

INTRODUCTION
Activating mutations in KIT are present in up to 20% of acral melanomas (AMs) or mucosal melanomas, and 5% of melanoma associated with chronic sun-damaged skin.1-5 In GI stroma tumor (GIST), small-molecule tyrosine kinase inhibitors (TKIs) that target aberrant KIT have revolutionized treatment, demonstrating markedly durable tumor responses.6,7 Early in vitro studies demonstrated TKI KIT inhibitors to be efficacious in KIT-mutant melanoma cell lines.8-10 However, KIT-mutant melanoma tumors tend to show a de novo resistance in most cases and a limited duration of response when response is achieved,10-13 suggesting that coactivated pathway(s) may drive resistance in the primary treatment setting of KIT-mutant melanoma.
We report a case of metastatic KIT-mutant AM that had a long-term clinical response to treatment with the combination of TKIs targeting KIT and MET. We show that KIT inhibition markedly decreased cell viability in melanoma cell lines with distinct KIT mutations; however, this effect was countered in the presence of hepatocyte growth factor (HGF), the ligand for MET. Addition of a MET-inhibiting TKI reversed the HGF-driven resistance for all KIT mutants.
CASE REPORT
In 2010, a 47-year-old white woman was referred with the diagnosis of metastatic melanoma to the breast and left inguinal lymph nodes of unknown primary origin, with no evidence of brain metastasis. A physical examination revealed a raised, erythematous, and well-circumscribed cutaneous lesion on her left great toe, which the patient attributed to trauma that occurred several months earlier. Histopathologic analysis revealed the left toe lesion to be AM (Data Supplement). Staging computed tomography (CT) and positron emission tomography scans revealed left inguinal and left external iliac lymphadenopathy; two soft-tissue densities in the left breast (one biopsy proven to be melanoma); numerous subcentimeter, hypodense liver lesions; and multiple subcentimeter, subcutaneous nodules, suggestive of metastatic disease. The patient underwent a left-breast segmental mastectomy and concomitant left great toe amputation. Tumor-infiltrating lymphocytes and melanoma cells from the tumor in the breast were collected. Gene mutation analysis of the metastatic melanoma tissue did not reveal mutations in BRAF or NRAS.
After two cycles of biochemotherapy (Data Supplement), restaging CT scans revealed overall stable disease and a mixed tumor response in the lymph nodes. However, the restaging brain magnetic resonance imaging showed three new punctate lesions consistent with brain metastases. The patient received whole-brain radiation therapy (30 Gy in 10 fractions), then stereotactic radiosurgery, followed by combination treatment with temozolomide, cisplatin, and vinblastine.
Subsequent restaging showed new mediastinal lymphadenopathy and lung nodules. The patient received two cycles of a carboplatin-paclitaxel regimen before evidence of disease progression in the left inguinal lymph nodes, liver, and brain. The patient was then treated with ipilimumab, which had just received US Food and Drug Administration approval for metastatic melanoma, 3 mg/kg for four cycles. There was evidence of stable disease response to ipilimumab, with tumor reduction of 30% by immune-related response criteria overall with disease response in the brain, liver, and lymph nodes. After 19 months of disease control with ipilimumab treatment, a brain magnetic resonance imaging demonstrated a left frontal operculum metastasis with perilesional edema (Fig 1A, upper panel) and positron emission tomography/CT scanning showed new hypermetabolic activity in the mediastinal lymph nodes, which was verified to be metastatic melanoma by interventional radiology-guided fine-needle aspiration (data not shown). Molecular profiling of the patient’s melanoma tumor revealed an A>T nucleotide transition at position 2464 in exon 17 of KIT, resulting in an amino acid change (N822Y) within the kinase domain (Fig 1B).
Fig 1.
