

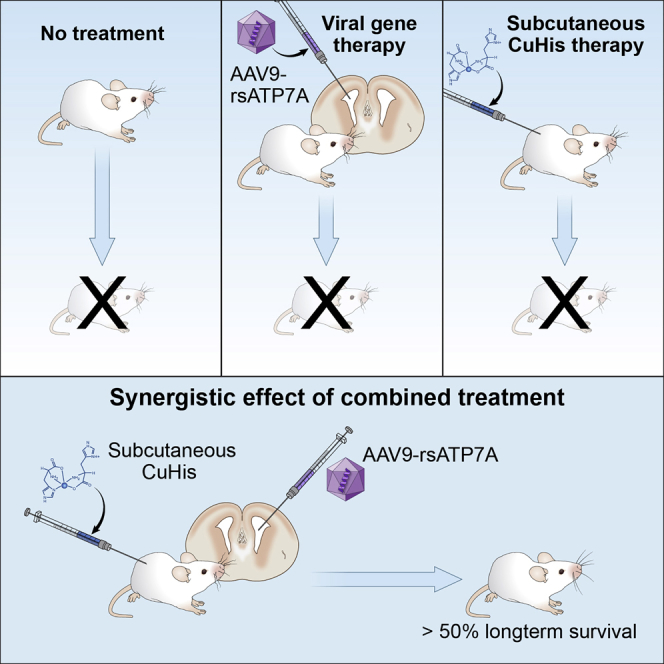
Abstract
Menkes disease is a lethal neurodegenerative disorder of copper metabolism caused by mutations in an evolutionarily conserved copper transporter, ATP7A. Based on our prior clinical and animal studies, we seek to develop a therapeutic approach suitable for application in affected human subjects, using the mottled-brindled (mo-br) mouse model that closely mimics the Menkes disease biochemical and clinical phenotypes. Here, we evaluate the efficacy of low-, intermediate-, and high-dose recombinant adeno-associated virus serotype 9 (rAAV9)-ATP7A delivered to the cerebrospinal fluid (CSF), in combination with subcutaneous administration of clinical-grade copper histidinate (sc CuHis, IND #34,166). Mutant mice that received high-dose (1.6 × 1010 vg) cerebrospinal fluid-directed rAAV9-rsATP7A plus sc copper histidinate showed 53.3% long-term (≥300-day) survival compared to 0% without treatment or with either treatment alone. The high-dose rAAV9-rsATP7A plus sc copper histidinate-treated mutant mice showed increased brain copper levels, normalized brain neurochemical levels, improvement of brain mitochondrial abnormalities, and normal growth and neurobehavioral outcomes. This synergistic treatment effect represents the most successful rescue to date of the mo-br mouse model. Based on these findings, and the absence of a large animal model, we propose cerebrospinal fluid-directed rAAV9-rsATP7A gene therapy plus subcutaneous copper histidinate as a potential therapeutic approach to cure or ameliorate Menkes disease.
Keywords: adeno-associated virus, Menkes disease, ATP7A, copper, dopamine-beta-hydroxylase, choroid plexus epithelia
Graphical Abstract
Introduction
Menkes disease is an X-linked recessive infantile-onset neurodegenerative disorder of copper transport initially described more than 50 years ago.1, 2 The illness is caused by mutations in a highly conserved copper-transporting ATPase, ATP7A.3, 4, 5, 6 The condition has an estimated incidence of one in 100,000 live births and is associated with high under-age-three mortality.7, 8 There are currently no US Food and Drug Administration (FDA)-approved treatments for this rare disease.
ATP7A is needed for copper absorption from the gastrointestinal tract, transport of copper across the blood-cerebrospinal fluid and blood-brain barriers, and metalation of various copper-dependent enzymes.7, 9 Affected Menkes infants manifest a distinctive clinical phenotype related to copper deficiency with sparse, depigmented hair, seizures, hypotonia, gross motor delays, failure to thrive, and connective tissue problems.1, 7 Plasma and cerebrospinal fluid (CSF) catecholamine levels are sensitive and specific biomarkers of the disease in early infancy, reflecting low activity of dopamine-beta-hydroxylase, a copper-dependent enzyme.10, 11, 12
A major obstacle to successful treatment of this illness is identifying mechanisms to transport sufficient copper to the developing brain and enable proper utilization by neurons. ATP7A normally traffics to the plasma membrane of polarized epithelial cells to mediate copper transport across membrane bilayers, including the gastrointestinal mucosa-blood, blood-brain, and blood-cerebrospinal fluid barriers.9 There have been numerous careful clinical efforts to modify the natural history of this difficult illness by various copper treatment regimens.2, 13, 14, 15, 16 Early (≤28 days postnatal) subcutaneous injections of copper histidinate (sc CuHis) can bypass the defect in gastrointestinal copper absorption, normalize blood copper levels, and reduce under-age-three mortality compared to untreated subjects.12, 17 Moreover, sc copper histidinate injections are especially effective when commenced in the early, pre-symptomatic phase and when the ATP7A molecular defect does not completely abrogate copper transport.12 Certain Menkes disease patients whose ATP7A mutations were associated with residual copper transport not only averted premature death but attained normal neurodevelopmental outcomes in response to early sc copper histidinate treatment.12, 17, 18, 19, 20, 21 However, affected patients harboring severe loss-of-function ATP7A mutations often respond less successfully to early intervention with sc copper histidinate alone.12, 17, 18, 22, 23 The presence of a rudimentary level of ATP7A activity in the brains of such patients would clearly optimize the probability of avoiding the most devastating neurological effects of this illness.6, 18 Recent progress in viral gene therapy makes it plausible to consider brain-directed gene addition approaches to accomplish this aim.24, 25
A naturally occurring mouse model of Menkes disease has provided a useful tool for detailed experimental therapeutic investigations.25, 26, 27, 28, 29, 30 We previously used cerebrospinal fluid (lateral brain ventricle) injections of recombinant AAV serotype 5 (rAAV5) and cerebrospinal fluid cupric chloride to rescue the mottled-brindled (mo-br) mice, which faithfully recapitulates the Menkes disease phenotype.25, 29, 30 The fact that recombinant AAV5 transduced only choroid plexus epithelia in the mouse brain ventricles, while increasing brain copper levels and mediating long-term survival in 22% of mutants, underscored the crucial role of these structures in normal mammalian brain copper homeostasis.25, 31, 32, 33, 34 While overall median survival in mutant mo-br mice was extended (from 13 days to 43 days), neurobehavioral and pathological assessments revealed significant abnormalities.25 These results were consistent with experimental evidence of the roles of ATP7A in neuronal transmission, synaptogenesis, and motor neuron function, in addition to choroid plexus-mediated copper delivery.9, 35, 36, 37
Since Atp7a is widely expressed in murine brain cells (choroid plexus epithelia, neurons, and glia) under normal conditions,34 we sought to refine our gene therapy regimen, utilizing an AAV serotype with broad tropism, rAAV9, that would target neurons as well as choroid plexus epithelia. We designed a new experimental therapeutic regimen that could be readily translated to clinical application, combining cerebrospinal fluid-directed rAAV9-rsATP7A with subcutaneous copper injections. For the latter, we used clinical grade copper histidinate, the investigational new drug (IND #34,166) used in our current clinical trial (ClinicalTrials.gov: NCT00001262). In contrast, our prior gene therapy study with this mouse model involved direct cerebrospinal fluid injections of cupric chloride.25
Results
Recombinant AAV9 Shows Broad Neuronal Tropism in Mouse Brain
To evaluate the transduction pattern of rAAV9 in C57BL/6 mouse brain following direct administration to the cerebrospinal fluid, we utilized a construct harboring the GFP cDNA. Based on GFP immunoreactivity, rAAV9 displayed broad tropism following administration to the mouse brain lateral ventricles on day 2, including choroid plexus epithelia, neuronal cells, and some glia (Figures 1A–1E). As expected, untreated mouse brains showed no GFP expression on immunohistochemical analysis (Figures 1A and 1B).
