

The mechanism for initiating DNA cleavage by DDE-family enzymes, including the RAG endonuclease that initiates V(D)J recombination, is not well understood. Here we report six zebrafish RAG structures in complex with one or two intact recombination signal sequences (RSSs) at up to 3.9 Å resolution, which surprisingly reveal DNA melting at the heptamer of the RSSs, resulting in corkscrew-like rotation of coding-flank DNA and positioning of the scissile phosphate in the active site. Substrate binding is associated with dimer opening and a piston-like movement in RAG1, first outward to accommodate unmelted DNA and then inward to wedge melted DNA. These pre-cleavage complexes show limited base-specific contacts of RAG at the conserved terminal CAC/GTG sequence of the heptamer, suggesting conservation based on a propensity to unwind. CA and TG overwhelmingly dominate terminal sequences in transposons and retrotransposons, implicating a universal mechanism for DNA melting during the initiation of retroviral integration and DNA transposition.
Transposable elements are present in all kingdoms of life and the transposases that they encode to enable the mobility of these elements represent the most abundant genes known1. Collectively, transposases promote the generation of new genetic traits, and help to drive evolution and interaction between organisms. Many DNA transposases, as well as retroviral integrases that may have been co-opted from the hosts of these viruses, belong to the large DDE family of polynucleotide transferases2,3. DDE enzymes utilize the triad of three conserved acidic residues Asp, Asp and Glu in their RNase H-like domain (RNH) active sites to perform phosphodiester bond hydrolysis2. In vertebrates, the lymphocyte specific recombination-activating gene 1 and 2 (RAG1−RAG2)2 endonuclease complex (RAG)4,5 is a DDE family member with RAG1 serving as the catalytic subunit (Fig. 1a). The RAG complex mediates combinatorial synapsis and cleavage of non-contiguous variable (V), diversity (D) and joining (J) gene segments during lymphocyte development to initiate the V(D)J recombination reaction that generates the large repertoire of immunoglobulin (Ig) and T-cell receptor genes for adaptive immunity6,7. RAG1 and RAG2 mutations that compromise this diversity are associated with severe combined and other forms of immunodeficiency8.
Fig. 1. Cryo-EM structure determination.
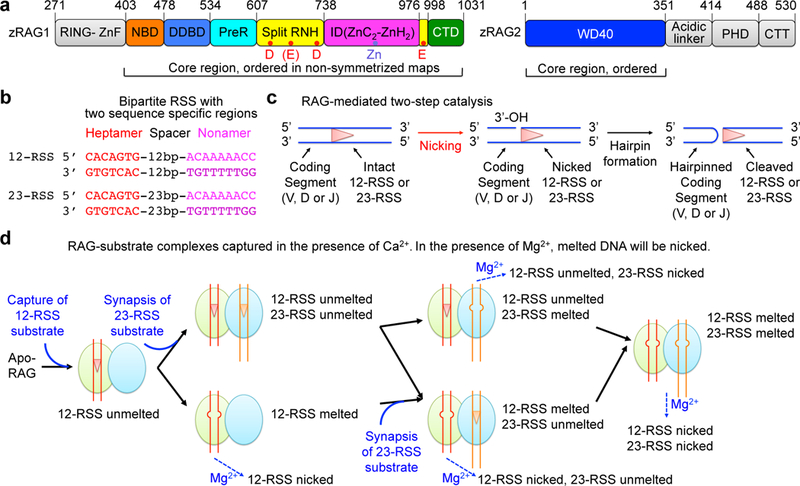
a, Domain organization of zebrafish RAG1 and RAG2 (zRAG1 and zRAG2). RING-ZnF: RING domain and zinc finger domain; NBD: nonamer binding domain; DDBD: dimerization and DNA binding domain; PreR: pre-RNaseH domain; RNH: RNase H-like domain; ID: insertion domain, which can be further divided into two parts, ZnC2 and ZnH2; CTD: C-terminal domain; WD40: tryptophan-aspartic acid repeat domain; PHD: plant homeodomain; CTT: C-terminal tail. Approximate domain boundaries are shown by residue numbers. Potential catalytic residues are indicated as red dots in RNH and the zinc ion is indicated as a slate dot in ID. The core regions that are ordered in the cryo-EM maps are shown in colors whereas disordered regions are shown in gray. b, Schematic displays of 12-RSS and 23-RSS. The consensus sequences of heptamer and nonamer are shown in red and magenta respectively. c, Schematic representation of RAG mediated catalysis that involves two consecutive reactions, nicking and hairpin formation. DNA strands are indicated as blue lines and the RSS is represented by a red triangle between the lines. d, Overview of different states of RAG-intact DNA complexes captured in the presence of Ca2+. RAG dimer is shown by double ovals colored in light green and light blue. The unmelted and melted RSSs are shown respectively as lines with a triangle and lines with a bubble in the middle. In the presence of Mg2+, melted DNA will be nicked.
Recombination signal sequences (RSSs) flanking the V, D, and J coding segments direct RAG for specific binding and catalysis (Supplementary Fig. 1a). Each RSS is composed of a heptamer, a spacer of either 12 or 23 base pairs, and a conserved nonamer, designated as either 12-RSS or 23-RSS9,10 (Fig. 1b and Supplementary Fig. 1b). Synapsis occurs preferentially between one coding segment flanked by a 12-RSS and another segment flanked by a 23-RSS, a phenomenon known as the 12–23 rule11,12 (Fig. 1b and Supplementary Fig. 1a). Since V, D and J segments are flanked by different RSSs, the 12–23 rule ensures recombination between V, D and J, but not within the same type of gene segments. Through structural studies of RAG in synaptic complexes with both 12-RSS and 23-RSS, we and others have previously shown that RSS binding induces a tilt of the nonamer-binding domain (NBD) dimer of RAG1 such that only one 12-RSS and one 23-RSS can be accommodated, providing a molecular explanation for the 12–23 rule13,14.
RAG-mediated cleavage occurs exactly at the junction between a coding segment and an RSS and proceeds through two consecutive steps of catalysis, nicking (strand cleavage) and hairpin formation (strand transfer), without substrate dissociation (Fig. 1c). The resulting coding end hairpins and blunt signal ends are passed on to proteins in the classical nonhomologous end joining (NHEJ) DNA repair pathway for processing and joining to complete the recombination reaction15. While the second catalytic step of hairpin formation has been revealed from RAG structures in complex with nicked 12-RSS and 23-RSS in which base flipping and backbone distortion help to position the 3’-OH and the scissile phosphate precisely for the strand transfer reaction13,14, how the first catalytic step of nicking is executed remains elusive. Notably, the recently reported structure of RAG in complex with intact 12-RSS and 23-RSS showed that the scissile phosphates for nicking are ~20 Å away from the catalytic sites and therefore represent a pre-reaction state14.
The initial nicking step is of particular interest because it is the only consensus step in the various mechanisms of cleavage and integration utilized by transposases and integrases of the DDE family to which RAG belongs3. Due to lack of structural information, nicking remains the least understood step for the entire family, likely reflecting the intrinsic conformational dynamics in this catalytic step. Here, through extensive classification in single-particle cryo-electron microscopy (cryo-EM) analysis, we determined structures of six different states of RAG in complex with either one or two intact DNA substrates (Fig. 1d). Surprisingly, these structures contain bound DNA substrates not only in unmelted, double-stranded form, which is similar to that in the reported pre-reaction state14, but also in melted, distorted form. This DNA melting, which occurs at the terminal CAC/GTG sequence of the heptamer, positions the scissile phosphate bond in the active site for catalysis. A piston-like, out-and-in movement of the insertion domain of RAG1 facilitates DNA melting, while the RNase H-like domain (RNH) active site remains stationary to engage differently distorted DNA in its two steps of catalysis. In contrast to dimer closure upon synapsis of nicked RSSs that is required for hairpin formation13, binding of either one or two intact DNA substrates opens the dimer and allows nicking with or without synapsis. Strikingly, despite being the most conserved sequence in the RSS, CAC/GTG of the heptamer is not extensively recognized by RAG in the intact DNA complexes, suggesting that its tendency to unwind is instead the critical factor in this nicking step. Because of the prevalence of CA and TG terminal nucleotides in repeat sequences of transposases and integrases16, we propose that nicking by other DDE family enzymes proceeds through an analogous DNA melting mechanism.
