

Abstract
Introduction
For the past decade, the focus of complex disease research has been the genotype. From technological advancements to the development of analysis methods, great progress has been made. However, advances in our definition of the phenotype have remained stagnant. Phenotype characterization has recently emerged as an exciting area of informatics and machine learning. The copious amounts of diverse biomedical data that have been collected may be leveraged with data-driven approaches to elucidate trait-related features and patterns.
Areas covered
In this review, the authors discuss the phenotype in traditional genetic associations and the challenges this has imposed. The authors address approaches for phenotype refinement that can aid in the more accurate characterization of traits. Further, the authors highlight promising machine learning approaches for establishing a phenotype and the challenges of electronic health record (EHR) derived data.
Expert Commentary
The authors hypothesize that through unsupervised machine learning, data-driven approaches can be used to define phenotypes rather than relying on expert clinician knowledge, which may be inaccurate. Through the use of machine learning and an unbiased set of features extracted from clinical repositories, researchers will have the potential to further understand complex traits and identify patient subgroups. This knowledge may lead to more preventative and precise clinical care.
Keywords: cluster analysis, complex traits, dimensionality reduction, electronic health records (EHR), heterogeneity, machine learning, missing data, phenotype, topological analysis, unsupervised analysis
1. Introduction
The genetic architecture of complex human disease is likely influenced by multiple components including both common and rare genetic variants, structural variants, and gene-gene and gene-environment interactions. Additional confounding variables such as genetic locus heterogeneity and phenotypic or trait heterogeneity are also likely at play. A better understanding of both the genetic and phenotypic complexities of these traits is required to fully elucidate their intricate etiologies and ultimately progress toward more predictive and precise clinical care.
Given the technological advances of genetic platforms, many studies have placed a large focus on the genotype. Considerable progress has been made in how we are defining and handling genetic data in our studies. This includes moving beyond common variants to examine the influence of other forms of sequence[1–4] and structural variation[3,5], considering the environment, and integrating variousomics data modules such as genomic, transcriptomic and metabolomic data so as to identify effective models that predict phenotypic outcome[6]. However, advancement in how we are defining and handling the phenotype in our studies has largely remained stagnant, and not kept pace with developments in studying the genotype. Appropriate phenotype assignment plays a crucial role when trying to resolve the genetic architecture of complex human traits. Regardless of what statistical method is employed, accurate phenotyping is essential in identifying a true, replicable genotype-phenotype association.
2. The phenotype in traditional genetic studies
Traditionally, association analyses have been performed by selecting participants from a given cohort, and dichotomizing them into one of two broad categories, cases or controls, using some clinical criterion. For example, in an obesity study, researchers may use body mass index (BMI) as the main determinant of obesity status. Subjects with a BMI greater than 30 kg/m2 will be classified as cases while all other subjects with a BMI below 30 kg/m2 would be labeled as controls. However, the clinical profile of obesity is extensive, and this categorization may be over-simplistic as it fails to account for the intrinsic complexities of this trait. Patients may exhibit a wide range of symptoms, including fatigue, polydipsia, hyperhidrosis or insulin resistance, and any number of comorbidities, such as arthritis, sleep apnea, type II diabetes, and hypertension[7,8], as highlighted in Figure 1a. Not all patients present with the same clinical characteristics, and so we must question whether lumping together all patients with a BMI above 30 kg/m2, regardless of variability in clinical manifestation, is creating too heterogeneous of a phenotype. This implicit phenotypic heterogeneity is likely mitigating our ability to detect the genetic effects of obesity. This scenario does not only apply to obesity. In fact, many diseases have been shown to exhibit phenotypic heterogeneity, including diabetes mellitus[9], autism spectrum disorder[10], and obstructive lung disorders like COPD and asthma[11]. While some traits may be more multifarious than others, heterogeneity is inherent in the nature of complex polygenic disorders, and addressing it may help further illuminate the genetic landscape of these traits.
Figure 1. Heterogeneous Landscape of Obesity.
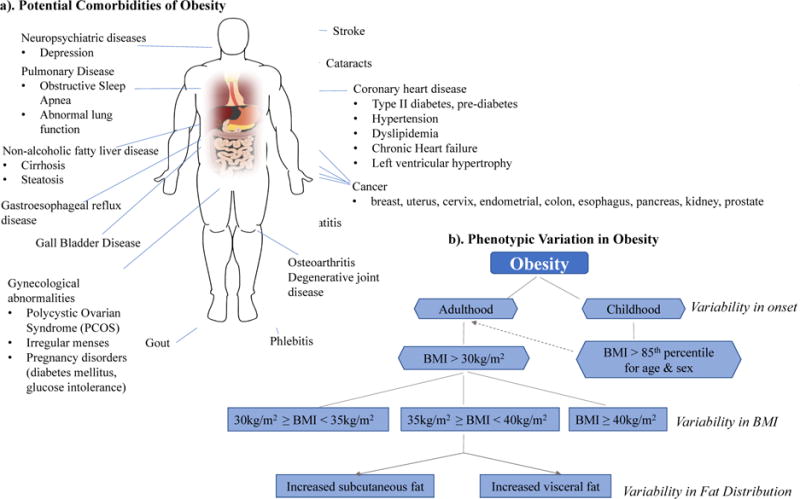
a). Illustrates some of the potential comorbidities of obesity. Patients with a BMI > 30 kg/m2 often exhibit heterogeneity in comorbidities. b). Identifies several factors that are involved in creating the spectrum of phenotypic variability in obesity. The dashed arrow indicates increased risk of developing obesity in adulthood for individuals who suffer from obesity in their childhood. Both a). and b). act in creating a heterogeneous phenotypic landscape.