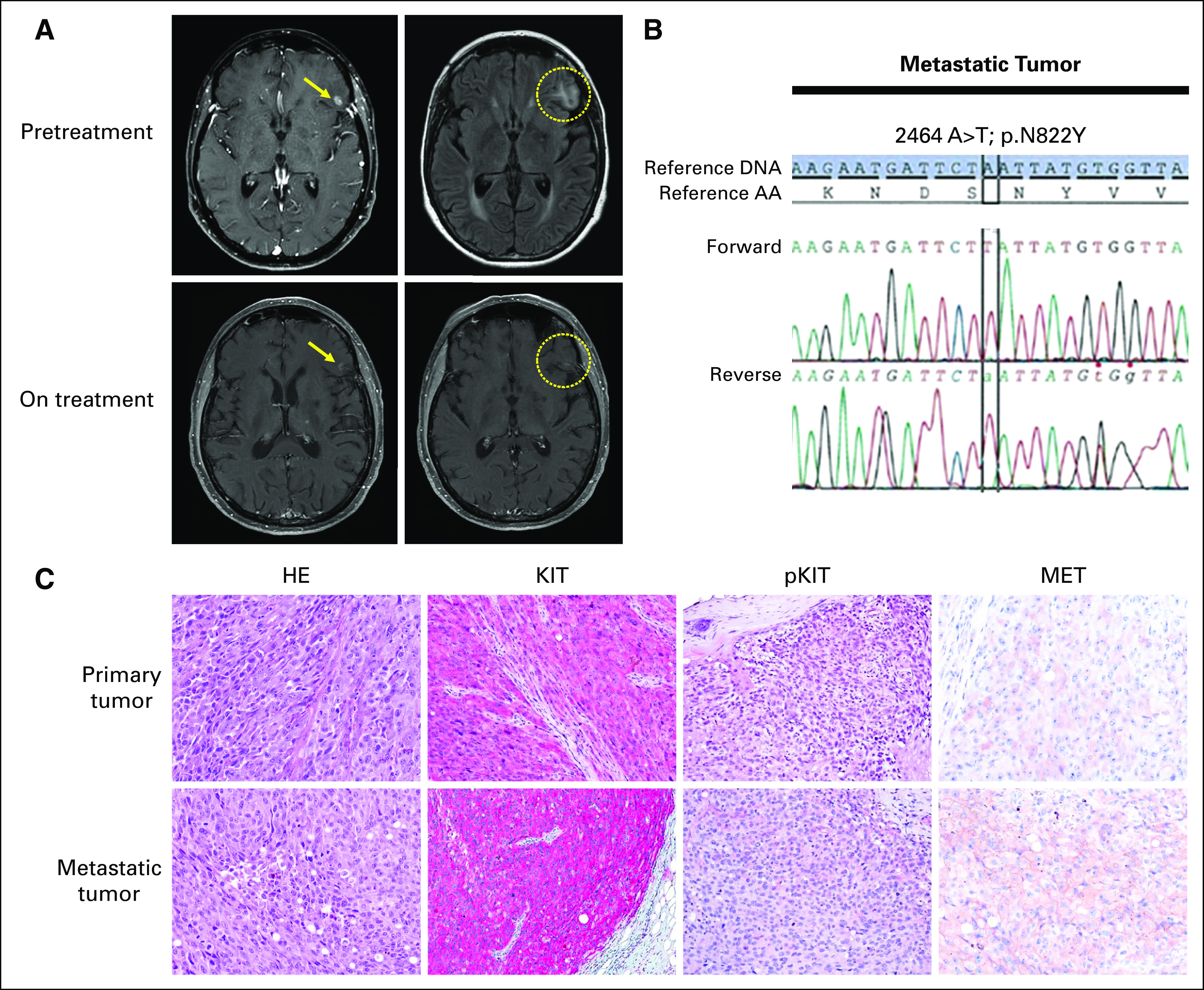
Clinical tumor and KIT mutation analysis. (A) Representative brain magnetic resonance images show left frontal operculum metastasis with perilesional edema pretreatment (top panel) and the on-treatment combination dasatinib and crizotinib therapy effect (lower panel). (B) Polymerase chain reaction–based DNA sequencing chromatogram showing a KIT nucleotide transition (2464 A>T) at codon 822 (AAT to TAT) in exon 17 from the patient’s acral melanoma metastatic tumor. (C) Hematoxylin and eosin (HE) and immunohistochemical staining for total KIT and phosphorylated KIT (p936) and total MET in the patient’s primary (upper panel) and metastatic (lower panel) acral melanoma tumors (original magnification, ×20).