Results
Multiple conformational states defined by unmelted and melted RSSs
Unlike nicked and cleaved RSSs that formed stable complexes with the RAG1−RAG2 dimer13, intact RSSs dissociated from RAG when subjected to purification by gel filtration chromatography. We therefore directly plunge-froze grids from samples that contained the RAG1−RAG2 dimer, intact 12-RSS, intact 23-RSS and HMGB1 in an approximate 1:1:1:2 molar ratio (Supplementary Fig. 1b for the 12-RSS and 23-RSS sequences used). The samples contained Ca2+ and were incubated at either 37 °C or 21 °C for 1 hour before freezing. We used Ca2+ because this ion often supports DNA substrate binding but inhibits enzymatic activities in nucleases and polymerases, and possesses coordination geometries indistinguishable from Mg2+, the physiological ion17. We have also previously used Ca2+ to inhibit RAG-mediated hairpin formation from nicked RSS substrates and revealed similar coordination geometry as predicted for Mg2+ 13. The cryo-EM data for the 37 °C and 21 °C samples were collected respectively on FEI Polara and FEI Titan Krios microscopes, both coupled with a Gatan K2 Summit direct detection detector (Table 1 and Supplementary Fig. 2–4).
Table 1.
Cryo-EM data collection, refinement and validation statistics
| 12I + 23I (EMD-7849, PDB 6DBT) |
12I + 23I (EMD-7850, PDB 6DBU) |
12I + 23I (EMD-7851, PDB 6DBV) |
12I + 23I (EMD-7847, PDB 6DBQ) |
12I + 23I (EMD-7848, PDB 6DBR) |
12I + 23I (EMD-7845, PDB 6DBL) |
12I + 23I (EMD-7846, PDB 6DBO) |
12I (EMD-7853, PDB 6DBX) |
12I (EMD-7852, PDB 6DBW) |
12N + 23N (EMD-7843, PDB 6DBI) |
12N + 23N (EMD-7844, PDB 6DBJ) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA states | Doubly unmelted |
Doubly unmelted |
12-RSS melted 23-RSS unmelted |
12-RSS unmelted 23-RSS melted |
One RSS melted The other unmelted |
Doubly melted | Doubly melted | Singly unmelted | Singly melted | Doubly nicked | Doubly nicked |
| Data collection & processing | |||||||||||
| Microscope | Titan Krios | Titan Krios | Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios & Polara combined |
Titan Krios | Titan Krios |
| Magnification | 130,000 | 130,000 | 13,000/31,000 | 13,000/31,000 | 13,000/31,000 | 13,000/31,000 | 13,000/31,000 | 13,000/31,000 | 13,000/31,000 | 130,000 | 130,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 71.49 | 71.49 | 71.49/47 | 71.49/47 | 71.49/47 | 71.49/47 | 71.49/47 | 71.49/47 | 71.49/47 | 40 | 40 |
| Defocus range (μm) | 0.7 – 2.0 | 0.7 – 2.0 | 0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
0.7 – 2.0/ 1.2 – 3.0 |
1.2 – 2.5 | 1.2 – 2.5 |
| Pixel size (Å) | 1.08 | 1.08 | 1.23 | 1.23 | 1.23 | 1.23 | 1.23 | 1.23 | 1.23 | 1.064 | 1.064 |
| Symmetry imposed | C1 | C2 | C1 | C1 | C1 | C1 | C2 | C1 | C1 | C1 | C2 |
| Initial particle images (no.) | 249,279 | 249,279 | 934,894 | 934,894 | 934,894 | 934,894 | 934,894 | 934,894 | 934,894 | 364,274 | 364,274 |
| Final particle images (no.) | 67,194 | 67,194 | 45,159 | 48,002 | 69,753 | 23,176 | 19,344 | 62,942 | 48851 | 53,109 | 196,652 |
| Map resolution (Å) | 4.3 | 3.9 | 4.3 | 4.2 | 4.0 | 5.0 | 4.4 | 4.2 | 4.7 | 3.4 | 3.0 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 4.1 – 8.6 | 3.9 – 7.3 | 4.1 – 7.7 | 4.1 – 7.7 | 3.9 – 7.0 | 4.6 – 9.3 | 4.3 – 7.0 | 4.0 – 7.4 | 4.3 – 8.5 | 3.3 – 8.7 | 2.9 – 5.8 |
| Refinement | |||||||||||
| Initial model used | PDB 3JBW | PDB 3JBY | PDB 3JBW | PDB 3JBW | PDB 3JBY | PDB 3JBW | PDB 3JBY | PDB 3JBW | PDB 3JBW | PDB 3JBW | PDB 3JBY |
| Model resolution (Å) | 4.4 | 4.2 | 4.6 | 4.5 | 4.3 | 5.1 | 4.7 | 4.5 | 5.0 | 3.4 | 3.1 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Model resolution range (Å) | 271.4 – 4.3 (4.454 – 4.3) |
271.4 – 3.9 (4.04 – 3.9) |
237.7 – 4.3 (4.445 – 4.292) |
237.7 – 4.2 (4.371 – 4.22) |
237.7 – 4.0 (4.143 – 4.0) |
237.7 – 5.0 (5.18 – 5.001) |
237.7 – 4.4 (4.609 – 4.45) |
271.4 – 4.2 (4.35 – 4.2) |
271.4 – 4.7 (4.868 – 4.7) |
272.4 – 3.4 (3.48 – 3.36) |
192.6 – 3.0 (3.097 – 2.99) |
| Map sharpening B factor (Å2) | −173.826271 | −190.937951 | −173.907798 | −180.056215 | −179.136839 | −160.678788 | −171.589046 | −173.792903 | −195.922158 | −91.510887 | −113.457233 |
| Model composition | |||||||||||
| Nonhydrogen atoms | 19,896 | 17,010 | 19,956 | 19,956 | 17,040 | 19,929 | 16,820 | 17,436 | 17,418 | 20,001 | 16,912 |
| Protein residues | 2,152 | 1,924 | 2,161 | 2,161 | 1,930 | 2,156 | 1,914 | 2,033 | 2,031 | 2,168 | 1,932 |
| Ligands (nucleotides) | 222 | 136 | 222 | 222 | 136 | 222 | 128 | 100 | 100 | 222 | 124 |
| B factors (Å2) | |||||||||||
| Protein | 136.25 | 84.52 | 102.96 | 133.83 | 102.66 | 237.04 | 163.44 | 126.09 | 168.92 | 89.38 | 73.93 |
| Ligand (DNA) | 172.79 | 83.67 | 129.31 | 220.07 | 91.56 | 225.11 | 204.95 | 158.71 | 228.40 | 48.08 | 58.08 |
| R.m.s. deviations | |||||||||||
| Bond lengths (Å) | 0.005 | 0.007 | 0.006 | 0.005 | 0.005 | 0.016 | 0.009 | 0.005 | 0.010 | 0.010 | 0.004 |
| Bond angles (°) | 0.806 | 1.197 | 0.842 | 1.058 | 1.161 | 2.213 | 1.345 | 0.837 | 1.484 | 1.506 | 0.794 |
| Validation | |||||||||||
| MolProbity score | 2.36 | 2.37 | 2.42 | 2.49 | 2.27 | 2.48 | 2.35 | 2.4 | 2.47 | 2.05 | 2.11 |
| Clashscore | 21.46 | 22.52 | 22.61 | 27.67 | 19.39 | 37.52 | 24.81 | 20.07 | 30.04 | 15.25 | 12.23 |
| Poor rotamers (%) | 0.18 | 0.19 | 0.18 | 0.18 | 0.51 | 0.18 | 0.13 | 0.24 | 0.53 | 0.82 | 1.7 |
| Ramachandran plot | |||||||||||
| Favored (%) | 90.62 | 90.93 | 89.06 | 89.53 | 92.04 | 93.50 | 92.45 | 87.98 | 91.35 | 94.79 | 95.17 |
| Allowed (%) | 9.17 | 8.84 | 10.78 | 10.37 | 7.85 | 5.67 | 6.82 | 11.86 | 8.60 | 5.11 | 4.83 |
| Disallowed (%) | 0.21 | 0.23 | 0.16 | 0.1 | 0.11 | 0.83 | 0.73 | 0.16 | 0.05 | 0.10 | 0.00 |
I: intact DNA; N: nicked DNA
Surprisingly, multiple rounds of three-dimensional (3D) classification and refinement revealed not only bound intact DNAs with double-stranded features along the entire length, but also those with a single-stranded bubble near the coding flank-RSS junction (Fig. 1d and Fig. 2a-c, Supplementary Video 1). Collectively, six different density maps were discerned, among which two are singly bound complexes with 12-RSS in unmelted and melted states respectively (4.2 and 4.7 Å resolutions), and four are doubly bound complexes with 12-RSS and 23-RSS in combinations of unmelted and melted states (4.3, 4.2, 4.3 and 5.0 Å resolutions) (Fig. 1d and Supplementary Fig. 2–5). Applying C2 symmetry to complexes doubly bound with unmelted or melted DNAs resulted in averaged maps at 3.9 Å and 4.4 Å resolutions, respectively (Fig. 2b and Supplementary Fig. 2a, 3a, 5f). To improve the density for the melted DNA in complexes with one unmelted and one melted DNA, we performed symmetry expansion and carried out signal subtraction 3D classification focused on the melted DNA regardless whether it is 12-RSS or 23-RSS. The subsequent 3D refinement of the particle images from a major 3D class resulted in a 4.0 Å resolution map containing one melted DNA and one unmelted DNA (Fig. 2c and Supplementary Fig. 2b). We also collected new cryo-EM data on the nicked RSS complex and improved its resolution to 3.4 Å without averaging and 3.0 Å with C2 averaging (Supplementary Fig. 3b, 4, 5c), which showed similar but improved details in comparison with our published structures13. For structural analysis, the highest resolution maps or models of the different states are used (Table 1). Detailed secondary structures determined from the 3.4 Å resolution RAG structure are shown on the aligned sequences of zebrafish, human and mouse RAG proteins (Supplementary Note 1).
