang Y. Discovery of a novel acetylcholinesterase inhibitor by structure-based virtual screening techniques. Bioorg. Med. Chem. Lett. 2012;22(9):3181–3187. doi: 10.1016/j.bmcl.2012.03.046. [http://dx.doi.org/10.1016/j.bmcl.2012.03. 046]. [PMID: 22472693]. [DOI] [PubMed] [Google Scholar]
- 93.Santos J., Lesnard A., Agondanou J.H., Dupont N. Godard. A. M.; Stiebing, S.; Rochais, C.; Fabis, F.; Dallemagne, P.; Bureau, R.; Rault, S. Virtual screening discovery of new acetylcholinesterase inhibitors issued from CERMN chemical library. J. Chem. Inf. Model. 2010;50:422–428. doi: 10.1021/ci900491t. [http://dx.doi.org/10.1021/ ci900491t]. [PMID: 20196555]. [DOI] [PubMed] [Google Scholar]
- 94.Patil S., Tyagi A., Jose J., Menon K.N., Mohan G. Integration of common feature pharmacophore modeling and in vitro study to identify potent AChE inhibitors. Med. Chem. Res. 2016;25:2965–2975. [http://dx.doi.org/10.1007/s00044-016-1716-6]. [Google Scholar]
- 95.Bottegoni G., Veronesi M., Bisignano P., Kacker P., Favia A.D., Cavalli A. Development and application of a virtual screening protocol for the identification of multitarget fragments. ChemMedChem. 2016;11(12):1259–1263. doi: 10.1002/cmdc.201500521. [http://dx.doi.org/10. 1002/cmdc.201500521]. [PMID: 26663255]. [DOI] [PubMed] [Google Scholar]
- 96.De Cesco S., Deslandes S., Therrien E., Levan D., Cueto M., Schmidt R., Cantin L.D., Mittermaier A., Juillerat-Jeanneret L., Moitessier N. Virtual screening and computational optimization for the discovery of covalent prolyl oligopeptidase inhibitors with activity in human cells. J. Med. Chem. 2012;55(14):6306–6315. doi: 10.1021/jm3002839. [http://dx.doi.org/10.1021/jm3002839]. [PMID: 22765237]. [DOI] [PubMed] [Google Scholar]
- 97.Chen D., Ranganathan A., IJzerman A.P., Siegal G., Carlsson J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A(2A) adenosine receptor. J. Chem. Inf. Model. 2013;53(10):2701–2714. doi: 10.1021/ci4003156. [http://dx.doi.org/10.1021/ci4003156]. [PMID: 23971943]. [DOI] [PubMed] [Google Scholar]
- 98.Mueller R. Dawson, E. S.; Meiler, J.; et, Al. Discovery of 2-(2-Benzoxazoyl amino)-4-Aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu5): from an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem. 2012;7:406–414. doi: 10.1002/cmdc.201100510. [http://dx. doi.org/10.1002/cmdc.201100510]. [PMID: 22267125]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lavecchia A., Di Giovanni C., Cerchia C., Russo A., Russo G., Novellino E. Discovery of a novel small molecule inhibitor targeting the frataxin/ubiquitin interaction via structure-based virtual screening and bioassays. J. Med. Chem. 2013;56(7):2861–2873. doi: 10.1021/jm3017199. [http://dx.doi.org/10.1021/jm3017199]. [PMID: 23506486]. [DOI] [PubMed] [Google Scholar]
- 100.Huang H.J., Chang T.T., Chen H.Y., Chen C.Y. Finding inhibitors of mutant superoxide dismutase-1 for amyotrophic lateral sclerosis therapy from traditional chinese medicine. Evid. Based Complement. Alternat. Med. 2014;2014:156276. doi: 10.1155/2014/156276. [http://dx.doi. org/10.1155/2014/156276]. [PMID: 24963318]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanno T., Tanaka K., Yanagisawa Y., Yasutake K., Hadano S., Yoshii F., Hirayama N., Ikeda J.E. A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2-ARE pathway: therapeutic implications for ALS. Free Radic. Biol. Med. 2012;53(11):2028–2042. doi: 10.1016/j.freeradbiomed.2012.09.010. [http://dx.doi.org/10.1016/j.freeradbiomed.2012.09.010]. [PMID: 23000247]. [DOI] [PubMed] [Google Scholar]
- 102.Rao S.P., Mujawar S., Chaudhary S., Thobias I.B. In-silico discovery of novel inhibitors against PrP protein (E196K, V203I and E211Q): creutzfeldt jakob disease. Int. J. Pharm. Sci. Rev. Res. 2015;32:288–292. [Google Scholar]
- 103.Hyeon J.W., Choi J., Kim Su. Y.; Govindaraj, R. G.; Hwang, K. J.; Lee, Y. S.; An, S. S.; Lee, M. K.; Joung, J. Y.; No, K. T.; Lee, J. Discovery of Novel Anti-prion Compounds Using In Silico and In Vitro Approaches. Sci. Rep. 2015;5:1–11. doi: 10.1038/srep14944. [http://dx.doi.org/10. 1038/srep14944]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ijjaali I., Barrere C., Nargeot J., Petitet F., Bourinet E. Ligand-based virtual screening to identify new T-type calcium channel blockers. Channels (Austin) 2007;1(4):300–304. doi: 10.4161/chan.4999. [http://dx.doi. org/10.4161/chan.4999]. [PMID: 18708747]. [DOI] [PubMed] [Google Scholar]
- 105.Lin C.H., Hsieh Y.S., Wu Y.R., Hsu C.J., Chen H.C., Huang W.H., Chang K.H., Hsieh-Li H.M., Su M.T., Sun Y.C., Lee G.C., Lee-Chen G.J. Identifying GSK-3β kinase inhibitors of Alzheimer’s disease: Virtual screening, enzyme, and cell assays. Eur. J. Pharm. Sci. 2016;89:11–19. doi: 10.1016/j.ejps.2016.04.012. [http://dx.doi.org/10.1016/j.ejps. 2016.04.012]. [PMID: 27094783]. [DOI] [PubMed] [Google Scholar]
- 106.Noeske T., Jirgensons A., Starchenkovs I., Renner S., Jaunzeme I., Trifanova D., Hechenberger M., Bauer T., Kauss V., Parsons C.G., Schneider G., Weil T. Virtual screening for selective allosteric mGluR1 antagonists and structure-activity relationship investigations for coumarine derivatives. ChemMedChem. 2007;2(12):1763–1773. doi: 10.1002/cmdc.200700151. [http://dx.doi.org/10.1002/cmdc.200700151]. [PMID: 17868161]. [DOI] [PubMed] [Google Scholar]
