

Abstract
Extracellular matrix stiffness (ECM) is one of the many mechanical forces acting on mammalian adherent cells and an important determinant of cellular function. While the effect of ECM stiffness on many aspects of cellular behavior has been studied previously, how ECM stiffness might mediate susceptibility of host cells to infection by bacterial pathogens is hitherto unexplored. To address this open question, we manufactured hydrogels of varying physiologically relevant stiffness and seeded human microvascular endothelial cells (HMEC-1) on them. We then infected HMEC-1 with the bacterial pathogen Listeria monocytogenes (Lm) and found that adhesion of Lm to host cells increases monotonically with increasing matrix stiffness, an effect that requires the activity of focal adhesion kinase (FAK). We identified cell surface vimentin as a candidate surface receptor mediating stiffness-dependent adhesion of Lm to HMEC-1 and found that bacterial infection of these host cells is decreased when the amount of surface vimentin is reduced. Our results provide the first evidence that ECM stiffness can mediate the susceptibility of mammalian host cells to infection by a bacterial pathogen.
INTRODUCTION
The extracellular environment of cells provides both chemical and mechanical stimuli to influence cell behavior and function (Chien et al., 2005; Geiger et al., 2009). Extracellular matrix stiffness (ECM), one of the many mechanical forces acting on cells, is an important determinant of cellular behavior for most adherent mammalian cells (Wells, 2008; Gattazzo et al., 2014). Cells can sense the stiffness of their matrix, which can vary over many orders of magnitude, and accordingly alter their motility, adhesion, growth, and differentiation (Discher et al., 2005; Birukova et al., 2013). Yet the exact pathways by which cells sense mechanical signals and transduce them to generate biological signal cascades and specific cellular responses are not yet fully understood (Trepat et al., 2008).
In the context of host–pathogen interactions, effects of matrix stiffness variation may be most interesting for infectious agents that have the capacity to infect many different kinds of tissues in the human body. One such agent is the food-borne facultative bacterial pathogen Listeria monocytogenes (Lm). After initial invasion of the intestinal epithelium, Lm is able to spread through the vasculature to distant organs, and can cause serious complications such as meningitis and late-term spontaneous abortion by virtue of its unusual ability to penetrate and cross a wide variety of endothelial barriers, including the blood–brain barrier and the placenta (Vazquez-Boland et al., 2001; Jackson et al., 2010). Lm has a broad range of susceptible host animals and uses multiple pathogenic strategies to achieve infection of a wide variety of tissues within each host. It can directly adhere to and invade intestinal epithelial cells and hepatocytes using bacterial surface proteins belonging to the internalin family, such as InlA, InlB, and InlF, which interact with host cell surface receptors (Mengaud et al., 1996; Shen et al., 2000; Kirchner and Higgins, 2008). Lm can also directly adhere to and invade the vascular endothelial cells (VECs) that line the inner lumen of blood vessels, using several distinct molecular mechanisms that may vary depending on the subtype of VEC being infected (Drevets et al., 1995; Greiffenberg et al., 1997, 1998; Parida et al., 1998; Wilson and Drevets, 1998; Rengarajan et al., 2016).
Vascular endothelial cells (VECs) are known to be highly mechanosensitive. The stiffness of the ECM surrounding blood vessels can vary significantly in space (location within the vascular tree), in time (aging), and with pathophysiological conditions (e.g., arteriosclerosis, cancer; Volkman et al., 2002; Chien et al., 2005; Krishnan et al., 2011; Yeh et al., 2012; Collins et al., 2014). These variations in stiffness can dramatically affect the gene expression and barrier integrity of VECs, thereby regulating the movement of vascular components from the bloodstream into underlying tissues, including the dissemination of bacterial pathogens (Lemichez et al., 2010). Clinical studies have shown that the local compliance of the basement membrane of VECs in the brain tissue is as soft as 1 kPa, while bigger vessels such as the aorta are as stiff as 20 kPa (Wells, 2008; Wood et al., 2010; Janmey and Miller, 2011; Onken et al., 2014; Kohn et al., 2015), and their stiffness can increase up to 70 kPa due to aging (Huynh et al., 2011) and in the context of cardiovascular diseases, such as atherosclerosis or hypertension (Blacher et al., 1999; Boutouyrie et al., 2002). VECs, like other adherent cell types, probe features of their environment, including its stiffness, through integrins, transmembrane receptors that allow attachment of the cells to their matrix through direct binding to ECM ligand proteins (Senger et al., 2002; Schwartz, 2010). Integrin binding to the ECM leads to recruitment of additional proteins and to formation of focal adhesions that in turn relay information to the actin cytoskeleton and to various signaling molecules, modulating cellular adhesion, shape, contractility, gene expression, and fate in general (Pelham and Wang, 1997). Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase that has been established as a key component of the signal transduction pathways triggered by integrins. The expression and/or activity of FAK is dependent on ECM rigidity, showing decreased expression and/or activation on softer matrices for certain cell types and can influence cell adhesion and motility (Khatiwala et al., 2006; Provenzano et al., 2009; Higuita-Castro et al., 2014; Du et al., 2016). However, no studies have previously addressed whether and how VEC matrix stiffness sensing through focal adhesions might affect host cells’ interactions with bacterial pathogens.
Bacterial adhesion to the surface of cells is typically the initial event in the pathogenesis of infection and can occur through receptor-mediated interactions between the host cell and the pathogen (Pizarro-Cerdá and Cossart, 2006). Vimentin is generally an intracellular cytoskeletal protein that forms intermediate filaments in many mesoderm-derived cells (Clarke and Allan, 2002). Vimentin can also be localized on the surfaces of cells for a variety of cell types, although the precise mechanism by which vimentin is delivered there is not yet fully understood (Mor-Vaknin et al., 2003; Päll et al., 2011; Rohrbeck et al., 2014; Mitra et al., 2015; Shigyo et al., 2015). Recent studies have shown that various bacteria, as well as viruses, use surface vimentin as an attachment receptor to facilitate entry into host cells (Garg et al., 2006; Zou et al., 2006; Bhattacharya et al., 2009; Du et al., 2014; Rohrbeck et al., 2014; Mak and Brüggemann, 2016; Yang et al., 2016; Yu et al., 2016; Ghosh et al., 2018). However, little is known about what modulates the amount of vimentin exposed on the surface of cells and whether regulation of its surface presentation potentially mediates bacterial uptake.
To assess whether ECM stiffness could mediate infection susceptibility of host cells, we used Lm as our model pathogen and examined in vitro Lm infection in endothelial cells derived from the microvasculature (human microvascular endothelial cells, HMEC-1) cultured on matrices of varying stiffness. Subendothelial stiffness depends on the blood vessel’s location within the body, on the size of the vessel, on age, and on physiological or pathophysiological conditions, and so we chose our substrates’ stiffness to span a wide range of stiffnesses, from 0.6 to 70 kPa (Stroka and Aranda-Espinoza, 2011; Kohn et al., 2015). We found a twofold increase in bacterial adhesion when cells reside on stiff rather than soft matrices. We also found that Tyr397 phosphorylation of FAK was higher for VECs residing on stiff matrices, and furthermore that knockdown of FAK or treatment of VECs with FAK inhibitors lead to a decrease in bacterial infection. When we searched for candidate VEC surface receptors differentially modulated depending on FAK activity, we identified surface vimentin as a candidate and found that decreasing the amount or the accessibility of host cell–surface vimentin leads to a concomitant decrease in Lm infection. Taken together, our findings provide evidence that environmental stiffness of VECs, a previously unappreciated factor, plays an important role in mediating infection susceptibility to Lm through differential activity of FAK, which in turn affects the amount of surface vimentin.
RESULTS
Uptake of Listeria monocytogenes by HMEC-1 is more efficient when cells reside on stiff substrates
Substrates on which vascular endothelial cells (VECs) are cultured in vitro, commonly glass or tissue culture (TC) polystyrene, are approximately six orders of magnitude stiffer than the natural ECM of human VECs (Sperling and Friedman, 1969; Dussurget et al., 2004). To be able to recapitulate in vivo conditions and determine in a systematic way the effect of the VECs’ matrix stiffness on the efficiency of Lm infection, we developed an assay based on manufacturing thin polyacrylamide hydrogels of varying stiffness ranging from 0.6 to 70 kPa on multiwell glass-bottom plates (Georges et al., 2006; Mih et al., 2011; Ahmed, 2015) (Supplemental Figure S1A). To enable cell adhesion, hydrogels were surface-coated with collagen I, using a consistent density of collagen independent of hydrogel stiffness, as reported in previous studies (Yeung et al., 2005; Tse and Engler, 2010; Huang et al., 2013). Human microvascular endothelial cells (HMEC-1) seeded for 24 h on the hydrogels formed monolayers with comparable densities regardless of substrate stiffness, similar to growth of these cells on TC polystyrene (Supplemental Figure S1B).
To measure the efficiency of bacterial infection quantitatively, we infected HMEC-1 with a strain of Lm lacking expression of the ActA protein, which is necessary for intra- and intercellular spread of this bacterium. In that way, we could attribute changes in the number of infected cells directly to initial invasion events and not to spread of the bacteria from cell to cell. We also confirmed that adhesion and invasion of wild-type Lm into HMEC-1 is similar to that of ΔactA Lm, consistent with previous studies on other host cell types (Kocks et al., 1992; Brundage et al., 1993), suggesting that ActA is not involved in adhesion or invasion (Supplemental Figure S1, D–F). The ΔactA Lm strain we used also expresses a fluorescent protein under a promoter that is activated several hours after exposure of the bacteria to the host cell cytosol (actAp::mTagRFP; Zeldovich et al., 2011), allowing reliable fluorescence-based detection of only the bacteria that have successfully invaded the host cells. We determined the fraction of HMEC-1 infected with Lm in each well using flow cytometry (Figure 1, A–D). At a constant multiplicity of infection (MOI) and constant host cell density, we found a monotonic increase (strictly increasing relationship) in the number of VECs infected by Lm with increasing hydrogel stiffness that was highly reproducible among biological replicates (Figure 1E; Supplemental Figure S1C). The overall efficiency of infection on the stiffest hydrogel substrates tested, 70 kPa, was comparable to the efficiency of infection for the same cells grown on TC polystyrene, suggesting that above a certain level of stiffness the infection efficiency does not change (Supplemental Figure S1C). Compared with this maximum level of infection for HMEC-1 cultured on the stiffest substrates, infection efficiency on the softest substrates decreased about twofold (Figure 1E; Supplemental Figure S1C).
