

ABSTRACT
Objective: Using electronic health records (EHRs) and biomolecular data, we sought to discover drug pairs with synergistic repurposing potential. EHRs provide real-world treatment and outcome patterns, while complementary biomolecular data, including disease-specific gene expression and drug-protein interactions, provide mechanistic understanding.
Method: We applied Group Lasso INTERaction NETwork (glinternet), an overlap group lasso penalty on a logistic regression model, with pairwise interactions to identify variables and interacting drug pairs associated with reduced 5-year mortality using EHRs of 9945 breast cancer patients. We identified differentially expressed genes from 14 case-control human breast cancer gene expression datasets and integrated them with drug-protein networks. Drugs in the network were scored according to their association with breast cancer individually or in pairs. Lastly, we determined whether synergistic drug pairs found in the EHRs were enriched among synergistic drug pairs from gene-expression data using a method similar to gene set enrichment analysis.
Results: From EHRs, we discovered 3 drug-class pairs associated with lower mortality: anti-inflammatories and hormone antagonists, anti-inflammatories and lipid modifiers, and lipid modifiers and obstructive airway drugs. The first 2 pairs were also enriched among pairs discovered using gene expression data and are supported by molecular interactions in drug-protein networks and preclinical and epidemiologic evidence.
Conclusions: This is a proof-of-concept study demonstrating that a combination of complementary data sources, such as EHRs and gene expression, can corroborate discoveries and provide mechanistic insight into drug synergism for repurposing.
Keywords: drug repurposing, drug interactions, drug discovery, breast cancer, electronic health records, combination therapies
INTRODUCTION
Electronic health records (EHRs) reflect real-world treatment patterns including polypharmacy, offering a unique opportunity to study drug-associated outcomes for drug safety and repurposing efforts.1–3 Molecular data, such as gene expression, and drug-protein interactions offer possible mechanistic insight into drug-disease relationships.4 These 2 types of data strongly complement each other, for example, in assessing the repurposing potential of existing drug combinations. Prior studies have mainly focused on discovering adverse effects of single or combined drugs (ie, drug-drug interactions5,6) or repurposing single drugs,2,7–9 such as metformin for breast cancer. Although there have been mixed results in replicating metformin’s apparent anticancer benefit,1,10–15 preliminary results from ongoing clinical trials16 appear promising.
Beyond repurposing individual drugs, combinations of drugs may yield adjuvant therapeutic effects or allow lower doses to achieve the same therapeutic effects while minimizing the undesirable side effects triggered at higher doses.17 Drugs can interact with each other such that the bioavailability of one drug is increased or prolonged (pharmacokinetic interaction) or the target receptor or pathway is modulated to elicit a stronger therapeutic response (pharmacodynamic interaction).18 Additionally, finding beneficial combinations of approved or investigational drugs can save considerable cost and time, because some safety assessments have already been performed.7,19 Such multidrug synergism is currently discovered experimentally through large-scale target screening20 and theoretically through reasoning based on known pharmacokinetic or pharmacodynamic interactions.21
This study demonstrates the novel use of both EHRs and molecular data to discover and validate pairs of drugs whose combined therapeutic effect on mortality among breast cancer patients appears to be greater than that of the individual drugs alone. Our approach for eliciting beneficial pairs of drugs is a first step toward discovering more complex multidrug combinations that can optimize the use of existing drugs.
METHODS
Analysis of electronic health records
EHR data sources
We used Oncoshare,22 a breast cancer database linking long-term survival outcomes from the California Cancer Registry with EHRs detailing patient, tumor, and treatment information from a tertiary cancer care center (Stanford Hospital) and a neighboring community health system (Palo Alto Medical Foundation, PAMF). Oncoshare followed 14 885 female patients (at least 18 years old) with a breast cancer diagnosis in the registry and sought treatment at Stanford Hospital or PAMF between January 2000 and April 2013. To determine 5-year mortality, we included only patients who were followed for at least 5 years starting from the index date of breast cancer diagnosis or who died within the follow-up period. By excluding patients who were followed <5 years (ie, diagnosed after April 2008), we minimized the loss to follow-up; this process was already facilitated through statewide mortality tracking by the California Cancer Registry. We extracted 1531 demographic, tumor (eg, stage, hormone subtype), and treatment (eg, prescriptions, radiotherapy, diagnostic imaging) variables. Data and methodological details on Oncoshare can be found in Kurian et. al.22 Individual drugs were analyzed as generic ingredients according to RxNORM23 as well as aggregated into 95 drug classes according to the Anatomical Therapeutic Chemical (ATC) system24 (Supplementary Table S1). Demographic and tumor variables, if missing and comprising at least 10% of the cohort, were coded as “unknown” and analyzed as a separate category, or otherwise (if <10% were missing) replaced by mode imputation (for categorical variables) or mean imputation (for continuous variables).
To examine concomitant drug exposures, we set up a data matrix in which each row is an exposure period for every unique drug combination (Figure 1A). This matrix contains 171 940 unique exposure periods derived from 9945 eligible patients. A cumulative exposure variable measures the duration patients were exposed to that drug combination during their follow-up time. Drugs and drug classes used for fewer than 14 days (cumulative) or present in less than 0.5% of the exposure periods were removed, leaving 294 drugs (Supplementary Table S1) for analysis of 43 071 possible pairwise combinations. Variables entered into the logistic regression included all demographic, tumor, and treatment variables. We examined for association at both the individual drug level and the ATC drug class level.
Figure 1.

Method overview of (A) scoring EHR-based synergistic drug pairs, (B) scoring gene expression–based synergistic drug pairs, and (C) gene set enrichment analysis–like analysis of enrichment of EHR-based drug class pairs among gene expression–based drug pairs.
Synergism score from EHR
To identify potentially interesting associations between 5-year mortality and pairwise combinations between drugs and drug classes, we used lasso25 regularization on a logistic regression model with pairwise interactions (Equation 1). Drug interactions were modeled as statistical interactions. Here, we used Group-Lasso INTERaction NETwork26 (glinternet), an overlap group lasso designed to select a pairwise interaction effect βi,j along with its constituting main effects βi and βj.
| Equation 1 |
A main effect βi refers to the contribution of an independent variable xi toward the response (log odds of 5-year mortality where p is the probability of 5-year mortality) while ignoring all other independent variables. An interaction effect βi,j arises when 2 independent variables, xi and xj, influence each other’s contribution toward the response. For example, although drug i and drug j are individually associated with an outcome by βi and βi, respectively, when used together, they may modify each other’s contribution toward the response such that the combined response (βi + βi + βi,j) is not simply the sum of their individual parts (βi + βi).