Fig. 2. Overview of cryo-EM maps and models.
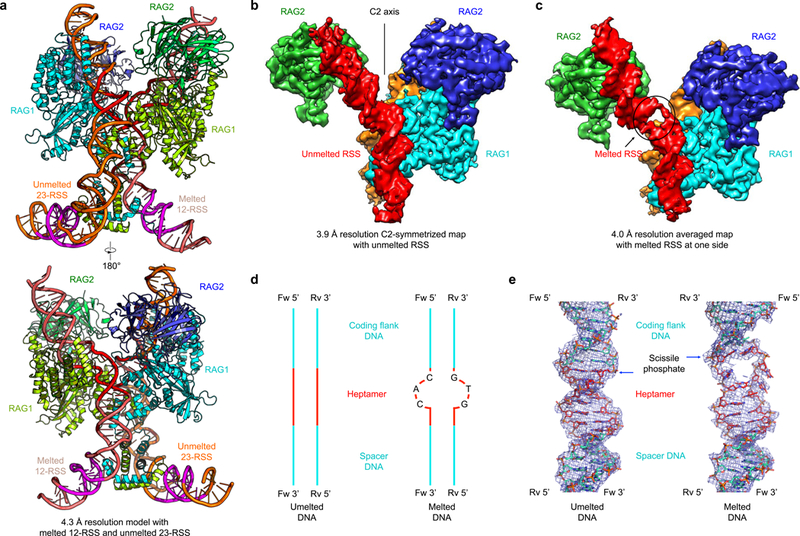
a, Ribbon diagrams in orthogonal views of the RAG model in complex with melted 12-RSS and unmelted 23-RSS at 4.3 Å resolution. RAG1: lemon green and cyan; RAG2: green and slate. 12-RSS and 23-RSS are shown in salmon and orange respectively, except that heptamers are highlighted in red and nonamers in magenta. b, Cryo-EM map of C2-symmetrized RAG in complex with two unmelted RSSs (red and orange) at 3.9 Å resolution. One RAG1 is omitted in order to present the bound DNAs (red and orange) inside the protein. c, Cryo-EM map of RAG in complex with one melted RSS (red) and one unmelted RSS (orange) at 4.0 Å resolution. The black circle indicates the melted region in the RSS. d, Schematic representations of unmelted and melted RSS DNAs in the models. The heptamer region is highlighted in red. Nucleotides AC and GT do not have base pairing in the melted state. e, Cryo-EM densities superimposed with the final unmelted and melted DNA models (shown in sticks) in the 3.9 Å C2-symmetrized and 4.0 Å averaged maps, respectively. The heptamer region is highlighted in red and the scissile phosphate position is indicated.
Heptamer melting is associated with a corkscrew rotation of DNA that positions the scissile phosphate bond for nicking
The bubble in the melted DNA spans the second and third positions of the heptamer at the AC/TG sequence (Fig. 2d,e). Strikingly, the coding flank of the melted DNA rotates by ~180° relative to the unmelted DNA so that the minor and major grooves are reversed (Fig. 3a,b). This is created by a corkscrew rotation of the coding flank initiated from the unwinding at the heptamer region near a kink in the DNA at the heptamer-coding flank border (Fig. 3a,b). Because coding flank recognition by RAG1 and RAG2 is mediated by non-specific interactions with sugar phosphate backbones13, the dramatic rotation is likely accommodated without a significant alteration of the binding energy. Importantly, the coding flank of the melted DNA superimposes well with that of nicked DNA (Fig. 3a), consistent with the idea that nicking occurs in the melted configuration (see below). Both unwinding and nicking cause the coding flank DNA to extend further out from the protein complex than with the unmelted DNA (Fig. 3b).
Fig. 3. DNA distortion and active sites in the RNH domains for unmelted, melted and nicked RSSs.
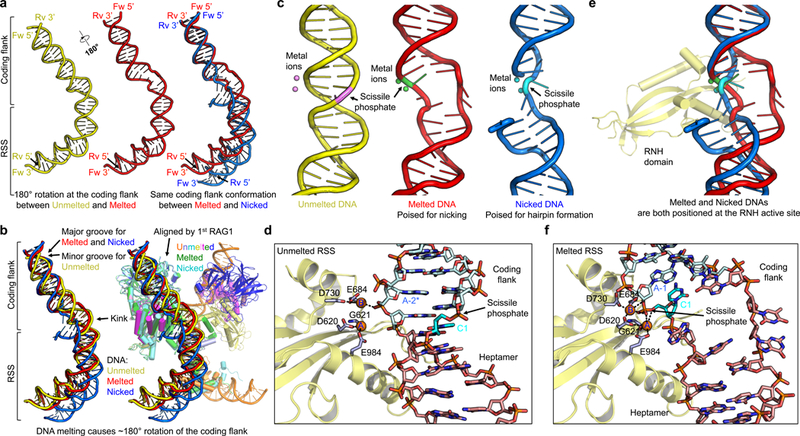
a,b, Comparison of the bound unmelted (yellow), melted (red) and nicked RSSs (blue) when the 1st RAG1 molecules in the complexes are superimposed. The 1st RAG1 is defined as the RAG1 subunit that performs the catalysis on a given bound RSS. The RAG dimer that binds to unmelted RSS is shown and colored by domains as in Fig.1a. The aligned 1st RAG1 and its associated RAG2 in complex with melted RSS and nicked RSS are colored in green and cyan, respectively. The positions of RSS and coding flank are indicated. c, Cartoon representation of unmelted, melted and nicked RSSs with the associated metal ions in the active sites. The unmelted, melted and nicked RSSs are shown in yellow, red and blue and the metal ions are shown in violet, green and cyan. d, Active site in the RNH regions (shown in yellow) for unmelted RSS (shown as sticks). The heptamers and coding flanks are shown in salmon and light cyan, respectively. The nucleotides to be nicked are highlighted in cyan. The metal ions (A and B) in the active centers are shown as orange spheres and residues that coordinate the metal ions are highlighted in light blue. Dashed black lines indicate potential coordination between metal ions and ligands. e, Superimposed positions of melted and nicked RSSs when the RNH domains are aligned. Only the RNH that binds the melted RSS is shown for clarity. f, Active site in the RNH regions (shown in yellow) for melted RSS (shown as sticks). The heptamers and coding flanks are shown in salmon and light cyan, respectively. The nucleotides to be nicked are highlighted in cyan. The metal ions (A and B) in the active centers are shown as orange spheres and residues that coordinate the metal ions are highlighted in light blue. Dashed black lines indicate potential coordination between metal ions and ligands.
The effect of heptamer unwinding became clear when we analyzed the RAG1 active sites in complex with unmelted and melted DNA (Fig. 3c). As a member of the DDE family, the RAG1 RNH catalytic domain contains the D(E)DE motif composed of residues D620, E684, D730 and E984 (Fig. 1a). In the unmelted DNA complex, these active site residues and the bound metal ions locate at the strand opposite that containing the scissile phosphate bond at C1 (Fig. 3c,d and Supplementary Fig. 6a), similar to that observed in the recently published RAG-DNA complex structure14. Additionally, despite the coordination of bound metal ions by a phosphate group in the unmelted DNA, detailed analysis shows that the active site components are not positioned for catalysis (Fig. 3d). In the melted DNA complex, the scissile phosphate bond between C1 and A-1 is at the active site (Fig. 3e,f and Supplementary Fig. 6b), and the bound metal ions are poised to perform nicking with metal ion A most likely activating a water molecule and metal ion B stabilizing the leaving group (Fig. 3f). In the nicked DNA complex we and others reported previously13,14, a similar catalytic complex is observed except that the roles of metal ions A and B are reversed with A stabilizing the leaving group and B activating the attacking 3’-OH of A-1 (Supplementary Fig. 6c).