Figure 1.

Recombinant AAV9 Tropism in Mouse Brain
Immunohistochemistry to illustrate reporter gene (GFP) expression after injection of rAAV9 into the cerebrospinal fluid of wild-type mice. (A) Low-power sagittal view of wild-type mouse brain on day 12 after cerebrospinal fluid administration of 1.6 × 1010 vg of rAAV9-GFP on day 2 (top). Staining with anti-GFP defines the transduction pattern of rAAV9 (brownish color) in these experiments since there is no reliable anti-ATP7A antibody for immunohistochemical applications. An uninjected mouse brain (bottom) shows no GFP expression. Scale bars, 1 mm. (B) Representative brain region images with GFP expression on day 12 after injection of 5 × 109 vg rAAV9-GFP into the cerebrospinal fluid of wild-type mice on day 2. CPLV, choroid plexus epithelia of lateral ventricle. Scale bars, 100 μm. (C–E) High-power views of cortex (C), cerebellum (D), and brainstem (E), respectively, from 12-day-old mice after cerebrospinal fluid injection of 5 × 109 vg of rAAV9-GFP on day 2, stained with anti-GFP on day 12. Closed arrows denote transduced neurons and open arrows, transduced glial cells. Scale bars, 25 μm.
To accommodate the packaging limits of rAAV, we employed a compact version of the human ATP7A cDNA in which the first four copper-binding motifs were removed (Figure 2A).25 This compact, reduced-size cDNA (rsATP7A) was used for the rAAV9 construct (Figure 2B). Other elements included flanking inverted terminal repeat (ITR) motifs, a cytomegalovirus (CMV) enhancer, chicken β-actin (CBA) promoter, and rabbit β-globin polyadenylation (poly(A)) signal. We previously demonstrated that the capacity of rsATP7A for functional copper transport was minimally reduced (≈80% compared to the full-length version in a yeast complementation assay) and localized properly to the trans-Golgi network under normal intracellular copper conditions.25 Here, we documented that rsATP7A also undergoes normal intracellular trafficking in transfected HEK293T cells in response to higher copper concentrations (Figure 2C), expresses a protein product of the predicted size (125 kDa; Figure 2D), and restores copper delivery to tyrosinase in a cell-based copper enzyme activation assay (Figure 2E).
Figure 2.

Reduced Size ATP7A (rsATP7A) for rAAV9 Gene Therapy
(A) Model of ATP7A including six copper (Cu)-binding domains in the N terminus and other functional domains, as indicated. The light blue-shaded area shows the segment removed to create rsATP7A, leaving only Cu-binding sites #5 and #6. Red asterisks indicate location of the two amino acids deleted in the mo-br mouse ortholog (Atp7a). (B) Diagram of the recombinant AAV9 construct used in this study. ITR, inverted terminal repeat of AAV serotype 2; CMV, cytomegalovirus (CMV) enhancer; CBA, chicken β-actin promoter; poly(A), rabbit β-globin polyadenylation signal. (C) Confocal microscopic images of HEK293T cells transfected with a Venus-tagged version of rsATP7A, showing normal trafficking (relocation from the perinuclear trans-Golgi network to plasma membrane) in response to increased concentration (from 0.5 μM to 200 μM) of copper. Scale bars, 10 μm. (D) Western blot of total protein from transfected HEK293T cells compared to non-transfected controls. Both rsATP7A protein (≈125 kDa) and endogenous full-length ATP7A (≈165 kDa) are detected in transfected cells. (E) Plasmid-mediated expression of rsATP7A in YSTT cells restores copper delivery to the trans-Golgi network and activates tyrosinase. This functional assay qualitatively assesses the physiological capacity of human copper-ATPases to transport copper into the trans-Golgi network (TGN) for biosynthetic incorporation into copper-dependent enzymes. YSTT cells lack endogenous ATP7A and are unable to transport copper into the TGN, thus rendering tyrosinase, a copper-dependent enzyme, inactive unless transfected with a functional version of ATP7A or ATP7B. Formation of black pigment [levo-3,4-dihydroxy-l-phenylalanine (L-DOPA) quinone] in the cells indicates activation of tyrosinase by both reduced size and full-length ATP7A. The assay was performed in triplicate, and representative images are shown. Scale bars, 50 μm.
Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus sc Copper Histidinate Enhances Survival in mo-br Mice
We identified mutant mo-br mice at birth by clinical appearance (male gender, short curly vibrissae) and confirmed their affected status by genotyping, as previously described.25 For viral gene therapy, we used rAAV9, a serotype with expected tropism for mouse choroid plexus epithelia as well as neurons throughout the brain (Figure 1). Low, intermediate, and high doses of rAAV9-rsATP7A contained a total of 1.6 × 109, 5 × 109, or 1.6 × 1010 vg, respectively. A total dose of 15 μg of clinical grade copper histidinate was administered by subcutaneous injection in 5-μg doses on postpartum days 4, 5, and 6 (see Materials and Methods for further details).
Treatment with rAAV9-rsATP7A alone, or with sc copper histidinate alone, was unable to rescue the mutant mice (Figure 3A; Table 1). Short-term (≥30 days) survival was achieved in 25%–75% treated with AAV9-rsATP7A combined with sc copper histidinate, depending on AAV dose (Table 1). Median survival was longest with intermediate- and high-dose rAAV9-rsATP7A in combination with sc copper histidinate (199 days and 300 days, respectively, Table 1).
Figure 3.

Results of Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus sc Copper Histidinate versus Individual Treatments
(A) Kaplan-Meier survival curve demonstrates superior survival with high-dose AAV9-rsATP7A plus sc copper histidinate (magenta) in comparison to untreated mutants (UT), rAAV9-rsATP7A alone (blue), and sc copper histidinate alone (brown) did not significantly enhance survival beyond that in untreated mutants (green) (p < 0.0001). (B) Viral genome quantification using real-time PCR to detect rsATP7A transgene in mo-br brain DNA after administration of 1.6 × 1010 viral particles of either AAV. Non-injected mutant (UT) and wild-type (WT) controls also shown. Error bars, SEM. (C) Western blot of total proteins from high-dose rAAV9-rsATP7A-injected wild-type mouse brain detects recombinant rsATP7A (≈125 kDa) as well as endogenous full-length murine ATP7A (≈165 kDa). Bottom, Beta-actin loading control. (D) Stable long-term expression of rsATP7A 597 days post-administration of 1.6 × 1010vg AAV9-rsATP7A, documented by RT-PCR from brain RNA extracted from a long-term surviving mutant male. −RT refers to a negative control for RT-PCR, with RNA but no reverse transcriptase included in the reaction. (E) Capillary DNA sequencing of the excised band verified that it represented the ATP7A transgene transcript (exon 5). Alignment to Homo sapiens ATP7A, sequence ID, gi|532691751|NM_001282224.1.
Table 1.
Short- and Long-Term Survival of mo-br Mice by Treatment Group
| Treatment |
None |
sc Copper Histidinate Alone |
AAV9 Alone |
AAV9 Plus sc Copper Histidinate |
||
|---|---|---|---|---|---|---|
| Dose | – | 15 μg | High | Low | Intermediate | High |
| n | 5 | 6 | 7 | 4 | 3 | 15 |
| % Short-term (≥30 days) survival | 0 | 0 | 0 | 25 | 67 | 75 |
| Median survival (days) | 15 | 21.5 | 15 | 20 | 199 | 300 |
| % Long-term (≥300 days) survival | 0 | 0 | 0 | 0 | 0 | 53 |
sc CuHis, subcutaneous copper histidinate. Low dose, 1.6 × 109 vg; intermediate dose, 5 × 109 vg; high dose, 1.6 × 1010 vg.