FIGURE 1:

Uptake of Lm by HMEC-1 depends on the stiffness of the matrix on which cells reside. HMEC-1 residing on PA hydrogels of varying stiffness coated with collagen I were infected with ΔactA Lm (actAp::mTagRFP). Infection was analyzed by flow cytometry 7–8 h postinfection. Bacteria were added at a multiplicity of infection (MOI) between 30 and 50 bacteria per host cell. (A–D) Histograms of the logarithm of bacterial fluorescence intensity per cell for HMEC-1 plated on 0.6-kPa (A), 3-kPa (B), 20-kPa (C), and 70-kPa (D) PA hydrogels. Histograms for N = 5 replicates are shown in different colors. The histogram of control uninfected cells is shown in purple. Based on the autofluorescence of the control group, a gate is defined (see black and red lines) showing what is considered uninfected (left, black line) and infected (right, red line). (E) Boxplots of percentage of HMEC-1 infected with ΔactA Lm vs. hydrogel stiffness for the data shown in panels A–D. Circles represent outliers, and the boxplots’ notched sections show the 95% confidence interval around the median (Wilcoxon–Mann–Whitney test; for details about boxplots see Materials and Methods). One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively; Wilcoxon rank-sum test).
HMEC-1 on soft hydrogels show decreased focal adhesion kinase activity and decreased focal adhesion kinase K activity leads to less efficient Lm infection
Focal adhesion kinase (FAK) has been established as a key component of the signal transduction pathways triggered by integrins binding to the ECM, and for some cell types FAK expression and/or phosphorylation on residue Y397 have been shown to depend on mechanical cues sensed by cells, such as shear stresses imposed by fluid flow and/or ECM rigidity (Khatiwala et al., 2006; Provenzano et al., 2009; Mathieu et al., 2010; Higuita-Castro et al., 2014; Du et al., 2016). We therefore sought to determine whether FAK expression or activity could be modulated depending on the substrate stiffness where HMEC-1 reside. To that end, HMEC-1 were grown on soft 3-kPa hydrogels, stiff 70-kPa hydrogels, and TC polystyrene substrates treated with vehicle control or PF573228 FAK inhibitor. Cells were then harvested and total amounts of FAK and pY397-FAK were determined using Western blot analysis. We found that increasing the stiffness of the underlying substrate from 3 to 70 kPa resulted in increasing levels of pY397-FAK, but the levels of total FAK remained largely unchanged (Figure 2, A–C). In addition, the levels of pFAK-Y397 were similar for cells residing on stiff 70-kPa hydrogels and TC polystyrene substrates (Figure 2, A–C). Treating cells residing on polystyrene with the specific FAK inhibitor PF573228 led to a significant decrease of pY397-FAK, while total FAK levels remained unchanged as compared with controls (Figure 2, A–C). Our results suggest that FAK phosphorylation at Y397 is modulated by the stiffness of the substrate on which HMEC-1 reside and therefore by different levels of cell–ECM interaction-induced focal adhesion signaling.
FIGURE 2:

FAK activity of HMEC-1 residing on soft PA hydrogels is decreased, as is Lm uptake. (A) Western blots from whole HMEC-1 lysates showing expression of phosphorylated FAK (Tyr397) and total FAK for cells residing on soft gels (3 kPa), stiff gels (70 kPa), and TC polystyrene substrates with or without 2 μM PF537228 FAK inhibitor. In each Western blot, equal quantities of protein were loaded and equal loading was confirmed in relation to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. In each case, the Western blots shown are representative of three independent experiments. (B, C) Normalized ratios of FAK/GAPDH (B) and pFAK (Tyr397)/GAPDH (C) for HMEC-1 residing on varying-stiffness substrates and treated or not with 2 μM PF537228 FAK inhibitor. Different color circles correspond to data from three independent experiments. Black bars represent the means of the three independent experiments. For each experiment, values have been normalized relative to the ratio for cells residing on polystyrene substrates. (D) Inhibition of bacterial uptake by FAK inhibitors. FAK-14, PF573228, or vehicle control was added 1 h before addition of bacteria to HMEC-1 residing on polystyrene substrates. Percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) as a function of inhibitor concentration (mean ± SD, N = 4 replicates). x = 0 corresponds to cells treated with vehicle control. Inset shows the same data with concentration on a log scale. Infection was analyzed by flow cytometry, 7–8 h after infection. MOI is 80. Representative data come from one of three independent experiments. (E) Boxplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) for cells treated either with nontargeting siRNA (siNT) or FAK siRNA (siFAK) (means ± SD, three independent experiments and N = 6 replicates per experiment). MOI is 60 (gray) or 20 (green). Circles represent outliers, and the boxplots’ notched sections show the 95% confidence interval around the median (Wilcoxon–Mann–Whitney test; for details about boxplots see Materials and Methods). One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively; Wilcoxon rank-sum test). (F) Normalized ratio of pFAK (Tyr397)/GAPDH for HMEC-1 residing on polystyrene substrates and treated for various amounts of time (min) with 100 nM Angiotensin-II. Different-colored circles correspond to Western blot data from three independent experiments. In each Western blot, equal quantities of protein were loaded and equal loading was confirmed in relation to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. Black bars represent the means of the three independent experiments. For each experiment, values have been normalized relative to the ratio for untreated cells (t = 0 min). (G) Boxplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) for cells pretreated for 2 h either with vehicle control or 100 nM angiotensin-II (means ± SD, three independent experiments and N = 4 replicates per experiment). One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively; Wilcoxon rank-sum test).
To address whether reduced Lm uptake as observed for HMEC-1 residing on soft matrices can be attributed at least in part to reduced FAK activity, we treated HMEC-1 residing on polystyrene substrates with FAK inhibitors FAK-14 or PF573228 for 1 h prior to infection. We then measured the efficiency of infection with Lm as described above, and found that both FAK inhibitors caused a similar dose-dependent inhibition of Lm infection (Figure 2D). We then transfected HMEC-1 with commercial nontargeting control small interfering RNA (siRNA) (siNT) or siRNA-targeting FAK (siFAK) (Supplemental Table S2), and total FAK expression was found to be about fivefold reduced for the FAK knockdown cells as compared with controls, as determined by immunofluorescence (Supplemental Figure S2A) and real-time quantitative PCR (RT-qPCR) (Supplemental Figure S2B). We found a significant, approximately twofold decrease in Lm infection efficiency for the FAK knockdown cells as compared with control cells (Figure 2E; Supplemental Figure S2, C and D). Taken together, these results are consistent with the hypothesis that decreased bacterial infection of HMEC-1 residing on highly compliant soft matrices may be due in part to decreased FAK activity.
To address whether elevating FAK activity can lead to increased bacterial uptake, we pretreated cells for various times with the vasoconstrictor peptide hormone Angiotensin II, which has been shown to induce an increase in phosphorylation of FAK in vivo in murine aortas (Louis et al., 2007) and also shown in vitro to lead to enhanced phosphorylation of FAK at Y397 for a variety of cell types (Greco et al., 2002; Weng and Shukla, 2002; Torsoni et al., 2005), including endothelial cells (Montiel et al., 2005). We first treated HMEC-1 with 100 nM Angiotensin II for various times and then lysed the cells and used Western blotting to assess FAK expression and phosphorylation of FAK at Y397 (Figure 2F; Supplemental Figure S2F). Interestingly, we found that, although FAK expression did not change upon treatment with Angiotensin II, phosphorylation of FAK at Y397 increased substantially for cells treated with Angiotensin II for more than 30 min. Because FAK activity was shown to be elevated for cells pretreated for more than 30 min with Angiotensin II, we then asked whether that would be sufficient to lead to increased infection susceptibility. When we infected HMEC-1 cells seeded on polystyrene substrates and pretreated with 100 nM of Angiotensin II, we observed a modest but significant 23% increase in infection susceptibility as compared with control cells (Figure 2G; Supplemental Figure S2E). We then seeded HMEC-1 cells on soft 3-kPa and stiff 70-kPa hydrogels and pretreated cells with or without 100 nM Angiotensin II for 2 h. We found a much stronger effect of Angiotensin II increasing infection for cells plated on soft substrates than on stiff substrates; indeed, treatment of cells on 3-kPa substrates with 100 nM Angiotensin II for 2 h was sufficient to raise the level of infection to be approximately equal to infection on 70-kPa substrates (Supplemental Figure S2G). This result further supports the hypothesis that a primary determinant of the relatively inefficient infection of HMEC-1 on soft substrates is the relatively low level of FAK activity under this environmental condition.
Adhesion but not invasion efficiency is increased when Lm infects HMEC-1 residing on stiff substrates
Decreased infection efficiency of Lm in HMEC-1 residing on soft matrices or treated with FAK inhibitors could be due to quantitative changes at several different steps in the infection process: for example, decreased bacterial adhesion on HMEC-1, or decreased invasion of the adhering bacteria into HMEC-1, or both. To help identify the exact step in infection that is sensitive to substrate stiffness, we used a constitutively GFP-expressing strain of Lm to infect HMEC-1 residing on either soft 3-kPa or stiff 70-kPa hydrogels and treated with vehicle control or PF573228. Samples were fixed shortly after infection, and bacteria that were attached to the cell surface but not yet internalized were specifically labeled with antibodies under nonpermeabilizing conditions. This inside/outside labeling method allows us to distinguish between bacteria that are adhered but not internalized (GFP-positive and labeled with the antibody) and those that are fully internalized by the HMEC-1 (GFP-positive but not labeled with the antibody). We found that both the total number of bacteria per host cell and the number of internalized bacteria per cell are significantly increased when HMEC-1 reside on stiff 70-kPa hydrogels as compared with soft 3-kPa hydrogels (Figure 3, A and B). However, importantly, the invasion efficiency (ratio of internalized bacteria to total bacteria) did not differ between HMEC-1 residing on stiff as compared with soft matrices, suggesting that it is specifically the adhesion of bacteria to the surface of host cells that is increased when the matrix stiffness is elevated and that leads to increased Lm infection efficiency for cells residing on stiffer matrices (Figure 3C). Interestingly, we found a decrease in both Lm adhesion and invasion efficiency irrespective of hydrogel stiffness for the cells treated with the FAK inhibitor (Figure 3), suggesting that FAK activity levels modulate both adhesion and invasion efficiency of Lm in HMEC-1, while substrate stiffness affects adhesion only.