To perform two successive catalytic steps by the single catalytic center, either the active site needs to move to accommodate the different DNA substrates or the DNA has to alter its conformation to be placed into the active site. Superposition of the RNH domains in RAG complexes with melted DNA and nicked DNA revealed the same arrangement of active site D(E)DE residues (D620, E684, D730 and E984) and the bound metal ions (Supplementary Fig. 6d). This structure comparison indicates that the catalytic RNH domain exhibits limited conformational changes. Therefore, it is the extensive DNA distortion that brings the scissile phosphate bond in either intact or nicked DNA into the catalytic center, allowing RAG to perform two successive catalytic steps using a single catalytic center (Fig. 3e).
Interestingly, 3D classification after combining the data sets of 37 °C and 21 °C samples revealed an overrepresentation of melted DNA complexes in the 37 °C sample over the 21 °C sample. For singly bound RAG, the ratio between particles with melted DNA and those with unmelted DNA is ~1 for the 37 °C sample and only ~0.5 for the 21 °C sample; for doubly bound RAG, the ratio between particles with at least one melted DNA and those with both unmelted DNA is ~2 for the 37 °C sample and only ~1 for the 21 °C sample. These data suggest that higher temperature facilitates unwinding. Both singly bound and doubly bound DNAs have a melted state, suggesting that nicking can occur without synapsis (Fig. 1d), consistent with previous biochemical studies12,18. This is in contrast to hairpin formation, which occurs preferentially in the synaptic complex12,13,18. We did not observe any singly bound states with 23-RSS, perhaps reflecting the prior observation that 12-RSS forms complexes with RAG and HMGB1 three to five times more efficiently than 23-RSS in gel shift analyses19. In this context and similar to our previous structures13, we observed additional low-resolution cryo-EM density compatible with bound HMGB1 at both 12-RSS and 23-RSS. For the 23-RSS, the additional density agrees with the locations of the two HMG boxes in the recently published RAG crystal structure14, but the connection between the two boxes could not be determined.
Piston movement and RAG dimer opening that facilitate intact DNA binding and melting
To understand how initial DNA binding and subsequent melting occur, we compared the structures of Apo-RAG and RAG bound to various forms of intact RSSs (Supplementary Video 2). Because each RSS interacts with both RAG1 molecules in the (RAG1−RAG2)2 complex, for a given RSS, we named the RAG1 that also mediates the phosphodiester bond hydrolysis of the RSS the first RAG (or 1st RAG), and the partner RAG1 that only binds the RSS the second RAG (or 2nd RAG). The structural comparison revealed a prominent movement in the DNA-interacting insertion domain (ID) of the first RAG1 (Fig. 4a-d and Supplementary Fig. 7a). The location of the ID in Apo-RAG is the most “in” as shown by the position of helix α15, while intact unmelted DNA interaction moves α15 outward by about ~13 Å in Cα positions (most “out”) (Fig. 4b,e,f and Supplementary Fig. 7b). In the melted DNA complex, helix α15 moves inward by ~10 Å, and the associated α15-α16 loop wedges into the melted DNA (Fig. 4c,g and Supplementary Fig. 7c). Because Apo-RAG assumes the most “in” conformation, we speculate that the most “out” conformation in the unmelted DNA complex has spring-loaded the ID to prime a piston-like inward movement to facilitate and stabilize DNA melting. Upon nicking, helix α15 occupies a similar location as in the melted DNA structure (Fig. 4d,h and Supplementary Fig. 7a). The movement of the ID is mostly rigid body as shown by the pairwise superposition of 0.8–1.0 Å among the different states (Fig. 4i). As expected, the NBD region changes its tilt in the transition from Apo-RAG to RAG bound to intact DNA (Fig. 4b). However, the NBD tilt changes again when the DNA becomes nicked (Fig. 4d), suggesting that the NBD orientation is molded to the form of the doubly bound DNAs.
Fig. 4. Structural comparisons of RAG monomers in different states.
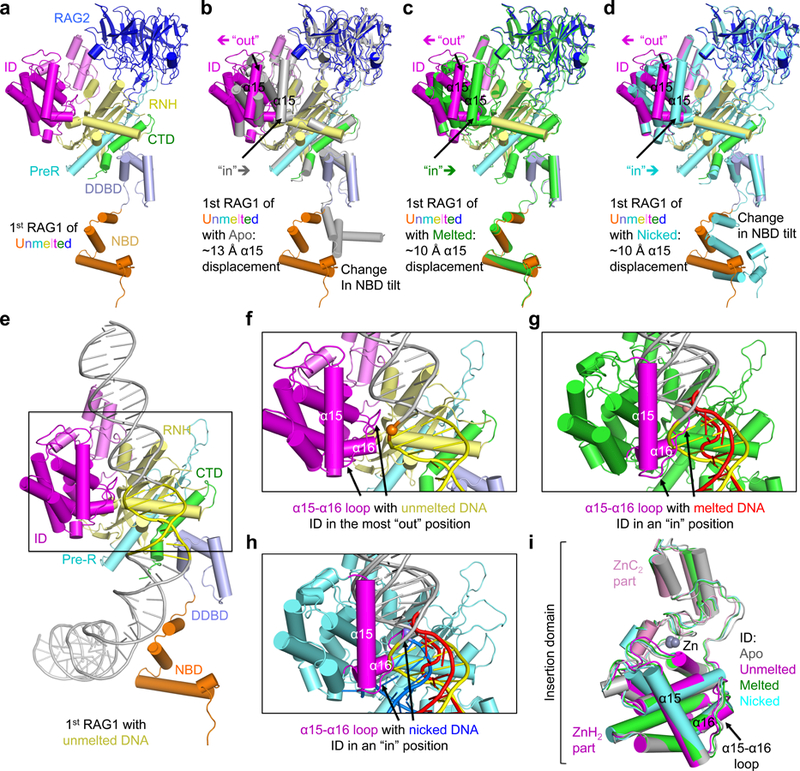
a, Cartoon representation for the conformation the 1st RAG1-RAG2 monomer that binds unmelted RSS. The 1st RAG1-RAG2 monomer is defined as containing the RAG1 subunit that performs the catalysis on a given bound RSS. The domains are colored using the scheme defined in Fig. 1a. The unmelted RSS is omitted. b-d, Superposition of the 1st RAG1-RAG2 monomer that binds unmelted RSS (colored as in Fig. 1a) with the crystal structure of Apo-RAG (4WWX, gray) (b), the complex that binds melted RSS (green) (c) and the complex that binds nicked RSS (cyan) (d). Helix α15 in the insertion domain (ID) and the NBD region are indicated, with the former exhibiting either an “in” or “out” position. e, Cartoon representation of the 1st RAG1 molecule (colored as in Fig. 1a) in complex with unmelted RSS. The heptamer region is shown in yellow. f, Zoom-in view of e showing the position of the α15-α16 loop in the ID relative to unmelted RSS. g, Zoom-in view of the 1st RAG1 molecule (green) in complex with melted RSS. The heptamer region is shown in red and the α15-α16 loop in magenta. The unmelted RSS as in f is also shown (gray and yellow). h, Zoom-in view of the 1st RAG1 molecule (cyan) in complex with nicked RSS. The heptamer region is shown in blue and the α15-α16 loop in magenta. The unmelted RSS (gray and yellow) and melted RSS (gray and red) as in g are also shown. The spheres colored in orange (f), salmon (g) and slate (h) indicate the scissile phosphates in the unmelted, melted and nicked DNA, respectively. i, Superposition of the insertion domains of the 1st RAG1 that bind unmelted (pink and magenta), melted (green) and nicked (cyan) RSSs respectively, and of Apo-RAG (gray). The regions of ZnC2, ZnH2 and the bound zinc ion are indicated.
We previously observed closure of the RAG dimer upon synapsis with nicked or cleaved 12-RSS and 23-RSS in comparison with the cryo-EM and crystal structures of Apo-RAG13,20. Here, although the different RAG structures in complex with intact RSSs exhibit slight differences in their dimer openness (Fig. 5a), they are more open than both the synapsed closed conformation (Fig. 5b) and the Apo-RAG conformation (Fig. 5c). These data suggest that binding to intact DNA pries open the RAG dimer. The reason for dimer opening may be explained by the clash of a bound intact unmelted RSS with both RAG1 monomers if the RAG dimer stays in the Apo conformation (Fig. 5c,d). On one side of a bound RSS, the ID α15-α16 loop of the first RAG1 needs to move out, and on the other side, the β4-β5 loop and the α23-α24 region of the second RAG1 needs to open more to avoid the clash (Fig. 5d). Therefore, the outward movement of the ID and the opening of the dimer together permit intact DNA binding, and the subsequent inward movement of the ID promotes melting.
Fig. 5. Opening of the RAG dimer upon RSS substrate binding.