An interaction effect is termed synergistically beneficial when the combined effect is more negatively correlated with mortality than the most negative association of individual drugs. An interaction effect is synergistically adverse when the combined effect is more positively associated with mortality than the maximum positive association of individual drugs. An interaction may also be antagonistic when the combined effect is closer to null than either drug’s effect. Note that these terms describe the net association of the drug combination relative to that of individual drugs instead of the sign of the interaction coefficient.
Modeling implementation
Interactions involving categorical and continuous variables were handled differently in the glinternet R package (version 3.1.0).26 For each categorical variable (eg, tumor stage), all possible levels (0 to IV, unknown) and their pairwise interactions with another variable (eg, received zoledronate or not) were considered in a group lasso.26 Modeling parameters were set to select up to 300 interaction terms for computational tractability.
We set aside a 10% hold-out set for model validation and a 10% tuning set for tuning the lasso penalty factor, λ. After obtaining the optimal λ from the 10% tuning set by 3-fold cross-validation, we trained the regression model on the full non–hold-out set. Finally, trained models were then validated on the 10% hold-out set. All reported performance measures (eg, sensitivity, specificity, area under curve) were from validating the models in the hold-out set.
We generated 95% confidence intervals (CIs) for the beta coefficients of the regression model by bootstrapping 500 times, fitting a regression model to each bootstrapped sample.27 Bootstrap resampling was performed at the patient level instead of the exposure period level to account for within-patient correlated periods. In other words, patients were randomly sampled with replacement such that all their exposure periods were also sampled together. Each bootstrap sample also maintained the case-control ratio (at the patient level) and had approximately the same number of periods as the original training sample. This generated 500 different values for each beta coefficient, where the 2.5th and 97.5th percentiles were taken as the limits of the 95% CI.
Analysis of gene expression data
Molecular data sources
From ArrayExpress28 and Gene Expression Omnibus,29 14 gene expression datasets from breast tissue of patients matched to healthy controls, or of tumor tissue matched to normal tissue within the same patient, were appropriately formatted for use in the analysis (Supplementary Table S2). Raw data were downloaded and normalized using Robust Multi-array Average (RMA) (R package Affy)30 after low-quality samples were removed by ArrayQualityMetrics.31 When raw data were unavailable, processed data were used instead. For microarray data, GeoDE32 was used to identify significant differentially expressed genes. For RNA-seq data, raw reads were downloaded and quality trimmed (trimGalore33), and transcripts quantified (kallisto34). Default settings were used for all packages.
Breast cancer association score of drug pairs from gene expression data
Differentially expressed genes were mapped to proteins using UniProt identifiers. Differentially expressed proteins in breast cancer, drugs linked by drug-protein interactions (DPIs; DrugBank.ca v4.035), and proteins linked by protein-protein interactions (PPIs; Dr PIAS36) were integrated in a network (Figure 1B). Inclusion of PPI data in this network captures potentially relevant drug-protein relationships in which a drug’s direct interacting protein, or target, may not itself be differentially expressed, but may have altered activity in breast cancer (eg, drug interacting with a transcriptional regulator). Additionally, including PPI can improve the reproducibility of molecular models of cancer.37,38 Drugs were scored according to: (1) the number of proteins differentially expressed in breast cancer with which that drug’s targets interact, (2) the confidence and directionality of those interactions, and (3) the consistency of differential protein expression across individual breast cancer datasets. Higher scores indicate increased molecular association with breast cancer and potential therapeutic efficacy. After scoring drugs individually, scores were assigned to over 10 million possible drug pairs. Synergistically beneficial pairs were defined as those in which the union of the 2 drugs’ protein interactions resulted in a higher association score compared to the maximum of either drug’s individual score. Scores for all nonsynergistic pairs were set to 0.
Enrichment of EHR-derived drug pairs among gene expression–derived drug pairs
Using a method similar to gene set enrichment analysis (GSEA),39 we determined the enrichment of drug pairs coming from classes identified as synergistically interacting from the EHR among the highest scoring drug pairs identified using gene expression.
First, all synergistic drug pairs identified using gene expression were ranked by their synergism score (Figure 1C, shaded area). Then, starting with the highest ranked gene expression–derived drug pair, a cumulative sum (Figure 1C, Enrichment score) was either increased (if the pair consisted of drugs present in EHR-derived synergistic class pairs) or decreased (if both drugs were not present in the EHR-derived synergistic drug classes). The value added to the cumulative sum was proportional to the drug pair’s breast cancer association score, while the value subtracted was dependent on the number of total drug pairs examined, such that the cumulative sum was normalized between −1 and 1. For drug pairs with tied synergism scores, the value computed for all tied pairs was added to or subtracted from the cumulative sum at the first drug pair in the tie. Subsequent pairs in the tie did not affect the cumulative sum.
A raw enrichment score was derived based on the maximum deviation of the cumulative sum from 0. To determine statistical significance, we obtained a median baseline from 10 000 bootstrap samples of random drug pairs. A normalized enrichment score (NES) ratio (ie, raw enrichment score divided by median baseline) greater than 1 with low P value indicates significant enrichment.
Results and Discussion
Study cohort
To discover synergistic drug combinations from EHRs, we used a final study cohort (Table 1 and Figure 1A) consisting of 9945 patients who either died within 5 years starting from the index date of breast cancer diagnosis (1212 cases) or were followed for at least 5 years (8733 controls).
Table 1.