Long-term (≥300 days) survival was only achieved with high dose rAAV9-rsATP7A plus sc copper histidinate (Figure 3A; Table 1), with Kaplan-Meier survival analysis highly statistically significant (p < 0.0001) (Figure 3A). Eight of 15 mice (53%) in this treatment cohort survived long-term, whereas lifespans in the other seven mutant mice were 16 days, 25 days, 26 days, 30 days, 40 days (2), and 56 days. Supplemental Videos S1 and S2 illustrate the dramatic synergistic effect of combined rAAV9-rsATP7A plus sc copper histidinate treatments in mutant mo-br mice versus sc copper histidinate alone.
To correlate with these survival data, western blots of total brain proteins from 12-day-old mice treated with high-dose rAAV9-rsATP7A on day 2 showed robust expression of the rsATP7A transgene at the expected size (≈125 kDa) (Figure 3B) at 12 days of age. Long-term expression of the transgene was confirmed by RT-PCR from the brain in a 599-day-old surviving mutant mouse rescued by rAAV9-rsATP7A plus sc copper histidinate (Figure 3C), sacrificed to evaluate this question. Western blots of brain proteins did not disclose a distinct band corresponding to rsATP7A in the older surviving mutant. This was related to a prominent background band in the 125-kDa region of the blots that obscured visualization of rsATP7A, in contrast to the western blots of brains from younger (12-day-old) animals using the same antibody. However, we detected the transgene transcript 597 days after rAAV9 administration via RT-PCR (Figure 3C), with DNA analysis confirming the human sequence from exon 5 of ATP7A (Figure 3D).
Cerebrospinal Fluid-Directed rAAV9-rsATP7A Gene Therapy Enables Brain Uptake of sc Copper Histidinate
The combination of cerebrospinal fluid-directed rAAV9-rsATP7A (1.6 × 1010 vg) with sc copper histidinate injections significantly increased brain copper levels in 12-day-old mutant mice (Figure 4A). X-ray fluorescence microscopy (XFM) confirmed the important role in this process of the choroid plexus, in which increased copper signal was evident after high-dose rAAV9-rsATP7A and sc copper histidinate in mo-br mutants compared to untreated mutants (Figure 4B). Our previous rescue of mo-br mutant mice was exquisitely dependent on rAAV5-rsATP7A transduction of choroid plexus epithelia.25 Those findings suggested a crucial role for choroid plexus in retention of copper in mammalian brain, a concept supported by the relatively high level of Atp7a expression in this tissue34 as well as the X-ray fluorescence microscopy results reported here.
Figure 4.

Cerebrospinal Fluid-Directed rAAV9-rsATP7A Gene Therapy Plus Subcutaneous Copper Histidinate Increases Brain Copper Levels in mo-br Mutant Mice
(A) Brain copper levels, measured by ICP-MS, are significantly higher in 12-day-old mutant mice treated with rAAV9-rsATP7A (1.6 × 1010 vg) plus sc copper histidinate (AAV9 + Cu) compared to untreated mutants (UT). A two-tailed, paired Student’s t test was used to calculate p. (B) X-ray fluorescence microscopy (XFM) of representative 12-day-old mo-br mouse brain sections from untreated (top) and high-dose rAAV9-rsATP7A plus sc copper histidinate-treated (AAV9 + Cu, bottom) mice. Left, H&E-stained tissue sections. Boxed areas in white (left) correspond to the X-ray fluorescence microscopy elemental maps for Cu (middle) and phosphorus (P, right), respectively, the latter obtained as control. The elemental map for Cu shows diffusely increased signals within the choroid plexus of the lateral ventricle (box indicates region of focus) and subventricular zone of the rAAV9-rsATP7A plus sc copper histidinate-treated brain, whereas the phosphorus signals (right) are comparable. Red temperature scale represents copper concentration (0–53 μg/g) and phosphorus concentration 0–10 mg/g or 0–12 mg/g, as noted. Error bars, SEM.
Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus sc Copper Histidinate Improves Neurochemical Ratios and Levels
ATP7A normally pumps copper into the secretory pathway of cells in the process of incorporating the metal as a cofactor for certain enzymes, including dopamine-beta-hydroxylase (DBH), a copper-dependent enzyme involved in catecholamine biosynthesis (Figure 5A).9, 10, 11, 12 Measurement of dopamine-beta-hydroxylase activity directly is not useful as a biomarker in mo-br mice (or in Menkes patients), since the enzymatic assay requires addition of endogenous copper, bioavailability of which represents the underlying defect. However, two proximal metabolites in the pathway, dopamine (DA) and dihydroxyphenylacetic acid (DOPAC), strikingly increase as a result of impaired copper incorporation into dopamine-beta-hydroxylase and two distal metabolites, norepinephrine (NE) and dihydroxyphenylglycol (DHPG), dramatically decrease in plasma and cerebrospinal fluid.10, 30 Measurement of these metabolites by high-performance liquid chromatography and calculation of proximal:distal metabolite ratios are excellent surrogates for dopamine-beta-hydroxylase activity in mo-br mice.25, 30
Figure 5.

Brain Neurochemical Measurements in rAAV9-rsATP7A Plus sc Copper Histidinate Combination-Treated mo-br Mice
(A) Summarized version of catecholamine biosynthetic pathway. Dopamine (DA) is converted to norepinephrine (NE) by the copper-dependent enzyme dopamine-beta-hydroxylase (DBH). (B) Markedly increased DA:NE and DOPAC:DHPG ratios in 12-day-old untreated mo-br mutants (UT) compared to mutant mice that received high-dose rAAV9-rsATP7A plus sc copper histidinate. A two-tailed, paired Student’s t test was used to calculate p. Error bars, SEM. Elevated ratios of proximal:distal metabolites in this pathway are sensitive and specific for diagnosing Menkes disease.10, 12, 30 (C) Individual brain neurochemical levels measured by high-performance liquid chromatography (HPLC). Proximal metabolites in the pathway (DA, DOPAC) are significantly lower in 12-day-old mutant mice treated with high-dose rAAV9-rsATP7A plus copper histidinate compared to untreated mutants (UT). Distal metabolites (NE, DHPG) are significantly higher. Two-tailed, paired Student’s t tests were used to calculate p values. Error bars, SEM. (D) Brain cytochrome c oxidase (CCO) activity in untreated mo-br (UT), high-dose rAAV9-rsATP7A plus copper histidinate-treated mo-br (AAV9 + Cu), and wild-type (WT) mice. Note progressive increase in cytochrome c oxidase activity with advancing age in long-term surviving rAAV9 plus sc copper histidinate-treated mutant mice (sacrificed at 256 days and 599 days). Results in the latter four animals reflect triplicate measurements in the four individual brains. Error bars, SEM.
To assess the impact of rAAV9-rsATP7A plus sc copper histidinate treatment on restoration of dopamine-beta-hydroxylase activity, we calculated proximal:distal metabolite ratios (Figure 5B) as well as individual brain catecholamine levels (Figure 5C). Untreated 12-day-old mo-br mice showed a 95-fold increase of DA:NE and 108-fold increase of DOPAC:DHPG compared to 12-day-old wild-type mice (Figure 5B), reflecting profound dopamine-beta-hydroxylase deficiency. In contrast, high-dose rAAV9-rsATP7A plus sc copper histidinate-treated 12-day-old mutant mice showed markedly lower ratios (Figure 5B) and significant improvement in individual neurochemical levels (Figure 5C), indicating restored dopamine-beta-hydroxylase activity.
Brain activity of cytochrome c oxidase (CCO), a mitochondrial copper enzyme for which metal cofactor acquisition does not rely on ATP7A, increased in 12-day-old rAAV9-rsATP7A plus sc copper histidinate-treated mutants (Figure 5D). Although mean cytochrome c oxidase activity rose from 21% of normal in untreated mutants to 32% in the combination-treated mice, this increase was not statistically significant. In older mo-br mutants rescued by rAAV9-rsATP7A plus sc copper histidinate, however, brain cytochrome c oxidase activity was near normal (Figure 5D).
Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus sc Copper Histidinate Normalizes Growth and Neurobehavior
Weight gain in rescued rAAV9-rsATP7A plus sc copper histidinate-treated mutants was slightly lower compared to normal littermates between 40 and 90 days of age (Figure 6A). However, by approximately 90 days of age, growth in the combination-treated mutant mice overlapped with that of WT mice.
Figure 6.

Growth and Neurobehavioral Outcomes in rAAV9-rsATP7A Plus sc Copper Histidinate-Treated mo-br Mice
(A) Weight gain in rAAV9 plus sc copper histidinate-treated mutant mice (blue line; n = 10 at start, n = 8 at end) was lower than in wild-type mice (red line; n = 21 at start, 18 at end), until 90–120 days of age, by which time “catch-up” growth was evident. Wire-hang (B) and rotarod (C) test results for mutant mice treated with rAAV9-rsATP7A plus sc copper histidinate combination treatment (blue; n = 10 at start, 8 at end) compared to wild-type (WT) male littermates (red). Error bars, SEM.
In rescued rAAV9-rsATP7A plus sc copper histidinate-treated mo-br mice, we assessed neurobehavioral function on a weekly basis beginning at age 40 days of age up to 300 days and compared with wild-type littermates. We used the wire-hang test to assess neuromuscular strength and constant speed rotarod to measure balance and motor coordination (Figures 6B and 6C). In our previous study with cerebrospinal fluid-directed rAAV5-rsATP7A plus cupric chloride (also intracerebroventricular), surviving mo-br mice manifested abnormal wire-hang and rotarod performances compared to normal littermates, particularly evident in the wire hang.25 In contrast, the current experimental treatment regimen of cerebrospinal fluid-directed high dose rAAV9-rsATP7A plus sc copper histidinate was associated with neuromuscular strength and coordination more comparable to the WT performance (Figures 6B and 6C).
Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus sc Copper Histidinate Treatment Corrects mo-br Brain Pathology
In correlation with improved survival, biochemical, and neurobehavioral outcomes, we also noted normalization of brain histology in cerebrospinal fluid-directed high-dose rAAV9-rsATP7A plus sc copper histidinate-treated 12-day-old mutants on H&E staining (Figure 7A). Untreated 12-day-old mutant mice showed a high number of pyknotic neurons (Figure 7A, black arrows) and other abnormally stained neurons consistent with an intermediate state of necrosis (Figure 7A, red arrow). Electron micrographs of cerebellum and hippocampus in 12-day-old mutant mice treated with cerebrospinal fluid-directed rAAV9-rsATP7A plus sc copper histidinate documented amelioration of the ultrastructural defects evident in untreated mutant brains. The latter included swollen dendrites with disorganized neurofilament patterns and pale mitochondria (Figure 7B, right). Quantitation of neuronal mitochondria in electron micrographs of brain cortex and cerebellum also disclosed significantly lower mitochondrial number in untreated 12-day-old old mo-br mutants compared to wild-type (Figures 7C and 7D). In contrast, mitochondrial counts in the rAAV9-rsATP7A plus sc copper histidinate-treated mutants did not differ significantly from wild-type (Figures 7C and 7D).
Figure 7.

Brain Pathology at 12 Day of Age in mo-br Mice Treated with rAAV9-rsATP7A Plus sc Copper Histidinate Compared to Untreated Controls
(A) H&E staining in 12-day-old wild-type (WT), high-dose rAAV9-rsATP7A plus sc copper histidinate-treated (AAV9 + Cu), and untreated mo-br (untreated mutant) mouse brains. The treated mutant brain (AAV9 + Cu) resembles the WT, whereas the untreated mutant manifests pathological abnormalities including pyknotic (black arrows) and dying (red arrow) neurons. (B) Electron micrographs of 12-day-old wild-type (WT), high-dose rAAV9-rsATP7A plus sc copper histidinate-treated, and untreated mo-br (untreated mutant) mouse brain. The untreated mo-br mouse brain shows palely stained, enlarged mitochondria (yellow arrow), swollen dendrites (red arrow), and reduced neurofilament density (black arrows); abnormalities not evident in the treated mutant or wild-type brains. (C and D) Brain mitochondria numbers in 12-day-old treated and untreated mo-br mutant mice brain, by treatment group. One-way ANOVA for the multigroup means was not statistically significant. There was no statistically significant difference between WT and mutant mice treated with AAV9-rsATP7A plus sc copper histidinate in either cerebral cortex (C) or cerebellum (D) on post-ANOVA Student’s t tests, suggesting modest mitochondrial treatment benefit. (E) H&E staining of cerebellar peduncles showing normal cerebellar Purkinje cell layers (arrows) in a 256-day-old rAAV9-rsATP7A plus sc Cu-treated mo-br mutant (left) and his wild-type littermate (right). Age-matched untreated mutant brain could not be shown since untreated mo-br mice typically die by 15 days of age. For comparison, the reader is referred to an abnormal cerebellum from a 300-day-old mo-br mouse treated with rAAV5 and cupric chloride depicted in an earlier publication (Figure 6C in Donsante et al.25).
Normal Cerebellar Purkinje Cells in AAV9-rsATP7A Plus sc Copper Histidinate-Treated mo-br Brain
We specifically evaluated cerebellar pathology in long-term surviving mice treated with high-dose rAAV9-rsATP7A plus sc copper histidinate, since Purkinje cell defects previously were observed in mo-br mutants rescued with AAV5-rsATP7A.25 Consistent with the improved (essentially normal) rotarod performance of the rAAV9-rsATP7A plus sc copper histidinate-treated cohort (Figure 6C), no Purkinje cell abnormalities were distinguishable by H&E staining in an adult mutant mouse rescued in the first week of life by high-dose rAAV9-rsATP7A plus sc copper histidinate compared to his wild-type littermate (Figure 7E).
Discussion
Using a mouse model of severe Menkes disease, we developed a new experimental therapeutic strategy that combines central nervous system viral gene therapy with peripheral substrate (copper histidinate) replacement. This regimen provided the most efficient rescue of the mo-br C57BL/6-Atp7amo-br mouse achieved to date. Our findings suggest that rAAV9-rsATP7A in combination with sc copper histidinate has a strong synergistic effect and holds promise for clinical translation to treatment of human subjects with Menkes disease.
We sought to optimize delivery of copper to the mutant mouse brain by administering copper histidinate to coincide with onset of rsATP7A expression in brain choroid plexus epithelia, estimated at 48 hr post-viral administration, based on our prior work.32 The three peripheral (subcutaneous) copper histidinate doses on days 4, 5, and 6 allowed for some leeway in onset of gene expression. Moreover, the regimen enabled direct determination of the impact of adding working copies of the human rsATP7A to the mutant mouse brain, since three subcutaneous copper histidinate doses alone failed to rescue mutant mice (Table 1).
We previously rescued the mo-br mouse by cerebrospinal fluid-directed administration of rAAV5-rsATP7A with cerebrospinal fluid-directed cupric chloride.25 The treatment regimen was associated with 22% long-term survival and median survival of 43 days, compared to 53% and 300 days, respectively, in the current study using high-dose rAAV9-rsATP7A and subcutaneous injections of copper histidinate. We found that the combination treatment was clearly superior to either rAAV9-rsATP7A treatment alone or sc copper histidinate alone (Table 1; Figure 3A). Although the vector doses and the form and route of copper administration differed, a rational interpretation of our results with these two serotypes in mo-br is that selective rAAV transduction of choroid plexus epithelia (rAAV5), without also adding working copies of rsATP7A to neuronal cells, is less than ideal. Recombinant AAV9 not only transduced the choroid plexus epithelia needed for copper delivery and retention in the cerebrospinal fluid and brain, but also enabled transgene delivery to numerous other neuronal cell types as well (Figure 1).32, 38, 39, 40 Egress from the cerebrospinal fluid led to transduction of liver and heart (Figure 3B), although no untoward effects of this off-target transduction were observed.