- 107.Daidone F., Montioli R., Paiardini A., Cellini B., Macchiarulo A., Giardina G., Bossa F., Borri V.C. Identification by virtual screening and in vitro testing of human DOPA decarboxylase inhibitors. PLoS One. 2012;7(2):e31610. doi: 10.1371/journal.pone.0031610. [http://dx.doi.org/ 10.1371/journal.pone.0031610]. [PMID: 22384042]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lepailleur A., Freret T., Lemaître S., Boulouard M., Dauphin F., Hinschberger A., Dulin F., Lesnard A., Bureau R., Rault S. Dual histamine H3R/serotonin 5-HT4R ligands with antiamnesic properties: pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 2014;54(6):1773–1784. doi: 10.1021/ci500157n. [http://dx. doi.org/10.1021/ci500157n]. [PMID: 24857631]. [DOI] [PubMed] [Google Scholar]
- 109.Ferreira R.J., dos Santos D.J.V.A., Ferreira M-J.U., Guedes R.C. Toward a better pharmacophore description of P-glycoprotein modulators, based on macrocyclic diterpenes from Euphorbia species. J. Chem. Inf. Model. 2011;51(6):1315–1324. doi: 10.1021/ci200145p. [http://dx.doi. org/10.1021/ci200145p]. [PMID: 21604687]. [DOI] [PubMed] [Google Scholar]
- 110.Lu S.H., Wu J.W., Liu H.L., Zhao J.H., Liu K.T., Chuang C.K., Lin H.Y., Tsai W.B., Ho Y. The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011;18:8. doi: 10.1186/1423-0127-18-8. [http://dx.doi.org/10.1186/1423-0127-18-8]. [PMID: 21251245]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patel H.M., Noolvi M.N., Sharma P., Jaiswal V., Bansal S., Lohan S., Sharad Kumar S., Abbot V., Dhiman S., Bhardwaj V. Quantitative structure-activity relationship (QSAR) studies as strategic approach in drug discovery. Med. Chem. Res. 2014;23:4991–5007. [http://dx.doi.org/10.1007/s00044-014-1072-3]. [Google Scholar]
- 112.Damale M.G., Harke S.N., Kalam Khan F.A., Shinde D.B., Sangshetti J.N. Recent advances in multidimensional QSAR (4D-6D): a critical review. Mini Rev. Med. Chem. 2014;14(1):35–55. doi: 10.2174/13895575113136660104. [http:// dx.doi.org/10.2174/13895575113136660104]. [PMID: 24195665]. [DOI] [PubMed] [Google Scholar]
- 113.Abdolmaleki A., Ghasemi J.B., Shiri F., Pirhadi S. Application of multivariate linear and nonlinear calibration and classification methods in drug design. Comb. Chem. High Throughput Screen. 2015;18(8):795–808. doi: 10.2174/1386207318666150803142158. [http://dx.doi.org/10.2174/1386207318666 150803142158]. [PMID: 26234507]. [DOI] [PubMed] [Google Scholar]
- 114.Xu J., Hagler A. chemoinformatics and drug discovery. Molecules. 2002;7:566–600. [http://dx.doi.org/10.3390/70800566]. [Google Scholar]
- 115.Mitchell J.B.O. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014;4(5):468–481. doi: 10.1002/wcms.1183. [http://dx.doi.org/10.1002/wcms.1183]. [PMID: 25285160]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luan F., Cordeiro M.N., Alonso N., García-Mera X., Caamaño O., Romero-Duran F.J., Yañez M., González-Díaz H. TOPS-MODE model of multiplexing neuroprotective effects of drugs and experimental-theoretic study of new 1,3-rasagiline derivatives potentially useful in neurodegenerative diseases. Bioorg. Med. Chem. 2013;21(7):1870–1879. doi: 10.1016/j.bmc.2013.01.035. [http://dx.doi.org/10.1016/j.bmc.2013.01. 035]. [PMID: 23415089]. [DOI] [PubMed] [Google Scholar]
- 117.Bautista-Aguilera O.M., Esteban G., Bolea I., Nikolic K., Agbaba D., Moraleda I., Iriepa I., Samadi A., Soriano E., Unzeta M., Marco-Contelles J. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014;75:82–95. doi: 10.1016/j.ejmech.2013.12.028. [http://dx.doi.org/10. 1016/j.ejmech.2013.12.028]. [PMID: 24530494]. [DOI] [PubMed] [Google Scholar]
- 118.Speck-Planche A., Kleandrova V.V., Luan F., Cordeiro M.N. Multi-target inhibitors for proteins associated with Alzheimer: in silico discovery using fragment-based descriptors. Curr. Alzheimer Res. 2013;10(2):117–124. doi: 10.2174/1567205011310020001. [http://dx.doi.org/10.2174/ 1567205011310020001]. [PMID: 22515494]. [DOI] [PubMed] [Google Scholar]
- 119.Prado-Prado F., García-Mera X., Escobar M., Alonso N., Caamaño O., Yañez M., González-Díaz H. 3D MI-DRAGON: new model for the reconstruction of US FDA drug- target network and theoretical-experimental studies of inhibitors of rasagiline derivatives for AChE. Curr. Top. Med. Chem. 2012;12(16):1843–1865. [http://dx.doi.org/10.2174/1568026611209061843]. [PMID: 23030618]. [PubMed] [Google Scholar]
- 120.Makhaeva G.F., Radchenko E.V., Baskin I.I., Palyulin V.A., Richardson R.J., Zefirov N.S. Combined QSAR studies of inhibitor properties of O-phosphorylated oximes toward serine esterases involved in neurotoxicity, drug metabolism and Alzheimer’s disease. SAR QSAR Environ. Res. 2012;23(7-8):627–647. doi: 10.1080/1062936X.2012.679690. [http:// dx.doi.org/10.1080/1062936X.2012.679690]. [PMID: 22587543]. [DOI] [PubMed] [Google Scholar]
- 121.Ambure P., Roy K. Exploring structural requirements of leads for improving activity and selectivity against CDK5/ p25 in Alzheimer’s disease: an in silico approach. RSC Advances. 2014;4:6702–6709. [http://dx.doi.org/10.1039/C3RA46861E]. [Google Scholar]
- 122.Araújo J.Q., de Brito M.A., Hoelz L.V., de Alencastro R.B., Castro H.C., Rodrigues C.R., Albuquerque M.G. Receptor-dependent (RD) 3D-QSAR approach of a series of benzylpiperidine inhibitors of human acetylcholinesterase (HuAChE). Eur. J. Med. Chem. 2011;46(1):39–51. doi: 10.1016/j.ejmech.2010.10.009. [http://dx.doi.org/10.1016/j.ejmech. 2010.10.009]. [PMID: 21074294]. [DOI] [PubMed] [Google Scholar]