Patients who died within (cases) or survived (controls) 5 years of breast cancer diagnosis
| Patient Characteristic |
Cases/Dead
(n = 1212) |
Controls/Alive
(n = 8733) |
Total
(n = 9945) |
|||
|---|---|---|---|---|---|---|
| N or mean | % or SD | N or mean | % or SD | N or mean | % or SD | |
| Agea | ||||||
| <40 | 121 | 10% | 787 | 9% | 908 | 9% |
| 40–49 | 221 | 18% | 2403 | 28% | 2,624 | 26% |
| 50–59 | 251 | 21% | 2490 | 29% | 2,741 | 28% |
| 60–69 | 219 | 18% | 1794 | 21% | 2,013 | 20% |
| ≥70 | 400 | 33% | 1259 | 14% | 1,659 | 17% |
| Year of diagnosis | ||||||
| 2000–2003 | 334 | 28% | 2988 | 34% | 3,322 | 33% |
| 2004–2006 | 366 | 30% | 3222 | 37% | 3,588 | 36% |
| 2007–2009 | 402 | 33% | 2523 | 29% | 2,925 | 29% |
| 2010–2011 | 110 | 9% | 0 | 0% | 110 | 1% |
| Race | ||||||
| White/unknown | 997 | 82% | 7109 | 81% | 8,106 | 82% |
| Black | 62 | 5% | 192 | 2% | 254 | 3% |
| Asian/Pacific islander | 152 | 13% | 1423 | 16% | 1,575 | 16% |
| Native American | <10 | 0.1% | <10 | 0.1% | <10 | 0.1% |
| Marrieda | 661 | 55% | 5813 | 67% | 6,474 | 65% |
| Socioeconomic statusa | ||||||
| Lowest 20% | 74 | 6% | 266 | 3% | 340 | 3% |
| 21st–40th percentile | 142 | 12% | 607 | 7% | 749 | 8% |
| 41st–60th percentile | 174 | 14% | 975 | 11% | 1,149 | 12% |
| 61st–80th percentile | 245 | 20% | 1739 | 20% | 1,984 | 20% |
| Top 20% | 577 | 48% | 5146 | 59% | 5,723 | 58% |
| Hormone receptor subtype | ||||||
| ER+ only | 98 | 8% | 612 | 7% | 710 | 7% |
| ER+/PR+ and HER2+ | 115 | 9% | 819 | 9% | 934 | 9% |
| HER2+ only | 101 | 8% | 362 | 4% | 463 | 5% |
| PR+ only | 420 | 35% | 4406 | 50% | 4,826 | 49% |
| TNBC | 264 | 22% | 589 | 7% | 853 | 9% |
| Unknown | 214 | 18% | 1945 | 22% | 2,159 | 22% |
| Stagea | ||||||
| Stage 0 | 49 | 4% | 1736 | 20% | 1,785 | 18% |
| Stage I | 219 | 18% | 3260 | 37% | 3,479 | 35% |
| Stage II | 351 | 29% | 2576 | 29% | 2,927 | 29% |
| Stage III | 262 | 22% | 548 | 6% | 810 | 8% |
| Stage IV | 252 | 21% | 89 | 1% | 341 | 3% |
| Unknown | 79 | 7% | 524 | 6% | 603 | 6% |
| Gradea | ||||||
| Grade I | 101 | 8% | 1714 | 20% | 1815 | 18% |
| Grade II | 321 | 26% | 3451 | 40% | 3772 | 38% |
| Grade III | 527 | 43% | 2031 | 23% | 2558 | 26% |
| Grade IV | 43 | 4% | 501 | 6% | 544 | 5% |
| Unknown | 220 | 18% | 1036 | 12% | 1256 | 13% |
| Ductal tumora | 1,033 | 85% | 7459 | 85% | 8492 | 85% |
| Behavior of tumora | ||||||
| In situ | 62 | 5% | 2058 | 24% | 2120 | 21% |
| Malignant | 1150 | 95% | 6675 | 76% | 7825 | 79% |
| Bilaterala | 1164 | 96% | 8586 | 98% | 9750 | 98% |
| Lymph vascular invasiona | 35 | 3% | <10 | 0.1% | 41 | 0.4% |
| Comorbiditiesa | ||||||
| Myocardial infarction | <10 | 0.7% | 17 | 0.2% | 26 | 0.3% |
| Congestive heart failure | 15 | 1.2% | 11 | 0.1% | 26 | 0.3% |
| Peripheral vascular disease | 26 | 2% | 28 | 0.3% | 54 | 0.5% |
| Cerebrovascular disease | 34 | 3% | 66 | 0.8% | 100 | 1% |
| Dementia | <10 | 0.1% | <10 | 0.01% | <10 | 0.02% |
| Chronic obstructive pulmonary disease | 74 | 6% | 215 | 2% | 289 | 3% |
| Rheumatic disorders | <10 | 0.6% | 15 | 0.2% | 22 | 0.2% |
| Peptic ulcer disease | <10 | 0.0% | <10 | 0.01% | <10 | 0.01% |
| Liver, mild | <10 | 0.7% | <10 | 0.08% | 16 | 0.2% |
| Liver, severe | <10 | 0.5% | <10 | 0.02% | <10 | 0.08% |
| Diabetes (uncomplicated) | 25 | 2% | 44 | 0.5% | 69 | 0.7% |
| Diabetes (complicated) | <10 | 0.7% | <10 | 0.09% | 17 | 0.2% |
| Plegia | <10 | 0.0% | <10 | 0.03% | <10 | 0.03% |
| Renal disease | 17 | 1.4% | <10 | 0.1% | 26 | 0.3% |
| Malignancy | 286 | 24% | 1584 | 18% | 1870 | 19% |
| Metastasis | 61 | 5% | 57 | 1% | 118 | 1% |
| HIV | <10 | 0.4% | 10 | 0.1% | 15 | 0.2% |
| Charlson Comorbidity Scorea | 2.4 | 2.6 | 1.1 | 1.4 | 1.2 | 1.7 |
aAt: time of diagnosis; ER: estrogen receptor; PR: progesterone receptor; HER2: human epidermal growth factor receptor 2; TNBC: triple-negative breast cancer.
Small values are stated as “<10” for privacy purposes, in accordance with California Cancer Registry guidelines.
Main factors associated with survival from EHR
Comparing cases against controls, our logistic models, using 5-year mortality as the binary response, achieved satisfactory classification performance (90% area under the curve, 40% sensitivity, 99% specificity) on a 10% hold-out validation set.
Consistent with well-established breast cancer prognostic factors,40 the main factors associated with lower mortality identified in our model (Figure 2 and Supplementary Table S3) are lower stage and living in a neighborhood or census block in the top 20% of socioeconomic status in California.41 In contrast, factors such as advanced stage, older age at diagnosis, and the triple-negative breast cancer subtype were associated with higher mortality.
Figure 2.

Odds ratios of factors (excluding pairwise interactions) most associated with 5-year mortality (see also Supplementary Table S3).