The requirement for ATP7A by most cells, related to its multiple and versatile roles,9 underscores the importance of employing a vector with broad neuronal tropism for treatment of this disorder. These tasks include transfer of copper into the lumen of the Golgi compartment and neurosecretory granules for activation of dopamine-beta-hydroxylase and peptidylglycine-amidating mono-oxygenase (PAM), whose activities depend on the metal cofactor. In addition, ATP7A pumps copper into melanosomes for metalation of tyrosinase, exports copper across the plasma membrane in response to increased concentration, contributes to axonal and synaptic development and participates in neuronal activation.9 As illustrated here (and previously25), the truncated, reduced-sized version of ATP7A (rsATP7A) appears to function as a suitable substitute for the full-length version, which is too large for rAAV packaging.
Dopamine-beta-hydroxylase is a copper-dependent enzyme that relies on ATP7A, to make this metal cofactor available to dopamine-beta-hydroxylase apoenzyme in the secretory pathway of dopaminergic cells. Therefore, the brain neurochemicals measured in this study represent pharmacodynamic response biomarkers to assess effects of cerebrospinal fluid-directed rsATP7A gene addition. However, ATP7A is not required for metalation of other copper-dependent enzymes, e.g., the mitochondrial cuproenzyme, cytochrome c oxidase. Based on data in 12-day-old combination-treated mice, it appears that cytochrome c oxidase activity does not rebound as robustly as dopamine-beta-hydroxylase in these mice and may contribute to the failure to attain 100% long-term survival.
Individual treatments with rAAV9-rsATP7A alone or 15 μg sc copper histidinate alone did not rescue mo-br mice, implying that combination treatments will likely also be needed to remedy the counterpart human illness. To more precisely model currently available human treatment, we administered copper subcutaneously instead of by intracerebroventricular administration as previously,25 and we substituted clinical grade copper histidinate for cupric chloride. We reasoned that copper provided subcutaneously would enter the circulation, and subsequently the mo-br brain, via working copies of rsATP7A conveyed by rAAV9 to choroid plexus epithelia and brain neurons. In this context, the present study embodies a novel therapeutic approach directly relevant to a first-in-human clinical trial for this devastating illness.
Recombinant AAV has emerged as one of the most valuable and promising vectors for gene transfer to the brain.24, 41, 42 In contrast to the capacity of wild-type AAV DNA to integrate into host genomes, the rAAVs used for gene therapy applications do not express non-structural (rep, cap) proteins required for replication and integration, rendering the rAAV safe.43 The vector genomes of rAAVs normally persist episomally for long periods of time after transduction of post-mitotic cells,44 as illustrated here, in which transgene expression in mo-br mouse brain was documented 597 days after rAA9-rsATP7A administration to the cerebrospinal fluid (Figure 3C).
In the current work, we sought to build on the theoretical concept that expressing a functional form of a missing gene directly within brain cells affords the best hope for correction and prevention of Menkes disease neuropathology. We introduce a novel mechanism of long-term treatment, which does not rely on substrate (copper) replacement alone, but rather on the ability of viral gene therapy to remodel both choroid plexus epithelia and neuronal cells in the CNS. This approach seeks to add functional ATP7A to relevant target cells and tissues so as to enable long-term normalization of brain copper homeostasis.
The reasons for which 7/15 (47%) mo-br mice treated with high-dose rAAV9-rsATP7A plus sc copper histidinate in this study did not attain long-term survival remain unclear. We were unable to formally correlate treatment success or failure in this cohort with biochemical outcomes or transduction efficiency, since survival of combination-treated mutant mice was not possible to predict in advance of death. We speculate that subtle variability in operative technique or efficiency of choroid plexus and/or neuronal transduction may account for incomplete uniformity in treatment response. We believe these technical aspects, e.g., brain lateral ventricle injection of small mouse pups with small drug (vector) volumes could result in variable biodistribution of viral particles and, therefore, variable transgene expression among treated animals. Translation to human subjects would be less impacted by these factors, given the ease of neurosurgical access to the brain lateral ventricles and larger rAAV solution volume to be used. We are also aware of phenotypic variability among human subjects despite the same ATP7A mutation45 and, in some instances, in their response to copper histidinate treatment (S.G.K., unpublished data). Therefore, the innate heterogeneity of disturbed copper metabolism may be a factor in treatment response for both mice and human with defects in this essential copper ATPase.
Our initial results with cerebrospinal fluid-directed rAAV5 gene addition plus cerebrospinal fluid-directed copper in an animal model provided an important proof of concept25 as to synergy between the large (DNA) and small (copper) molecule treatments. The present experiments, combining cerebrospinal fluid-directed rAAV9 with subcutaneous copper injections, represent an even more improved therapeutic approach, the success of which provides important considerations for determining safe and effective approaches for a first-in-human pilot study for Menkes disease. Since there is no large animal model of this illness, a hybrid pharmacology-toxicology-product fate study in mo-br mice, plus formal pre-clinical toxicology studies of rAAV9-rsATP7A in rats or rabbits, and evaluation in non-human primates represent rational next steps. As a rare pediatric illness with high morbidity and mortality, curative or ameliorative therapy for Menkes disease would be profoundly welcomed by the patients’ parents and families whom it impacts.7 In addition to enhancing knowledge on the intersection between copper transport and neuronal function, a future clinical trial will provide general insight as to viral gene therapy for neonatal-onset brain disorders.
Materials and Methods
Animal Care
Breeding pairs were obtained from the C57BL/6-Atp7amo-br colony at Jackson Laboratories (Bar Harbor, ME). Each pair consisted of a female heterozygous for the mutant allele (Atp7a+/mo-br) and a wild-type male (Atp7a+/y). Breeding mainly consisted of sibling crosses. Genotyping was performed as previously described.25 Mouse experimental procedures were approved by the NICHD Animal Care and Use Committee.
Brain Dissection
Mouse brain dissections were carried out in wild-type or mutant C57BL/6J-Atp7amo-br males. Sagittal bisected brain halves were snap frozen on dry ice and stored at −80°C for molecular and biochemical assays. For pathology, immunohistochemistry, and electron microscopy, brains were immersed in fixative or perfused fixed, as previously described.25 Mouse peripheral organs (liver, kidney, heart, muscle) were dissected and snap frozen, and DNA was isolated using a Wizard Genomic DNA purification kit following the manufacturer’s instructions (Promega; Madison, WI).
AAV Constructs
Reduced-size human ATP7A cDNA (rsATP7A) was amplified by from pAD-LOX-GFP-MNK as previously described.25 Recombinant AAV9-rsATP7A vector was produced by the University of Florida Vector Core Laboratory (Gainesville, FL) as previously described.25
Brain Lateral Ventricle Injections and Subcutaneous Copper Histidinate Administration
Recombinant AAV injections to the brain lateral ventricles were performed as previously described.25 Recombinant AAV9-rsATP7A doses were calculated based on the estimated cerebrospinal fluid volume in neonatal mice (20 μL) so as to provide 2.5 × 1011 vg per mL cerebrospinal fluid. This calculation produced the amount we used as intermediate dose (5 × 109 vg), which we bracketed with doses 0.5 log lower (1.6 × 109 vg) and higher (1.6 × 1010 vg), as low and high dose, respectively. copper histidinate (1 μg/μL) produced by the NIH Clinical Center Pharmaceutical Development Service was administered in a total dose of 15 μg by subcutaneous injection on the back, flank, or neck region, in three 5-μg doses on day 4, day 5, and day 6. The latter regimen was based on previous work28, 46, 47, 48 indicating the first 10 days of life as a critical window for copper treatment of mo-br (on a non-C57BL/6 background), using intraperitoneal copper doses of 10 μg/g body weight.28
GFP Immunohistochemistry
Paraffin-embedded tissues were cut to a thickness of 4 μm, placed on ProbeOn Plus slides (Thermo Fisher Scientific, Pittsburgh, PA), and de-paraffinized. Slides were pre-treated in citrate buffer (pH 6.0) (Cell Marque, Rocklin, CA) for 20 min in a steamer, allowed to cool to room temperature (RT), blocked with Dual Endogenous (DAKO, Carpentaria, CA), and then mouse anti-GFP (JL-8) (BD Biosciences Clontech; Palo Alto, CA) applied at 1:2,000 dilution for 60 min. After TBS rinses, DAB + secondary antibody (DAKO, Carpentaria, CA) was applied for 10 min. For negative controls, mouse monoclonal IgG was used instead of primary antibody.