The obesity example highlights two types of heterogeneity-related components that muddle complex trait analyses, trait heterogeneity and phenotypic variation[12,13]. Phenotype or trait heterogeneity exists when a given trait is defined with inadequate specificity such that it may actually represent multiple distinct traits[13]. Phenotypic variation describes the spectrum of variability in symptoms, severity, and age of onset in subjects who exhibit the same trait or disease. These two concepts often coincide and contribute toward creating a heterogeneous phenotypic landscape, and hence, this work will simply refer to both as trait/phenotype heterogeneity. Figure 1 provides an example of such a heterogeneous landscape as exemplified by obesity.
The role of trait heterogeneity on genetic studies has been known since the dawn of association studies, and it was even cited as a potential reason for the limitations of GWAS in fully explaining the genetic variance in complex traits[5]. Heterogeneity can lead to increased type II error probabilities resulting in substantially decreased statistical power or ability to detect a true association between a disease and a locus[14–17]. While the impact of phenotype heterogeneity is well chronicled, addressing the issue has proven quite difficult. Loss of statistical power may be combatted by one of two approaches, either increasing the sample size in the study or increasing the effect size of the association. Regarding the former, it is difficult to accurately estimate how much of a sample size increase is needed in the presence of trait heterogeneity. However, studies of phenotypic misclassification estimate that as much as a 39-fold increase in sample size is required in case/control studies when the misclassification rate is 5% and disease prevalence is 1%[17]. Although this value is but a rough estimate, and actual measures may not be as stark for circumstances of heterogeneity, increasing the sample size in most studies may not be feasible as it could necessitate recruitment of additional subjects. Thus, a more practical approach may be to focus on elevating the effect size of the association which could be accomplished by decreasing noise and redundancy in the data. For example, additional phenotypic and clinical information can be used to facilitate stratification of subjects. This was often not possible in the early days of GWAS as scarce detailed phenotypic information had been collected in conjunction with genetic data. However, with expansions in data collection and the establishment of electronic health record (EHR)-linked biorepositories[18–22], copious amounts of biomedical data have become available. These data may be used for phenotypic refinement in the development of phenotype algorithms or they may be leveraged in clustering approaches for the stratification of patients into more homogeneous subgroups. Both approaches aid in increasing the effect size and help address the influence of trait heterogeneity in association studies.
3. Phenotype Refinement
Large collaborative efforts have focused on the creation of sophisticated algorithms for better phenotypic classification in many complex traits. The main goal of these algorithms is to develop a robust EHR-based model for improved case definition of a given trait[23]. EHRs provide a rich source of both structured and unstructured data that can be integrated for research tasks such as cohort development, outcome ascertainment, and clinical translation when coupled to a biobank. Structured EHR data is largely made up of diagnosis or billing codes, including international classification of disease (ICD) codes and current procedural terminology (CPT) codes, as well as electronic prescriptions, and vital signs. The ICD codes most often seen in studies are the 9th and 10th editions of these codes, ICD-9 and ICD-10 codes, respectively. Unstructured EHR data is in the form of notes and reports. This includes clinician notes (e.g. family history, signs and symptoms, social history), and reports (e.g. radiology reports, discharge reports, and others), which are often in form of free text[24]. Most models to define a disease have focused on the use of diagnosis codes as this data is structured, readily available, and does not require sophisticated curation methods. Diagnosis codes provide a rich source of information and have been very useful in further elucidating the architecture of many traits[25–27] and characterizing comorbidity profiles[28,29], to name a few applications. Unfortunately, the accuracy of these models is limited by coding practice variations, use of multiple diagnosis codes, and the fact that these codes were developed for billing purposes often by administrative non-clinical staff[23].
Advanced phenotype algorithms use both structured and unstructured data to pull all informative components from the EHR and more accurately define a disease. To date, phenotype algorithms with high positive predictive values have been developed for many conditions, including diabetes mellitus[30], depression[31], Crohn’s disease and ulcerative colitis[23], and rheumatoid arthritis[32]. Further, databases like PheKB[33], accessible at https://phekb.org/, which contains over 60 finalized and multiple in-development publicly available algorithms, provide a catalog of phenotype models across a wide range of complex traits. Phenotype algorithms provide an efficient approach for addressing heterogeneity in our data. But while significant progress has been made in this area, phenotype algorithm development is a challenging task. It is time intensive, requires the use of natural language processing (NLP) techniques, as well as a multidisciplinary team with collaborative efforts from clinicians, bioinformaticians, EHR informaticians, and genomics researchers. The use of data-driven approaches to leverage EHR data and elucidate trait-related features and patterns may provide a more high-throughput and generalizable means of addressing heterogeneity in the phenotype. Furthermore, knowledge gained in this process can be integrated into the phenotype algorithm development pipeline to aid in more precise phenotyping.
4. Machine learning approaches for establishing a phenotype
Various classes of unsupervised, data-driven, machine learning algorithms have been implicated in undertaking the issue of trait heterogeneity. In this review, we will specifically focus on clustering approaches, topological methods, and dimensionality reduction techniques, as they all show promise in addressing this issue. It should be noted that these three categories are not mutually exclusive, and several algorithms often fall into multiple classes.
Clustering approaches are unsupervised machine learning algorithms that aim to produce homogeneous subsets of data when subgroup labels are unknown. Clustering is an effective method for grouping objects with similar attributes using a measure of distance or similarity[34]. There are over 100 different clustering techniques which may fall into a number of broad algorithmic categories. The most common distinction among clustering methods is hierarchical, or nested, versus partitional, or un-nested, approaches. Partitional methods, such as k-means, divide the data into non-overlapping subsets so that each subject falls into exactly one subgroup. Hierarchical approaches, like agglomerative clustering, allow clusters to have nested subclusters, which are often organized as a tree model and shown as a dendrogram[11]. Despite the differences, these methods aim to produce subsets which have high intra-group similarities (objects within a group are similar), and low inter-group similarities (objects between groups are more dissimilar). Cluster analysis has widely been used to assess microarray data[35] and has seen success in EHR applications[11,36–41]. Additionally, hierarchical clustering has been used to assess the contribution of obesity (a trait with known heterogeneity, as seen in Figure 1) to respiratory conditions[42,43]. Unfortunately, many clustering algorithms are often not robust to high dimensional data, and thus most EHR applications have been restricted to smaller or more homogenous datasets[34].