Synergistic interactions from EHRs
Variables that consistently formed synergistic interactions associated with lower mortality (nodes with mostly blue edges, Figure 3) coincided with the main effects associated with lower mortality described above (eg, being diagnosed at a lower stage, having a higher socioeconomic status). In contrast, variables that consistently formed synergistic interactions associated with higher mortality (nodes with mostly red edges, Figure 3) coincided with the main effects associated with higher mortality (eg, older age at diagnosis, advanced stage). In addition to patient and tumor characteristics that synergistically influence mortality, we identified drug pairs that are synergistically associated with lower mortality (Table 2 and Figure 3, bold blue edges).
Figure 3.

Variables (nodes) that synergistically interact such that they are associated with lower mortality (blue edges) or higher mortality (red edges, also see Table 2). Variable nodes that tend to have synergistically beneficial interactions (blue edges) also tend to be factors associated with lower mortality (eg, Stage I), while those with synergistically risky interactions (red) tend to be risk factors on their own (eg, Stage IV). Nodes are grouped together (eg, by categorical level, ATC class) to facilitate visual comparison within a group (eg, Stages I and II have many synergistically beneficial interactions while Stages III and IV have many synergistically risky interactions). Case studies described in the Discussion section are highlighted with thicker edges.
Table 2.
Synergistic drug pairs discovered
| Overall | ER or PR without HER2 expression | HER2 expression | TNBC |
|---|---|---|---|
|
|
|
|
Subgroup analysis by molecular subtype
We analyzed synergistic variable interactions in patients stratified by molecular subtype given their varied prognoses and drug utilizations (Table 2 and Supplementary Figure S1). In the estrogen receptor or progesterone receptor positive group, our model identified synergistic pairs of antiestrogens or aromatase inhibitors with antiemetics (eg, ondansetron, granisetron), possibly due to the increased tolerance afforded by the antiemetics.42 Among human epidermal growth factor receptor 2–positive patients, who often have worse prognoses than other hormone-sensitive subtypes, several synergistic pairs included phenazopyridine, which might have been prescribed to relieve urethral discomfort from aggressive estrogen suppression. Rediscovering such coprescription patterns known to alleviate side effects suggests that our approach can uncover beneficial combinations. Note that while these combinations are associated with reduced mortality, causality cannot be determined.
Several synergistic interactions were replicated in the molecular subtypes and the overall cohort. Lipid modifiers (C10, including statins, eg, simvastatin) paired with either anti-inflammatory agents (M01, which includes nonsteroidal anti-inflammatory drugs [NSAIDs], eg, naproxen) or drugs for obstructive airways (R03, eg, fluticasone) reduced mortality both in the overall cohort and the TNBC subtype group.
Synergistic drug pairs from gene expression data
In an orthogonal approach, we identified 8966 differentially expressed proteins from breast cancer gene expression data. These proteins were than associated with 7686 drugs via a DPI database (Drugbank35) These data were then used to construct a molecular network. From this network, a synergistic breast cancer association score was calculated for all possible pairs of drugs in the DPI data; these were then ranked in descending order (see the shaded histograms scaled to the right axis in Figures 4A and B).
Figure 4.
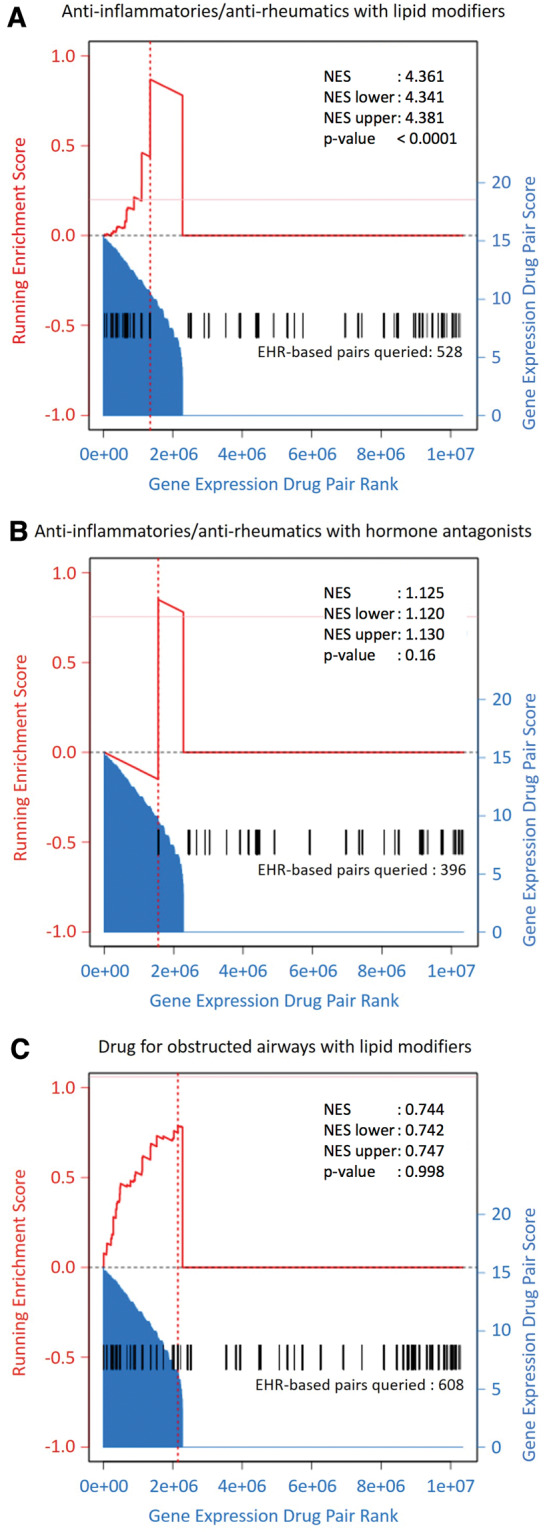
Enrichment analysis of EHR-based synergistic drug class pairs (A) anti-inflammatories/antirheumatics with lipid modifiers, (B) anti-inflammatories/antirheumatics with hormone antagonists, and (C) lipid modifiers and drugs for obstructed airways among gene expression–based synergistic drug pairs. All possible pairs of drugs from DrugBank v. 4.0 were scored on their association with genes differentially expressed in breast cancer (shaded area). A GSEA-based analysis was then performed to score the enrichment of pairs of drugs derived from the respective EHR-based classes (derived drug pairs represented by black vertical lines, running enrichment represented by red bold line) and compared to a randomly sampled null distribution (10 000 iterations) to assess significance and fold enrichment.