Although secondary mutations in exon 17 render GIST with primary exon 11 or 13 KIT-mutant tumors resistant to KIT inhibition, isolated exon 17 KIT mutations have been shown to be more sensitive to KIT inhibition.14,15 Specifically, preclinical studies have shown dasatinib to be a potent inhibitor of KIT activation loop mutants in exon 17.9,15 The patient was enrolled in a phase I clinical trial in which dasatinib (KIT TKI, 140 mg orally daily) was combined with crizotinib (MET/ALK/ROS1 TKI, 200 mg orally daily).16 Restaging studies showed a decrease in the size of the brain metastasis and resolution of the peritumoral edema over an approximately 5-month interval (Fig 1A, lower panel). Restaging imaging performed every 2 to 3 months continued to show disease control in all metastatic sites for a total of 34 months.
Histopathologic examination of the patient’s primary and metastatic melanoma tumors showed widespread expression of phosphorylated/total KIT protein in melanoma cells (Fig 1C). Given the response to combination dasatinib and crizotinib therapy, we also assessed the expression of the MET, for which crizotinib has high binding affinity, and observed positive staining in both primary and metastatic tumors, with moderately higher intensity in the metastatic tumor.
To better understand the mechanism by which combination therapy resulted in the observed clinical response, we performed a molecular analysis of the melanoma cell line we established from the patient’s metastatic tumor (cell line 2391), as well as three other melanoma cell lines shown to have endogenous KIT mutations (namely, MelMS, Ma-Mel-144, and M230).17-19 Mutation analysis confirmed the presence of the 2464 A>T nucleotide transition in KIT with a resultant N822Y amino acid change in the activation loop within 2391, the patient’s melanoma cell line (Data Supplement). We verified the presence of each specific KIT mutation within each cell line using targeted next-generation DNA sequencing (Table 1; Data Supplement).20 No BRAF or NRAS mutations, or secondary mutations in KIT were detected in any of the cell lines.
Table 1.
KIT Mutations and Copy Number Alterations in 4q (KIT) and 11q (CCND1) for the Melanoma Cell Lines