FIGURE 3:

Lm adhesion, but not invasion efficiency, is increased when HMEC-1 reside on stiff hydrogels. HMEC-1 residing on soft (3-kPa) or stiff (70-kPa) PA hydrogels and treated with vehicle control or 2 μM PF537228 FAK inhibitor were infected with Lm (constitutively expressing GFP) at an MOI between 1.5 and 15. At 30 min postinfection, samples were fixed and immunostained, and infection was analyzed by microscopy followed by image processing. Boxplots show (A) total bacteria per cell; (B) internalized bacteria per cell; (C) invasion efficiency (ratio of internalized bacteria to total bacteria); (D) cells in the field of view. Representative data come from one of three independent experiments. N = 800–1000 cells were analyzed for each condition. Two asterisks denote statistically significant differences between the medians of two distributions (<0.01; Wilcoxon rank-sum test).
InlB contributes to Lm infection of HMEC-1 but is not modulated by substrate stiffness
Our data indicate that lower FAK activity as it occurs when HMEC-1 are seeded on softer hydrogels leads to decreased Lm adhesion on the surface of HMEC-1. This finding led us to hypothesize that a receptor at the surface of cells could be differentially regulated depending on substrate stiffness, leading to decreased bacterial adhesion. To examine whether any of the Lm internalins known to play a role in adhesion or invasion of other cell types, including VECs, are implicated in infection of HMEC-1 and responsible for the matrix stiffness–dependent susceptibility to infection, we infected HMEC-1 with mutant strains deficient in each of the internalin-encoding genes inlA, inlB, and inlF (Figure 4A; Supplemental Table S1). We found a twofold decrease in bacterial infection when InlB was not present, suggesting that InlB is one of possibly multiple bacterial factors that contribute to Lm infection of HMEC-1. In contrast to the effect of the deletion in the inlB gene, deletion of inlF had no effect on bacterial infection of HMEC-1. Deletion of inlA resulted in very modest but reproducible reduction in infection, as reported previously for human brain microvascular endothelial cells infected with Lm (Greiffenberg et al., 1998).
FIGURE 4:

Infection of HMEC-1 by Lm is in part mediated by InlB in a manner independent of matrix stiffness. (A) Percentage of HMEC-1 infected with Lm as a function of the logarithm of MOI (mean ± SD, N = 4 replicates). HMEC-1 were infected with the indicated strains: wild type (circle); ΔinlA (square); ΔinlB (cross); ΔinlF (diamond; actAp::mTagRFP). The frequency of infected HMEC-1 was determined by flow cytometry 7–8 h postinfection. Representative data come from one of three independent experiments. (B) Percentage of HMEC-1 infected with Lm as a function of the logarithm of MOI (mean ± SD, N = 4 replicates). HMEC-1 were infected with the indicated strains: ΔactA (black circles); ΔactA/ΔinlB (gray squares; actAp::mTagRFP). The frequency of infected HMEC-1 was determined by flow cytometry 7–8 h postinfection. Representative data come from one of three independent experiments. (C) Boxplots of percentage of HMEC-1 infected with Lm as a function of substrate stiffness (N = 5–6 replicates). HMEC-1 were infected with the indicated Lm strains: ΔactA (gray); ΔactA/ΔinlB (black; actAp::mTagRFP) at an MOI of 20. Infection was analyzed by flow cytometry 7–8 h after infection. Representative data come from one of three independent experiments. One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively; Wilcoxon rank-sum test). (D–F) HMEC-1 residing on collagen I–coated glass substrates were infected with Lm (constitutively expressing GFP) or ΔinlB Lm at an MOI of 3.5. At 30 min postinfection, samples were fixed and, immunostained and infection was analyzed by microscopy followed by image processing. Boxplots show (D) total bacteria per cell; (E) internalized bacteria per cell; (F) invasion efficiency (ratio of internalized bacteria to total bacteria). For each condition, 500–550 cells were analyzed in total. (G) Percentage of HMEC-1 infected with Lm as a function of SGX-523 Met inhibitor concentration (mean ± SD, N = 4 replicates). SGX-523 or vehicle control was added 1 h before addition of bacteria. HMEC-1 were infected with the indicated strains: ΔactA (black circles); ΔactA/ΔinlB (gray squares; actAp::mTagRFP) at an MOI of 75. Infection was analyzed by flow cytometry 7–8 h after infection. Representative data come from one of three independent experiments. (H) Boxplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) for cells treated either with nontargeting siRNA (siNT) or with Met siRNA (siFAK; means ± -SD, three independent experiments and N = 6 replicates per experiment). MOI is 60 (gray) or 20 (green). Circles represent outliers, and the boxplots’ notched sections show the 95% confidence intervals around the medians (Wilcoxon–Mann–Whitney test; for details about boxplots see Materials and Methods). One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively; Wilcoxon rank-sum test). (I) Boxplots of percentage of HMEC-1 infected with Lm as a function of substrate stiffness (N = 5–6 replicates). HMEC-1 were treated with vehicle control or 1 μM SGX-523 Met inhibitor for 1 h prior to infection and then infected with ΔactA Lm (actAp::mTagRFP). Infection was analyzed by flow cytometry 7–8 h after infection. MOI is 20. Representative data come from one of three independent experiments. (J) Percentage of HMEC-1 infected with Lm as a function of PF537228 inhibitor concentration (mean ± SD, N = 4 replicates). PF537228 or vehicle control was added 1 h before addition of bacteria (see Supplemental Figure S1D). HMEC-1 were infected with the indicated strains: ΔactA (black circles); ΔactA/ΔinlB (gray squares; actAp::mTagRFP) at an MOI of 75. Infection was analyzed by flow cytometry 7–8 h after infection. Representative data come from one of three independent experiments. Inset shows the same data with concentration on a log scale.
The importance of InlB for initial invasion of Lm into HMEC-1 was further confirmed by infecting HMEC-1 with a different strain that had deletions in both inlB and actA so that bacteria could not spread from cell to cell. Consistent with our previous findings, we observed a twofold decrease in bacterial uptake for a series of different multiplicities of infection (MOI) when inlB was knocked out (Figure 4B). Importantly, bacteria carrying a deletion of inlB still exhibited a strong monotonic dependence of infection efficiency on substrate stiffness (Figure 4C). This finding indicates that, while InlB contributes to some step in the infection of HMEC-1 by Lm, it is not governing the step that is sensitive to substrate stiffness.
We confirmed this conclusion using four different lines of experimentation. First, we directly assessed the effect of inlB deletion on bacterial adhesion and invasion of HMEC-1 using inside/outside labeling as described above. We found that the total number of bacteria associated with HMEC-1 cells was the same for wild-type bacteria as for an isogenic ΔinlB strain (Figure 4D); that is, adhesion to the host cell surface is indeed unchanged. However, the number of internalized bacteria per cell and therefore the overall invasion efficiency was significantly decreased for the ΔinlB strain (Figure 4, E and F). Second, we examined the effect of inhibiting the host cell binding partner for InlB, the receptor tyrosine kinase Met (also known as the hepatocyte growth factor receptor), on bacterial invasion. We confirmed that HMEC-1 express Met (Supplemental Figure S3A) and then treated the cells with various concentrations of the highly selective Met inhibitor SGX-523 prior to infection (Buchanan et al., 2009). We found that only uptake of the control ΔactA strain of Lm decreased with increasing concentration of SGX-523, while uptake of the isogenic ΔactA/ΔinlB Lm did not change substantially as a function of SGX-523 concentration (Figure 4G). Third, when we then transfected HMEC-1 with commercial nontargeting control siRNA (siNT) or siRNA targeting Met (siMet) and then infected HMEC-1 with Lm, we found a significant twofold decrease in Lm infection efficiency for the Met knockdown cells as compared with control cells (Figure 4H; Supplemental Figure S3, B and C), while confirming that Met expression was fivefold reduced for the Met knockdown cells as compared with controls through RT-PCR (Supplemental Figure S3D; Supplemental Table S2). When we then infected transfected siNT and siMet HMEC-1 with ΔactA/ΔinlB Lm, we found no significant difference in infection Lm uptake between the two conditions, consistent with the previous results of cells pretreated or not with SGX-523 (Figure 4G; Supplemental Figure S3E). These results confirm that InlB most likely contributes to Lm invasion into HMEC-1 by interacting with host Met, as has been shown for other cell types (Parida et al., 1998; Bierne and Cossart, 2002). However, Lm invasion into HMEC-1 treated with SGX-523 still retains a strong dependence on substrate stiffness (Figure 4I), just as we found for the ΔinlB strain. Fourth, we found that infection of HMEC-1 with the ΔactA/ΔinlB strain of Lm is still highly sensitive to inhibition of FAK with PF573228, showing a dose–response curve that parallels that of the control ΔactA strain (Figure 4J).
So far, we can conclude that substrate stiffness affects the adhesion of Lm to the surface of HMEC-1 so that bacterial adhesion to host cells residing on soft substrates is about twofold less than that to host cells residing on stiff substrates. The well-characterized InlB–Met interaction appears to contribute to invasion of adherent bacteria into HMEC-1, and is responsible for about half of the invasion efficiency for Lm infecting these cells. However, this interaction does not govern bacterial adhesion per se, is not sensitive to substrate stiffness, and is orthogonal to the FAK signaling axis. We therefore sought to identify alternative possible host cell receptors for adhesion of Lm to the surface of HMEC-1 that might be responsible for the stiffness-dependent effects we consistently observe.