Next we determined whether this gene expression approach identified the same synergistic drug classes as the EHR (Table 2). To do so, we used a GSEA-based enrichment method to quantify the enrichment of EHR synergistic classes among gene expression synergistic pairs.39Figure 4A shows that the 528 drug pairs derived from one pair of synergistic classes identified using EHR (anti-inflammatories/antirheumatics paired with lipid modifiers, black bars) tended to also be high-scoring gene expression–based drug pairs (shaded histograms with scale on the right axis). Specifically, drug pairs derived from these EHR-based classes were 4.4 times more enriched among high-scoring gene expression–based pairs compared to 10 000 randomly selected sets of 528 drug pairs (P < 0.0001). In Figure 4B, anti-inflammatories/antirheumatics paired with hormone antagonists also received high gene expression–based scores, driving a slight enrichment (about 1.1-fold over random sets of 396 drug pairs, P = 0.164). Finally, although many drug pairs derived from the synergistic EHR classes of lipid modifiers with drugs for obstructive airways also scored high based on gene expression, a large number of drug pairs corresponding to these classes were not synergistic based on gene expression, resulting in no enrichment (NES < 1, Figure 4C).
Therefore, for 2 out of 3 EHR-based synergistic drug class pairs, NES > 1 suggests that these pairs also tended to be high scoring based on breast cancer gene expression association. Furthermore, the molecular networks comprising gene expression, drug-protein, and protein-protein interactions used to derive gene expression–based scores provide mechanistic hypotheses for the observed synergism of the EHR-based pairs. These pairs are also supported by preclinical and epidemiologic studies. The third drug class pair discovered using EHR (lipid modifiers and drugs for obstructive airways) was not enriched among gene expression–based pairs. As this study focuses on synergistically beneficial interactions, we will only discuss in detail the 2 synergistically beneficial drug class pairs uncovered by both EHR and gene expression data.
Synergistically beneficial pair 1: anti-inflammatory agents and lipid modifiers
Drugs belonging to the first pair of synergistic drug classes identified using EHR data (anti-inflammatory agents, especially NSAIDs, paired with lipid modifiers, especially statins) have been proposed as a general regimen for chemoprevention.43 While no benefit specifically against breast cancer has been reported for this combination, there is a growing body of epidemiological evidence supporting a synergistic anticancer benefit of NSAIDs with statins, especially against colorectal and prostate cancer.43–46 In addition, preclinical studies suggest plausible anticancer mechanisms of these drugs individually, with NSAIDs functioning as aromatase inhibitors and the inhibitory effects of statins on breast cancer cell growth and proliferation.43,47,48 It has been suggested that, in combination, NSAIDs and statins inhibit cell growth and promote apoptosis, possibly by inducing the tumor suppressor RhoB and inhibiting the Akt pathway, key targets in tumorigenesis.43,49
Using our breast cancer network model, we identified frequent protein interactions with this pair of drug classes that corroborate this epidemiological and preclinical evidence (Supplementary Table S4). Both transcription factor AP-1 (which interacts with several anti-inflammatories/antirheumatics50 and sequence CCAAT/enhancer-binding protein beta (which interacts with several lipid modifiers) influence breast cancer cell senescence and apoptosis.51,52 A drug combination that targets these proteins simultaneously may therefore elicit stronger effects on cell death or proliferation.
Drug synergism could also be achieved when one drug influences the efficacy of a second drug. For example, expression levels of insulin receptor substrate 1 (which interacts with several anti-inflammatory/antirheumatic drugs,53 can predict patient responses to chemotherapeutic or hormonal breast cancer therapies.54,55 Inhibiting AP-1 also potentiates hormonal therapies.56 These associations suggest that proteins targeted by anti-inflammatory agents may participate in synergistic combinations with hormone antagonists (see below) and possibly other anticancer therapies.
Synergistically protective pair 2: anti-inflammatory agents and anticancer hormone antagonists
While anti-inflammatory drugs are known to help patients better tolerate hormone therapy’s undesirable side effects until endocrine responsiveness is elicited, there is evidence in the literature suggesting joint anticancer action between anti-inflammatory agents and hormone antagonists. Many anti-inflammatories inhibit cyclooxygenase-2, which in turn inhibits aromatase that is otherwise required for estrogen production.47,57–59 By combining a cyclooxygenase-2 inhibitor (NSAID or coxib) with a hormone antagonist like an aromatase inhibitor, synergistic regulation of hormone production may halt or slow mammary tumorigenesis.59,60 Clinical trials have shown that the combination of celecoxib and exemestane is slightly better or equivalent to exemestane monotherapy.59,61 Benefits include longer periods of stable disease (tumor shrinkage or no new lesions)59 and reduced tumor expression of proliferation-associated genes.61 However, the increased cardiovascular risk associated with celecoxib has raised concerns about its risk-benefit ratio.
Although synergistic pairs of anti-inflammatory/antirheumatic agents and hormone antagonists were only slightly enriched among all synergistic pairs identified based on gene expression, analysis of proteins that frequently interact with drugs in these classes suggests possible molecular mechanisms to explain their observed synergy (Supplementary Table S5). For example, genetic knock-down of caveolin-1, which interacts with several anti-inflammatory drugs, renders breast tumors hypersensitive to estrogen.62,63 Simultaneous inhibition of caveolin-1 may therefore enhance the efficacy of antiestrogen therapies. Another protein linked to multiple anti-inflammatory drugs, tristetraprolin, interacts with progesterone, estrogen, and androgen receptors.64 Reducing protein levels of tristetraprolin in breast cancer cell lines augments hormonal effects on cell growth and proliferation, possibly rendering cells more sensitive to hormone antagonist therapies.65 These molecular links could support the predicted synergistic efficacy of anti-inflammatory/antirheumatic drugs and hormone antagonists in breast cancer treatment.