Dimensionality reduction algorithms are another set of unsupervised techniques that can be used to find patterns and structure in the data. These methods facilitate the embedding of data in a lower dimension and aim to maximize the variance with the goal of removing noise and elucidating features[44,45]. Dimensionality reduction techniques are often classified as linear, such as principal component analysis (PCA), or non-linear, such as multi-dimensional scaling (MDS). These methods have the advantage of handling high dimensional, noisy data sets; and they have shown success in sub-phenotyping applications[37,46,47]. One of the properties that can be used to divide dimensionality reduction approaches is the parametric nature of the mapping between high-dimensional and low-dimensional space. Many dimensionality reduction techniques (including isomap, LLE, and Laplacian Eigenmaps) are non-parametric in nature, meaning that they do not specify a mapping from high to low dimension. This can make it impossible to obtain insight into how much high dimensional information was retained in a lower dimensional embedding. Conversely, parametric techniques also present certain challenges. These algorithms include the presence of free parameters that influence the cost function and need to be tuned for algorithm performance[45]. While these parameters provide flexibility in the method, optimization can often be challenging. Further, techniques that have non-convex functions, such as t-distributed stochastic neighbor embedding (t-SNE)[48], have additional parameters that are imposed including iterations, and learning rate.
Topology-based methods hold a lot of promise for addressing challenges imposed by phenotype heterogeneity. These methods provide a geometric approach to perform pattern recognition within large, multidimensional datasets[49,50]. They aim to extract the “shape” and “connectivity” of complex data in order to find existing structures. Overall, topological data analysis (TDA) is a very broad collection of methods which include the aforementioned nonlinear dimensionality reduction, but also ridge estimation, manifold learning, and persistent homology[51]. Some have even categorized clustering as a TDA method as density-based clustering approaches rely on the connection of data objects to elucidate patterns. Application of TDA to phenotype subgrouping is an exciting and growing area of research, and multiple studies highlight its promise[9,52,53]. However, given its infancy in EHR applications, there are likely many factors that still need to be considered. This includes the challenge of choosing appropriate tuning parameters for topological algorithms. This choice often requires its own set of methods, such as using bootstrap approaches to assess the number of significant topological features[51]. Further, some TDA applications to biomedical data have been made used professional, non-open source software, which is often financially infeasible for many researchers.
Clustering, nonlinear dimensionality reduction, and TDA are promising approaches with demonstrated successes in addressing phenotypic heterogeneity. However, they each have strengths and weaknesses, and will need to be thoroughly evaluated for the application of elucidating subgroups in EHR data. These approaches, along with strengths, limitations, and examples of successful biomedical applications are described in Table 1.
Table 1.
Summary of machine learning approaches that are promising in addressing issues imposed by trait heterogeneity. The Approaches column lists examples of algorithms in the respective method category. Strengths and Limitations are described in terms of EHR-derived data applications. The Biomedical Applications column lists traits for which subgroups have been identified using the respective method.
| Method Category | Approaches | Strengths | Limitations | Biomedical Applications |
|---|---|---|---|---|
| Cluster analysis | Hierarchical, k-means | Wide range of applications; easy interpretation | Not robust to highly dimensional data or large datasets; most approaches restricted to one data type; some approaches require number of clusters | COPD[11,37], Fibromyalgia[39], Tinnitus[40], Diabetes[41], Obesity[42,43] |
| Topological approaches | TDA, manifold learning algorithms | Able to handle highly dimensional and noisy data; does not require knowledge of number of clusters; sensitive to global and local structure | Optimization of free parameters; computational cost; deep knowledge of topological methods for correct application | T2D[9], Breast cancer [53], Attention deficit [52] |
| Dimensionality Reduction | Linear (PCA), Non-linear (MDS, t-SNE, Isomap, LLE) | Able to handle highly dimensional, noisy data; does not require knowledge of number of clusters | Optimization of free parameters; Many methods are non-parametric and do not provide information on how dimensionality was reduced; projection loss; result inconsistency; computational cost | COPD[37,46], Changes during anesthesia[54], temporal lobe epilepsy[55] |
5. Challenges
For a machine learning algorithm to be appropriate in undertaking trait heterogeneity and elucidating homogeneous patient subgroups, it must address key challenges associated with EHR-derived data and analyses. These include the handling of heterogeneous data types, robustness to missing data and high dimensionality, as well as computational feasibility, to name a few[56]. Data extracted from the EHR is heterogeneous in data type as variables may be continuous, such as clinical lab measures and BMI, or categorical, such as comorbidities and race. Ideal methods would be robust to handling mixed data types as each value may contain meaningful information. Alternatively, data may be altered to a single type by using methods such as the categorization of continuous variables, and dummy or one-hot encoding[57] of continuous variables. However, the drawback of this approach is that the type of variable encoding used has been shown to affect results[11].