We also determined the global chromosomal copy number alteration status within each KIT-mutant melanoma cell line (Fig 2A; Table 1). Cell lines 2391 and Ma-Mel-144 had chromosome 4q copy number elevation, which corresponds with the location of the KIT locus, consistent with initial observations that KIT mutations co-occur with KIT amplifications.1,2 Three of the four cell lines also had chromosome 11q copy number elevation, which corresponds with the location of the CCND1 locus, also well described to be amplified in AM tumors.21,22 Thus, the patient’s cell line 2391 and others in our panel had characteristic genetic features observed in AM tumors.
Fig 2.
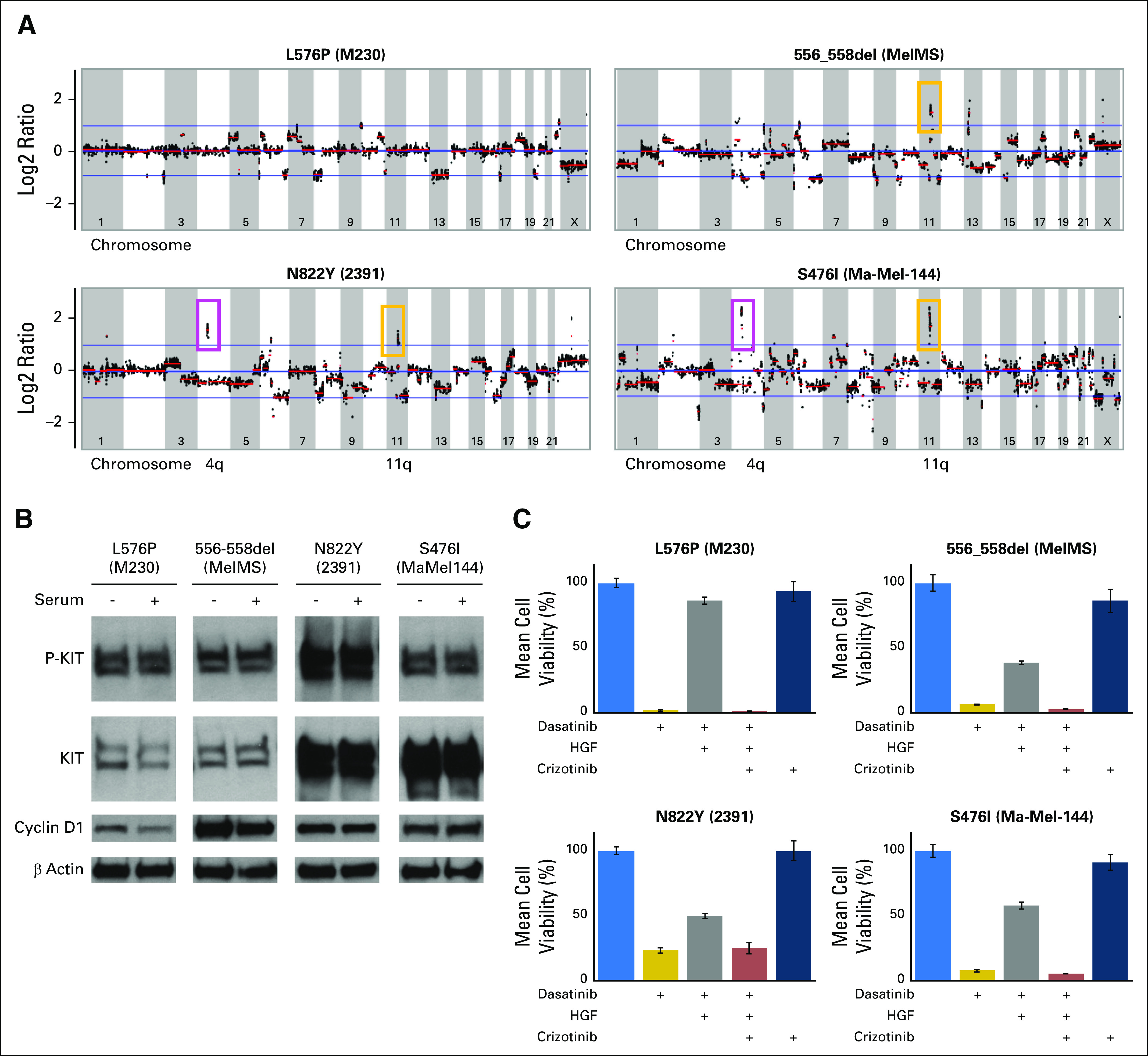
Characterization and HGF-driven resistance in KIT-mutant melanoma cell lines. (A) Copy number alteration status in the four KIT-mutant cell lines. Cell lines 2391 (KITN822Y) and Ma-Mel-144 (KITS476I) show copy number increase at chromosome 4q (location of KIT allele). Cell lines 2391 (KITN822Y), MelMS (KIT556-558del), and Ma-Mel-144 (KITS476I) show amplification at 11q (location of CCND1 allele). (B) Western blot analysis for phospho-KIT, total KIT, and cyclin D1 in the KIT-mutant melanoma cell lines. Highest expression of phospho-KIT protein is observed in the 2391 (KITN822Y) cell line. The level of total KIT protein corresponds with 4q amplification status. (C) KIT-mutant cell lines exhibit resistance to KIT inhibition in the presence of HGF, reversed by MET inhibition. Cell viability assay of M230 (KITL576P), MelMS (KIT556-558del), 2391 (KITN822Y), and Ma-Mel-144 (KITS476I) melanoma cell lines: no treatment, dasatinib (50 nM), dasatinib plus HGF (100 ng/mL), dasatinib plus HGF plus crizotinib (5 μM), and crizotinib alone.
To determine the endogenous constitutive activity of each KIT mutation, we assessed the total and phosphorylated levels of KIT in each melanoma cell line. Western blot analysis showed phosphorylated KIT to be present in all the KIT-mutant melanoma cell lines in the presence or absence of serum (Fig 2B); this was not observed in those without a KIT mutation (Data Supplement). Each of the cell lines showed sensitivity to gene-specific KIT knockdown and to multiple TKIs that share KIT as a target but have many nonoverlapping targets (eg, dasatinib targets Src-family kinases), whereas imatinib does not (data not shown). Thus, KIT mutations in melanoma, as observed in the patient’s 2391 cell line, were specific and sufficient to drive robust activation of the protein.