HEK293T Cell Transfection and ATP7A Trafficking Studies
The rsATP7A cDNA was inserted between the SacI and ApaI sites of pEYFP-C1 plasmid and used for transfection of HEK293T cells in 0.5 μM or 200 μM copper concentrations, as previously described.37, 49 Cells were viewed by confocal microscopy (Zeiss 510; Carl Zeiss Microimaging, Thornwood, NY) and images captured using META software. Transfections were performed in duplicate with several hundred cells examined.
Tyrosinase Activation Assay
YSTT cells (generous gift from S. Lutsenko) were grown on glass coverslips, transfected with 1.5 μg of tyrosinase expression plasmid as previously described,50 and co-transfected with 0.75 μg reduced size ATP7A (rsATP7A), full-length ATP7A expression plasmid, or an empty plasmid vector using Lipofectamine LTX and PLUS transfection reagent (Thermo Fisher Scientific). Transfected cells were fixed for 30 s in cold acetone-methanol (1:1) at −20°C 24 hr post-transfection. Fixed cells were then incubated in 0.1 M sodium phosphate buffer (pH 6.8) containing 0.4 mg/mL 3,4-dihydroxy-L-phenylalanine (L-DOPA) at 37°C for 4 hr. Coverslips were mounted on slides, and formation of the L-DOPA chrome pigment was detected by Nikon ECLIPSE E600 microscope.
RT-PCR
Total RNA in brain samples from AAV9-treated and wild-type mice was extracted using RNeasy Mini Kit (QIAGEN, Hilden, Germany). First-strand cDNA synthesis was performed using Enhanced Avian First Strand Synthesis Kit (Sigma, St. Louis, MO). We used components for cDNA synthesis with enhanced AMV reverse transcriptase and oligo d(T)23 for synthesis of mRNA. RT-PCR was performed using primers (forward, 5′-CACCAGTTCAAGACAAGGAGG-3′; and reverse 5′-CTTACTTCTGCCTTGCCAGC-3′). RT-PCR products (166 bp) were analyzed on a 1% agarose gel, the bands excised, and purified using QIAGEN (Germantown, MD, USA) gel extraction kit. The purified DNA underwent capillary DNA sequencing (FDA Facility for Biotechnology Resources, NIH).
Protein Extraction and Western Blotting
Total proteins were extracted from transfected HEK293T cells using radioimmunoprecipitation assay (RIPA) buffer and from sagittal bisected mouse brains of wild-type (WT) or hemizygous mutant males using Mammalian Protein Extraction Reagent buffer (Life Technologies, Carlsbad, CA). Supernatants from lysed tissues were collected and total proteins denatured by adding 4× NuPage, LDS sample buffer (Invitrogen, Life Technologies, NY, USA) with 50 μM DTT and heating at 95°C for 5 min. Total proteins (30–70 μg) from each sample were electrophoresed through precast 4%–12% NOVEX Tris-glycerin SDS polyacrylamide gels (Invitrogen) for 1 hr at 170 V and transferred to polyvinylidine fluoride (PVDF) membranes for 2 hr at 25 V (150 mA). Membranes were incubated at 4°C overnight in blocking solution containing 0.1% Tween 20 (Phosphate buffered saline supplemented with Tween 20 at 0.1 % [PBST]) and 5% non-fat milk (Bio-Rad, Hercules, CA). Blots were washed with PBST and incubated for 2 hr at room temperature with a rabbit carboxyl-terminus anti-ATP7A antibody diluted 1:1,000, or anti-β-actin monoclonal antibody (1:5,000) (Abcam, Cambridge, UK). Membranes were washed three times with PBST and incubated with a goat anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP)-conjugated secondary antibody (1:2,000) (Santa Cruz Biotechnology, Dallas, TX) for 1 hr at room temperature. Membranes were incubated in SuperSignal West Pico Luminol/Enhancer Solution (Thermo Fisher Scientific, Waltham, MA), exposed to a Kodak BioMax MR autoradiography film and developed using a Kodak X-OMAT 2000A processor.
Viral Genome Quantitation
With primers specific for ATP7A (forward, 5′-GGTGTTTTGGAACTTGTTGTGA-3′ and reverse, 5′-GCCACGGAGCAGTATAGGAT-3′) qPCR was used to determine AAV9 viral genome copies in brain and peripheral organs (kidney, muscle, heart, liver). Genomic DNA was extracted using the Wizard Genomic DNA purification kit (Promega, Madison, WI) and used as template for Real Time PCR (Opticon; MJ Research, Waltham, MA), following the manufacturer’s instructions. Final results were expressed as AAV vector genomes per diploid mouse genome, as previously described.32
Brain Copper Measurement
Copper levels in fresh-frozen anterior brain thirds were determined by graphite furnace atomic absorption and confirmed by inductively coupled plasma mass spectrometry (ICP-MS), as previously described.22, 25
X-Ray Fluorescence Microscopy
For X-ray fluorescence microscopy, brains from saline-perfused animals were embedded in optimal cutting temperature (OCT) compound and flash-frozen in dry ice-cooled isopentane. 10-μm sagittal sections were transferred to Ultralene, an X-ray fluorescence microscopy-compatible window material, and mounted on Lucite sample holders. The tissue sections were subsequently thawed, air-dried, and quality confirmed by light microscopy. X-ray fluorescence microscopy data were collected on beam line 2-ID-E at the Advanced Photon Source (APS), Argonne National Laboratory (Argonne, IL). Sample holders were mounted onto kinematic sample holders and the target areas for imaging determined using a high-resolution scanning light microscope (Leica, Buffalo Grove, IL) equipped with a high precision, motorized x,y stage (Ludl Electronic Products, Hawthorne, NY). The incident X-ray energy for the beam line microprobe was tuned to 10 keV using a Si-monochromator. The monochromatic beam was focused to 350 × 350 nm using three-stacked Fresnel zone plates (APS). The sample was placed at 19° to the incident X-ray beam and the resulting X-ray fluorescence was collected at 90° using an energy dispersive four-element detector (Vortex ME-4, SII Nanotechnology, Northridge, CA). Raster scans were collected in fly-scan mode, scanning parameter were 2 μm × 2 μm step size with 50 ms dwell per point. Two-dimensional maps for Cu and P were created by extracting, background subtracting, and fitting fluorescence photon counts using the program MAPS.41 Fluorescent counts were translated into μg/cm2 using calibrated X-ray standards (AXO products, Dresden, Germany).