Another challenge is the amount of missing data in the EHR. To date, many approaches require exclusion of subjects with missing values, and thus the majority of studies have been conducted on complete datasets. This tactic, however, may be biasing results by restricting analysis to patients who for some reason have complete data. It could be that these patients are the sickest which would bias analyses towards more extreme phenotype subsets. Or, there may be socioeconomic reasons for patients being complete across their data attributes, which would again impose a bias. Regardless of the reason, missingness in the EHR is meaningful, but appropriately accounting for this missingness is a challenge. As an alternative to using complete datasets, imputation strategies may be employed to aid in estimating missing values. There is large debate whether imputation of phenotype data is suitable[58], and careful considerations need to be made when choosing to impute. First, characterization of the type of missingness is needed. Knowledge of whether missing data is at random (MAR) or missing not at random (MNAR) is important as each imputation method assumes a specific model of missingness, and a violation of these assumptions may impose downstream analytical biases[59]. Accurate characterization of missingness may prove difficult, especially in the EHR, as data are rarely entirely MAR or MNAR. Instead, they likely fall somewhere in the middle, with variable components of missingness. Second, considerations for the choice of imputation strategy need to be made. While there is no such thing as best imputation method, comparisons of error, bias, and implementation difficulty can be leveraged in making a knowledgeable choice[60]. This, however, comes with the caveat that such conclusions may not be generalizable between different datasets. Although efforts can be made to minimize its occurrence, missing data in the EHR is unavoidable. Promising machine learning methods would ideally be robust to handling missing data. If not, then researchers must choose if imputation or restriction to complete data is most appropriate, with an understanding of the limitations and biases that are imposed by this decision.
A further challenge facing researchers is the presence of inaccurate or incorrect data in the EHR. There are multiple reasons for data inaccuracies in the EHR; some of these include incorrect entry or miskeying of information (incorrect data is mistakenly entered), miscommunication with a patient (inaccurate information is provided by the patient or there is a miscommunication between patient and clinician), and timeliness of data entry (medical professional may chart data much later than it was originally observed due to time constraints)[61]. Incorrect data in the EHR is a very difficult challenge to address as it may not be clear which data elements are correct and which are inaccurate. Currently, the most common means of limiting data inaccuracy is the use of various quality control assessments. According to Weiskopf et al.[62], comparison of EHR data to a “gold standard” is the most frequently used method for assessing data accuracy. These gold standards are often of various types including physical paper records[63,64], contact with treating clinician[65], and patient interviews[66]. However, the use of such gold standards may not be feasible due to limited access to patients and/or clinicians, and the time-consuming nature of these tasks. Alternatively, data verification within the EHR can be used as a “gold standard”, in which agreement between data elements within the EHR can be used to assess data accuracy. This can be performed computationally, and it may involve diagnosis verification by examining associated laboratory tests, medications, and procedures[67–69]. Additionally, NLP has also been used to analyze written or unstructured texts in the EHR, and compared with structured elements for agreement[23,71]. Inaccurate data is a concern for all researchers working with EHRs, and computation approaches can be leveraged to limit the inaccuracies.
Additional considerations that need to be made include the computational attractiveness of the chosen method. Data extracted from the EHR are often highly dimensional and longitudinal; they may include hundreds to thousands of features measured across thousands of subjects. Potential algorithms should be computationally sophisticated for handling such large data. Additionally, advanced platforms, such as Hadoop, Spark, and MongoDB or cloud-computing services, like Amazon Web Services (AWS), and Google Cloud Platform, which make the analysis and storage of large-scale data feasible, should be considered[72]. While they provide significant aid in computational performance, these platforms are often quite costly and may not be financially feasible for many research groups, therefore, computational robustness within a given algorithm is a key advantage. Even if computational needs are met, dimensionality can pose an analytical challenge. As EHR data are high dimensional, successful methods will either need to be coupled to or contain internal feature selection techniques to help ensure that resultant clusters are meaningful. Clustering on all data attributes may lead to subsets that contain redundant information and are clinically irrelevant[73]. The EHR contains a wealth of valuable data that can be leveraged for patient subgrouping, however, for an analytical approach to be successful in this setting, it must address a host of challenges imposed by the nature of EHR data.
6. Conclusion
Unsupervised machine learning approaches can be applied to rich phenotypic data from the EHR to create homogeneous patient subsets with more consistent underlying factors contributing to disease. These new homogeneous subgroups can then be examined for genetic, environmental, as well as other contributing factors that may associate and predict disease susceptibility. This may help uncover important biological insights, identify biomarkers, as well as inform clinical care and drug treatment. However, given their incomplete, inaccurate, highly complex, dimensional and biased nature, EHR data present analytical difficulties[56]. Thus, many studies have been restricted to using data that is complete (i.e. not accounting for missingness), of a single data type (e.g. categorical or continuous), and limited to a small sample size. To successfully mine and extract meaningful information from the EHR, machine learning approaches must overcome the challenges imposed by these data. Overall, the EHR provides an invaluable resource of information that can be leveraged to better understand the phenotypic complexity of many traits and may aid in progressing to a more precise treatment of disease.
7. Expert Commentary
Phenotype characterization is emerging as one of the most exciting areas of informatics and machine learning. For the past decade, the primary focus of genetic research has been on the development of methods and technologies for theomics component of the research. Many new high-throughput, cost-effective molecular technologies have been developed and made available to the community. This has led to a wealth of data and an enormous number of genome-wide studies exploring the genetic architecture of common, complex traits. However, the definition of the trait or phenotype has largely been overly simplistic. Now that we have dense, comprehensive molecular data to characterize the genomic aspect of the equation, the future is defining the phenotypic component of the equation.
Rich, dense, clinical data are becoming increasingly more available for research through large EHR databases, clinical data repositories, and clinical trials data sources. With these longitudinal clinical data, accurate and specific phenotypes can be defined for participant groups in genomics projects. For decades, we have relied on expert knowledge from the clinical community to guide our development of phenotype algorithms. A key weakness of this strategy is that clinical experts and our current knowledgebase of what constitutes a disease or trait could be inaccurate. Perhaps there are other clinical features or symptoms that comprise the true definition of the trait. By using our current state of knowledge, we could be missing important features and incorrectly defining traits.