We next used the four KIT-mutant cell lines to better understand the mechanism of resistance to single-agent TKI KIT inhibition. Preclinical studies have shown isolated exon 17 KIT mutations (including codon 822) in nonmelanoma cells are sensitive to TKI KIT inhibitors (eg, imatinib or dasatinib) in vitro.14,15 However, clinical reports have shown only rare responses in exon 17 KIT-mutant melanoma tumors (none with codon N822 mutations) to any of the front-line TKI KIT inhibitors.10,11,13,23-26 This discrepancy suggests that KIT-targeting TKIs may be sufficient to extinguish that activation state of the KIT protein, but their ultimate effect on the tumor may be insufficient given coactivated pathways.27,28
Using dasatinib, the same TKI KIT inhibitor used to treat our patient, we observed that all four melanoma cell lines with distinct KIT mutations displayed growth inhibition at low nanomolar concentrations (1 to 5 nM; Fig 2C). Given the high levels of MET receptor expression in our patient’s tumor (Fig 1C) and the marked and durable clinical response observed when the MET-targeting TKI crizotinib was coadministered, we posited that the concurrent inhibition of MET may enhance the effect of single-agent dasatinib in the tumor setting. Thus, we tested if the presence of HGF, the only known ligand for MET, in the extracellular environment can modulate the reduction in cell viability observed with dasatinib treatment (Fig 2C). The effect of dasatinib was significantly reduced in all four KIT-mutant cell lines in the presence of HGF. Importantly, the addition of crizotinib reversed the HGF-mediated resistance to dasatinib in each cell line, whereas no effect was observed with crizotinib alone.
DISCUSSION
In this report, we describe a patient with metastatic AM who achieved a marked tumor response and disease control after treatment with the combination of dasatinib (a KIT inhibitor) and crizotinib (a MET inhibitor). Analysis of the patient’s tumor and cell line (and three other KIT-mutant cell lines) suggests that the HGF-MET axis may be a mechanism of de novo resistance in KIT-mutant melanomas.
The emergence of secondary mutations in KIT is a common mechanism of clinical resistance to KIT inhibition in KIT-mutant GIST.29 However, nearly all resistance observed in KIT-mutant melanoma occurs in the initial treatment setting, before the clonal evolution of secondary mutations can emerge,10-13,23-26 suggesting that the presence of coactivating pathways may play a role in this primary resistance.
The observed efficacy of the combination dasatinib and crizotinib could, in part, be due to the inhibition of multiple kinases; however, the melanoma cell line studies indicate the primacy of the KIT and MET receptors to explain the observed results. Furthermore, crizotinib alone was only effective in the presence of extracellular HGF, the only known MET ligand, indicating crizotinib’s modulation of the well-described HGF-MET axis, for which it was initially designed.30 These data are consistent with those from preclinical studies showing that MET inhibition increases the effect of imatinib in KIT-mutant GIST models.31
It is intriguing to speculate that the response to combination dasatinib and crizotinib may have been immunologically enhanced, given the patient’s prior tumor response to ipilimumab therapy. We have shown that the ability of dasatinib to markedly decrease tumor volumes and prolong survival in a syngeneic KIT-mutant mastocytosis mouse model is mediated by a selective decrease in regulatory CD4+ T cells and an enhanced antigen-specific CD8+ T-cell response.9
Despite the clear capacity of small-molecule TKIs to target and deactivate mutant KIT in melanoma cells in vitro,8-10 only infrequent tumor responses of typically minimal durability have been observed in clinical trials treating patients with KIT-mutant melanoma.10-13,23-26 The studies in this report indicate that the combined inhibition of KIT and MET may be a next-step effective clinical strategy in KIT-mutant melanoma.
ACKNOWLEDGMENT
We thank the Center for Environmental and Molecular Carcinogenesis Anderson Cancer Center, Smithville, TX, for antibody optimization.