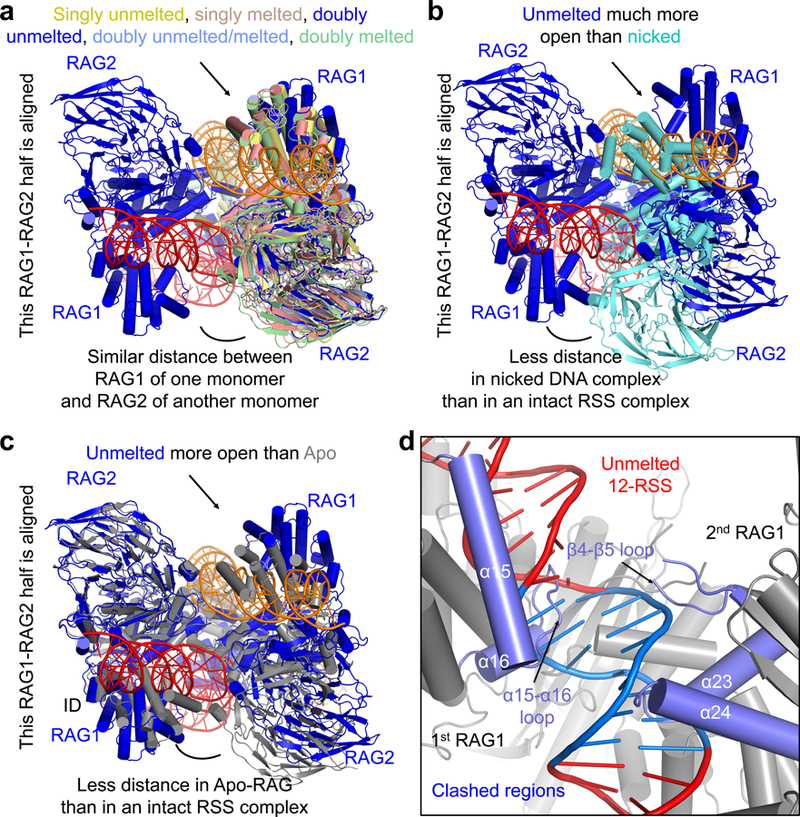
a, Superimposed conformations of RAG dimers singly or doubly bound to different forms of intact DNAs. One RAG monomer (left) from each complex is used for the superposition. Only the RAG monomer (blue) that is doubly bound to the unmelted RSSs is shown on the left. The positions of the RAG monomers at the right show the similar openness of these states. b, Superimposed conformations of the RAG dimer doubly bound to unmelted RSS (blue) and the RAG dimer doubly bound to nicked RSS (cyan). The RAG monomers at the left from the dimers are superimposed, showing the different positions of the RAG monomers at the right and the much more closed state of the nicked RSS complex. c, Superimposed conformations of the RAG dimer doubly bound to unmelted RSS (blue) and crystal structure of Apo-RAG (4WWX, gray) by aligning one RAG1-RAG2 monomer at the left and displaying the relative positions of the monomers at the right. d, If unmelted RSS is bound to RAG in its Apo conformation, there is clash between the RSS and both monomers of the RAG dimer (zoomed-in view). The heptamer in the unmelted RSS is highlighted in blue and the secondary structures in Apo-RAG that clash with unmelted RSS are highlighted in slate. The 1st and 2nd RAG1 molecules shown in the cartoon designate the catalytic RAG1 subunit for a given RSS and its partner RAG1, respectively.
The terminal CAC/GTG of the heptamer is placed for unwinding by measuring from the nonamer
The first three base pairs (CAC/GTG) of the heptamer are almost perfectly conserved in RSS sequences and functionally vital for V(D)J recombination10,21. It was therefore unanticipated that there are few base-specific interactions for CAC/GTG in the intact DNA complexes, either when the DNA is unmelted as in our structures and the published RAG-DNA crystal structures14, or when the DNA is melted (Fig. 6a,b). In contrast, the CAC/GTG region is extensively recognized by base-specific interactions in the nicked or cleaved RSS complex13,14 (Fig. 6c), and explains the previous observation that RAG in complex with nicked RSS possesses slower off rate than with intact RSS22. Previous footprinting studies supported a lack of strong contacts at the 5’ end of the heptamer in intact DNA substrates18, fully consistent with our structures. Because CAC/GTG base pairs contain alternating purine-pyrimidine tracts, which tend to display bending, distortion and even lack of base stacking, especially at the weaker A-T base pair position23, we hypothesize that the CAC/GTG sequence is essential in early steps of the reaction for its tendency to unwind. Indeed, even the intact unmelted DNA exhibits a kink near the coding flank-RSS junction (Fig. 3a-b).
Fig. 6. Structural comparisons of RSS recognition and key conserved RSS interactions.

a-c, Schematic depictions of the detailed interactions between RAG and the unmelted (a), melted (b) and nicked (c) RSSs in the heptamer region. Residues from the ID, RNH and CTD of the 1st RAG1 molecules, which perform catalysis on the given RSS, are shown as magenta, yellow and green ovals. Residues from the DDBD, RNH and CTD of the 2nd RAG1 molecules, which only bind to the given RSS, are shown as light blue, yellow and green ovals with a shaded pattern. Letters and numbers in the ovals indicate the residue names and numbers in zRAG1. Heptamer bases that are specifically recognized by RAG are highlighted in red. Blue, black and orange arrows indicate respectively specific interactions at bases, non-specific interactions at phosphate backbones and Van der Waals contacts with bases. Conserved interactions at the last three base pairs of the heptamer are highlighted within the red squares. d, Superposition of the 2nd RAG1 molecules that bind to but do not perform catalysis on the unmelted (colored as in Fig. 1a), melted (green) and nicked (cyan) RSSs. The heptamer of unmelted, melted and nicked RSSs are highlighted in yellow, red and blue, respectively. The zoom-in view below shows the β4-β5 loops in the RNH domain. e, Conserved recognition of nonamer, spacer and last three base pairs of heptamer mainly by the 2nd RAG1 molecule that binds to but does not perform catalysis on the given RSS. Such interactions may dictate intact DNA binding, which places the CAC/GTG base pairs with unwinding tendency to the active site.
For a given bound RSS, the two RAG1−RAG2 monomers in the RAG dimer provide different functions to the bound DNA, with the first monomer cradling the coding flank and the heptamer and executing catalysis, and the second monomer interacting with the heptamer and the spacer (Fig. 4e and Fig. 6d). The two NBDs together interact with the nonamer (Fig. 2a). In the different DNA complexes, the most constant interactions are mediated by the NBD dimer and by RAG1 in the second RAG1−RAG2 monomer (Fig. 6d). The dimerization and DNA binding domain (DDBD) and the C-terminal domain (CTD) of the second RAG1 interact similarly with the last three base pairs of the heptamer and the spacer in the unmelted, melted and nicked complexes (Fig. 6a-d), and the β4-β5 loop of its RNH domain molds its way into the respective DNA forms (Fig. 6d and Supplementary Fig. 8a,b). Therefore, our structures suggest that the nonamer, spacer and the second part of the heptamer are important for initial binding, which is more mediated by the second RAG1 instead of the first RAG1. These interactions away from the coding flank-heptamer junction may act as a ruler to position the CAC/GTG sequence at the active site for melting and nicking (Fig. 6e). This structural analysis is consistent with the requirement of the nonamer and the last four positions of the heptamer for the ability of an RSS to be an effective competitor for catalysis24 and for efficient formation of a shifted complex of RAG with 12-RSS25. In contrast, changes in the first three positions of the heptamer had little or no effect in these assays24,25 supporting the structural observation that these positions are involved in cleavage, rather than binding. Because its interactions with DNA do not change significantly during the RAG catalytic cycle, the second RAG1 is able to clamp down on the distal components of the RSS as the first three positions of the heptamer and the coding flank go through the conformational gymnastics in the different catalytic steps.
Discussion
Conformational transitions in the RAG catalytic pathway
Our RAG structures containing melted DNA substrates reveal the molecular mechanism for the first step of DNA cleavage and fill an important gap in our understanding of the RAG catalytic machinery13,14. Four major moving parts are illustrated: relative dimer orientation, positioning of the ID (especially the α15-α16 loop), conformation of the β4-β5 loop of the RNH domain, and the tilt of the NBD dimer (Fig. 7a). Upon intact DNA binding, the RAG dimer assumes an even more open conformation compared to Apo-RAG. There are several aspects that contribute to the opening of the RAG dimer when intact DNA substrates are engaged, including a cooperative rotation between the two RAG monomers and an outward movement of the ID in the RAG monomer. All of these movements are required to avoid a steric clash with intact RSS substrates. Subsequently, the intact RSS substrates are melted at the coding flank-heptamer junction, which is driven by the piston-like inward movement of the ID without closure of the RAG dimer and is facilitated by the intrinsic unwinding tendency of the CAC/GTG base pairs in the heptamer and engagement by the β4-β5 loop. RSS melting and corkscrew DNA rotation lead to positioning of the scissile phosphate in the active site for nicking. Upon nicking of both RSSs, the RAG dimer assumes a fully closed conformation through relative rotation of the two monomers. The nicked DNAs are highly distorted with CAC/GTG of the heptamer extensively recognized and poised to undergo hairpin formation at the coding flank. Throughout these steps, the NBD dimer shows flexibility to accommodate the movements in the RAG monomer, the ID and the bound DNA.