- 123.Saracoglu M., Kandemirli F. The investigation of structure-activity relationships of tacrine analogues: electronic-topological method. Open Med. Chem. J. 2008;2:75–80. doi: 10.2174/1874104500802010075. [http://dx.doi.org/ 10.2174/1874104500802010075]. [PMID: 19662147]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dastmalchi S., Hamzeh-Mivehroud M., Asadpour-Zeynali K. Comparison of different 2D and 3D-QSAR methods on activity prediction of histamine H3 receptor antagonists. Iran. J. Pharm. Res. 2012;11(1):97–108. [PMID: 25317190]. [PMC free article] [PubMed] [Google Scholar]
- 125.Gupta S., Fallarero A., Vainio M.J., Saravanan P., Santeri Puranen J., Järvinen P., Johnson M.S., Vuorela P.M., Mohan C.G. Molecular docking guided comparative GFA, G/PLS, SVM and ANN models of structurally diverse dual binding site acetylcholinesterase inhibitors. Mol. Inform. 2011;30(8):689–706. doi: 10.1002/minf.201100029. [PMID: 27467261]. [DOI] [PubMed] [Google Scholar]
- 126.Ajmani S., Janardhan S., Viswanadhan V.N. Toward a general predictive QSAR model for gamma-secretase inhibitors. Mol. Divers. 2013;17(3):421–434. doi: 10.1007/s11030-013-9441-2. [http://dx.doi.org/10.1007/s11030-013-9441-2]. [PMID: 23612850]. [DOI] [PubMed] [Google Scholar]
- 127.dastmalchi, S.; Hamzeh-Mivehroud, M.; Ghafourian, T.; Hamzeiy, H. Molecular modeling of histamine H3 receptor andQSAR studies on arylbenzofuran derived H3 antagonists. J. Mol. Graph. Model. 2008;26:834–844. doi: 10.1016/j.jmgm.2007.05.002. [http://dx.doi.org/10.1016/j.jmgm.2007.05.002]. [PMID: 17561422]. [DOI] [PubMed] [Google Scholar]
- 128.Nicolotti O., Pellegrini-Calace M., Altomar C., Carotti A., Carrieri A., Sanz F. Ligands of neuronal nicotinic acetylcholine receptor (nAChR): inferences from the Hansch and 3-D quantitative structure-activity relationship (QSAR) Models. Curr. Med. Chem. 2002;9(1):1–29. doi: 10.2174/0929867023371463. [http://dx.doi.org/10.2174/0929867023371463]. [PMID: 11864064]. [DOI] [PubMed] [Google Scholar]
- 129.Nicolotti O., Altomare C., Pellegrini-Calace M., Carotti A. Neuronal nicotinic acetylcholine receptor agonists: pharmacophores, evolutionary QSAR and 3D-QSAR models. Curr. Top. Med. Chem. 2004;4(3):335–360. doi: 10.2174/1568026043451384. [http://dx.doi.org/10.2174/1568026043451384]. [PMID: 14754450]. [DOI] [PubMed] [Google Scholar]
- 130.Asadabadi E.B., Abdolmaleki P., Barkooie S.M.H., Jahandideh S., Rezaei M.A. A combinatorial feature selection approach to describe the QSAR of dual site inhibitors of acetylcholinesterase. Comput. Biol. Med. 2009;39(12):1089–1095. doi: 10.1016/j.compbiomed.2009.09.003. [http://dx.doi.org/ 10.1016/j.compbiomed.2009.09.003]. [PMID: 19854437]. [DOI] [PubMed] [Google Scholar]
- 131.Bitencourt M., Freitas M.P., Rittner R. The MIA-QSAR method for the prediction of bioactivities of possible acetylcholinesterase inhibitors. Arch. Pharm. (Weinheim) 2012;345(9):723–728. doi: 10.1002/ardp.201200079. [http:// dx.doi.org/10.1002/ardp.201200079]. [PMID: 22674790]. [DOI] [PubMed] [Google Scholar]
- 132.Liu S., Fu R., Cheng X., Chen S.P., Zhou L.H. Exploring the binding of BACE-1 inhibitors using comparative binding energy analysis (COMBINE). BMC Struct. Biol. 2012;12:21. doi: 10.1186/1472-6807-12-21. [http:// dx.doi.org/10.1186/1472-6807-12-21]. [PMID: 22925713]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Manoharan P., Vijayan R.S.K., Ghoshal N. Rationalizing fragment based drug discovery for BACE1: insights from FB-QSAR, FB-QSSR, multi objective (MO-QSPR) and MIF studies. J. Comput. Aided Mol. Des. 2010;24(10):843–864. doi: 10.1007/s10822-010-9378-9. [http://dx.doi.org/ 10.1007/s10822-010-9378-9]. [PMID: 20740315]. [DOI] [PubMed] [Google Scholar]
- 134.Park H.R., Kim M.K., Kim D.W. 3D QSAR CoMFA Study on Phenylthiazolylhydrazide (PTH) Derivataives as Tau Protein Aggregation Inhibitors. Bull. Korean Chem. Soc. 2010;31:3838–3841. [http://dx.doi.org/10.5012/bkcs.2010.31.12.3838]. [Google Scholar]
- 135.Fang J., Huang D., Zhao W., Ge H., Luo H.B., Xu J. A new protocol for predicting novel GSK-3β ATP competitive inhibitors. J. Chem. Inf. Model. 2011;51(6):1431–1438. doi: 10.1021/ci2001154. [http://dx.doi.org/ 10.1021/ci2001154]. [PMID: 21615159]. [DOI] [PubMed] [Google Scholar]
- 136.Zheng F., Zhan M., Huang X., Abdul H.M.D., Zhan C.G. Modeling in vitro inhibition of butyrylcholinesterase using molecular docking, multi-linear regression and artificial neural network approaches. Bioorg. Med. Chem. 2014;22(1):538–549. doi: 10.1016/j.bmc.2013.10.053. [http://dx. doi.org/10.1016/j.bmc.2013.10.053]. [PMID: 24290065]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fernández M., Carreiras M.C., Marco J.L., Caballero J. Modeling of acetylcholinesterase inhibition by tacrine analogues using Bayesian-regularized Genetic Neural Networks and ensemble averaging. J. Enzyme Inhib. Med. Chem. 2006;21(6):647–661. doi: 10.1080/14756360600862366. [http:// dx.doi.org/10.1080/14756360600862366]. [PMID: 17252937]. [DOI] [PubMed] [Google Scholar]
- 138.Simeon S., Anuwongcharoen N., Shoombuatong W., Malik A.A., Prachayasittikul V., Wikberg J.E.S., Nantasenamat C. Probing the origins of human acetylcholinesterase inhibition via QSAR modeling and molecular docking. PeerJ. 2016;4:e2322. doi: 10.7717/peerj.2322. [http://dx.doi.org/10.7717/peerj.2322]. [PMID: 27602288]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Subramanian G., Ramsundar B., Pande V., Denny R.A. Computational Modeling of β-Secretase 1 (BACE-1) Inhibitors Using Ligand Based Approaches. J. Chem. Inf. Model. 2016;56(10):1936–1949. doi: 10.1021/acs.jcim.6b00290. [http://dx.doi.org/10.1021/acs.jcim.6b00290]. [PMID: 27689393]. [DOI] [PubMed] [Google Scholar]