Brain Neurochemical Assay
Fresh-frozen posterior brain thirds were weighed and immediately homogenized in 5–10 volumes of 0.4 N perchloric acid containing 0.1% ethylene diamine tetra-acetic acid. The homogenates were centrifuged and supernatant frozen at −80°C until assay. Concentrations of brain neurochemicals dihydroxyphenylalanine (DOPA), DOPAC, DA, NE, and DHPG were determined by high-performance liquid chromatography with electrochemical detection, as previously described.25, 30
Cytochrome c Oxidase Activity
The middle thirds of sagittal divided half-brains were used for cytochrome c oxidase determination. Mitochondrial proteins were isolated using the mitochondria isolation kit MITOISO1 from Sigma-Aldrich (St. Louis, MO). Cytochrome c oxidase (COX) activity was measured spectrophotometrically via loss of ferrocytochrome c at 550 nm (CYTOCOX1, Sigma-Aldrich, St. Louis). Reaction rate was calculated by measuring changes in optical density/min at 550 nm using a molar extinction coefficient of 21.84 for reduced-oxidized cytochrome c. Results were expressed in units per mg of protein.
Neurobehavioral Testing
Wild-type and AAV9-rsATP7A plus sc copper histidinate copper histidinate-treated mo-br mutant mice were tested weekly using two neurobehavioral tests (wire hang, rotarod) to evaluate muscle strength, balance, coordination, and locomotor function, as described previously.25 Testing was performed beginning at 40 days of age and until the mice reached 180 days or expired. Untreated mo-br mutant mice were unavailable for testing because these mice typically died by 15 days of age.
Brain Pathology and Electron Microscopy
Mice were euthanized with CO2 gas, decapitated, brains promptly removed, and one hemisphere fixed in 10% neutral buffered formalin for 48 hr at 4°C. After paraffin embedding, 5- to 15-micron-thick sections were stained with H&E (Histoserve, Germantown, MD), as previously described.25 For electron microscopy, mice were deeply anesthetized and perfused-fixed with 1% glutaraldehyde and 4% paraformaldehyde in PBS prior to brain removal, processing, and imaging, as described.25
Analysis of Mitochondrial Number
Mitochondrial number was quantitated and morphology assessed in 27 electron micrographs of brains from rAAV9-rsATP7A plus sc copper histidinate -treated mutants, untreated mutants, and wild-type control mice.
Statistics
Kaplan-Meier survival analysis and one-way ANOVA (multiple group brain mitochondria counts) were performed with GraphPad Prism 6 software. Biochemical parameters and brain mitochondria counts (post-ANOVA) were evaluated for between group statistical significance (p < 0.05) by two-tailed Student’s t tests.
Author Contributions
S.G.K. and M.R.H. contributed to the conception and design of the study; M.R.H., E.-Y.C., L.Y., P.M.Z., D.M., P.S., L.R.B., M.R., J.A.C., and D.S.G. contributed to data acquisition and analysis; S.G.K., M.R.H., L.Y., and E.-Y.C. contributed to drafting the text and preparing the figures.
Conflicts of Interest
S.G.K. reports a cooperative research and development agreement (CRADA) with Cyprium Therapeutics, Inc., New York, NY, during the conduct of the study. In addition, S.G.K. reports a patent based in part on this work (U.S. Patent Application No. 15/769,294 filed April 18, 2018) with royalties paid to the NIH.
Acknowledgments
We thank the Animal Facility staff who contributed to the care and monitoring of the mouse colony, especially D. Abebe. We acknowledge the NICHD Confocal Microscopy Facility and V. Schram for support, L. Holtzclaw for assistance in mouse perfusions, J. Miller for preparing mouse brains for X-ray fluorescence microscopy, D. Vine and O. Antipova for support and assistance with X-ray fluorescence microscopy data collection at Advanced Photon Source, and Svetlana Lutsenko for YSTT cells. Work reported here was funded by the NIH (Z01 HD008768 and Z01 HD008927 to S.G.K. and R01 GM090016 to M.R.). The Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract DE-AC02-06CH11357. The opinions and/or assertions expressed herein are the private views of the authors and shall not be construed as official or as reflecting the views of the U.S. Department of Health and Human Services, the NIH, the U.S. Food and Drug Administration, or the U.S. Federal Government.
Footnotes
Supplemental Information includes two videos and can be found with this article online at https://doi.org/10.1016/j.omtm.2018.07.002.
Supplemental Information
The mo-br coat color is characterized by incomplete pigmentation. This mutant animal lived for 199 days.
This mutant animal died at 25 days of age.
References
- 1.Menkes J.H., Alter M., Steigleder G.K., Weakley D.R., Sung J.H. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- 2.Danks D.M., Campbell P.E., Walker-Smith J., Stevens B.J., Gillespie J.M., Blomfield J., Turner B. Menkes’ kinky-hair syndrome. Lancet. 1972;1:1100–1102. doi: 10.1016/s0140-6736(72)91433-x. [DOI] [PubMed] [Google Scholar]
- 3.Vulpe C., Levinson B., Whitney S., Packman S., Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 4.Chelly J., Tümer Z., Tønnesen T., Petterson A., Ishikawa-Brush Y., Tommerup N., Horn N., Monaco A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 5.Mercer J.F., Livingston J., Hall B., Paynter J.A., Begy C., Chandrasekharappa S., Lockhart P., Grimes A., Bhave M., Siemieniak D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 6.Kaler S.G., Gallo L.K., Proud V.K., Percy A.K., Mark Y., Segal N.A., Goldstein D.S., Holmes C.S., Gahl W.A. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat. Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- 7.Kaler S.G. Menkes disease. Adv. Pediatr. 1994;41:263–304. [PubMed] [Google Scholar]
- 8.Tümer Z., Møller L.B. Menkes disease. Eur. J. Hum. Genet. 2010;18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaler S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaler S.G., Goldstein D.S., Holmes C., Salerno J.A., Gahl W.A. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann. Neurol. 1993;33:171–175. doi: 10.1002/ana.410330206. [DOI] [PubMed] [Google Scholar]
- 11.Kaler S.G., Gahl W.A., Berry S.A., Holmes C.S., Goldstein D.S. Predictive value of plasma catecholamine levels in neonatal detection of Menkes disease. J. Inherit. Metab. Dis. 1993;16:907–908. doi: 10.1007/BF00714295. [DOI] [PubMed] [Google Scholar]
- 12.Kaler S.G., Holmes C.S., Goldstein D.S., Tang J., Godwin S.C., Donsante A., Liew C.J., Sato S., Patronas N. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadal D., Baerlocher K. Menkes’ disease: long-term treatment with copper and D-penicillamine. Eur. J. Pediatr. 1988;147:621–625. doi: 10.1007/BF00442477. [DOI] [PubMed] [Google Scholar]
- 14.Sherwood G., Sarkar B., Kortsak A.S. Copper histidinate therapy in Menkes’ disease: prevention of progressive neurodegeneration. J. Inherit. Metab. Dis. 1989;12(Suppl 2):393–396. doi: 10.1007/BF03335432. [DOI] [PubMed] [Google Scholar]
- 15.Kodama H. Recent developments in Menkes disease. J. Inherit. Metab. Dis. 1993;16:791–799. doi: 10.1007/BF00711911. [DOI] [PubMed] [Google Scholar]
- 16.Christodoulou J., Danks D.M., Sarkar B., Baerlocher K.E., Casey R., Horn N., Tümer Z., Clarke J.T. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am. J. Med. Genet. 1998;76:154–164. [PubMed] [Google Scholar]
- 17.Kaler S.G. Neurodevelopment and brain growth in classic Menkes disease is influenced by age and symptomatology at initiation of copper treatment. J. Trace Elem. Med. Biol. 2014;28:427–430. doi: 10.1016/j.jtemb.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaler S.G. Menkes disease mutations and response to early copper histidine treatment. Nat. Genet. 1996;13:21–22. doi: 10.1038/ng0596-21. [DOI] [PubMed] [Google Scholar]