Inhibiting FAK activity leads to a reduced amount of cell surface vimentin for HMEC-1
Because the decreased FAK activity associated with growth of HMEC-1 on softer hydrogels leads to decreased Lm adhesion on the surfaces of host cells, we attempted to identify candidate host surface receptors whose amount is modulated by FAK activity and that mediate bacterial adhesion. Because manufacturing soft and stiff PA hydrogels with large dimensions is both technically challenging and costly, we instead grew cells on large TC polystyrene plates and treated them with vehicle control or the FAK inhibitor PF573228. We then biotinylated the cell surface–exposed proteins using membrane-impermeable EZ-Link sulfo-NHS-SS-biotin and isolated them by affinity chromatography with streptavidin-agarose beads (Fujimoto et al., 1992; Nunomura et al., 2005). Isolated surface proteins were then run in two-dimensional (2D)-PAGE gels to allow separation of proteins according to both their isoelectric points and their molecular weight (Elia, 2012). Gels were silver-stained and comparison between gels was performed by marking the spots that differed in intensity (Figure 5, A and B). We found a spot at ∼55 kDa consistently differing in two independent experiments between the control sample and the sample treated with the FAK inhibitor PF573228 (see circles in Figure 5, A and B). In a third replicate experiment of surface protein isolation, 2D-PAGE gels were Coomassie stained and the specific 55 kDa spot that differed as a function of FAK inhibitor treatment was excised and further analyzed by nanoliquid chromatography tandem mass spectrometry (nanoLC-MS/MS). We identified this spot as the protein vimentin (accession number: gi|340219, Mascot identification score: 788, number of identified peptides: 87, mass: 53,752, isoelectric point: 5.05, sequence coverage: 61%). Vimentin is a major component of the cytoskeleton, forming intermediate filaments, but exists also in soluble form and has been previously found to localize at the cell surface for a wide variety of cells including VECs (Fuchs and Weber, 1994; Zou et al., 2006; Du et al., 2014; Mitra et al., 2015; Yu et al., 2016).
FIGURE 5:
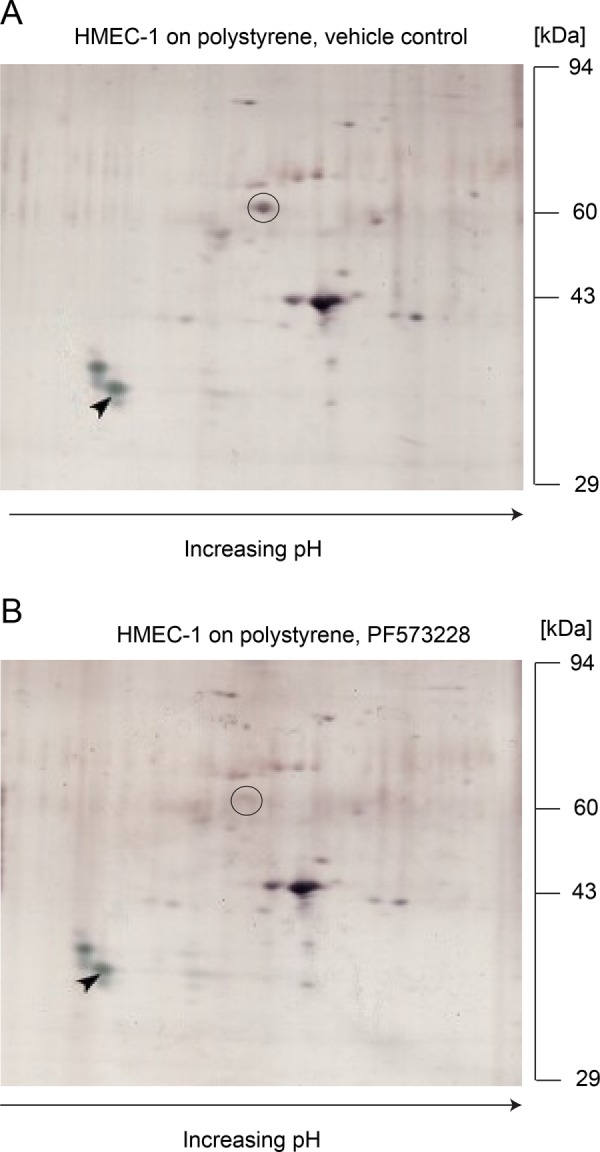
Lower FAK activity leads to reduced amount of cell surface vimentin. (A, B) 2D-PAGE gels of plasma membrane proteins of HMEC-1 grown on TC polystyrene substrates treated for 1 h with vehicle control (A) or 2 µM PF537228 FAK inhibitor (B). pH increases from left to right. Gels were silver-stained and one isoelectric point marker (tropomyosin), added to each sample as an internal standard, is marked with a black arrow. The one spot that differed consistently between three independent experiments is indicated with a black circle and corresponds to vimentin (55 kDa).
Given the high validity of identification, we sought to confirm whether vimentin is expressed at the surface of HMEC-1 by performing immunostaining of nonpermeabilized HMEC-1 seeded on collagen I–coated glass substrates followed by epifluorescence imaging (Figure 6). For negative controls, cells were incubated with secondary antibody alone. The anti-vimentin H-84 antibody stained the vimentin intermediate filaments of permeabilized cells very well (Figure 6, A and B). Nonpermeabilized cells showed surface staining of vimentin near cell–cell junctions, and very low background staining was observed for cells incubated with secondary antibody alone (Figure 6, C and D). This localization confirmed that cell surface vimentin could be a plausible candidate responsible for adhesion of Lm to HMEC-1.
FIGURE 6:
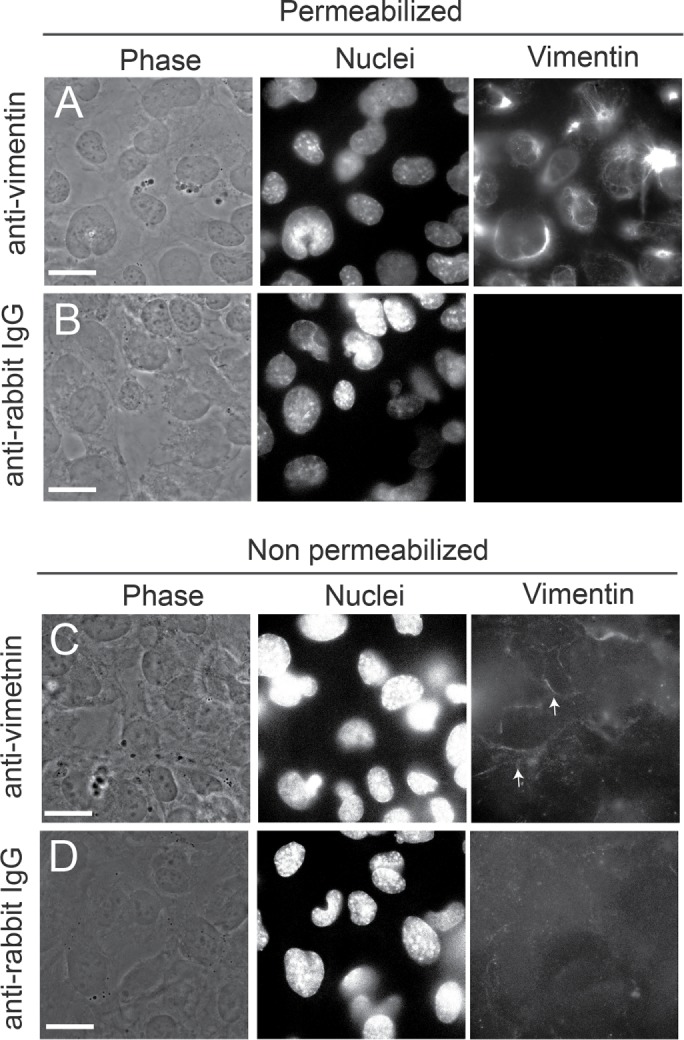
Surface vimentin is localized along the periphery of HMEC-1. (A–D) Cells were stained for vimentin using the rabbit anti-vimentin H-84 antibody. For negative controls, cells were stained with secondary anti-rabbit IgG antibody alone. Representative phase image of cells (left column), image of the nuclei (middle column), and H-84 anti-vimentin antibody fluorescence (right column) are shown for (A) permeabilized HMEC-1 strained for intracellular vimentin; (B) permeabilized HMEC-1 incubated with anti-rabbit IgG alone as negative controls; (C) nonpermeabilized HMEC-1 stained for surface vimentin; (D) nonpermeabilized HMEC-1 incubated with anti-rabbit IgG alone as negative controls. Scale bar shown in white is 20 μm. White arrows point at the localization of surface vimentin at cell–cell junctions.
HMEC-1 surface vimentin contributes to Lm uptake
To determine whether surface vimentin specifically contributes to Lm adhesion and thereby infection of HMEC-1, we used three independent methods to decrease the availability of cell surface vimentin for bacterial adhesion. First, we pretreated HMEC-1 with various concentrations of H-84 anti-vimentin antibody prior to infection with Lm and found a dose-dependent decrease in infection efficiency (Figure 7A). We found a maximum twofold decrease in Lm infection when cells with 80 μg/ml anti-vimentin antibody were incubated, while increasing the concentration of the antibody further did not lead to further decrease in Lm uptake (Supplemental Figure S3A).
FIGURE 7:

Surface vimentin of HMEC-1 is implicated in Lm uptake. (A) Decrease in bacterial uptake after blocking HMEC-1 with anti-vimentin antibody H-84. Barplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) as a function of antibody concentration (means ± SD and N = 6 replicates per experiment). Representative data come from one of three independent experiments. Infection was analyzed by flow cytometry, 7–8 h after infection. (B) Barplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) for cells treated either with nontargeting siRNA (siNT) or with vimentin siRNA (siVIM) (means ± SD, and N = 6 replicates per experiment). Representative data come from one of three independent experiments. MOI is 50 (black barplots) and 17 (gray barplots). (C) Decreased uptake of Lm when HMEC-1 are treated with withaferin that captures soluble vimentin 30 min prior to infection. Barplots of percentage of HMEC-1 infected with ΔactA Lm (actAp::mTagRFP) as a function of withaferin concentration (means ± SD and N = 6 replicates per experiment). Representative data come from one of three independent experiments. Infection was analyzed by flow cytometry 7–8 h after infection. (D) Percentage of HMEC-1 infected with Lm as a function of the logarithm of MOI (mean ± SD, N = 4 replicates). HMEC-1 were infected with the indicated strains: ΔactA (black), ΔactA/ΔinlB (gray; actAp::mTagRFP), and HMEC-1 were treated with vehicle control (circle) or withaferin (diamond) for 30 min prior to infection. The frequency of infected HMEC-1 was determined by flow cytometry 7–8 h postinfection. Representative data come from one of three independent experiments. MOI ranged from 50 to120. Two asterisks denote statistically significant differences between the medians of infection fraction of control vs. all other groups (p < 0.01; Wilcoxon rank-sum test).
Second, HMEC-1 were transfected with commercial nontargeting control siRNA (siNT) or siRNA targeting vimentin (siVIM), and total vimentin expression was found to be fivefold reduced for the vimentin knockdown cells as compared with controls through RT-PCR. Reduced total vimentin expression was also confirmed through immunofluorescence (Supplemental Figure S4, A and B; Supplemental Table S2). We found a significant decrease (∼30%) in Lm infection efficiency for the vimentin knockdown cells as compared with control cells, while confirming that cell confluency under both conditions was similar (Figure 7B; Supplemental Figure S4, C–F).