Our hypothesis is that through the use of machine learning algorithms, we will be able to rely on data-driven approaches to define phenotypes, rather than assuming our expert knowledge about traits is accurate. It is conceivable that there are features that are important to define more precise and accurate phenotypes, that are not yet known to clinicians. Thus, through machine learning in unsupervised approaches, we can identify relevant and important features not yet known. The ultimate goal is to make use of all relevant clinical data to define accurate, specific, phenotypes for research purposes.
These machine learning approaches will also enable the community to identify research participant subgroups of specific traits – identifying participants who share clinical features and who present differently from other participants. Clustering of participant groups to handle trait heterogeneity has been done in the past, however, this was mostly based on known clinical features[12,13,36,37]. We propose that through the use of unsupervised machine learning, and an unbiased set of clinical features, we will have the potential to learn more about complex traits and identify patient subgroups simultaneously. The promise of these approaches is based on how well these methods have done in other areas of research[49,74–76]. If these methods work well for financial data, sports predictions, oceanography, and cosmology, we believe they will work well for clinical data.
8. Five-year View
In our opinion, the next three to five years will be a very exciting time for phenotype informatics. Large clinical data sets will continue to become publicly available such as UK Biobank[21] and the All of Us Cohort Program[77]. These data, along with the data sets from health systems and academic medical centers, such as those in the eMERGE network[20,78], and government studies like the Million Veterans Program (MVP)[22], will lead to an enormous amount of clinical data in the public domain. This will facilitate the community of machine learning, computer science, and data science experts the opportunity to develop and deploy novel algorithms on these rich, longitudinal data sources to define more robust phenotypes, identify patient subgroups, and further our understanding of many complex traits. This knowledge has the potential to influence more precise clinical care of patients. Phenotype informatics will be emerging as a stimulating, innovative area for years to come.
Key issues.
The primary focus of complex disease research has been the development of methods and technologies for studying the genotype. However, advances in studying the phenotype have remained stagnant. Appropriate phenotype characterization plays a crucial role whe n trying to resolve the genetic architecture of complex human traits.
Phenotype characterization in traditional genetic studies has not accounted for trait/phenotype heterogeneity which reduces statistical power and mitigates the ability to detect shared genetic effects.
Electronic Health Records (EHRs) provide an invaluable resource of information that can be leveraged to better understand the phenotypic complexity of many traits and may aid in progressing to a more precise treatment of disease.
Phenotype informatics has recently emerged as an innovative, and exciting area of informatics and machine learning. The copious amounts of diverse biomedical data that have been collected in EHRs may be utilized with data-driven, machine learning approaches to elucidate trait-related features and patterns.
For a machine learning approach to be appropriate in undertaking trait heterogeneity and elucidating homogeneous patient subgroups, it must address key challenges associated with EHR-derived data and analyses including the handling of heterogeneous, robustness to missing data, high dimensionality, and computational feasibility.
The public availability of large clinical data sets along with the data from health systems and academic medical centers will lead to an enormous amount of clinical data in the public domain. This will facilitate the community of machine learning, computer science, and data science experts the opportunity to develop novel algorithms on these rich, longitudinal data sources, to define more robust phenotypes.
Data-driven approaches can be used to define phenotypes rather than relying on expert clinician knowledge, which may be inaccurate. Through the use of machine learning and an unbiased set of features extracted from clinical repositories, researchers will have the potential to further understand complex traits and identify patient subgroups. This knowledge may lead to more preventative and precise clinical care.
Acknowledgments
Funding
This project is funded, in part, under a grant with the Pennsylvania Department of Health (#SAP4100070267). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work is also supported by the Biomedical Big Data to Knowledge (B2D2K) Pre-doctoral Training Program from the National Library of Medicine (1T32 LM012415-01).
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Do R, Kathiresan S, Abecasis GR. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum Mol Genet. 2012;21:R1–9. doi: 10.1093/hmg/dds387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner MJ. Rare-variant genome-wide association studies: a new frontier in genetic analysis of complex traits. Pharmacogenomics. 2013;14:413–24. doi: 10.2217/pgs.13.36. [DOI] [PubMed] [Google Scholar]