Study limitations
We acknowledge that the effect sizes of the synergism discussed above are small (beta coefficients of interaction terms: 0.004–0.05) despite statistical significance (P < 0.05) in EHR and enrichment in molecular data and supporting evidence from the literature. Some of the drug pairs discovered (Figure 3 and Table 2) may represent intentional concurrent usage rather than actual mechanistic synergism. Disambiguating between the 2 is challenging when using observational data, as the intent is not stated. For example, deliberate concurrent use may be in order to overcome resistance to a single therapy or to relieve side effects (eg, venlafaxine to improve tolerance to aromatase inhibitors) or to treat coincidental conditions (eg, venlafaxine for psychological distress, prevalent in 30–50% of breast cancer patients66).
Nevertheless, one key advantage of our approach of formulating pairwise interaction effects is the simultaneous discovery of multiple interaction effects. This generates multiple hypotheses for in-depth evaluation by drug screening in cell lines and animal models, as well as by subsequent observational studies and clinical trials. Our approach also discovered synergistically adverse drug pairs (ie, adverse drug-drug interactions, Figure 3), which we did not discuss in detail, given our focus here on the synergistically beneficial ones that could potentially be repurposed. One disadvantage is the risk of false discoveries, especially when correlated pairs could be falsely detected as interaction effects. To minimize false discoveries, we bootstrapped samples and reran our models 500 times to empirically generate 95% CI, in an attempt to address the variance associated with the beta estimates but not necessarily the bias inherent in penalized regressions such as glinternet.67 While there are sophisticated bootstrapping procedures designed to reduce the bias, estimating the CIs for penalized regression remains an active area of research.68
The purported breast cancer benefit of metformin could also be used as a positive control for testing our method on monotherapy drugs.1,10,47 Metformin, on its own, was not associated with lower mortality. We did, however, find a borderline benefit (Hazard ratio: 0.86 [0.52–1.00]) in a separate lasso Cox regression survival analysis without pairwise interactions (Supplementary Table S6). While a Cox regression model with the same overlap group lasso (as used in glinternet here) was an attractive alternative, the current implementation of glinternet supports only logistic regression. A survival analysis setup with time-varying exposures could also account for the temporal sequence of the drug exposures, which was not considered here. However, Cox regression models are prone to time-dependent biases (eg, immortal bias), and some studies have indeed questioned whether metformin’s benefit could have arisen from such biases.10
Patients might also have received care, including the drugs studied, outside of Stanford Hospital and PAMF. Such instances and other supporting information, undocumented in our data source, may result in unmeasured confounding, a known limitation of EHR-based studies.69 Nevertheless, we tried to obtain the most comprehensive clinical details possible in our choice of Oncoshare, which links EHRs from Stanford University and the neighboring PAMF community health service with the California Surveillance, Epidemiology, and End Results Program registry and other supporting services such as Oncotype DX.22,70
Another limitation is the use of predefined drug classes, which may be overly broad and heterogeneous (eg, drugs for obstructive airways, R03). We repeated the analysis (Supplementary Table S7), aggregating drugs to ATC drug classes of various granularities and pair 1 between anti-inflammatory agents and, in particular, aromatase inhibitors was replicated. On the other hand, overly specific subclasses containing rarely used drugs may also pose problems, as we did not always observe synergism among the more granular subclasses (Supplementary Table S7).
Our molecular analysis was limited primarily by gene expression and drug data availability. In terms of gene expression, only 14 datasets were of sufficient quality and contained appropriate case and control samples for differential gene expression analysis. This precluded us from performing separate meta-analyses of breast cancer molecular subtypes. Information on drugs is similarly limited to those with reported protein interactions, which may be additionally restricted to anticipated interactions based on a drug’s class and approved indications. For example, many specific pairs of EHR-based synergistic drugs lack reported protein interaction information in DrugBank, but have protein interaction information in DrugBank at the drug class level. Although DrugBank is one of several widely used sources of drug information,71 alternate sources could have been explored. Similarly, protein-protein interactions may not be fully documented or validated, or may vary in their biological relevance (eg, some interactions were discovered in yeast 2-hybrid assays that are less relevant to breast cancer pathology). Despite these limitations, gene expression–based ranking of synergistic drug pairs provides an alternative data source to validate pairs discovered from EHRs. The consistency of the results from multiple data sources and analysis methods should increase the robustness of our findings.
Conclusions and Implications
This is a proof-of-concept study demonstrating that searching for statistical interactions can discover drug pairs that moderate each other’s effects. Such an approach has also been used to discover epistatic interactions among genes.26,72 Much of the published literature on drug interactions has focused on adverse drug-drug interactions instead of potentially beneficial interactions for drug repurposing. Here, we report 3 synergistically therapeutic pairs of drug classes associated with lower 5-year mortality in patients with breast cancer. Of the 3 synergistically protective pairs, 2 were supported by analysis of gene expression data of breast cancer patients, biological plausibility, preclinical models, and epidemiologic evidence in the literature. The glinternet analysis of EHRs we presented is scalable to drug combinations of 2 or more. As demonstrated, coupling with orthogonal analysis of gene expression data can corroborate the EHR-based findings and reveal protein interactions that may relate to the mechanism driving drug synergism. This study further demonstrates the translational potential of existing data sources such as real-world patient EHRs and gene expression databases. The multidrug combinations uncovered can be computationally prioritized to help direct preclinical research and, if promising, undergo clinical trial validation, repurposing, and optimizing of existing drugs for maximum therapeutic benefit.
Funding
This work was supported in part by National Library of Medicine grant R01 LM011369 (to NS), National Institute of General Medical Sciences grant R01 GM101430 (to NS), National Science Foundation grant DMS-1407548 (to TH), and National Institutes of Health grant RO1-EB001988-15 (to TH). This work was also supported by the Breast Cancer Research Foundation (to AK, GS, NS); the Susan and Richard Levy Gift Fund (to AK); the Suzanne Pride Bryan Fund for Breast Cancer Research (to AK); the Regents of the University of California’s California Breast Cancer Research Program (16OB-0149 and 19IB-0124) (to AK); and the Stanford University Developmental Research Fund (to AK). The project was supported by National Institutes of Health Clinical and Translational Science award number UL1 RR025744. The collection of cancer incidence data used in this study was supported by the California Department of Health Services as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103 885; the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program under contracts HHSN261201000140C (to the Cancer Prevention Institute of California), HHSN261201000035C (to the University of Southern California), and HHSN261201000034C (to the Public Health Institute); and the Centers for Disease Control and Prevention’s National Program of Cancer Registries, under agreement 1U58 DP000807-01 (to the Public Health Institute). The ideas and opinions expressed herein are those of the authors, and endorsement by the University or State of California, the California Department of Health Services, the National Cancer Institute, or the Centers for Disease Control and Prevention or their contractors and subcontractors is not intended nor should be inferred. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Contributions
Conceived and designed the experiments: YL, NS, AK, GS. Analyzed the data: YL, AD, WC. Contributed data: TS, PK, SG, SW, CT, PY, AD. Contributed tools: MD, ML, TH, MM, AD, AR, CF. Wrote the paper: YL, NS, AK, AD, ES, MD.