Third, we treated HMEC-1 cells with various concentrations of withaferin, a natural product known to bind the soluble form of vimentin (Chi et al., 2010; Bargagna-Mohan et al., 2013, 2014). We found a dose-dependent decrease in infection efficiency for Lm in withaferin-treated cells, again finding a maximum twofold decrease when cells were treated with 5–10 μM of withaferin prior to infection (Figure 7C). Higher concentrations of withaferin disrupted HMEC-1 cell–substrate adhesion. Overall, these results are consistent with the hypothesis that cell surface vimentin is responsible for about half of the adhesion of Lm to the HMEC-1 surface and that vimentin presentation on the cell surface is sensitive to substrate stiffness.
As an additional control to confirm that anti-vimentin antibodies and withaferin treatment inhibit Lm uptake due to effects on surface vimentin and not to nonspecific effects, we transfected HMEC-1 with commercial nontargeting control siRNA (siNT) or siRNA targeting vimentin (siVIM) and then blocked cells for 1 h with the H-84 anti-vimentin antibody. We then infected the host cells with constitutively GFP-expressing Lm and fixed samples shortly after infection. Through quantitative microscopy, we observed no significant change in infection susceptibility for vimentin knockdown cells blocked with anti-vimentin antibodies prior to infection as compared with vimentin knockdown alone, confirming that the antibodies inhibit uptake was due to effects on vimentin and not to nonspecific effects (Supplemental Figure S4G). Similarly, when HMEC-1 transfected with siNT or siVIM were pretreated with or without withaferin, we found a significant decrease in bacterial adhesion only for the control cells treated with withaferin and not for the vimentin knockdown cells (Supplemental Figure S4H). Finally, we hypothesized that if vimentin localizes at cell–cell junctions, and given that perturbing the amount of surface vimentin leads to up to a twofold decrease in infection susceptibility, then we should expect that when HMEC-1 are infected with Lm, half of the bacteria should adhere at cell–cell junctions and half could adhere anywhere else. Indeed, when we infected HMEC-1 with Lm, fixed the samples, and then immunostained for VE-cadherin as a proxy for cell–cell junctions (Supplemental Figure S5A), we observed that quite often bacteria were found adhering at cell–cell junctions. This was not the case for vimentin knockdown cells, where bacteria were found all across the apical surfaces of the host cells (Supplemental Figure S5B).
To examine any possible interaction between the contribution of vimentin-based adhesion and the contribution of InlB/Met-based invasion of Lm infecting HMEC-1, we pretreated HMEC-1 with withaferin or vehicle control and then infected them with either ΔactA or ΔactA/ΔinlB Lm over a range of MOIs. Consistent with our results described above, we found a twofold decrease in Lm uptake when cells were pretreated with withaferin and infected with ΔactA Lm or when cells were not treated with withaferin but were infected with ΔactA/ΔinlB Lm (Figure 7D). When cells were both pretreated with withaferin and infected with ΔactA/ΔinlB Lm, infection was decreased fourfold relative to controls (Figure 7D); that is, InlB/Met and vimentin contribute independently to infection efficiency. However, under all conditions, there was still residual infection, suggesting that Lm uses additional strategies to achieve uptake by HMEC-1, other than interactions with surface vimentin for bacterial adhesion and with Met for increased bacterial internalization.
Uptake of Listeria innocua but not carboxylated latex beads by HMEC-1 depends on cell surface vimentin
Our results demonstrate that the vimentin-dependent adhesion of Lm to HMEC-1 is sensitive to substrate stiffness and is not mediated by the bacterial internalins InlA, InlB, and InlF. To gain insight into whether vimentin-based bacterial adhesion is a result of a nonspecific stickiness or a specific host–bacterium interaction, we measured the influence of cell surface vimentin on adhesion of other bacteria and of nonbiological particles. L. innocua (Li) is a bacterium closely related to Lm that is considered nonpathogenic in that it lacks the putative internalin family members and other virulence factors that Lm carries (Glaser et al., 2001; Lauer et al., 2002). When we infected HMEC-1 with comparable loads of Lm and Li, we found that adhesion of both bacteria is comparable (Figure 8A). We also found that treating HMEC-1 with the FAK inhibitor PF537228 or blocking HMEC-1 with the anti-vimentin antibody H-84 reduces adhesion of both Lm and Li onto HMEC-1 by similar amounts (Figure 8A). Next, to examine whether a bacterial factor common to Lm and Li could be responsible for surface vimentin–mediated adhesion, we blocked HMEC-1 with anti-vimentin antibody and exposed cells to 2-μm carboxylated latex beads, which are efficiently taken up by VECs. We found that uptake of beads did not vary with antibody concentration and was identical to uptake of cells treated with isotype control (Figure 8B). Our results suggest that cell surface vimentin is a common receptor for both Lm and Li in the context of adhesion to HMEC-1, but uptake of nonbiological particles such as carboxylated latex beads does not depend on surface vimentin.
FIGURE 8:

Blocking HMEC-1 with anti-vimentin antibody reduces Li adhesion onto HMEC-1 but not uptake of beads. (A) Boxplots showing the number of bacteria per cell, for HMEC-1 residing on glass substrates and treated with vehicle control, 2 μM PF537228 FAK inhibitor, or 80 μg/ml H-84 anti-vimentin antibody prior to infection. Cells were infected with Lm or Li at an MOI of 4. At 30 min postinfection, samples were fixed and immunostained and adhesion of bacteria was analyzed by microscopy followed by image processing. For each condition, 2300–2600 cells were analyzed in total and data refer to one of two independent experiments. Two asterisks denote statistically significant differences between the median values of control cells vs. all other groups (<0.01; Wilcoxon rank-sum test). (B) HMEC-1 residing on TC polystyrene substrates and blocked for 1 h with various concentrations of H-84 anti-vimentin antibody or isotype control were “infected” with 2 μm beads at different concentrations. The frequency of microbead uptake by HMEC-1 was determined by flow cytometry 2 h post–addition of beads. Plot shows percentage of cells that internalized beads as a function of the beads/cell added for different H-84 antibody concentrations (mean ± SD, N = 4 replicates).
DISCUSSION
Bacterial infection of VECs depends on subendothelial stiffness
Our study provides the first evidence that ECM stiffness is a crucial determinant in modulating susceptibility of host cells to infection by bacterial pathogens. Using HMEC-1 as model adherent host cells and Lm as model bacterial pathogen, we demonstrated that host cell susceptibility to Lm infection increases with increasing ECM stiffness. Mechanosensitive cells, such as VECs, can “feel” a variety of physical cues such as fluid shear stresses and matrix stiffness and respond to them by altering their behavior and gene expression (Reinhart-King, 2008; Kohn et al., 2015). Interestingly, many signaling pathways involved in mechanotransduction are common irrespective of the exact mechanical stimulus (Li et al., 1997; Zebda et al., 2012). For instance, exposure of VECs to shear flow leads to recruitment of FAK at focal adhesions and to an increase in the phosphorylation at Y397 (Li et al., 1997). Consistent with FAK activity being sensitive to mechanical stimuli, we found that FAK activity is increased when cells reside on stiffer matrices. Concurrently, we observe a decrease in Lm uptake when FAK activity is reduced. It is possible that enhanced FAK activity for VECs exposed to shear stresses, might also lead to increased Lm uptake, but this still remains to be investigated (Li et al., 1997). In our study, we focused only on the effect of ECM stiffness on host cell infection and did not consider in our model additional mechanical cues that host cells experience (Shi and Tarbell, 2011). On the basis of our findings and on recent studies on shear flow-exposed VECs infected with pathogens, we speculate that it is plausible that additional mechanical cues could also play an important role in mediating susceptibility to infection, similarly to matrix stiffness (Claes et al., 2014; Niddam et al., 2017). The development of organotypic models for studying bacterial infection that take into account all of the different in vivo mechanical cues sensed by cells is therefore pertinent.
Our findings suggest that, together with biochemical cues, the ECM stiffness where cells reside is an additional variable worth considering, since it affects cellular behavior and susceptibility to bacterial infection. In traditional studies of bacterial infection of host cells, ECM stiffness is not taken into account, and such studies are typically performed on nonphysiological glass or polystyrene surfaces. Our study on how HMEC-1 interact with Lm at varying subendothelial stiffness was designed with the intent to mimic variations that occur under physiological conditions depending on the anatomic location of the vessel, in aging, and during pathophysiological conditions (Zieman et al., 2005; Acerbi et al., 2015). Our lower range of subendothelial matrix stiffness (0.6 kPa) could be relevant for the brain microvasculature, where the blood vessel microenvironment is very soft, whereas higher values are more relevant for bigger vessels or aged/diseased vasculature (Klein et al., 2009; Huynh et al., 2011; Stroka and Aranda-Espinoza, 2011; Kothapalli et al., 2012; Kohn et al., 2015). We showed that subendothelial stiffness changes the amount of surface vimentin expressed by VECs, therefore affecting critically how much host cells are susceptible to bacterial infection. That is, aged or diseased vessels might be more susceptible to infection based on our findings, while VECs lining healthy and softer vessels should be less susceptible to infection by Lm. It is plausible that low stiffness might be a host mechanism to protect against infection. For the purpose of the present study, we just examined how subendothelial stiffness modulates bacterial adhesion on previously uninfected VECs. However, it is very possible that once bacteria infect their hosts they might have developed ways to circumvent such a host mechanism by somehow altering matrix stiffness, that is, by reprogramming host cells to alter their production of ECM proteins or matrix-degrading enzymes, which could lead to alterations in subendothelial stiffness and subsequently VEC biomechanics, critically impacting infection. Future studies should address how host cell biomechanics changes upon infection and how bacterial infection changes the susceptibility of host cells to further infection.