- 140.Salum L.B., Andricopulo A.D. Fragment-based QSAR: perspectives in drug design. Mol. Divers. 2009;13(3):277–285. doi: 10.1007/s11030-009-9112-5. [http://dx.doi.org/10.1007/s11030-009-9112-5]. [PMID: 19184499]. [DOI] [PubMed] [Google Scholar]
- 141.Leal F.D., da Silva Lima C.H., de Alencastro R.B., Castro H.C., Rodrigues C.R., Albuquerque M.G. Hologram QSAR models of a series of 6-arylquinazolin-4-amine inhibitors of a new Alzheimer’s disease target: dual specificity tyrosine-phosphorylation-regulated kinase-1A enzyme. Int. J. Mol. Sci. 2015;16(3):5235–5253. doi: 10.3390/ijms16035235. [http://dx.doi.org/10.3390/ijms16035235]. [PMID: 25756379]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumar R., Långström B., Darreh-Shori T. Novel ligands of Choline Acetyltransferase designed by in silico molecular docking, hologram QSAR and lead optimization. Sci. Rep. 2016;6:31247. doi: 10.1038/srep31247. [http://dx.doi.org/10.1038/srep31247]. [PMID: 27507101]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bhayye S.S., Roy K., Saha A. Pharmacophore generation, atom-based 3D-QSAR, HQSAR and activity cliff analyses of benzothiazine and deazaxanthine derivatives as dual A2A antagonists/MAO-B inhibitors. SAR QSAR Environ. Res. 2016;27:1–20. doi: 10.1080/1062936X.2015.1136840. [http://dx. doi.org/10.1080/1062936X.2015.1136840]. [PMID: 26873265]. [DOI] [PubMed] [Google Scholar]
- 144.Zambre V.P., Hambarde V.A., Petkar N.N., Patela C.N. ; Sawanta S.D. Structural investigations by in silico modeling fordesigning NR2B subunit selective NMDA receptorantagonists. RSC Advances. 2015;5:23922–23940. [http://dx.doi.org/10.1039/ C5RA01098E]. [Google Scholar]
- 145.Nikolic K., Mavridis L., Bautista-Aguilera O.M., Marco-Contelles J., Stark H., do Carmo Carreiras M., Rossi I., Massarelli P., Agbaba D., Ramsay R.R., Mitchell J.B. Predicting targets of compounds against neurological diseases using cheminformatic methodology. J. Comput. Aided Mol. Des. 2015;29(2):183–198. doi: 10.1007/s10822-014-9816-1. [http://dx.doi.org/10.1007/s10822-014-9816-1]. [PMID: 25425329]. [DOI] [PubMed] [Google Scholar]
- 146.Goyal M., Dhanjal J.K., Goyal S., Tyagi C., Hamid R., Grover A. Development of dual inhibitors against Alzheimer’s disease using fragment-based QSAR and molecular docking. BioMed Res. Int. 2014;2014:979606. doi: 10.1155/2014/979606. [http://dx.doi.org/10.1155/2014/979606]. [PMID: 25019089]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Polychronopoulos P., Magiatis P., Skaltsounis A.L., Myrianthopoulos V., Mikros E., Tarricone A., Musacchio A., Roe S.M., Pearl L., Leost M., Greengard P., Meijer L. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004;47(4):935–946. doi: 10.1021/jm031016d. [http://dx.doi.org/10.1021/jm 031016d]. [PMID: 14761195]. [DOI] [PubMed] [Google Scholar]
- 148.Ain Q.U., Méndez-Lucio O., Ciriano I.C., Malliavin T., van Westen G.J.P., Bender A. Modelling ligand selectivity of serine proteases using integrative proteochemometric approaches improves model performance and allows the multi-target dependent interpretation of features. Integr. Biol. 2014;6(11):1023–1033. doi: 10.1039/c4ib00175c. [http://dx.doi.org/10.1039/C4IB00175C]. [PMID: 25255469]. [DOI] [PubMed] [Google Scholar]
- 149.Firoozpour L., Sadatnezhad K., Dehghani S., Pourbasheer E., Foroumadi A., Shafiee A., Amanlou M. An efficient piecewise linear model for predicting activity of caspase-3 inhibitors. Daru. 2012;20(1):31. doi: 10.1186/2008-2231-20-31. [http://dx.doi.org/10.1186/2008-2231-20-31]. [PMID: 23351435]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Oloff S., Mailman R.B., Tropsha A. Application of validated QSAR models of D1 dopaminergic antagonists for database mining. J. Med. Chem. 2005;48(23):7322–7332. doi: 10.1021/jm049116m. [http://dx.doi.org/10. 1021/jm049116m]. [PMID: 16279792]. [DOI] [PubMed] [Google Scholar]
- 151.Bolisetty V., Rao K.N., Manaiah V. QSAR and docking studies of aphorphine derivatives as efficacious partial antagonists for parkinson’s disease. Int. J. Pharma Bio Sci. 2014;5:465–480. [Google Scholar]
- 152.Mueller R., Dawson E.S., Meiler J., Rodriguez A.L., Chauder B.A., Bates B.S., Felts A.S., Lamb J.P., Menon U.N., Jadhav S.B., Kane A.S., Jones C.K., Gregory K.J., Niswender C.M., Conn P.J., Olsen C.M., Winder D.G., Emmitte K.A., Lindsley C.W. Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu5): from an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem. 2012;7(3):406–414. doi: 10.1002/cmdc.201100510. [http://dx.doi.org/10.1002/cmdc.201100510]. [PMID: 22267125]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chtitaa S., Larifb M., Ghamalia M., Bouachrinec M., Lakhlifia T. Quantitative structure-activity relationship studies of dibenzo [a,d]cycloalkenimine derivatives for non-competitive antagonists of N-methyl-d-aspartate based on density functional theory with electronic and topological descriptors. J. Taibah Univ. Sci. 2015;9:143–154. [http://dx.doi.org/10.1016/j.jtusci.2014.10.006]. [Google Scholar]