Data Sharing
Availability of patient data is subject to the Institutional Data Access/Ethics Committees of Stanford University, Palo Alto Medical Foundation, and the California Cancer Registry for researchers. Supplementary materials include the following: drugs and their ATC drug classes considered for analysis (Supplementary Table S1), gene expression datasets (Supplementary Table S2), main effects and interactions effects significantly associated with 5-year mortality and their odds ratios (Supplementary Table S3), breast cancer–related proteins that most frequently interact with lipid/NSAID pairs (Supplementary Table S4), breast cancer–related proteins that most frequently interact with NSAID/hormone antagonist pairs (Supplementary Table S5), results of Cox regression survival analysis (Supplementary Table S6), and results of alternative analysis with more granular ATC drug classes (Supplementary Table S7), drug utilization profiles by molecular subtypes (Supplementary Figure S1).
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
Supplementary Material
References
- 1. Xu H, Aldrich MC, Chen Q et al. . Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J. Am. Med. Inform. Assoc. 2015;22:179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jung K, LePendu P, Chen WS et al. . Automated detection of off-label drug use. PLoS One. 2014;9:e89324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yao L, Zhang Y, Li Y et al. . Electronic health records: Implications for drug discovery. Drug Discov. Today. 2011;16:594–99. [DOI] [PubMed] [Google Scholar]
- 4. Sirota M, Dudley JT, Kim J et al. . Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 2011;3:96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iyer S V, Harpaz R, LePendu P et al. . Mining clinical text for signals of adverse drug-drug interactions. J. Am. Med. Inform. Assoc. 2014;21:353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tatonetti NP, Ye PP, Daneshjou R et al. . Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012;4:125ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hurle MR, Yang L, Xie Q et al. . Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther. 2013;93:335–41. [DOI] [PubMed] [Google Scholar]
- 8. Shameer K, Readhead B, Dudley JT. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem. 2015;15:5–20. [DOI] [PubMed] [Google Scholar]
- 9. Issa NT, Byers SW, Dakshanamurthy S. Drug repurposing: translational pharmacology, chemistry, computers and the clinic. Curr. Top. Med. Chem. 2013;13:2328–36. [DOI] [PubMed] [Google Scholar]
- 10. Lega IC, Austin PC, Gruneir A et al. . Association between metformin therapy and mortality after breast cancer: a population-based study. Diabetes Care. 2013;36:3018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noto H, Goto A, Tsujimoto T et al. . Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Col NF, Ochs L, Springmann V et al. . Metformin and breast cancer risk: a meta-analysis and critical literature review. Breast Cancer Res. Treat. 2012;135:639–46. [DOI] [PubMed] [Google Scholar]
- 13. Dowling RJO, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bo S, Benso A, Durazzo M et al. . Does use of metformin protect against cancer in Type 2 diabetes mellitus? J. Endocrinol. Invest. 2012;35:231–35. [DOI] [PubMed] [Google Scholar]
- 15. DeCensi A, Puntoni M, Goodwin P et al. . Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010;3:1451–61. [DOI] [PubMed] [Google Scholar]
- 16. National Cancer Institute. Metformin: can a diabetes drug help prevent cancer? 2013. http://www.cancer.gov/cancertopics/research-updates/2013/metformin. Accessed April 23, 2015.
- 17. Berenbaum MC. What is synergy? Pharmacol. Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 18. Schmider W, Boulenc X. Drug-Drug Interaction Studies. In: Drug Discovery and Evaluation: Methods in Clinical Pharmacology. Springer: Berlin Heidelberg; 2011: 119–31. [Google Scholar]
- 19. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3:673–83. [DOI] [PubMed] [Google Scholar]
- 20. Cokol M, Chua HN, Tasan M et al. . Systematic exploration of synergistic drug pairs. Mol. Syst. Biol. 2011;7:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guimerà R, Sales-Pardo M. A network inference method for large-scale unsupervised identification of novel drug-drug interactions. PLoS Comput. Biol. 2013;9:e1003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurian AW, Mitani A, Desai M et al. . Breast cancer treatment across health care systems: linking electronic medical records and state registry data to enable outcomes research. Cancer. 2013;120:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bennett CC. Utilizing RxNorm to support practical computing applications: Capturing medication history in live electronic health records. J. Biomed. Inform. 2012;45:634–41. [DOI] [PubMed] [Google Scholar]
- 24. World Health Organization Collaborating Centre for Drug Statistics Methodology. Guidelines for ATC Classification and DDD Assignment. Oslo: 2015. [Google Scholar]
- 25. Tibshirani R. Regression shrinkage and selection via the lasso. J.R. Stat. Soc. Ser. B. 1996;58:267–88. [Google Scholar]
- 26. Lim M, Hastie T. Learning interactions via hierarchical group-lasso regularization. J. Comput. Graph Stat. 2015;24:627–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat. Sci. 1986;1:54–75. [Google Scholar]
- 28. Brazma A, Parkinson H, Sarkans U et al. . ArrayExpress: a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolstad BM, Irizarry RA, Astrand M et al. . A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. [DOI] [PubMed] [Google Scholar]
- 31. Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics: a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clark NR, Hu KS, Feldmann AS et al. . The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics. 2014;15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trim Galore! Version 0.4.1. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed February 5, 2016.