Fig. 7. Conformational transitions in the RAG catalytic pathway and implications on DNA transposases and retroviral integrase of the DDE family.
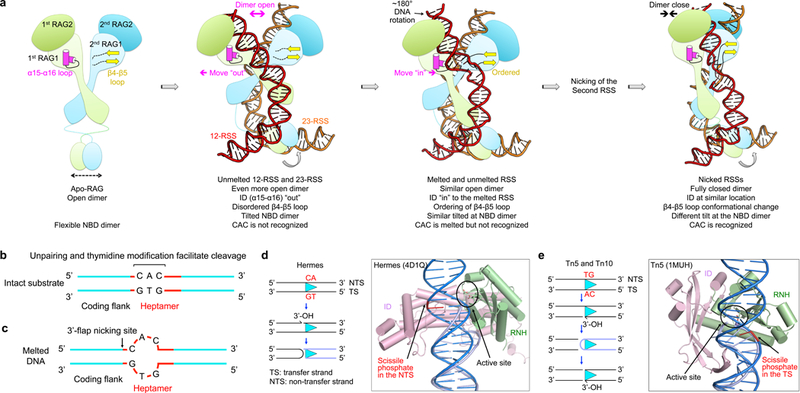
a, Structure-derived insights on RAG-mediated catalytic pathway in V(D)J recombination. RAG1: light green and light cyan; RAG2: green and cyan; α15-α16 loop in the 1st RAG1: magenta; β4-β5 loop in the RNH of 2nd RAG1: yellow; 12-RSS and 23-RSS: red and orange. The 1st and 2nd RAG1-RAG2 complexes are referred to relative to the 12-RSS that will be bound, in which the 1st RAG1 binds and performs the catalysis on the 12-RSS and the 2nd RAG1 only mediates the binding. b, Schematic representation of biochemical data that support distortion at the coding-RSS junction. c, Schematic representation of the cleavage site for the 3’-flap endonuclease activity of RAG at the coding flank-heptamer junction. d,e, Intact DNAs cannot reach the active sites of Hermes (d) or Tn5 (e) without melting or distortion. Left: schematic representations of the catalyzed reactions. The cyan triangles in the reaction schemes represent inverted repeat sequences that are recognized by the transposases, and the conserved di-nucleotides adjacent to the nicking sites are labeled in red. Right: ribbon diagrams in complex with a cleaved inverted repeat (light blue) for Hermes (d) and a hairpin intermediate (light blue) for Tn5 (e). An intact dsDNA (blue) is superimposed into each of the structures, showing that the proposed nicking site is far from the active site in the absence of DNA distortion. For the proteins, only the RNH (light green) and ID (pink) domains are shown.
The observation that DNA unwinding is required for the nicking activity of RAG is supported by classical biochemical experiments (Fig. 7b). First, sequence alterations that facilitate unpairing of the RSS sequence near the coding flank activate the cleavage reaction, suggesting that DNA distortion is critical for V(D)J recombination24,26. Second, the RAG1 core domain has been shown to exhibit more robust affinity for single-stranded DNA than double-stranded DNA27. Third, thymidine residues modified by potassium permanganate near the coding flank-heptamer junction are overrepresented in RAG-bound versus unbound DNA, suggesting that the modification has imparted flexibility or other perturbations that RAG prefers near the nicking site18,28. Finally and importantly, RAG possesses 3’-flap, but not 5’-flap, endonuclease activity with nicking preferentially at one nucleotide 5’ to the beginning of the 3’-flap29. In the RAG-intact DNA complex structures, the unwinding is centered at the AC/TG base pairs, with nicking immediately before the adjacent 5’ C (Fig. 7c), which agrees precisely with the 3’-flap endonuclease activity.
Implications for other DDE transposases and integrases
In light of the extensive mechanistic similarities between RAG and other DDE family transposases and retroviral integrases3, all of which initiate their reactions with phosphodiester bond hydrolysis (nicking), our cryo-EM structures of RAG in complex with intact DNA substrates provide the first glimpse of how the initial step of catalysis occurs and how sequential catalytic steps are executed for this enzyme family. Structural analysis suggests that DNA melting and conformational changes may be general features of the universal first step, despite the diversified pathways these enzymes use to generate their DNA products. For example, for the Mu transpososome, the duplex at the junction between the Mu DNA and the flanking DNA upon transpososome assembly is deformed as shown by hypersensitivity to nucleases and chemical reagents as well as by enhanced fluorescence of 2-amino purine (2-AP), a fluorescent purine analog whose fluorescence is quenched in double-stranded DNA30. For two DDE family members, the Hermes hAT transposase31,32 and the bacterial transposase Tn5 33,34, for which both Apo and cleaved DNA intermediate structures are available, when an intact unmelted DNA is superimposed onto the proteins, the scissile phosphate of the nicking site is located away from the active site, suggesting the need for a structural reconfiguration consistent with DNA unwinding (Fig. 7d,e). When the Apo and DNA intermediate structures are compared, both Hermes and Tn5 exhibit a change of the dimer orientation and movement of the ID upon DNA interaction (Supplementary Fig. 9a,b), suggesting that dimer opening and piston-like ID movement may also be utilized for the initial catalytic step by these enzymes.
A survey of substrates of DDE family members showed the ubiquitous presence of CA/GT sequences near the site of nicking (Supplementary Fig. 9c). These base pairs are characterized by their greater conformational flexibility23, which might facilitate DNA melting. In particular, examination of activity of substrates carrying mismatched termini in the transposable phage Mu supported the flexibility hypothesis35. In the human genome, statistical analyses of the highly abundant long terminal repeat (LTR) retrotransposons and DNA transposons also revealed a high conservation of their terminal two to three nucleotides, with the most abundant species being 5’-TG…CA-3’ at the ends of LTR retrotransposons and 5’-CAG…CTG-3’ for DNA transposons16. Thus, conformational dynamics of DNA near the site of nicking, perhaps similar to those illustrated for RAG in our study, might be a universal feature of the first step of catalysis by DDE-family enzymes.
Methods
Cloning, protein expression and purification.
Based on our previous biochemical and structural studies13, we continued employing zebra fish RAG (zRAG) proteins in this study. Briefly, the coding region of zRAG1 comprising residues from 271 to 1031 was subcloned to the modified pFastBac 1 vector which contains the coding region of 6xHis-MBP tag in the upstream of the zRAG1 coding region with an Human Rhinovirus (HRV) 3C protease-cleavable site (LEVLFQ|GP, where ‘|’ indicates the cutting site) in between. The full-length zRAG2 gene was subcloned to the similarly modified vector except that the HRV3C cleavage site was replaced by the tobacco etch virus (TEV) protease cleavage site (ENLYFQ|G). The plasmids containing the genes of interest were confirmed by Sanger sequencing and transformed into the DH10Bac competent cells. The recombinant bacmid DNAs were isolated and verified according to the instructions of the Bac-to-Bac® baculovirus expression system manual (Life Technologies). Monolayer Spodoptera frugiperda 9 (Sf9) insect cells were transfected using Cellfectin® II (Life Technologies) to generate the recombinant baculovirus and the titer of the baculovirus was amplified by infecting suspension Sf9 insect cells. In order to reduce the aggregation possibly caused by inappropriate protein folding, RAG1 and RAG2 proteins with an N-terminal 6xHis-MBP tag were co-expressed by co-infection of Sf9 cells with the recombinant baculovirus at 23 °C for 60 hours when the cell density reaches to 4 million per ml. Human HMGB1 (constructs 1–166), which is nearly identical with zebra fish HMGB1, was subcloned into vector pET26b with an uncleavable C-terminal 6xHis tag and the protein was overexpressed in BL21 (DE3) RIPL cells by induction using 0.2 mM IPTG at 30 °C for 3 hours when the OD600 reached 0.8.