Surface vimentin is a receptor for Listeria infection of HMEC-1
We found that lower FAK activity such as occurs when HMEC-1 are seeded on softer hydrogels leads to decreased Lm and Li adhesion onto the surface of HMEC-1 that is attributable to lower levels of cell surface vimentin. The role of cell surface vimentin as an attachment receptor facilitating bacterial or viral entry has been documented previously (Garg et al., 2006; Zou et al., 2006; Bhattacharya et al., 2009; Du et al., 2014; Rohrbeck et al., 2014; Mak and Brüggemann, 2016; Yang et al., 2016; Yu et al., 2016). We speculate that the matrix stiffness of VECs might possibly also affect uptake of the above pathogens, since the amount of surface vimentin is reduced when FAK activity is decreased, as in the case of cells residing on compliant matrices. Most importantly, our findings corroborate recent findings (Ghosh et al., 2018) showing that vimentin knockout mice show decreased Lm colonization of the brain. This same report also demonstrated that cell surface vimentin can contribute to Lm adhesion to or invasion of several rodent-derived cell lines, including mouse embryonic fibroblasts, mouse endothelial cells, and rat lung epithelial cells, as well as human microvascular endothelial cells, and suggested that the Lm surface protein InlF can serve as a binding partner for vimentin in the rodent model systems.
Bacterial adhesion to the surfaces of cells is commonly required as a precursor to invasion (Lebrun et al., 1996; Lecuit et al., 2000), and our results indicate that surface vimentin contributes specifically to adhesion. In contrast to the findings of Ghosh et al. (2018), we found that Lm infection of HMEC-1 is independent of InlF. Instead, we demonstrate that a decreased amount of cell surface vimentin leads to decreased adhesion of both pathogenic Lm and its nonpathogenic relative Li on HMEC-1, suggesting that the possible bacterial binding partner of vimentin might be expressed in both bacterial species. Surface vimentin has been shown to possess lectin-like activity binding N-acetylglucosamine (GlcNAc), a sugar that is also commonly found as a component of bacterial cell envelopes (Ise et al., 2010; Konopka, 2012). Specifically, GlcNAc is found decorating the wall teichoic acids of Lm serotype 1/2a strains, such as the 10403S strain used in this study (Fiedler, 1988). This implies that bacterial carbohydrate binding to host cell surface vimentin might also be a plausible specific interaction.
Lm uses multiple pathogenic strategies to infect VECs, some of which are independent of ECM stiffness
We found that the Lm virulence factor InlB contributes to invasion of HMEC-1 cells, most likely via its well-characterized interaction with the host cell receptor Met. Similarly, Lm uptake is decreased twofold when Met is inhibited. However, deletion of inlB and decrease of Met activity by siRNA knockdown or by pharmacological inhibition do not affect the ability of the infection process to respond to changes in substrate stiffness. When we infected HMEC-1 with ΔactA/ΔinlB Lm after treating host cells with withaferin that binds to soluble vimentin, we found a fourfold decrease in infection relative to controls, indicating that the vimentin-dependent and InlB-dependent pathways contribute to infection independent of one another; however, even when both pathways were blocked, there was still residual Lm infection. This residual Lm infection suggests that there are additional InlB-independent and vimentin-independent mechanisms that Lm leverage to infect HMEC-1.
Increased Lm adhesion on HMEC-1 residing on stiff matrices could be attributed to multiple factors. In this work, we investigated the possibility of a receptor at the surfaces of host cells being differentially regulated when FAK activity is increased, as in stiff matrices, and identified cell surface vimentin as a well-supported candidate receptor. However, it is also possible that additional factors might also lead to the decreased adhesion of Lm on HMEC-1 residing on softer matrices. It is plausible that the cell surface glycans of HMEC-1 might be differentially regulated depending on the stiffness of the environment. For example, previous studies have suggested both a positive correlation between FAK activity and the heparan sulfate portion of the glycocalyx (Liu et al., 2002; Gopal et al., 2010) and between the heparan sulfate portion of the glycocalyx and Lm adhesion on enterocytes (Henry-Stanley et al., 2003). Another possibility could be the differential regulation of biophysical properties of the host cells depending on matrix stiffness (e.g., cell cortical stiffness, surface roughness). For instance, studies on adhesion of bacteria on hydrogel surfaces have shown that adherence is increased on stiffer hydrogels (Kolewe et al., 2015), suggesting that if cells become stiffer on stiffer hydrogels, cellular stiffness alone could lead to increased bacterial adhesion (Tang et al., 2012; Higuita-Castro et al., 2014).
VEC origin and physical cues might explain discrepancies among previous studies on VEC infection by Lm
The endothelial lining of blood vessels displays remarkable spatiotemporal heterogeneity, and the basis of these morphological and functional differences is still incompletely characterized (Ingram et al., 2004; Aird, 2007, 2012). It has been documented that mechanical and chemical cues relayed to VECs by their environment can alter function and gene expression of VECs found at different tissues (Aird, 2012; Kohn et al., 2015). However, even when VECs from different origins are placed in vitro in the same environment, they can still exhibit unique behavior intrinsic to the cells themselves and not determined by differential culture or environmental conditions (Craig et al., 1998; Stroka and Aranda-Espinoza, 2011; Ostrowski et al., 2014; Ye et al., 2014). For example, the response of human umbilical cord endothelial cells (HUVEC) to changes in curvature or shear stress is completely distinct from that of human brain microvascular endothelial cells (HBMEC; Ye et al., 2014). In accordance with the differences these two cell types exhibit in response to geometrical constraints or mechanical stimuli, studies have also shown differences with respect to Lm infection. Past studies suggest that HBMEC infection by Lm depends on InlB (Greiffenberg et al., 1998), while HUVEC uptake of Lm appears to be independent of both the internalins InlA and InlB (Greiffenberg et al., 1997; Rengarajan et al., 2016). Similarly, it was recently reported that an interaction between vimentin and InlF contributes to the spatial distribution of Lm adhesion on the surface of a mouse brain endothelial cell line (Ghosh et al., 2018), whereas we showed that Lm infection of HMEC-1 is independent of InlF. These studies are not necessarily conflicting, but suggest that bacterial infection of VECs depends on both the environment and the particular origin of VECs, and therefore conclusions should refer to the exact cell type and not to endothelial cells in general. Because many pathological processes occur at the microvascular level of organs, we chose in this study to use HMEC-1 as our model for VECs. However, extrapolation from results obtained for HMEC-1 infection by Lm to other VECs should be performed with caution. Similarly, although Lm uptake by HMEC-1 increases with increasing subendothelial stiffness, that relationship could be different if HMEC-1 were infected with different bacterial pathogens. We speculate that the relationship between ECM stiffness and susceptibility to infection is likely host cell— and pathogen-specific.
In conclusion, our results provide the first strong evidence that the local mechanical environment and in particular the matrix stiffness of VECs regulates susceptibility of VECs to Lm infection. The novel mechanosensitive pathway we identified suggests that increased ECM stiffness sensed by VECs leads to enhanced FAK activity that augments the amount of vimentin exposed at the surfaces of VECs. The increased amount of surface vimentin in turn increases Lm adhesion and subsequent uptake by VECs. Our studies on the effect of ECM stiffness on bacterial uptake of VECs can have other significant implications, since they provide an example of how mechanotransduction can be exploited in biology and medicine, facilitating the development of therapeutic interventions against bacterial infections and other diseases.
MATERIALS AND METHODS
Fabrication of thin two-layered polyacrylamide (PA) hydrogels on 24-well glass-bottom dishes
PA hydrogel fabrication was done as previously described with a few modifications for the specific experiments (Georges et al., 2006; Bastounis et al., 2011, 2014; Vincent et al., 2013). Glass-bottom plates with 24 wells (MatTek; P24G-1.5-13-F) were incubated for 1 h with 500 μl of 2 M NaOH. Wells were rinsed with distilled water, and 500 μl of 2% 3-aminopropyltriethoxysilane (Sigma; 919-30-2) in 95% ethanol was added to each well for 5 min. Wells were rinsed again with water, and 500 μl of 0.5% gluteraldehyde was added to each well for 30 min. Wells were rinsed with water and dried at 60°C. To prepare hydrogels of varying stiffness, mixtures containing 3–10% acrylamide (Sigma; A4058) and 0.06–0.6% bis-acrylamide (Fisher; BP1404-250) were prepared. Specifically, 0.6-kPa hydrogels contained 3% acrylamide and 0.06% bis-acrylamide, 3-kPa hydrogels contained 5% acrylamide and 0.1% bis-acrylamide, 20-kPa hydrogels contained 8% acrylamide and 0.26% bis-acrylamide, and 70-kPa hydrogels contained 10% acrylamide and 0.6% bis-acrylamide. For each stiffness, two mixtures were prepared, the second of which contained 0.03% 0.1 μm–diameter fluorescent beads (Invitrogen, F8803). Mixtures were then degassed for 15 min to remove oxygen, which inhibits acrylamide polymerization.
First, 0.06% APS and 0.43% TEMED were added to the solutions containing no beads to initiate polymerization. Then, 3.6 µl of the mixture was added at the center of each well, capped with 12-mm untreated circular glass coverslips, and allowed to polymerize for 20 min. The coverslips were then lifted with a syringe needle with a small hook at its tip, and then 2.4 µl of the mixture containing tracer beads was added, sandwiched again with a 12-mm untreated circular glass coverslip, gently pressed downward with forceps, and allowed to polymerize for 20 min. Next, 50 mM HEPES at pH 7.5 was added to the wells, and coverslips were removed using the syringe needle and forceps. PA hydrogels were UV-sterilized for 1 h. Hydrogels were then activated by adding 200 µl of 0.5% wt/vol heterobifunctional cross-linker Sulfo-SANPAH (ProteoChem; c1111) in 1% dimethyl sulfoxide (DMSO) and 50 mM HEPES, pH 7.5, on the upper surface of the hydrogels and exposing them to UV light for 10 min. After activation, the hydrogels were washed with 50 mM HEPES at pH 7.5 to remove excess cross-linker and were coated with 200 µl of 0.25 mg/ml rat tail collagen I (Sigma-Aldrich; C3867) in 50 mM HEPES overnight at room temperature. Prior to seeding cells, hydrogels were incubated with cell media to allow equilibration for 30 min.
HMEC-1 culture and infection with Listeria monocytogenes
HMEC-1 (generous gift from the Welch lab, University of California, Berkeley) were maintained in MCDB 131 medium (Fisher Scientific; 10372-019) supplemented with 10% FBS (GemBio; 900-108), 10 ng/ml epidermal growth factor (Sigma; E9644), 1 μg/ml hydrocortisone (Sigma; H0888), and 2 mM l-glutamine (Sigma; 56-85-9).