- 3.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-Variant Association Analysis: Study Designs and Statistical Tests. Am J Hum Genet. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher B. Personal genomes: The case of the missing heritability. Nat News. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 6.Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. 2015;16:85–97. doi: 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin T. Metabolic heterogeneity of obesity: role of adipose tissue. Int J Obes Suppl. 2012;2:S8–10. doi: 10.1038/ijosup.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownell KD, Wadden TA. The heterogeneity of obesity: fitting treatments to individuals. Behav Ther. 1991;22:153–77. doi: 10.1016/j.beth.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 9**.Li L, Cheng W-Y, Glicksberg BS, Gottesman O, Tamler R, Chen R, et al. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci Transl Med. 2015;7:311ra174–311ra174. doi: 10.1126/scitranslmed.aaa9364. This reference is one of the first successful applications of a topological machine learning approach to identify disease sub-groups in a large cohort using EHR-derived data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georgiades S, Szatmari P, Boyle M, Hanna S, Duku E, Zwaigenbaum L, et al. Investigating phenotypic heterogeneity in children with autism spectrum disorder: a factor mixture modeling approach: ASD factor mixture model. J Child Psychol Psychiatry. 2013;54:206–15. doi: 10.1111/j.1469-7610.2012.02588.x. [DOI] [PubMed] [Google Scholar]
- 11.Burgel P-R, Paillasseur J-L, Roche N. Identification of Clinical Phenotypes Using Cluster Analyses in COPD Patients with Multiple Comorbidities. Bio Med ResInt. 2014;2014:e420134. doi: 10.1155/2014/420134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thornton-Wells TA, Moore JH, Haines JL. Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004;20:640–7. doi: 10.1016/j.tig.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 13**.Thornton-Wells TA, Moore JH, Haines JL. Dissecting trait heterogeneity: a comparison of three clustering methods applied to genotypic data. BMC Bioinformatics. 2006:7. doi: 10.1186/1471-2105-7-204. This reference was one of the first manuscripts to characterize trait heterogeneity, and prescribe potential methodologies for addressing this heterogeneity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon D, Yang Y, Haynes C, Finch SJ, Mendell NR, Brown AM, et al. Increasing power for tests of genetic association in the presence of phenotype and/or genotype error by use of double-sampling. Stat Appl Genet Mol Biol. 2004:3. doi: 10.2202/1544-6115.1085. Article26. [DOI] [PubMed] [Google Scholar]
- 15.Ji F, Yang Y, Haynes C, Finch SJ, Gordon D. Computing asymptotic power and sample size for case-control genetic association studies in the presence of phenotype and/or genotype misclassification errors. Stat Appl Genet Mol Biol. 2005:4. doi: 10.2202/1544-6115.1184. Article37. [DOI] [PubMed] [Google Scholar]
- 16.Edwards BJ, Haynes C, Levenstien MA, Finch SJ, Gordon D. Power and sample size calculations in the presence of phenotype errors for case/control genetic association studies. BMC Genet. 2005;6:18. doi: 10.1186/1471-2156-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buyske S, Yang G, Matise TC, Gordon D. When a case is not a case: effects of phenotype misclassification on power and sample size requirements for the transmission disequilibrium test with affected child trios. HumHered. 2009;67:287–92. doi: 10.1159/000194981. [DOI] [PubMed] [Google Scholar]
- 18.Roden D, Pulley J, Basford M, Bernard G, Clayton E, Balser J, et al. Development of a Large-Scale De-Identified DNA Biobank to Enable Personalized Medicine. Clin Pharmacol Ther. 2008;84:362–9. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18:906–13. doi: 10.1038/gim.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, Larson EB, et al. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–23. doi: 10.1016/j.jclinepi.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 23*.Ananthakrishnan AN, Cai T, Savova G, Cheng S-C, Chen P, Perez RG, et al. Improving case definition of Crohn’s disease and ulcerative colitis in electronic medical records using natural language processing: a novel informatics approach. Inflamm Bowel Dis. 2013;19:1411–20. doi: 10.1097/MIB.0b013e31828133fd. This reference provides a successful example of using natural language processing (NLP) to improve phenotype characterization of Crohn’s disease and ulcerative colitis using patient data extracted from an EHR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao KP, Cai T, Savova GK, Murphy SN, Karlson EW, Ananthakrishnan AN, et al. Development of phenotype algorithms using electronic medical records and incorporating natural language processing. The BMJ [Internet] 2015:350. doi: 10.1136/bmj.h1885. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707569/ [DOI] [PMC free article] [PubMed]
- 25.Verma A, Leader JB, Verma SS, Frase A, Wallace J, Dudek S, et al. INTEGRATING CLINICAL LABORATORY MEASURES AND ICD-9 CODE DIAGNOSES IN PHENOME-WIDE ASSOCIATION STUDIES. Pac Symp Biocomput Pac Symp Biocomput. 2016;21:168–79. [PMC free article] [PubMed] [Google Scholar]