The Sf9 cells that expressed RAG1−RAG2 complex proteins were harvested by centrifugation at 2,000 rpm for 20 min. The cell pellets were re-suspended in a lysis buffer containing 20 mM HEPES at pH 7.5, 500 mM NaCl, 10% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) and a protease inhibitor cocktail, and homogenized by ultra-sonication. The cell lysate was clarified by ultracentrifugation at 42,000 rpm at 4 °C for 2 hours. The supernatant containing the RAG complex was incubated with amylose resin (New England Biolabs) that was pre-equilibrated with the lysis buffer for 1 hour at 4 °C. After incubation, the resin-supernatant mixture was poured into a column and the resin was washed with the lysis buffer for 20 bed volumes. The resin was further washed by the buffer containing 20 mM HEPES at pH 7.5, 150 mM NaCl, 10% glycerol, 0.5 mM TCEP for 2 bed volumes before elution. The proteins were then eluted by the above 150 mM salt buffer supplemented with 20 mM maltose, and further purified by HiTrap Heparin HP affinity columns (GE Healthcare). The proteins were concentrated and loaded onto a Superdex 200 10/300 GL column (GE Healthcare) that was pre-equilibrated with the buffer containing 20 mM HEPES at pH 7.5, 150 mM NaCl, 0.5 mM TCEP and divalent metal ions Ca2+. After elution from the gel filtration column, the 6xHis-MBP tag on RAG2 protein was removed by incubation of the RAG complex with TEV protease at 16 °C for overnight. After TEV protease cleavage, the resulting 6xHisMBP-RAG1−RAG2 complex (Apo-RAG) was concentrated and applied to the gel filtration column again to remove TEV protease and the cleaved 6xHisMBP tag from RAG2. The Peak fractions from the gel filtration were collected, concentrated and quantified by the A280 method.
The E. coli cells expressing human HMGB1 with a C-terminal 6xHis tag were harvested by centrifugation at 3,500 rpm for 30 min. Cell pellets were re-suspended by the buffer containing 50 mM Na2HPO4 and 10 mM KH2PO4 at pH 7.4, 500 mM NaCl, and 2.7 mM KCl. The cells were lysed by ultra-sonication and cell debris was removed by centrifugation at 17,000 rpm at 4 °C for half an hour. The supernatant was applied to a column pre-packed with Ni-NTA resin (Qiagen) that pre-equilibrated with the lysis buffer. The column was subsequently washed sequentially using lysis buffer supplemented with 20 mM and 50 mM imidazole, respectively. The protein was eluted using lysis buffer containing 300 mM imidazole. The eluted protein was further purified by ion exchange and finally homogenized by gel filtration chromatography in buffer containing 20 mM HEPES at pH 7.5, 150 mM NaCl and 0.5 mM TCEP.
The DNAs used in this study were synthesized as oligos (Integrated DNA Technologies) with sequences shown in Supplementary Table 1. Double stranded DNA substrates were generated by mixing the corresponding oligos with the same molar ratio in buffer containing 20 mM HEPES at pH 7.5 and 150 mM NaCl, annealed by a temperature gradient and further purified by gel filtration chromatography with the same buffer. The purified DNA duplexes were concentrated and quantified by the A260 method.
To reconstitute the RAG complex with 12- and 23-RSS substrates for cryo-EM studies, Apo-RAG was combined with 12- and 23-RSS substrates and HMGB1 protein in the buffer containing 20 mM HEPES at pH 7.5, 50 mM KCl, 0.5 mM TCEP, 10 mM Ca2+ and 10 mM L-lysine, with the approximate Apo-RAG (regard as a dimer, Mw 380.67 Kd): 12-RSS: 23-RSS: HMGB1 molar ratio of 1: 1: 1: 2. The final total molar concentration for RAG protein is 1 μM (mass concentration is 0.38 mg/ml). The complex was incubated for 1 hour at either 21 °C or 37 °C prior to cryo-EM grid frozen in different batches.
Cryo-EM sample preparation and data acquisition.
For cryo-EM, 2.5 μl of above reconstituted RAG complex at a concentration of ~0.38 mg/ml was applied to a glow-discharged C-flat carbon grid (1.2/1.3, 400 mesh). Grids were blotted for 3.5 s with ~90 % humidity and plunge-frozen in liquid ethane using a Cryoplunge 3 System (Gatan). Cryo-EM data were collected on a Polara electron microscope (FEI) at Harvard Medical School (HMS), or on a Titan Krios electron microscope (FEI) at New York Structural Biology Center (NYSBC), or on a Titan Krios electron microscope on National Cancer Institute (NCI), all equipped with a K2 Summit direct electron detector (Gatan). The Polara at HMS was operated at 300 kV and cryo-EM movies were manually recorded in super-resolution counting mode using the program UCSFImage4. Specifically, images were acquired at a nominal magnification of 31,000x, corresponding to a calibrated pixel size of 1.23 Å on the specimen level and 0.615 Å for super-resolution images. The dose rate was set to be 8.2 counts per physical pixel per second. The total exposure time of each movie was 7.2 s, leading to a total accumulated dose of 47 electrons per Å2, fractionated into 36 frames (200 ms per frame). All movies were recorded using a defocus ranging from −1.2 to −3.0 µm. For data collected on Titan Krios at NYSBC, images were automatically acquired with Leginon with counting mode at a nominal magnification of 130,000x, corresponding to a calibrated pixel size of 1.06 Å on the specimen level, with a total dose of 72 electrons per Å2 and a defocus ranging from −0.7 to −2 µm. For data collected on Titan Krios at NCI, images were recorded with super resolution mode, at a nominal magnification of 130,000x, corresponding to a calibrated pixel size of 1.06 Å and 0.532 Å for super-resolution. The dose rate was set to be 5.8 e-/s/pixel, with 12 second and 40 frames in total with defocus ranging from −1.5 to −2.5 µm.
Image processing.
Dose-fractionated super-resolution movies collected using the K2 Summit direct electron detector were binned over 2 × 2 pixels, yielding a pixel size of 1.23 Å (data from Polara) or 1.06 Å (data from Titan Krios), and then subjected to motion correction using the program MotionCor2. A sum of all frames of each image stack was calculated following a dose-weighting scheme, and used for all image-processing steps except for defocus determination. The program CTFFIND4 was used to calculate defocus values of the summed images from all movie frames without dose weighting. Particles were boxed with 192 × 192 pixels for data collected on Polara, or 224 × 224 pixels for data collected on Titan Krios. To merge data from different sources, particles boxed with 224 × 224 pixels were rescaled to 192 × 192 box size with Fourier interpolation. Particle picking was performed using a semi-automated procedure. 2D classification of selected particle images was carried out either by ‘samclasscas.py’, which uses SPIDER operations to run 10 cycles of correspondence analysis, K-means classification and multi-reference alignment, or by RELION 2D classification. Initial 3D models were generated with previous RAG maps with low pass filtered to 30 Å. 3D classification and refinement were carried out in RELION. The masked 3D classification focusing on certain parts of the RAG complex with residual signal subtraction was done following a previously described procedure36. The orientation parameters of the homogenous set of particle images in the selected 3D classes were iteratively refined to yield higher resolution maps using the ‘auto-refine’ procedure. All refinements followed the gold-standard procedure, in which two half data sets are refined independently. RELION ‘post-processing’ was used to estimate resolution based on the Fourier shell correlation (FSC) = 0.143 criterion, after correcting for the effects of a soft shape mask using high-resolution noise substitution37. The final model was cross validated as previously described38. Briefly, 0.1 Å noise was randomly added to the final model with the PDB tools in Phenix, and refined the noise-added model against the first half map (Half1) generated from onehalf of the particles during refinement by RELION. The refinement of the coordinate file was performed in phenix refine by one round of coordinate and B-factor refinement. The refined model was then correlated with the two half-maps (Half1 and Half2) in Fourier space to produce two FSC curves: FSCwork (model versus Half1 map) and FSCfree (model versus Half2 map), respectively. A third FSC curve was calculated between the refined final model and the sum of two half-maps produced from all particles.
Model building and refinement.
As the structural models of the zRAG1−RAG2 complex (PDB ID: 3JBX, 3JBY and 3JBW)13 and the NBD−DNA complex (PDB ID: 3GNA)39 were available, we first fitted these protein models into B-factor sharpened non-symmetrized, C2 symmetrized or averaged EM maps in UCSF Chimera40 and manually adjusted in COOT41. The unambiguous fitting of the DNA sequences was facilitated by several factors. First, the nonamer sequences need to be bound to the NBDs, which fixed the orientation of the bound DNAs in these structures. Second, because the conserved interaction between the last three base pairs in the heptamer and the residues R999 and Q1000 in the α23-α24 loop in all the high- and low-resolution models, only one DNA registration can accommodate signal end region. Third, for the unmelted DNA models, the density from signal end to coding end is continuous, which can be fitted with ideal B-form DNA generated in COOT and the phosphate backbone fitted well with the bulges in the trajectory of DNA density. Fourth, for the melted DNA models, the density in the melted DNA region is clear to fit all the melted nucleotides and the density for coding ends superimpose well with the previous PC map, so the coding end region can be fitted when half of the RAG model is superimposed. All the junctions between DNA fragments converged well with the experimental DNA sequences and lengths. However, due to the limits in resolutions, we could not distinguish purine bases from pyrimidine bases in the DNA structures.