- 154.Dong X., Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors. Curr. Chem. Genomics. 2008;2:29–39. doi: 10.2174/1875397300802010029. [http://dx.doi.org/ 10.2174/1875397300802010029]. [PMID: 20161841]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sinha S., Tyagi C., Goyal S., Jamal S., Somvanshi P., Grover A. Fragment based G-QSAR and molecular dynamics based mechanistic simulations into hydroxamic-based HDAC inhibitors against spinocerebellar ataxia. J. Biomol. Struct. Dyn. 2016;34(10):2281–2295. doi: 10.1080/07391102.2015.1113386. [http://dx.doi.org/10.1080/07391102.2015.1113386]. [PMID: 26510381]. [DOI] [PubMed] [Google Scholar]
- 156.Ringsted T., Nikolov N., Jensen G.E., Wedebye E.B., Niemelä J. QSAR models for P450 (2D6) substrate activity. SAR QSAR Environ. Res. 2009;20(3-4):309–325. doi: 10.1080/10629360902949195. [http://dx.doi.org/10.1080/ 10629360902949195]. [PMID: 19544194]. [DOI] [PubMed] [Google Scholar]
- 157.Gharaghani S., Khayamian T., Keshavarz F. Docking, molecular dynamics simulation studies, and structure-based QSAR model on cytochrome P450 2A6 inhibitors. Struct. Chem. 2012;23:341–350. [http://dx.doi.org/10.1007/s11224-011-9874-0]. [Google Scholar]
- 158.Mandi P., Nantasenamat C., Srungboonmee K., Isarankura-Na-Ayudhya C., Prachayasittikul V. QSAR study of anti-prion activity of 2-aminothiazoles. EXCLI J. 2012;11:453–467. [PMID: 27418919]. [PMC free article] [PubMed] [Google Scholar]
- 159.Cosentino U., Pitea D., Moro G., Saracino G.A., Caria P., Varì R.M., Colombo L., Forloni G., Tagliavini F., Salmona M. The anti-fibrillogenic activity of tetracyclines on PrP 106-126: a 3D-QSAR study. J. Mol. Model. 2008;14(10):987–994. doi: 10.1007/s00894-008-0348-2. [http://dx.doi.org/10.1007/s00894-008-0348-2]. [PMID: 18629550]. [DOI] [PubMed] [Google Scholar]
- 160.Amin S.A., Adhikari N., Jha T., Gayen S. First molecular modeling report on novel arylpyrimidine kynurenine monooxygenase inhibitors through multi-QSAR analysis against Huntington’s disease: A proposal to chemists! Bioorg. Med. Chem. Lett. 2016;26(23):5712–5718. doi: 10.1016/j.bmcl.2016.10.058. [http://dx.doi.org/10.1016/j.bmcl.2016.10.058]. [PMID: 27838184]. [DOI] [PubMed] [Google Scholar]
- 161.Leone S., Mutti C., Kazantsev A., Sturlese M., Moro S., Cattaneo E., Rigamonti D., Contini A. SAR and QSAR study on 2-aminothiazole derivatives, modulators of transcriptional repression in Huntington’s disease. Bioorg. Med. Chem. 2008;16(10):5695–5703. doi: 10.1016/j.bmc.2008.03.067. [http://dx.doi.org/10.1016/j.bmc.2008.03.067]. [PMID: 18406155]. [DOI] [PubMed] [Google Scholar]
- 162.Helguera A.M., Perez-Castillo Y., Cordeiro D.S.M.N., Tejera E. Paz-Y-Miño, C.; Sánchez-Rodríguez, A.; Teijeira, M.; Ancede-Gallardo, E.; Cagide, F.; Borges, F.; Cruz-Monteagudo, M. Ligand-based virtual screening using tailored ensembles: A prioritization tool for dual A2A adenosine receptor antagonists / monoamine oxidase B inhibitors. Curr. Pharm. Des. 2016;22:3082–3096. doi: 10.2174/1381612822666160302103542. [http://dx.doi.org/10.2174/1381612822666160302103542]. [PMID: 26932160]. [DOI] [PubMed] [Google Scholar]
- 163.Helguera A.M., Pérez-Garrido A., Gaspar A., Reis J., Cagide F., Vina D., Cordeiro M.N., Borges F. Combining QSAR classification models for predictive modeling of human monoamine oxidase inhibitors. Eur. J. Med. Chem. 2013;59:75–90. doi: 10.1016/j.ejmech.2012.10.035. [http://dx.doi.org/ 10.1016/j.ejmech.2012.10.035]. [PMID: 23207409]. [DOI] [PubMed] [Google Scholar]
- 164.Zhu Y.Q., Pei J.F., Liu Z.M., Lai L.H., Cui J.R., Li R.T. 3D-QSAR studies on tripeptide aldehyde inhibitors of proteasome using CoMFA and CoMSIA methods. Bioorg. Med. Chem. 2006;14(5):1483–1496. doi: 10.1016/j.bmc.2005.10.003. [http://dx.doi.org/10.1016/j.bmc.2005.10.003]. [PMID: 16256351]. [DOI] [PubMed] [Google Scholar]
- 165.Fresqui M.A.C., Ferreira M.M., Trsic M. The influence of R and S configurations of a series of amphetamine derivatives on QSAR models. Anal. Chim. Acta. 2013;759:43–52. doi: 10.1016/j.aca.2012.11.004. [http://dx.doi.org/ 10.1016/j.aca.2012.11.004]. [PMID: 23260675]. [DOI] [PubMed] [Google Scholar]
- 166.Bharate S.B., Yadav R.R., Vishwakarma R.A. QSAR and pharmacophore study of Dyrk1A inhibitory meridianin analogs as potential agents for treatment of neurodegenerative diseases. Med. Chem. 2013;9(1):152–161. doi: 10.2174/157340613804488459. [http://dx.doi.org/10.2174/ 157340613804488459]. [PMID: 22920091]. [DOI] [PubMed] [Google Scholar]
- 167.Tong W., Collantes E.R., Chen Y., Welsh W.J. A comparative molecular field analysis study of N-benzylpiperidines as acetylcholinesterase inhibitors. J. Med. Chem. 1996;39(2):380–387. doi: 10.1021/jm950704x. [http://dx.doi.org/10.1021/jm950704x]. [PMID: 8558505]. [DOI] [PubMed] [Google Scholar]
- 168.Ponmary D., Latha P., Jeya D., Sharmila S. QSAR study for the prediction of Half Maximal Inhibitory Concentration of Compounds structurally similar to Glycerol. Turk. J. Biochem. 2010;35:287–292. [Google Scholar]
- 169.Jung M., Tak J., Lee Y., Jung Y. Quantitative structure-activity relationship (QSAR) of tacrine derivatives against acetylcholinesterase (AChE) activity using variable selections. Bioorg. Med. Chem. Lett. 2007;17(4):1082–1090. doi: 10.1016/j.bmcl.2006.11.022. [http://dx.doi.org/10.1016/j. bmcl.2006.11.022]. [PMID: 17158047]. [DOI] [PubMed] [Google Scholar]