Footnotes
Supported by the University of Texas System Rising Star Award, National Cancer Institute (NCI) Specialized Programs of Research Excellence (SPORE) in Melanoma (P50 CA093459 to D.S.H.); Developmental Research Project Award, National Institutes of Health/NCI Cancer Center Support Grant No. P30 CA016672, Faculty Award, the University of Texas MD Anderson Cancer Center Melanoma Moon Shot Program (all to S.E.W.); Dr Miriam and Sheldon G. Adelson Medical Research Foundation, Aim at Melanoma Foundation and the Miriam and Jim Mulva Foundation (all to E.A.G.).
This study was presented in part at 2015 ASCO Annual Meeting, Chicago, IL, May 29 to June 2, 2015.
AUTHOR CONTRIBUTIONS
Conception and design: Junna Oba, David S. Hong, Scott E. Woodman
Financial support: Elizabeth A. Grimm, David S. Hong, Scott E. Woodman
Administrative support: Junna Oba
Provision of study material or patients: Xiaoxing Yu, Chantale Bernatchez, Alexander J. Lazar, David S. Hong
Collection and assembly of data: Junna Oba, Sun-Hee Kim, Wei-Lien Wang, Mariana Macedo, Fernando Carapeto, Meredith A McKean, John Van Arnam, Agda K. Eterovic, Shiraj Sen, Charuta R. Kale, Xiaoxing Yu, Cara L. Haymaker, Mark Routbort, Chantale Bernatchez, Alexander J. Lazar, David S. Hong, Scott E. Woodman
Data analysis and interpretation: Junna Oba, Sun-Hee Kim, Meredith A McKean, Agda K. Eterovic, Shiraj Sen, Lauren E. Haydu, Chantale Bernatchez, Alexander J. Lazar, Elizabeth A. Grimm, David S. Hong, Scott E. Woodman
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Junna Oba
No relationship to disclose
Sun-Hee Kim
No relationship to disclose
Wei-Lien Wang
No relationship to disclose
Mariana P. Macedo
No relationship to disclose
Fernando Carapeto
No relationship to disclose
Meredith A. McKean
No relationship to disclose
John Van Arnam
No relationship to disclose
Agda K. Eterovic
No relationship to disclose
Shiraj Sen
No relationship to disclose
Charuta R. Kale
No relationship to disclose
Xiaoxing Yu
No relationship to disclose
Cara L. Haymaker
Research Funding: Nektar, Idera
Mark Routbort
No relationship to disclose
Lauren E. Haydu
No relationship to disclose
Chantale Bernatchez
Stock and Other Ownership Interests: Lexicon (I)
Research Funding: Nektar, Idera, Iovance Biotherapeutics, MedImmune, Pfizer, EMD Serono
Patents, Royalties, Other Intellectual Property: Patent pending on BTLA as a marker for better CD8 T cells for adoptive immunotherapy
Alexander J. Lazar
Employment: GE Healthcare (I)
Leadership: Beta Cat Pharmaceuticals, Archer Biosciences
Stock and Other Ownership Interests: Archer Biosciences, Beta Cat Pharmaceuticals
Honoraria: Novartis, Bristol-Myers Squibb, Janssen Oncology, Roche
Consulting or Advisory Role: Novartis, Illumina, GE Healthcare
Research Funding: MedImmune, AstraZeneca, Roche, Novartis
Patents, Royalties, Other Intellectual Property: Elsevier
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis
Elizabeth A. Grimm
No relationship to disclose
David S. Hong
Stock and Other Ownership Interests: MolecularMatch, Oncorena
Honoraria: Adaptimmune, Baxter, Merrimack, Bayer
Consulting or Advisory Role: Baxter, Bayer, Guidepoint Global, Janssen
Research Funding: Novartis, Genentech, Eisai, AstraZeneca, Pfizer, miRNA Therapeutics, Amgen, Daiichi Sankyo, Merck, Mirati Therapeutics, Eli Lilly, Adaptimmune, Abbvie, Bayer, Bristol-Myers Squibb, Genmab, Ignyta, Infinity Pharmaceuticals, Kite Pharma, Kyowa Hakko Kirin, Loxo, MedImmune, Molecular Templates, Takeda
Travel, Accommodations, Expenses: Loxo, miRNA Therapeutics
Other Relationship: Oncorena
Scott E. Woodman
No relationship to disclose
REFERENCES
- 1.Curtin JA, Busam K, Pinkel D, et al. : Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 24:4340-4346, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Beadling C, Jacobson-Dunlop E, Hodi FS, et al. : KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 14:6821-6828, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Torres-Cabala CA, Wang WL, Trent J, et al. : Correlation between KIT expression and KIT mutation in melanoma: A study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol 22:1446-1456, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrido MC, Bastian BC: KIT as a therapeutic target in melanoma. J Invest Dermatol 130:20-27, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodman SE, Davies MA: Targeting KIT in melanoma: A paradigm of molecular medicine and targeted therapeutics. Biochem Pharmacol 80:568-574, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demetri