HMEC-1 were infected as previously described with the following modifications (Rengarajan et al., 2016). The day prior to infection, host cells were seeded at a density of 4 × 105 cells/well for cells residing on wells of 24-well plates or at a density of 1 × 105 cells/well for cells residing on wells of 48-well plates and grown for 24 h. Lm liquid cultures were started from a plate colony and grown overnight, while being shaken, at 30°C in brain heart infusion (BHI) media (BD; 211059) supplemented with 200 μg/ml streptomycin and 7.5 μg/ml chloramphenicol. The day of the infection, the O.D.600 of the overnight cultures was measured and diluted to 0.1. Samples were incubated for 2 h while being shaken in the dark at 30°C in BHI media supplemented with 200 μg/ml streptomycin and 7.5 μg/ml chloramphenicol to allow the O.D. 600 to reach 0.2–0.3 and let bacteria enter the logarithmic growth phase. Bacteria were then washed three times with phosphate-buffered saline (PBS) to remove any soluble factors, and infections were performed in normal growth media (Reed et al., 2014).
To synchronize invasion, Lm diluted in normal growth media were added to the HMEC-1 cells and samples were spun for 10 min at 200 × g prior to incubation. After 30 min of incubation at 37°C, samples were washed four times in PBS, and after an additional 30 min, media were replaced with media supplemented with 20 μg/ml gentamicin. Multiplicity of infection (MOI) was determined by plating bacteria at different dilutions on BHI agar plates with 200 μg/ml streptomycin and 7.5 μg/ml chloramphenicol and measuring the number of colonies formed 2 d postinfection. A similar approach was followed when HMEC-1 were infected with Li, except that BHI plates with no antibiotics were used. All bacterial strains used in these studied are indicated in Supplemental Table S1.
Analysis of infection via flow cytometry was performed 7–8 h after exposure, unless otherwise stated. For drug exposure experiments, unless otherwise indicated, media were removed from the cells and replaced with media containing either the drug or vehicle control 1 h prior to infection or 30 min prior to infection for withaferin. Cells were cultured in the drug-containing media for 1 h after infection. At 1 h postinfection, cells were washed four times, and drug-free gentamicin-containing media were added to the cells.
Antibodies and reagents
Hoechst (Thermofisher; D1306) was dissolved at 1 mg/ml in DMSO and used at 1:1000. Drugs were dissolved in DMSO (Sigma; D2650) at the stock concentrations indicated: 10 mM FAK inhibitor-14 (FAK-14; Tocris Bioscience; 3414), 100 mM PF573228 (Tocris Bioscience; 3239), 20 mM SGX 523 (Tocris Bioscience; 5356), 42 mM withaferin (Sigma-Aldrich; W4394). Primary antibody used for staining of extracellular Lm was rabbit polyclonal anti-Listeria genus-specific antibody (ABCAM; ab35132). Primary antibody used for staining Li was goat polyclonal BacTrace anti-Listeria genus-specific antibody (SeraCare Life Sciences; 01-90-90). For incubation of cells with vimentin antibodies prior to infection or immunostaining of surface vimentin, rabbit polyclonal H-84 vimentin antibody (Santa Cruz Biotechnologies; sc-5565) was used. For isotype controls, cells were incubated with rabbit immunoglobulin G (IgG) (Sigma; I5006). For Western blotting, the following antibodies were used: rabbit polyclonal phospho-FAK Tyr397 (ThermoFisher; 44-624G), mouse monoclonal anti-FAK (EMD Millipore, 05-182), rabbit monoclonal anti-Met (cMet) antibody (Abcam; ab51067), and anti-GAPDH antibody (Cell Signaling; 140C10).
Flow cytometry of HMEC-1 cells infected with Lm
At 7–8 h postinfection, infected HMEC-1 cells were detached from the substrate by incubating them for 10 min with a mixture of 200 μl of 0.25% trypsin-EDTA and 0.05% collagenase (Sigma; C0130). Solutions in each well were pipetted up and down six times to ensure single-cell suspensions, and 200 μl of complete media was added to inactivate trypsin in each well. Solutions were transferred into 5-ml polystyrene tubes with a 35-μm cell strainer cap (Falcon; 352235, and then samples were immediately analyzed by flow cytometry on the Scanford FACScan analyzer (Custom Stanford and Cytek upgraded FACScan). From 10 to 20,000 cells were analyzed per replicate. To ensure analysis of single cells, the bulk of the distribution of cell counts was gated using the forward versus side scatterplot. This gating strategy ensures that single cells are analyzed and debris or cell doublets or triplets are eliminated from the analysis. A second gating step was then performed to exclude cells that autofluoresce by measuring the fluorescence of control-uninfected cells and gating the population of infected cells to exclude autofluorescence.
Immunostaining of extracellular adherent bacteria
HMEC-1 cells residing on either PA hydrogel substrates or glass collagen I–coated coverslips were infected as described above with an Lm strain that constitutively expresses GFP. At 20 min postinfection, 1 μg/ml Hoechst (Thermofisher; D1306) was added in each well to stain the cells’ nuclei. At 30 min postinfection, cells were washed four times in PBS and fixed with a nonpermeabilizing fixative for differential immunostaining for 20 min at room temperature (Yam and Theriot, 2004). Fixative contained 0.32 M sucrose, 10 mM MES at pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 4% formaldehyde of EM grade. Following a wash with PBS, samples were blocked for 30 min with 5% BSA in PBS and then incubated with anti-Lm primary antibody (Abcam ab35132) diluted 1:100 in PBS containing 2% BSA for 1 h. Samples were washed in PBS three times and then incubated with Alexa Fluor 546 goat anti-rabbit secondary antibody (Invitrogen A-11035) diluted 1:250 in PBS containing 2% BSA for 1 h. Samples were washed three times in PBS and stored in 1 ml PBS for imaging. N > 1000 cells were analyzed per condition. For imaging, we used an inverted Nikon Diaphot 200 with a charge-coupled device (CCD) camera (Andor Technologies) and a 40× air Plan Fluor NA 0.60 or a 100× oil objective. The microscope was controlled by the MicroManager software package (Edelstein et al., 2014). For differential immunostaining, all “green” bacteria associated with individual cells were counted as adherent; bacteria that were both “green” and “red” (due to antibody binding) were counted as noninternalized. Nuclei number was identified by running a custom-made script in MATLAB (Mathworks), and the CellC software was used for enumeration of the bacteria (Selinummi et al., 2005). For characterization of total bacteria adhering to host cells (Lm or Li), the same procedure was followed, with the exception that cells were permeabilized for 5 min in 0.2% Triton X-100 in PBS. The primary antibody used was goat polyclonal BacTrace anti-Listeria genus-specific antibody.
Immunostaining of surface vimentin
HMEC-1 cells residing on glass collagen I–coated coverslips were incubated at 37°C for 30 min with H-84 anti-vimentin antibody at 1:10 dilution in media. Cells were then washed three times in PBS and fixed in 4% formaldehyde (EM grade) in PBS for 10 min at room temperature. A blocking step with 5% FBS in PBS was followed for 30 min and then cells were incubated with secondary goat anti-rabbit AlexaFluor-546 antibody diluted 1:250 in PBS containing 2% BSA for 1 h. For negative controls to ensure that the label is specific to primary antibody, cells with no primary antibody were fixed and incubated for 1 h with secondary goat anti-rabbit AlexaFluor-546 antibody. As an additional control, cells were also fixed, permeabilized, and stained for intracellular vimentin. For imaging, we used an inverted Nikon Diaphot 200 with a CCD camera (Andor Technologies) and a 100× oil objective.
HMEC-1 cell transfection with siRNA
For each well of a 24-well plate, 6 × 104 HMEC-1 cells suspended in serum-free media were reverse transfected with siRNAs at 20 nM final concentration using 0.25 μl lipofectamine RNAiMAX (Invitrogen 13778075). The transfection mix was replaced with full media 8 h later. Synthetic siRNA pools (including four distinct siRNA sequences for each gene) to target vimentin, Met, and FAK were purchased from Dharmacon (Supplemental Table S2). HMEC-1 cells were treated with control (nontargeting, siGLO, and Kif11) or experimental siRNA in accordance with the manufacturer’s instructions. Specifically, to demonstrate that transfection performed was sufficient to get siRNAs into the cells, we transfected cells with synthetic siRNA, siGLO, which makes cells exposed to it fluorescent 24 h posttransfection (Supplemental Table S2). In addition, to track a cell cycle phenotype to verify that knockdown had occurred with our protocol, we transfected cells with siKif11, which results in substantial cell death of transfected cells ∼24–48 h posttransfection and can be verified on the TC microscope. Bacterial infections were performed ∼72 h after transfection.
RT-qPCR
HMEC-1 cells were treated with control or experimental siRNA as described above. mRNA was harvested using the RNeasy Micro Kit (Qiagen; 74004), and cDNA was prepared using the Superscript III First-strand Synthesis SuperMix (ThermoFisher; 18080–400). RT-qPCR was performed using TaqMan PreAmp Master (Thermofisher; 4331182). Genes of interest were amplified using primers Hs00958113_g1 for Vimentin (ThermoFisher; 4331182) and Hs99999905_m1 for GAPDH (ThermoFisher; 4333764) on a StepOnePlus real-time PCR system. Normalized relative quantity (NRQ) and error were calculated as previously described (Hellemans et al., 2007). GAPDH was used as control gene.
Western blotting of FAK and Met for HMEC-1 lysates coming from cells residing on different substrates
To assess FAK and Met phosphorylation and expression levels, cells were seeded on different substrates for 24 h, and then lysed with a buffer containing 1% Nonidet P-40, 0.5% sodium deoxycholate, and a protease inhibitor mixture (phenylmethylsulfonyl fluoride [PMSF], leupeptin, aprotinin, and sodium orthovanadate). The total cell lysate was separated by SDS–PAGE (10% running, 4% stacking) and transferred onto a nitrocellulose membrane (Immobilon P, 0.45-µm pore size). The membrane was then incubated with the designated antibodies. Immunodetection was performed using the Western-Light chemiluminescent detection system (Applied Biosystems).