- 170.Chen N., Liu C.K., Zhao L.Z., Zhang H.B. 3D-QSAR study of multi-target-directed AchE inhibitors based on autodocking. Med. Chem. Res. 2012;21:245–256. [http://dx.doi.org/10.1007/s00044-010-9516-x]. [Google Scholar]
- 171.Hoeglund A.B., Bostic H.E., Howard A.L., Wanjala I.W., Best M.D., Baker D.L., Parrill A.L. Optimization of a pipemidic acid autotaxin inhibitor. J. Med. Chem. 2010;53(3):1056–1066. doi: 10.1021/jm9012328. [http://dx.doi.org/10.1021/jm9012328]. [PMID: 20041668]. [DOI] [PubMed] [Google Scholar]
- 172.Recanatini M., Cavalli A., Hansch C. A comparative QSAR analysis of acetylcholinesterase inhibitors currently studied for the treatment of Alzheimer’s disease. Chem. Biol. Interact. 1997;105(3):199–228. doi: 10.1016/s0009-2797(97)00047-1. [http://dx.doi.org/10.1016/S0009-2797(97)00047-1]. [PMID: 9291997]. [DOI] [PubMed] [Google Scholar]
- 173.Jain P., Jadhav H.R. Quantitative structure activity relationship analysis of aminoimidazoles as BACE-I inhibitors. Med. Chem. Res. 2013;22:1740–1746. [http://dx.doi.org/10.1007/s00044-012-0166-z]. [Google Scholar]
- 174.Debord J., N’Diaye P., Bollinger J.C., Fikri K., Penicaut B., Robert J.M., Robert-Piessard S., Le Baut G. Cholinesterase inhibition by derivatives of 2-amino-4,6-dimethylpyridine. J. Enzyme Inhib. 1997;12(1):13–26. doi: 10.3109/14756369709027660. [http://dx.doi.org/10.3109/ 14756369709027660]. [PMID: 9204379]. [DOI] [PubMed] [Google Scholar]
- 175.Huang D., Liu Y., Shi B., Li Y., Wang G., Liang G. Comprehensive 3D-QSAR and binding mode of BACE-1 inhibitors using R-group search and molecular docking. J. Mol. Graph. Model. 2013;45:65–83. doi: 10.1016/j.jmgm.2013.08.003. [http://dx.doi.org/10.1016/j.jmgm.2013.08.003]. [PMID: 24004830]. [DOI] [PubMed] [Google Scholar]
- 176.Hossain T., Islam M.A., Saha R.P. Exploring structural requirement andbinding interactions ofb-amyloidcleavage enzyme inhibitors usingmolecular modeling techniques. Med. Chem. Res. 2013;22:4766–4774. [http://dx.doi.org/10.1007/s00044-013-0481-z]. [Google Scholar]
- 177.Dessalew N., Patel D.S., Bharatam P.V. 3D-QSAR and molecular docking studies on pyrazolopyrimidine derivatives as glycogen synthase kinase-3beta inhibitors. J. Mol. Graph. Model. 2007;25(6):885–895. doi: 10.1016/j.jmgm.2006.08.009. [http://dx.doi.org/10.1016/j.jmgm.2006.08.009]. [PMID: 17018257]. [DOI] [PubMed] [Google Scholar]
- 178.García I., Fall Y., Gómez G., González-Díaz H. First computational chemistry multi-target model for anti-Alzheimer, anti-parasitic, anti-fungi, and anti-bacterial activity of GSK-3 inhibitors in vitro, in vivo, and in different cellular lines. Mol. Divers. 2011;15(2):561–567. doi: 10.1007/s11030-010-9280-3. [http://dx.doi.org/10.1007/s11030-010-9280-3]. [PMID: 20931280]. [DOI] [PubMed] [Google Scholar]
- 179.Bhadoriya K.S., Sharma M.C., Sharma S. An approach to design potent anti- Alzheimer’s agents by 3D-QSAR studies on fused 5, 6-bicyclic heterocycles as beta-secretase modulators using kNNMFA methodology. Arab. J. Chem. 2014;7:924–935. [http://dx.doi.org/ 10.1016/j.arabjc.2013.02.002]. [Google Scholar]
- 180.Barreca M.L., Gitto R., Quartarone S., De Luca L., De Sarro G., Chimirri A. Pharmacophore modeling as an efficient tool in the discovery of novel noncompetitive AMPA receptor antagonists. J. Chem. Inf. Comput. Sci. 2003;43(2):651–655. doi: 10.1021/ci025625q. [http://dx.doi.org/ 10.1021/ci025625q]. [PMID: 12653534]. [DOI] [PubMed] [Google Scholar]
- 181.Valasani K.R., Hu G., Chaney M.O., Yan S.S. Structure-based design and synthesis of benzothiazole phosphonate analogues with inhibitors of human ABAD-Aβ for treatment of Alzheimer’s disease. Chem. Biol. Drug Des. 2013;81(2):238–249. doi: 10.1111/cbdd.12068. [http://dx.doi. org/10.1111/cbdd.12068]. [PMID: 23039767]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Koutsoukas A., Paricharak S., Galloway W.R., Spring D.R., Ijzerman A.P., Glen R.C., Marcus D., Bender A. How diverse are diversity assessment methods? A comparative analysis and benchmarking of molecular descriptor space. J. Chem. Inf. Model. 2014;54(1):230–242. doi: 10.1021/ci400469u. [http://dx.doi.org/10.1021/ci400469u]. [PMID: 24289493]. [DOI] [PubMed] [Google Scholar]
- 183.Zhou A., Hu J., Wang L., Zhong G., Pan J., Wu Z., Hui A. Combined 3D-QSAR, molecular docking, and molecular dynamics study of tacrine derivatives as potential acetylcholinesterase (AChE) inhibitors of Alzheimer’s disease. J. Mol. Model. 2015;21(10):277. doi: 10.1007/s00894-015-2797-8. [http://dx.doi.org/10.1007/s00894-015-2797-8]. [PMID: 26438408]. [DOI] [PubMed] [Google Scholar]
- 184.Pourbasheer E., Shokouhi T.S., Masand V.H., Aalizadeh R., Ganjali M.R. 3D-QSAR and docking studies on adenosine A2A receptor antagonists by the CoMFA method. SAR QSAR Environ. Res. 2015;26(6):461–477. doi: 10.1080/1062936X.2015.1049666. [http://dx.doi.org/10.1080/1062936X. 2015.1049666]. [PMID: 26055215]. [DOI] [PubMed] [Google Scholar]
- 185.Dinata D.I., Rendrika R., Pryanda H. Computational study of 2-aminothiazole as antiprion lead compound in the treatment of creutzfeldt-jakob disease using an approach model of hansch quantitativ structure activity relationship andtoxicity prediction. Int. J. Pharm. Teach. Pract. 2013;4:797–805. [Google Scholar]
- 186.Hajimahdi Z., Safizadeh F., Zarghi A. QSAR Analysis for Some 1, 2-Benzisothiazol-3-one Derivatives as Caspase-3 Inhibitors by Stepwise MLR Method. Iran. J. Pharm. Res. 2016;15(2):439–448. [PMID: 27642314]. [PMC free article] [PubMed] [Google Scholar]