The map was then placed into an artificial unit cell with P1 symmetry and converted to MTZ format using phenix.map_to_structure_factors42. The resulting reflection file was used to perform maximum likelihood phased refinement using PHENIX42 with secondary structure restraints, X-ray and stereochemistry weight restraints, X-ray and atomic displacement parameter weight restraints and Ramachandran restraints or reference model restraints iteratively. The final structures were validated using MolProbity43. While most side chain densities were clear, some acidic side chains were not as well defined in the cryo-EM maps, likely contributed by their radiation sensitivity44,45. All molecular representations were generated in PyMOL (http://www.pymol.org)46 and UCSF Chimera40.
Data availability.
The atomic coordinates and cryo-EM maps are available in the PDB and EMDB databases, under accession numbers PDB 6DBT and EMD-7849 (12I + 23I doubly unmelted with I for intact RSS); PDB 6DBU and EMD-7850 (12I + 23I doubly unmelted and 2-fold averaged); PDB 6DBV and EMD-7851 (12I melted + 23I unmelted); PDB 6DBQ and EMD-7847 (12I unmelted + 23I melted); PDB 6DBR and EMD-7848 (averaged map with one RSS melted and one RSS unmelted); PDB 6DBL and EMD-7845 (12I + 23I doubly melted); PDB 6DBO and EMD-7846 (12I + 23I doubly melted and 2-fold averaged); PDB 6DBX and EMD-7853 (12I unmelted); PDB 6DBW and EMD-7852 (12I melted); PDB 6DBI and EMD-7843 (12N + 23N doubly nicked with N for nicked); and PDB 6DBJ and EMD-7844 (12N + 23N doubly nicked and 2-fold averaged). The datasets generated during the current study are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
Some of this work was performed with the assistance of Hui Wei at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310). The work was supported by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research. We also acknowledge research funding from the National Institutes of Health (AI125535 to H.W. and M.L., AI0240067–36 to F.W.A. and AI032524 to D.G.S.), and the Irvington Postdoctoral Fellowship from the Cancer Research Institute (H.R.). F.W.A. is an HHMI investigator.
Footnotes
Competing interests
The authors declare no competing interests.
Reporting Summary. Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
References
- 1.Aziz RK, Breitbart M & Edwards RA Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res 38, 4207–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hickman AB, Chandler M & Dyda F Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol 45, 50–69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montano SP & Rice PA Moving DNA around: DNA transposition and retroviral integration. Curr Opin Struct Biol 21, 370–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schatz DG, Oettinger MA & Baltimore D The V(D)J recombination activating gene, RAG-1. Cell 59, 1035–48 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Oettinger MA, Schatz DG, Gorka C & Baltimore D RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248, 1517–23 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Tonegawa S Somatic generation of antibody diversity. Nature 302, 575–81 (1983). [DOI] [PubMed] [Google Scholar]
- 7.Fanning L, Connor A, Baetz K, Ramsden D & Wu GE Mouse RSS spacer sequences affect the rate of V(D)J recombination. Immunogenetics 44, 146–50 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Ngo VN et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akira S, Okazaki K & Sakano H Two pairs of recombination signals are sufficient to cause immunoglobulin V-(D)-J joining. Science 238, 1134–8 (1987). [DOI] [PubMed] [Google Scholar]
- 10.Ramsden DA, Baetz K & Wu GE Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res 22, 1785–96 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schatz DG & Ji Y Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11, 251–63 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Schatz DG & Swanson PC V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45, 167–202 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Ru H et al. Molecular Mechanism of V(D)J Recombination from Synaptic RAG1-RAG2 Complex Structures Cell 163, 1138–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MS et al. Cracking the DNA Code for V(D)J Recombination. Mol Cell (2018). [DOI] [PMC free article] [PubMed]
- 15.Lieber MR The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79, 181–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee I & Harshey RM Patterns of sequence conservation at termini of long terminal repeat (LTR) retrotransposons and DNA transposons in the human genome: lessons from phage Mu. Nucleic Acids Res 31, 4531–40 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang W, Lee JY & Nowotny M Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell 22, 5–13 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Swanson PC The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol Rev 200, 90–114 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Swanson PC Fine structure and activity of discrete RAG-HMG complexes on V(D)J recombination signals. Mol Cell Biol 22, 1340–51 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MS, Lapkouski M, Yang W & Gellert M Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 518, 507–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesse JE, Lieber MR, Mizuuchi K & Gellert M V(D)J recombination: a functional definition of the joining signals. Genes Dev 3, 1053–61 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Grawunder U & Lieber MR A complex of RAG-1 and RAG-2 proteins persists on DNA after single-strand cleavage at V(D)J recombination signal sequences. Nucleic Acids Res 25, 1375–82 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel DJ, Shapiro L & Hare D Nuclear magnetic resonance and distance geometry studies of DNA structures in solution. Annu Rev Biophys Biophys Chem 16, 423–54 (1987). [DOI] [PubMed] [Google Scholar]
- 24.Ramsden DA, McBlane JF, van Gent DC & Gellert M Distinct DNA sequence and structure requirements for the two steps of V(D)J recombination signal cleavage. EMBO J 15, 3197–206 (1996). [PMC free article] [PubMed] [Google Scholar]
- 25.Hiom K & Gellert M A stable RAG1-RAG2-DNA complex that is active in V(D)J cleavage. Cell 88, 65–72 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Cuomo CA, Mundy CL & Oettinger MA DNA sequence and structure requirements for cleavage of V(D)J recombination signal sequences. Mol Cell Biol 16, 5683–90 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peak MM, Arbuckle JL & Rodgers KK The central domain of core RAG1 preferentially recognizes single-stranded recombination signal sequence heptamer. J Biol Chem 278, 18235–40 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Swanson PC & Desiderio S V(D)J recombination signal recognition: distinct, overlapping DNA-protein contacts in complexes containing RAG1 with and without RAG2. Immunity 9, 115–25 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Santagata S et al. The RAG1/RAG2 complex constitutes a 3’ flap endonuclease: implications for junctional diversity in V(D)J and transpositional recombination. Mol Cell 4, 935–47 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Yanagihara K & Mizuuchi K Progressive structural transitions within Mu transpositional complexes. Mol Cell 11, 215–24 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Hickman AB et al. Structural basis of hAT transposon end recognition by Hermes, an octameric DNA transposase from Musca domestica. Cell 158, 353–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickman AB et al. Molecular architecture of a eukaryotic DNA transposase. Nat Struct Mol Biol 12, 715–21 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Davies DR, Mahnke Braam L, Reznikoff WS & Rayment I The three-dimensional structure of a Tn5 transposase-related protein determined to 2.9-A resolution. J Biol Chem 274, 11904–13 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Davies DR, Goryshin IY, Reznikoff WS & Rayment I Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science 289, 77–85 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Lee I & Harshey RM The conserved CA/TG motif at Mu termini: T specifies stable transpososome assembly. J Mol Biol 330, 261–75 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Bai XC, Rajendra E, Yang G, Shi Y & Scheres SH Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan Z et al. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat Struct Mol Biol 23, 217–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin FF et al. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat Struct Mol Biol 16, 499–508 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–12 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allegretti M, Mills DJ, McMullan G, Kuhlbrandt W & Vonck J Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. Elife 3, e01963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartesaghi A, Matthies D, Banerjee S, Merk A & Subramaniam S Structure of beta-galactosidase at 3.2-A resolution obtained by cryo-electron microscopy. Proc Natl Acad Sci U S A 111, 11709–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delano, WL The PyMol Molecular Graphics System. (2002).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and cryo-EM maps are available in the PDB and EMDB databases, under accession numbers PDB 6DBT and EMD-7849 (12I + 23I doubly unmelted with I for intact RSS); PDB 6DBU and EMD-7850 (12I + 23I doubly unmelted and 2-fold averaged); PDB 6DBV and EMD-7851 (12I melted + 23I unmelted); PDB 6DBQ and EMD-7847 (12I unmelted + 23I melted); PDB 6DBR and EMD-7848 (averaged map with one RSS melted and one RSS unmelted); PDB 6DBL and EMD-7845 (12I + 23I doubly melted); PDB 6DBO and EMD-7846 (12I + 23I doubly melted and 2-fold averaged); PDB 6DBX and EMD-7853 (12I unmelted); PDB 6DBW and EMD-7852 (12I melted); PDB 6DBI and EMD-7843 (12N + 23N doubly nicked with N for nicked); and PDB 6DBJ and EMD-7844 (12N + 23N doubly nicked and 2-fold averaged). The datasets generated during the current study are available from the corresponding authors upon reasonable request.