- 34. Bray NL, Pimentel H, Melsted P et al. . Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–27. [DOI] [PubMed] [Google Scholar]
- 35. Law V, Knox C, Djoumbou Y et al. . DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugaya N, Furuya T Dr. PIAS: an integrative system for assessing the druggability of protein-protein interactions. BMC Bioinformatics. 2011;12:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taylor IW, Linding R, Warde-Farley D et al. . Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009;27:199–204. [DOI] [PubMed] [Google Scholar]
- 38. Chuang HY, Lee E, Liu YT et al. . Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subramanian A, Tamayo P, Mootha VK et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Desantis C, Ma J, Bryan L et al. . Breast cancer statistics, 2013. CA Cancer J. Clin. 2014;64:52–62. [DOI] [PubMed] [Google Scholar]
- 41. Yost K, Perkins C, Cohen R et al. . Socioeconomic status and breast cancer incidence in California for different race/ethnic groups. Cancer Causes Control. 2001;12:703–11. [DOI] [PubMed] [Google Scholar]
- 42. Yeo W, Chan. Antiemetic therapy options for chemotherapy-induced nausea and vomiting in breast cancer patients. Breast Cancer Targets Ther. 2011;2011:151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao H, Yang CS. Combination regimen with statins and NSAIDs: A promising strategy for cancer chemoprevention. Int. J. Cancer. 2008;123:983–90. [DOI] [PubMed] [Google Scholar]
- 44. Lochhead P, Chan AT. Statins and colorectal cancer. Clin. Gastroenterol. Hepatol. 2013;11:109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoffmeister M, Chang-Claude J, Brenner H. Individual and joint use of statins and low-dose aspirin and risk of colorectal cancer: a population-based case-control study. Int. J. Cancer. 2007;121:1325–30. [DOI] [PubMed] [Google Scholar]
- 46. Moon H, Hill MM, Roberts MJ et al. . Statins: protectors or pretenders in prostate cancer? Trends Endocrinol. Metab. 2014;25:188–96. [DOI] [PubMed] [Google Scholar]
- 47. Holmes MD, Chen WY. Hiding in plain view: the potential for commonly used drugs to reduce breast cancer mortality. Breast Cancer Res. 2012;14:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hiľovska L, Jendželovsky R, Fedoročko P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy (Review). Mol. Clin. Oncol. 2015;3:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. [DOI] [PubMed] [Google Scholar]
- 50. Qiao Y, He H, Jonsson P et al. . AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016;291:5068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Atwood AA, Sealy L. Regulation of C/EBPbeta1 by Ras in mammary epithelial cells and the role of C/EBPbeta1 in oncogene-induced senescence. Oncogene. 2010;29:6004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Duan L, Sterba K, Kolomeichuk S et al. . Inducible overexpression of c-Jun in MCF7 cells causes resistance to vinblastine via inhibition of drug-induced apoptosis and senescence at a step subsequent to mitotic arrest. Biochem. Pharmacol. 2007;73:481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reiss K, Del Valle L, Lassak A et al. . Nuclear IRS-1 and cancer. J. Cell Physiol. 2012;227:2992–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Porter HA, Perry A, Kingsley C et al. . IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer Lett. 2013;338:239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Migliaccio I, Wu MF, Gutierrez C et al. . Nuclear IRS-1 predicts tamoxifen response in patients with early breast cancer. Breast Cancer Res. Treat. 2010;123:651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Malorni L, Giuliano M, Migliaccio I et al. . Blockade of AP-1 potentiates endocrine therapy and overcomes resistance. Mol. Cancer Res. 2016;14:470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao Y, Zhu S, Li X et al. . Association between NSAIDs use and breast cancer risk: a systematic review and meta-analysis. Breast Cancer Res. Treat. 2009;117:141–50. [DOI] [PubMed] [Google Scholar]
- 58. Arun B, Goss P. The role of COX-2 inhibition in breast cancer treatment and prevention. Semin. Oncol. 2004;31:22–29. [DOI] [PubMed] [Google Scholar]
- 59. Falandry C, Canney PA, Freyer G et al. . Role of combination therapy with aromatase and cyclooxygenase-2 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2009;20:615–20. [DOI] [PubMed] [Google Scholar]
- 60. Liu H, Talalay P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc. Natl. Acad. Sci. U.S.A. 2013;110:19065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brandão RD, Veeck J, Van de Vijver KK et al. . A randomised controlled phase II trial of pre-operative celecoxib treatment reveals anti-tumour transcriptional response in primary breast cancer. Breast Cancer Res. 2013;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mercier I, Casimiro MC, Zhou J et al. . Genetic ablation of caveolin-1 drives estrogen-hypersensitivity and the development of DCIS-like mammary lesions. Am. J. Pathol. 2009;174:1172–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang R, He W, Li Z et al. . Caveolin-1 functions as a key regulator of 17beta-estradiol-mediated autophagy and apoptosis in BT474 breast cancer cells. Int. J. Mol. Med. 2014;34:822–27. [DOI] [PubMed] [Google Scholar]
- 64. Barrios-Garcia T, Gomez-Romero V, Tecalco-Cruz A et al. . Nuclear tristetraprolin acts as a corepressor of multiple steroid nuclear receptors in breast cancer cells. Mol. Genet. Metab. Rep 2016;7:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barrios-Garcia T, Tecalco-Cruz A, Gomez-Romero V et al. . Tristetraprolin represses estrogen receptor alpha transactivation in breast cancer cells. J. Biol. Chem. 2014;289:15554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gold M, Dunn LB, Phoenix B et al. . Co-occurrence of anxiety and depressive symptoms following breast cancer surgery and its impact on quality of life. Eur. J. Oncol. Nurs. 2016;20:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goeman JJ. L1 penalized estimation in the Cox proportional hazards model. Biometrical J. 2010;52:70–84. [DOI] [PubMed] [Google Scholar]
- 68. Sartori S. Penalized Regression: Bootstrap Confidence Intervals and Variable Selection for High Dimensional Data Sets. 2011. PhD dissertation from Universitá degli Studi di Milano. [Google Scholar]
- 69. Overhage JM, Overhage LM. Sensible use of observational clinical data. Stat. Methods Med. Res. 2013;22:7–13. [DOI] [PubMed] [Google Scholar]
- 70. Thompson CA, Kurian AW, Luft HS. Linking electronic health records to better understand breast cancer patient pathways within and between two health systems. Generating Evid. Methods to Improv Patient Outcomes. 2015;3:Article 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Williams AJ, Ekins S, Tkachenko V. Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov. Today. 2012;17:685–701. [DOI] [PubMed] [Google Scholar]
- 72. Moore JH, Gilbert JC, Tsai C-T et al. . A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J. Theor. Biol. 2006;241:252–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.