- 187.Raha K., Peters M.B., Wang B., Yu N., Wollacott A.M., Westerhoff L.M., Merz K.M., Jr The role of quantum mechanics in structure-based drug design. Drug Discov. Today. 2007;12(17-18):725–731. doi: 10.1016/j.drudis.2007.07.006. [http://dx.doi.org/10.1016/j.drudis.2007.07.006]. [PMID: 17826685]. [DOI] [PubMed] [Google Scholar]
- 188.Lewars E.G. 2011. [Google Scholar]
- 189.Rahman A., Ali M.T., Shawan M.M.A.K., Sarwar M.G., Khan M.A.K., Halim M.A. Halogen-directed drug design for Alzheimer’s disease: a combined density functional and molecular docking study. Springerplus. 2016;5(1):1346. doi: 10.1186/s40064-016-2996-5. [http://dx.doi. org/10.1186/s40064-016-2996-5]. [PMID: 27588239]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.da Silva C.H., Carvalho I., Taft C.A. Virtual screening, molecular interaction field, molecular dynamics, docking, density functional, and ADMET properties of novel AChE inhibitors in Alzheimer’s disease. J. Biomol. Struct. Dyn. 2007;24(6):515–524. doi: 10.1080/07391102.2007.10507140. [http://dx. doi.org/10.1080/07391102.2007.10507140]. [PMID: 17508773]. [DOI] [PubMed] [Google Scholar]
- 191.Cisek K., Jensen J.R., Honson N.S., Schafer K.N., Cooper G.L., Kuret J. Ligand electronic properties modulate tau filament binding site density. Biophys. Chem. 2012;170:25–33. doi: 10.1016/j.bpc.2012.09.001. [http://dx. doi.org/10.1016/j.bpc.2012.09.001]. [PMID: 23072817]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Barbault F., Maurel F. Simulation with quantum mechanics/molecular mechanics for drug discovery. Expert Opin. Drug Discov. 2015;10(10):1047–1057. doi: 10.1517/17460441.2015.1076389. [http://dx.doi.org/10.1517/ 17460441.2015.1076389]. [PMID: 26289577]. [DOI] [PubMed] [Google Scholar]
- 193.da Cunha E.F.F., Resende J.E., Franca T.C.C., Gonçalves M.A., de Souza F.R., Santos-Garcia L., Ramalho T.C. Molecular modeling studies of piperidine derivatives as new acetylcholinesterase inhibitors against neurodegenerative diseases. J. Chem. 2013;2013:1–7. [http://dx.doi.org/10.1155/2013/278742]. [Google Scholar]
- 194.Gleeson D., Gleeson M.P. Application of QM/MM and QM methods to investigate histone deacetylase 8. MedChemComm. 2015;6:477–485. [http://dx.doi.org/10.1039/C4MD00471J]. [Google Scholar]
- 195.Lucas J., Gutierrez Exequiel E., Guisasola B., Peruchena N., Enriz R.D. A QM/MM study of the molecular recognition site of bapineuzumab toward the amyloid-β peptide isoforms. Mol. Simul. 2016;42:196–207. [http://dx.doi.org/10.1080/08927022.2015. 1032276]. [Google Scholar]
- 196.Malamas M.S., Erdei J., Gunawan I., Barnes K., Johnson M., Hui Y., Turner J., Hu Y., Wagner E., Fan K., Olland A., Bard J., Robichaud A.J. Aminoimidazoles as potent and selective human β-secretase (BACE1) inhibitors. J. Med. Chem. 2009;52(20):6314–6323. doi: 10.1021/jm9006752. [http://dx.doi.org/10.1021/jm9006752]. [PMID: 19757823]. [DOI] [PubMed] [Google Scholar]
- 197.Alonso H., Bliznyuk A.A., Gready J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006;26(5):531–568. doi: 10.1002/med.20067. [http://dx.doi.org/10.1002/med.20067]. [PMID: 16758486]. [DOI] [PubMed] [Google Scholar]
- 198.Kerrigan J.E. Molecular dynamics simulations in drug design. Methods Mol. Biol. 2013;993:95–113. doi: 10.1007/978-1-62703-342-8_7. [http://dx.doi.org/10.1007/ 978-1-62703-342-8_7]. [PMID: 23568466]. [DOI] [PubMed] [Google Scholar]
- 199.Bernardi R.C., Melo M.C.R., Schulten K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta. 2015;1850(5):872–877. doi: 10.1016/j.bbagen.2014.10.019. [http://dx. doi.org/10.1016/j.bbagen.2014.10.019]. [PMID: 25450171]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jorgensen W.L., Thomas L.L. Perspective on free-energy perturbation calculations for chemical equilibria. J. Chem. Theory Comput. 2008;4(6):869–876. doi: 10.1021/ct800011m. [http://dx.doi.org/10.1021/ct800011m]. [PMID: 19936324]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Isralewitz B., Gao M., Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001;11(2):224–230. doi: 10.1016/s0959-440x(00)00194-9. [http://dx.doi.org/10.1016/S0959-440X(00) 00194-9]. [PMID: 11297932]. [DOI] [PubMed] [Google Scholar]
- 202.Laio A., Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [http://dx.doi.org/10. 1073/pnas.202427399]. [PMID: 12271136]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ciordia M., Pérez-Benito L., Delgado F., Trabanco A.A., Tresadern G. Application of Free Energy Perturbation for the Design of BACE1 Inhibitors. J. Chem. Inf. Model. 2016;56(9):1856–1871. doi: 10.1021/acs.jcim.6b00220. [http://dx.doi.org/10.1021/acs.jcim.6b00220]. [PMID: 27500414]. [DOI] [PubMed] [Google Scholar]
- 204.Braun G.H., Jorge D.M., Ramos H.P., Alves R.M., da Silva V.B., Giuliatti S., Sampaio S.V., Taft C.A., Silva C.H. Molecular dynamics, flexible docking, virtual screening, ADMET predictions, and molecular interaction field studies to design novel potential MAO-B inhibitors. J. Biomol. Struct. Dyn. 2008;25(4):347–355. doi: 10.1080/07391102.2008.10507183. [http://dx.doi.org/10.1080/07391102.2008.10507183]. [PMID: 18092829]. [DOI] [PubMed] [Google Scholar]
- 205.Distinto S., Yáñez M., Alcaro S., Cardia M.C., Gaspari M., Sanna M.L., Meleddu R., Ortuso F., Kirchmair J., Markt P., Bolasco A., Wolber G., Secci D., Maccioni E. Synthesis and biological assessment of novel 2-thiazolylhydrazones and computational analysis of their recognition by monoamine oxidase B. Eur. J. Med. Chem. 2012;48:284–295. doi: 10.1016/j.ejmech.2011.12.027. [http://dx.doi.org/10.1016/ j.ejmech.2011.12.027]. [PMID: 22222137]. [DOI] [PubMed] [Google Scholar]