GD, von Mehren M, Blanke CD, et al. : Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347:472-480, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Demetri GD, van Oosterom AT, Garrett CR, et al. : Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 368:1329-1338, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Woodman SE, Trent JC, Stemke-Hale K, et al. : Activity of dasatinib against L576P KIT mutant melanoma: Molecular, cellular, and clinical correlates. Mol Cancer Ther 8:2079-2085, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Liu C, Peng W, et al. : Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood 120:4533-4543, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodi FS, Corless CL, Giobbie-Hurder A, et al. : Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 31:3182-3190, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carvajal RD, Antonescu CR, Wolchok JD, et al. : KIT as a therapeutic target in metastatic melanoma. JAMA 305:2327-2334, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minor DR, Kashani-Sabet M, Garrido M, et al. : Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res 18:1457-1463, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Guo J, Si L, Kong Y, et al. : Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29:2904-2909, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Heinrich MC, Corless CL, Demetri GD, et al. : Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21:4342-4349, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Schittenhelm MM, Shiraga S, Schroeder A, et al. : Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res 66:473-481, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Kato S, Jardim DL, Johnson FM, et al. : Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Invest New Drugs 10.1007/s10637-017-0513-5 [epub ahead of print on October 19, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang R, Wallace AR, Schadendorf D, et al. : The phosphatidyl inositol 3-kinase pathway is central to the pathogenesis of Kit-activated melanoma. Pigment Cell Melanoma Res 24:714-723, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Todd JR, Scurr LL, Becker TM, et al. : The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma. Oncogene 33:236-245, 2014 [DOI] [PubMed] [Google Scholar]
- 19.von Euw E, Atefi M, Attar N, et al. : Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer 11:22, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K, Meric-Bernstam F, Zhao H, et al. : Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem 61:544-553, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtin JA, Fridlyand J, Kageshita T, et al. : Distinct sets of genetic alterations in melanoma. N Engl J Med 353:2135-2147, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Sauter ER, Yeo UC, von Stemm A, et al. : Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res 62:3200-3206, 2002 [PubMed] [Google Scholar]
- 23.Carvajal RD, Lawrence DP, Weber JS, et al. : Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 21:2289-2296, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SJ, Kim TM, Kim YJ, et al. : Phase II trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: A multicenter trial of Korean Cancer Study Group (UN10-06). Oncologist 20:1312-1319, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo J, Carvajal RD, Dummer R, et al. : Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann Oncol 28:1380-1387, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalinsky K, Lee S, Rubin KM, et al. : A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 123:2688-2697, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Straussman R, Morikawa T, Shee K, et al. : Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487:500-504, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hochart A, Leblond P, Le Bourhis X, et al. : MET receptor inhibition: Hope against resistance to targeted therapies? [in French]. Bull Cancer 104:157-166, 2017 [DOI] [PubMed] [Google Scholar]
- 29.Antonescu CR, Besmer P, Guo T, et al. : Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 11:4182-4190, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Rodig SJ, Shapiro GI: Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr Opin Investig Drugs 11:1477-1490, 2010 [PubMed] [Google Scholar]
- 31.Cohen NA, Zeng S, Seifert AM, et al. : Pharmacological inhibition of KIT activates MET signaling in gastrointestinal stromal tumors. Cancer Res 75:2061-2070, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]