- 19.Kaler S.G., Das S., Levinson B., Goldstein D.S., Holmes C.S., Patronas N.J., Packman S., Gahl W.A. Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small In-frame deletion. Biochem. Mol. Med. 1996;57:37–46. doi: 10.1006/bmme.1996.0007. [DOI] [PubMed] [Google Scholar]
- 20.Kaler S.G., Tang J., Donsante A., Kaneski C.R. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann. Neurol. 2009;65:108–113. doi: 10.1002/ana.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang J., Donsante A., Desai V., Patronas N., Kaler S.G. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol. Genet. Metab. 2008;95:174–181. doi: 10.1016/j.ymgme.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haddad M.R., Macri C.J., Holmes C.S., Goldstein D.S., Jacobson B.E., Centeno J.A., Popek E.J., Gahl W.A., Kaler S.G. In utero copper treatment for Menkes disease associated with a severe ATP7A mutation. Mol. Genet. Metab. 2012;107:222–228. doi: 10.1016/j.ymgme.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaler S.G., Buist N.R., Holmes C.S., Goldstein D.S., Miller R.C., Gahl W.A. Early copper therapy in classic Menkes disease patients with a novel splicing mutation. Ann. Neurol. 1995;38:921–928. doi: 10.1002/ana.410380613. [DOI] [PubMed] [Google Scholar]
- 24.Murlidharan G., Samulski R.J., Asokan A. Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 2014;7:76. doi: 10.3389/fnmol.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donsante A., Yi L., Zerfas P.M., Brinster L.R., Sullivan P., Goldstein D.S., Prohaska J., Centeno J.A., Rushing E., Kaler S.G. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol. Ther. 2011;19:2114–2123. doi: 10.1038/mt.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunt D.M. A study of copper treatment and tissue copper levels in the murine congenital copper deficiency, mottled. Life Sci. 1976;19:1913–1919. doi: 10.1016/0024-3205(76)90124-7. [DOI] [PubMed] [Google Scholar]
- 27.Mann J.R., Camakaris J., Danks D.M. Copper metabolism in mottled mouse mutants: distribution of 64Cu in brindled (Mobr) mice. Biochem. J. 1979;180:613–619. doi: 10.1042/bj1800613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagara H., Yajima K., Suzuki K. The effect of copper supplementation on the brindled mouse: a clinico-pathological study. J. Neuropathol. Exp. Neurol. 1981;40:428–446. doi: 10.1097/00005072-198107000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Reed V., Boyd Y. Mutation analysis provides additional proof that mottled is the mouse homologue of Menkes’ disease. Hum. Mol. Genet. 1997;6:417–423. doi: 10.1093/hmg/6.3.417. [DOI] [PubMed] [Google Scholar]
- 30.Donsante A., Sullivan P., Goldstein D.S., Brinster L.R., Kaler S.G. L-threo-dihydroxyphenylserine corrects neurochemical abnormalities in a Menkes disease mouse model. Ann. Neurol. 2013;73:259–265. doi: 10.1002/ana.23787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donsante A., Johnson P., Jansen L.A., Kaler S.G. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am. J. Med. Genet. A. 2010;152A:2529–2534. doi: 10.1002/ajmg.a.33632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haddad M.R., Donsante A., Zerfas P., Kaler S.G. Fetal Brain-directed AAV Gene Therapy Results in Rapid, Robust, and Persistent Transduction of Mouse Choroid Plexus Epithelia. Mol. Ther. Nucleic Acids. 2013;2:e101. doi: 10.1038/mtna.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hladky S.B., Barrand M.A. Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS. 2016;13:19. doi: 10.1186/s12987-016-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo Y.M., Gitschier J., Packman S. Developmental expression of the mouse mottled and toxic milk genes suggests distinct functions for the Menkes and Wilson disease copper transporters. Hum. Mol. Genet. 1997;6:1043–1049. doi: 10.1093/hmg/6.7.1043. [DOI] [PubMed] [Google Scholar]
- 35.Schlief M.L., Craig A.M., Gitlin J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005;25:239–246. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Meskini R., Crabtree K.L., Cline L.B., Mains R.E., Eipper B.A., Ronnett G.V. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol. Cell. Neurosci. 2007;34:409–421. doi: 10.1016/j.mcn.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi L., Donsante A., Kennerson M.L., Mercer J.F., Garbern J.Y., Kaler S.G. Altered intracellular localization and valosin-containing protein (p97 VCP) interaction underlie ATP7A-related distal motor neuropathy. Hum. Mol. Genet. 2012;21:1794–1807. doi: 10.1093/hmg/ddr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cearley C.N., Vandenberghe L.H., Parente M.K., Carnish E.R., Wilson J.M., Wolfe J.H. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol. Ther. 2008;16:1710–1718. doi: 10.1038/mt.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLean J.R., Smith G.A., Rocha E.M., Hayes M.A., Beagan J.A., Hallett P.J., Isacson O. Widespread neuron-specific transgene expression in brain and spinal cord following synapsin promoter-driven AAV9 neonatal intracerebroventricular injection. Neurosci. Lett. 2014;576:73–78. doi: 10.1016/j.neulet.2014.05.044. [DOI] [PubMed] [Google Scholar]
- 40.Schuster D.J., Dykstra J.A., Riedl M.S., Kitto K.F., Belur L.R., McIvor R.S., Elde R.P., Fairbanks C.A., Vulchanova L. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front. Neuroanat. 2014;8:42. doi: 10.3389/fnana.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biffi A., Aubourg P., Cartier N. Gene therapy for leukodystrophies. Hum. Mol. Genet. 2011;20(R1):R42–R53. doi: 10.1093/hmg/ddr142. [DOI] [PubMed] [Google Scholar]
- 42.Sands M.S., Davidson B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Chiorini J.A., Wiener S.M., Yang L., Smith R.H., Safer B., Kilcoin N.P., Liu Y., Urcelay E., Kotin R.M. The roles of AAV Rep proteins in gene expression and targeted integration. Curr. Top. Microbiol. Immunol. 1996;218:25–33. doi: 10.1007/978-3-642-80207-2_2. [DOI] [PubMed] [Google Scholar]
- 44.Schnepp B.C., Clark K.R., Klemanski D.L., Pacak C.A., Johnson P.R. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 2003;77:3495–3504. doi: 10.1128/JVI.77.6.3495-3504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donsante A., Tang J., Godwin S.C., Holmes C.S., Goldstein D.S., Bassuk A., Kaler S.G. Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome. J. Med. Genet. 2007;44:492–497. doi: 10.1136/jmg.2007.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawasaki H., Yamano T., Iwane S., Shimada M. Golgi study on macular mutant mouse after copper therapy. Acta Neuropathol. 1988;76:606–612. doi: 10.1007/BF00689600. [DOI] [PubMed] [Google Scholar]
- 47.Fujii T., Ito M., Tsuda H., Mikawa H. Biochemical study on the critical period for treatment of the mottled brindled mouse. J. Neurochem. 1990;55:885–889. doi: 10.1111/j.1471-4159.1990.tb04574.x. [DOI] [PubMed] [Google Scholar]
- 48.Bhadhprasit W., Kodama H., Fujisawa C., Hiroki T., Ogawa E. Effect of copper and disulfiram combination therapy on the macular mouse, a model of Menkes disease. J. Trace Elem. Med. Biol. 2012;26:105–108. doi: 10.1016/j.jtemb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 49.Yi L., Kaler S.G. Direct interactions of adaptor protein complexes 1 and 2 with the copper transporter ATP7A mediate its anterograde and retrograde trafficking. Hum. Mol. Genet. 2015;24:2411–2425. doi: 10.1093/hmg/ddv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta A., Bhattacharjee A., Dmitriev O.Y., Nokhrin S., Braiterman L., Hubbard A.L., Lutsenko S. Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein. Proc. Natl. Acad. Sci. USA. 2011;108:5390–5395. doi: 10.1073/pnas.1014959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The mo-br coat color is characterized by incomplete pigmentation. This mutant animal lived for 199 days.
This mutant animal died at 25 days of age.