- 206.Palakurti R., Vadrevu R. Identification of abelson tyrosine kinase inhibitors as potential therapeutics for Alzheimer’s disease using multiple e-pharmacophore modeling and molecular dynamics. J. Biomol. Struct. Dyn. 2017;35:883–896. doi: 10.1080/07391102.2016.1166454. [http://dx.doi.org/10.1080/ 07391102.2016.1166454]. [PMID: 26982633]. [DOI] [PubMed] [Google Scholar]
- 207.Pang Y.P., Kozikowski A.P. Prediction of the binding sites of huperzine A in acetylcholinesterase by docking studies. J. Comput. Aided Mol. Des. 1994;8(6):669–681. doi: 10.1007/BF00124014. [http://dx.doi.org/10.1007/ BF00124014]. [PMID: 7738603]. [DOI] [PubMed] [Google Scholar]
- 208.Cavalli A., Bottegoni G., Raco C., De Vivo M., Recanatini M. A computational study of the binding of propidium to the peripheral anionic site of human acetylcholinesterase. J. Med. Chem. 2004;47(16):3991–3999. doi: 10.1021/jm040787u. [http://dx.doi.org/10.1021/jm040787u]. [PMID: 15267237]. [DOI] [PubMed] [Google Scholar]
- 209.Kacker P., Masetti M., Mangold M., Bottegoni G., Cavalli A. Combining dyad protonation and active site plasticity in BACE-1 structure-based drug design. J. Chem. Inf. Model. 2012;52(5):1079–1085. doi: 10.1021/ci200366z. [http://dx.doi.org/10.1021/ci200366z]. [PMID: 22313091]. [DOI] [PubMed] [Google Scholar]
- 210.Bochicchio A., Rossetti G., Tabarrini O., Krauß S., Carloni P. Molecular view of ligands specificity for CAG repeats in anti-Huntington therapy. J. Chem. Theory Comput. 2015;11(10):4911–4922. doi: 10.1021/acs.jctc.5b00208. [http://dx.doi.org/10.1021/acs.jctc.5b00208]. [PMID: 26574279]. [DOI] [PubMed] [Google Scholar]
- 211.Biarnés X., Bongarzone S., Vargiu A.V., Carloni P., Ruggerone P. Molecular motions in drug design: the coming age of the metadynamics method. J. Comput. Aided Mol. Des. 2011;25(5):395–402. doi: 10.1007/s10822-011-9415-3. [http://dx.doi.org/10.1007/s10822-011-9415-3]. [PMID: 21327922]. [DOI] [PubMed] [Google Scholar]
- 212.Decherchi S., Berteotti A., Bottegoni G., Rocchia W., Cavalli A. The ligand binding mechanism to purine nucleoside phosphorylase elucidated via molecular dynamics and machine learning. Nat. Commun. 2015;6:6155. doi: 10.1038/ncomms7155. [http://dx.doi.org/10.1038/ncomms7155]. [PMID: 25625196]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Srinivasan E., Rajasekaran R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Advances. 2016;6:102744–102753. [http://dx.doi.org/10.1039/C6RA21927F]. [Google Scholar]
- 214.Padhi A.K., Jayaram B., Gomes J. 2013. [Google Scholar]
- 215.Poongavanam V., Kongsted J., Wüstner D. Computational analysis of sterol ligand specificity of the Niemann Pick C2 protein. Biochemistry. 2016;55(36):5165–5179. doi: 10.1021/acs.biochem.6b00217. [http://dx.doi.org/10.1021/acs. biochem.6b00217]. [PMID: 27533706]. [DOI] [PubMed] [Google Scholar]
- 216.Mashamba-Thompson T., Soliman M.E.S. Insight into the binding theme of CA-074Me to cathepsin B: molecular dynamics simulations and scaffold hopping to identify potential analogues as anti-neurodegenerative diseases. Med. Chem. Res. 2015;24:701–713. [http://dx.doi.org/10.1007/s00044-014-1145-3]. [Google Scholar]
- 217.Sattin S., Tao J., Vettoretti G., Moroni E., Pennati M., Lopergolo A., Morelli L., Bugatti A., Zuehlke A., Moses M., Prince T., Kijima T., Beebe K., Rusnati M., Neckers L., Zaffaroni N., Agard D.A., Bernardi A., Colombo G. Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry. 2015;21(39):13598–13608. doi: 10.1002/chem.201502211. [http://dx.doi.org/10.1002/chem.201502211]. [PMID: 26286886]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vettoretti G., Moroni E., Sattin S. Tao, Jiahui.; Agard, D. A.; Bernardi, A.; Colombo, G. Molecular dynamics simulations reveal the mechanisms of allosteric activation of Hsp90 by designed ligands. Sci. Rep. 2016;6:1–13. doi: 10.1038/srep23830. [http://dx.doi.org/10.1038/srep23830]. [PMID: 28442746]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ran T., Zhang Z., Liu K., Lu Y., Li H., Xu J., Xiong X., Zhang Y., Xu A., Lu S., Liu H., Lu T., Chen Y. Insight into the key interactions of bromodomain inhibitors based on molecular docking, interaction fingerprinting, molecular dynamics and binding free energy calculation. Mol. Biosyst. 2015;11(5):1295–1304. doi: 10.1039/c4mb00723a. [http://dx.doi.org/10.1039/C4MB00723A]. [PMID: 25758752]. [DOI] [PubMed] [Google Scholar]
- 220.Morra G., Neves M.A.C., Plescia C.J., Tsustsumi S., Neckers L., Verkhivker G., Altieri D.C., Colombo G. Dynamics-based discovery of allosteric inhibitors: selection of new ligands for thec-terminal domain of hsp90. J. Chem. Theory Comput. 2010;6(9):2978–2989. doi: 10.1021/ct100334n. [http://dx.doi.org/10.1021/ct100334n]. [PMID: 26616092]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bejugam P.R., Kuppili R.R., Singh N., Gadewal N., Chaganti L.K., Sastry G.M., Bose K. Allosteric regulation of serine protease HtrA2 through novel non-canonical substrate binding pocket. PLoS One. 2013;8(2):e55416. doi: 10.1371/journal.pone.0055416. [http://dx.doi.org/10.1371/journal. pone.0055416]. [PMID: 23457469]. [DOI] [PMC free article] [PubMed] [Google Scholar]