- 26.Pendergrass SA, Ritchie MD. Phenome-Wide Association Studies: Leveraging Comprehensive Phenotypic and Genotypic Data for Discovery. Curr Genet Med Rep. 2015;3:92–100. doi: 10.1007/s40142-015-0067-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma A, Basile AO, Bradford Y, Kuivaniemi H, Tromp G, Carey D, et al. Phenome-Wide Association Study to Explore Relationships between Immune System Related Genetic Loci and Complex Traits and Diseases. PLOS ONE. 2016;11:e0160573. doi: 10.1371/journal.pone.0160573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L-T, Gersing KR, Swartz MS, Burchett B, Li T-K, Blazer DG. Using electronic health records data to assess comorbidities of substance use and psychiatric diagnoses and treatment settings among adults. J Psychiatr Res. 2013;47:555–63. doi: 10.1016/j.jpsychires.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schildcrout JS, Basford MA, Pulley JM, Masys DR, Roden DM, Wang D, et al. An analytical approach to characterize morbidity profile dissimilarity between distinct cohorts using electronic medical records. J Biomed Inform. 2010;43:914–23. doi: 10.1016/j.jbi.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richesson RL, Rusincovitch SA, Wixted D, Batch BC, Feinglos MN, Miranda ML, et al. A comparison of phenotype definitions for diabetes mellitus. J Am Med Inform Assoc JAMIA. 2013;20:e319–26. doi: 10.1136/amiajnl-2013-001952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perlis RH, Iosifescu DV, Castro VM, Murphy SN, Gainer VS, Minnier J, et al. Using electronic medical records to enable large-scale studies in psychiatry: treatment resistant depression as a model. Psychol Med. 2012;42:41–50. doi: 10.1017/S0033291711000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao KP, Cai T, Gainer V, Goryachev S, Zeng-treitler Q, Raychaudhuri S, et al. Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res. 2010;62:1120–7. doi: 10.1002/acr.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Kirby JC, Speltz P, Rasmussen LV, Basford M, Gottesman O, Peissig PL, et al. PheKB: a catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc JAMIA. 2016;23:1046–52. doi: 10.1093/jamia/ocv202. This reference catalogs the workflow for establishing phenotype algorithms in PheKB, which includes numerous advance phenotype algorithms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Singh N, Garg N, Pant J. A Comprehensive Study of Challenges and Approaches for Clustering High Dimensional Data. Int J Comput Appl. 2014;92:7–10. This reference provides a concise description of the challenges of working with high-dimensional data, such as that from the EHR, and it also describes approaches for clustering this type of data. [Google Scholar]
- 35.Kafieh R, Mehridehnavi A. A Comprehensive Comparison of Different Clustering Methods for Reliability Analysis of Microarray Data. J Med Signals Sens. 2013;3:22–30. [PMC free article] [PubMed] [Google Scholar]
- 36.Fens N, van Rossum AGJ, Zanen P, van Ginneken B, van Klaveren RJ, Zwinderman AH, et al. Subphenotypes of Mild-to-Moderate COPD by Factor and Cluster Analysis of Pulmonary Function, CT Imaging and Breathomics in a Population-Based Survey. COPD J Chronic Obstr Pulm Dis. 2013;10:277–85. doi: 10.3109/15412555.2012.744388. [DOI] [PubMed] [Google Scholar]
- 37.Burgel P-R, Paillasseur J-L, Caillaud D, Tillie-Leblond I, Chanez P, Escamilla R, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses. Eur Respir J. 2010;36:531–9. doi: 10.1183/09031936.00175109. [DOI] [PubMed] [Google Scholar]
- 38.Deliu M, Sperrin M, Belgrave D, Custovic A. Identification of Asthma Subtypes Using Clustering Methodologies. Pulm Ther. 2016;2:19–41. doi: 10.1007/s41030-016-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Docampo E, Collado A, Escaramís G, Carbonell J, Rivera J, Vidal J, et al. Cluster Analysis of Clinical Data Identifies Fibromyalgia Subgroups. PLOS ONE. 2013;8:e74873. doi: 10.1371/journal.pone.0074873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Berge MJC, Free RH, Arnold R, de Kleine E, Hofman R, van Dijk JMC, et al. Cluster Analysis to Identify Possible Subgroups in Tinnitus Patients. Front Neurol [Internet] 2017:8. doi: 10.3389/fneur.2017.00115. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5377919/ [DOI] [PMC free article] [PubMed]
- 41.Ahlqvist E, Storm P, Karajamaki A, Martinell M, Dorkhan M, Carlsson A, et al. Clustering of adult-onset diabetes into novel subgroups guides therapy and improves prediction of outcome. bioRxiv. 2017:186387. [Google Scholar]
- 42.Green MA, Strong M, Razak F, Subramanian SV, Relton C, Bissell P. Who are the obese? A cluster analysis exploring subgroups of the obese. J Public Health Oxf Engl. 2016;38:258–64. doi: 10.1093/pubmed/fdv040. [DOI] [PubMed] [Google Scholar]
- 43.Sutherland ER, Goleva E, King TS, Lehman E, Stevens AD, Jackson LP, et al. Cluster Analysis of Obesity and Asthma Phenotypes. PLOS ONE. 2012;7:e36631. doi: 10.1371/journal.pone.0036631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunningham JP, Ghahramani Z. Linear Dimensionality Reduction: Survey, Insights, and Generalizations. J Mach Learn Res. 2015;16:2859–900. [Google Scholar]
- 45.van der Maaten LJP, Postma EO, van den Herik HJ. Dimensionality Reduction: A Comparative Review. 2008 [Google Scholar]
- 46.Paoletti M, Camiciottoli G, Meoni E, Bigazzi F, Cestelli L, Pistolesi M, et al. Explorative data analysis techniques and unsupervised clustering methods to support clinical assessment of Chronic Obstructive Pulmonary Disease (COPD) phenotypes. J Biomed Inform. 2009;42:1013–21. doi: 10.1016/j.jbi.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 47.Xu H, Ma ’ayan A. Inf Qual E-Health [Internet] Springer; Berlin, Heidelberg: 2011. Visualization of Patient Samples by Dimensionality Reduction of Genome-Wide Measurements; pp. 15–22. [cited 2017 Oct 10] Available from: https://link.springer.com/chapter/10.1007/978-3-642-25364-5_2. [Google Scholar]
- 48.van deer Maaten L. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2505. [Google Scholar]
- 49.Lum PY, Singh G, Lehman A, Ishkanov T, Vejdemo-Johansson M, Alagappan M, et al. Extracting insights from the shape of complex data using topology. Sci Rep [Internet] 2013:3. doi: 10.1038/srep01236. [cited 2015 Jul 15] Available from: http://www.nature.com/srep/2013/130207/srep01236/full/srep01236.html. [DOI] [PMC free article] [PubMed]
- 50.Carlsson Gunnar. Topology and data. Bull Am Math Soc. 2009;46:255–308. [Google Scholar]
- 51.Wasserman L. Topological Data Analysis. ArXiv160908227 Stat [Internet] 2016 Available from: http://arxiv.org/abs/1609.08227.
- 52.Kyeong S, Kim J-J, Kim E. Novel subgroups of attention-deficit/hyperactivity disorder identified by topological data analysis and their functional network modular organizations. PLOS ONE. 2017;12:e0182603. doi: 10.1371/journal.pone.0182603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicolau M, Levine AJ, Carlsson G. Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proc Natl Acad Sci. 2011;108:7265–70. doi: 10.1073/pnas.1102826108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kortelainen J, Vayrynen E, Seppanen T. Isomap Approach to EEG-Based Assessment of Neurophysiological Changes During Anesthesia. IEEE Trans Neural Syst Rehabil Eng. 2011;19:113–20. doi: 10.1109/TNSRE.2010.2098420. [DOI] [PubMed] [Google Scholar]
- 55.Cantor-Rivera D, Khan AR, Goubran M, Mirsattari SM, Peters TM. Detection of temporal lobe epilepsy using support vector machines in multi-parametric quantitative MR imaging. Comput Med Imaging Graph Off J Comput Med Imaging Soc. 2015;41:14–28. doi: 10.1016/j.compmedimag.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 56.Pathak J, Kho AN, Denny JC. Electronic health records-driven phenotyping: challenges, recent advances, and perspectives. J Am Med Inform Assoc JAMIA. 2013;20:e206–11. doi: 10.1136/amiajnl-2013-002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cassel M, Lima F. Evaluating one-hot encoding finite state machines for SEU reliability in SRAM-based FPGAs. 12th IEEE Int. -Line Test. Symp. IOLTS06; 2006. p. 6. [Google Scholar]
- 58.Hormozdiari F, Kang EY, Bilow M, Ben-David E, Vulpe C, McLachlan S, et al. Imputing Phenotypes for Genome-wide Association Studies. Am J Hum Genet. 2016;99:89–103. doi: 10.1016/j.ajhg.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beaulieu-Jones BK, Moore JH. MISSING DATA IMPUTATION IN THE ELECTRONIC HEALTH RECORD USING DEEPLY LEARNED AUTOENCODERS. Pac Symp Biocomput Pac Symp Biocomput. 2016;22:207–18. doi: 10.1142/9789813207813_0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Beaulieu-Jones BK, Lavage DR, Snyder JW, Moore JH, Pendergrass SA, Bauer CR. Characterizing and Managing Missing Structured Data in Electronic Health Records. bioRxiv. 2017:167858. doi: 10.2196/medinform.8960. This reference categorizes and describes missing data structures in the EHR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hersh WR, Weiner MG, Embi PJ, Logan JR, Payne PRO, Bernstam EV, et al. Caveats for the use of operational electronic health record data in comparative effectiveness research. Med Care. 2013;51:S30–37. doi: 10.1097/MLR.0b013e31829b1dbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiskopf NG, Weng C. Methods and dimensions of electronic health record data quality assessment: enabling reuse for clinical research. J Am Med Inform Assoc JAMIA. 2013;20:144–51. doi: 10.1136/amiajnl-2011-000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lo Re V, Haynes K, Forde KA, Localio AR, Schinnar R, Lewis JD. Validity of The Health Improvement Network (THIN) for epidemiologic studies of hepatitis C virus infection. Pharmacoepidemiol Drug Saf. 2009;18:807–14. doi: 10.1002/pds.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roukema J, Los RK, Bleeker SE, van Ginneken AM, van der Lei J, Moll HA. Paper versus computer: feasibility of an electronic medical record in general pediatrics. Pediatrics. 2006;117:15–21. doi: 10.1542/peds.2004-2741. [DOI] [PubMed] [Google Scholar]
- 65.Lewis JD, Brensinger C. Agreement between GPRD smoking data: a survey of general practitioners and a population-based survey. Pharmacoepidemiol Drug Saf. 2004;13:437–41. doi: 10.1002/pds.902. [DOI] [PubMed] [Google Scholar]
- 66.Porter SC, Mandl KD. Data quality and the electronic medical record: a role for direct parental data entry. Proc AMIA Symp. 1999:354–8. [PMC free article] [PubMed] [Google Scholar]
- 67.Faulconer ER, de Lusignan S. An eight-step method for assessing diagnostic data quality in practice: chronic obstructive pulmonary disease as an exemplar. Inform Prim Care. 2004;12:243–54. [PubMed] [Google Scholar]
- 68.de Burgos-Lunar C, Salinero-Fort MA, Cárdenas-Valladolid J, Soto-Díaz S, Fuentes-Rodríguez CY, Abánades-Herranz JC, et al. Validation of diabetes mellitus and hypertension diagnosis in computerized medical records in primary health care. BMC Med Res Methodol. 2011;11:146. doi: 10.1186/1471-2288-11-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Linder JA, Kaleba EO, Kmetik KS. Using electronic health records to measure physician performance for acute conditions in primary care: empirical evaluation of the community-acquired pneumonia clinical quality measure set. Med Care. 2009;47:208–16. doi: 10.1097/MLR.0b013e318189375f. [DOI] [PubMed] [Google Scholar]
- 70.Margulis AV, García Rodríguez LA, Hernández-Díaz S. Positive predictive value of computerized medical records for uncomplicated and complicated upper gastrointestinal ulcer. Pharmacoepidemiol Drug Saf. 2009;18:900–9. doi: 10.1002/pds.1787. [DOI] [PubMed] [Google Scholar]
- 71.Botsis T, Hartvigsen G, Chen F, Weng C. Secondary Use of EHR: Data Quality Issues and Informatics Opportunities. Summit Transl Bioinforma. 2010;2010:1–5. [PMC free article] [PubMed] [Google Scholar]
- 72.Hashem IAT, Yaqoob I, Anuar NB, Mokhtar S, Gani A, Ullah Khan S. The rise of “big data” on cloud computing: Review and open research issues. Inf Syst. 2015;47:98–115. [Google Scholar]
- 73.Paul R, Hoque ASML. Clustering medical data to predict the likelihood of diseases. 2010 Fifth Int Conf Digit Inf Manag ICDIM. 2010:44–9. [Google Scholar]
- 74.Gidea M, Katz Y. Topological Data Analysis of Financial Time Series: Landscapes of Crashes. ArXiv170304385 Phys Q-Fin [Internet] 2017 [cited 2017 Oct 31]; Available from: http://arxiv.org/abs/1703.04385.
- 75.Rosenblum L. Oceanographic Data Profile Analysis Using Interactive Computer Graphics Techniques. OCEANS 1984. 1984:100–4. [Google Scholar]
- 76.van de Weygaert R, Vegter G, Edelsbrunner H, Jones BJT, Pranav P, Park C, et al. Alpha, Betti and the Megaparsec Universe: on the Topology of the Cosmic Web. ArXiv13063640 Astro-Ph [Internet] 2013 [cited 2017 Oct 31]; Available from: http://arxiv.org/abs/1306.3640.
- 77.Precision Medicine – Prevent Health Disparities | All of Us [Internet] [cited 2017 Oct 31]. Available from: https://www.joinallofus.org/
- 78.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, et al. The Electronic Medical Records and Genomics (eMERGE) Network: Past, Present and Future. Genet Med Off J Am Coll Med Genet. 2013;15:761–71. doi: 10.1038/gim.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]