Cell surface protein isolation
The isolation of cell surface proteins was adapted according to Roesli et al. (2008) and Karhemo et al. (2012). Cells were plated on 10 × 15 cm cell culture dishes until they reached confluency for each condition. Cells from 10 dishes were incubated with vehicle control (DMSO)–containing media and cells from 10 other dishes with 2 μM PF537228 FAK inhibitor for 1 h. Cells were then washed three times with DPBS+ (PBS, 0.901 mM CaCl2, 0.492 mM MgCl2, 2.667 mM KCl, 1.47 mM KH2PO4, 137.931 mM NaCl, 8.060 mM Na2HPO4-7H2O, 5.555 mM d-glucose, 0.327 mM sodium pyruvate) and incubated for 30 min with 0.5 mM EZ-Link sulfo-NHS-SS-biotin (APExBIO, 21331) in DPBS+ at 4°C. To quench any nonreacted biotinylation, 1 M Tris, pH 8.0, at a final concentration of 50 mM was added to each plate for 10 min at 4°C. The solution was then discarded, and plates were rinsed twice in 1× TBS (50 mM Tris-Cl, pH 7.2, 150 mM NaCl). A quantity of 2 ml of TBS, pH 7.2, with 1× protease inhibitor cocktail (1 mM EDTA, 10 μM E-64, 2 μg/ml leupeptin, 15 μg/ml benzamidine, 0.1 mM PMSF) and with 1× phosphatase inhibitor cocktail (1 mM Na3VO4, 1 mM NaF) was added to each dish and cells were scraped into 40-ml Falcon tubes. Tubes were spun at 400 × g for 5 min and the supernatant was discarded. A quantity of 10 ml of lysis buffer (50 mM Tris-HCl at pH 8.5, 150 mM NaCl, 2% NP-40, 0.25% deoxycholate, 2× protease inhibitor cocktail, 1× phosphatase inhibitor cocktail) was added to each single pellet obtained from 10 × 15 cm plates, and cells were passed three times through a 25 G needle attached to a 10-ml syringe. Lysates were then sonicated for 5 min in a bioruptor with a 30-s on/off cycle. A quantity of 1 mM MgCl2 and 50 U/ml benzonase (Sigma; 9025-65-4) was added to the samples, which were then incubated at 4°C for 2 h on a rotator. EDTA to a final concentration of 5 mM was added to each sample and samples were spun in an ultracentrifuge (Rotor type 70.1 Ti; 33,000 rpm) at 100,000 × g for 60 min at 4°C. Supernatants were then transferred into fresh 15-ml Falcon tubes and protein concentration was estimated by BCA protein assay (Thermo Fisher Scientific, Pierce Rockford). Equal amounts of protein (∼5 mg) from each extract were used for cell surface protein isolation.
Streptavidin agarose beads (Pierce, 20347) were washed three times with lysis buffer. A quantity of 50 µl per 5 mg/lysate was added for 2 h or overnight at 4°C on a rotator. Samples were then transferred to 2-ml gravity flow columns (BioRad) preequilibrated with 2 ml wash buffer I (50 mM Tris-HCl, pH 8.5, 150 mM NaCl, 2% NP-40, 0.25% deoxycholate, 0.5% SDS). The flowthrough was collected and stored at -80°C. Columns were washed once with 2 ml lysis buffer (excluding protease and phosphatase inhibitors), then with 2 ml wash buffer I, followed by a 2-ml wash with wash buffer II (50 mM Tris-HCl at pH 8.5, 300 mM NaCl, 2% NP-40, 0.25% deoxycholate, 0.5% SDS) and then again with 2 ml wash buffer I. Beads were then transferred to a fresh 1.5-ml Eppendorf tube, 3× volume of elution buffer (100 mM Tris-HCl, pH 8.5, 2% SDS, 100 mM DTT) was added, and samples were incubated at 37°C for 1 h on a thermomixer. Beads were then spun down, and supernatant was collected and 5% aliquoted for gel analysis (2.5% elution for Western blot and 2.5% elution for silver staining of 1D-PAGE gels) and the rest was put in a fresh 1.5-ml Eppendorf tube and stored at –80°C.
2D SDS–PAGE electrophoresis and staining
Two-dimensional electrophoresis was performed according to the carrier ampholyte method of isoelectric focusing (O’Farrell, 1975; Burgess-Cassler et al., 1989) by Kendrick Labs as follows: Isoelectric focusing was carried out in a glass tube of inner diameter 2.3 mm using 2% pH 3–10 Isodalt Servalytes (Serva, Heidelberg, Germany) for 9600 V-h. A quantity of 1 µg of an IEF internal standard, tropomyosin, was added to the sample. This protein migrates as a doublet with a lower polypeptide spot of MW 33,000 and pI 5.2. The enclosed tube gel pH gradient plot for this set of Servalytes was determined with a surface pH electrode. After equilibration for 10 min in Buffer O (10% glycerol, 50 mM dithiothreitol (DTT), 2.3% SDS, and 0.0625 M Tris at pH 6.8), each tube gel was sealed to the top of a stacking gel that overlaid a 10% acrylamide slab gel (0.75 mm thick). SDS slab gel electrophoresis was carried out for ∼4 h at 15 mA/gel. The following proteins (Sigma Chemical and EMD Millipore) were used as molecular weight standards: myosin (220,000), phosphorylase A (94,000), catalase (60,000), actin (43,000), carbonic anhydrase (29,000), and lysozyme (14,000). These standards appear along the basic edge of the silver-stained (Oakley et al., 1980) 10% acrylamide slab gel. The gels were dried between sheets of cellophane with the acid edge to the left.
Manual comparisons of patterns of silver-stained 2D gels
Each gel for comparison was overlaid with a transparent sheet for labeling polypeptide spot differences without marking the original gel. Two experienced analysts manually compared protein patterns, and polypeptide spots that were unique to or differed between controls and PF537228 samples were outlined.
Protein digestion and peptide extraction
Proteins that were separated by SDS–PAGE/2D-PAGE and stained by Coomassie dye were excised and washed and the proteins from the gel were treated according to published protocols (Shevchenko et al., 1996; Darie et al., 2011; Sokolowska et al., 2012c). Briefly, the gel pieces were washed in high-purity, high-performance liquid chromatography (HPLC)–grade water, dehydrated, cut into small pieces, and destained by incubating in 50 mM ammonium bicarbonate, 50 mM ammonium bicarbonate/50% acetonitrile, and 100% acetonitrile under moderate shaking, followed by drying in a speed-vac concentrator. The gel bands were then rehydrated with 50 mM ammonium bicarbonate. The procedure was repeated twice. The gel bands were then rehydrated in 50 mM ammonium bicarbonate containing 10 mM DTT and incubated at 56°C for 45 min. The DTT solution was then replaced with 50 mM ammonium bicarbonate containing 100 mM iodoacetamide for 45 min in the dark, with occasional vortexing. The gel pieces were then reincubated in 50 mM ammonium bicarbonate/50% acetonitrile and 100% acetonitrile under moderate shaking, followed by drying in a peed-vac concentrator. The dry gel pieces were then rehydrated using 50 mM ammonium bicarbonate containing 10 ng/µl trypsin and incubated overnight at 37°C under low shaking. The resulting peptides were extracted twice with 5% formic acid/50 mM ammonium bicarbonate/50% acetonitrile and once with 100% acetonitrile under moderate shaking. The peptide mixture was then dried in a speed-vac and solubilized in 20 µl of 0.1% formic acid/2% acetonitrile.
Liquid chromatography and tandem mass spectrometry
The peptide mixture was analyzed by reverse-phase liquid chromatography (LC) and MS (LC-MS/MS) using a NanoAcuity UPLC (Micromass/Waters) coupled to a Q-TOF Ultima API MS (Micromass/Waters) according to published procedures (Spellman et al., 2008; Darie et al., 2011; Sokolowska et al., 2012a,b). Briefly, the peptides were loaded onto a 100 µm × 10 mm NanoAquity BEH130 C18 1.7 µm UPLC column (Waters) and eluted over a 150-min gradient of 2–80% organic solvent (ACN containing 0.1% FA) at a flow rate of 400 nl/min. The aqueous solvent was 0.1% FA in HPLC water. The column was coupled to a Picotip Emitter Silicatip nanoelectrospray needle (New Objective). MS data acquisition involved survey MS scans and automatic data-dependent analysis (DDA) of the top three ions with the highest-intensity ions with the charge of 2+, 3+, or 4+. The MS/MS was triggered when the MS signal intensity exceeded 10 counts/s. In survey MS scans, the three most intense peaks were selected for collision-induced dissociation (CID) and fragmented until the total MS/MS ion counts reached 10,000 for up to 6 s each. The entire procedure used was previously described (Darie et al., 2011; Sokolowska et al., 2012a,b). Calibration was performed for both precursor and product ions using 1 pmol GluFib (Glu1-Fibrinopeptide B) standard peptide with the sequence EGVNDNEEGFFSAR and the monoisotopic doubly charged peak with m/z of 785.84.
LC-MS/MS data processing and protein identification
The raw data were processed using ProteinLynx Global Server (PLGS, version 2.4) software as previously described (Sokolowska et al., 2012a). The following parameters were used: background subtraction of polynomial order 5 adaptive with a threshold of 30%, two smoothings with a window of three channels in Savitzky–Golay mode and centroid calculation of top 80% of peaks based on a minimum peak width of four channels at half height. The resulting pkl files were submitted for database search and protein identification to the public Mascot database search (www.matrixscience.com, Matrix Science) using the following parameters: databases from NCBI (bacteria), parent mass error of 1.3 Da, product ion error of 0.8 Da, enzyme used: trypsin, one missed cleavage, propionamide as cysteine-fixed modification, and methionine oxidized as variable modification. To identify false-negative results, we used additional parameters such as different databases or organisms, a narrower error window for the parent mass error (1.2 and then 0.2 Da) and for the product ion error (0.6 Da), and up to two missed cleavage sites for trypsin. In addition, the pkl files were also searched against in-house PLGS database version 2.4 (www.waters.com) using searching parameters similar to the ones used for the Mascot search. The Mascot and PLGS database search provided a list of proteins for each gel band. To eliminate false positive results, for the proteins identified by either one peptide or a mascot score lower than 25, we verified the MS/MS spectra that led to identification of a protein.
Atomic force microscopy force–distance measurements for hydrogel stiffness characterization
AFM force–distance beating was performed on PA hydrogel samples in 50 mM HEPES, pH 7.5, buffer with a Park NX-10 AFM (Park Systems, Santa Clara, CA) using commercial silicon nitride cantilevers CP-PNP-SiO with a sphere tip (sQube, 0.08 N/m stiffness, sphere radius ∼1 μm) and gold coating on the reflective side. Temperature was kept at 37°C throughout the experiment. Tip calibration curves were determined on glass substrate surface, which was considered infinitely hard for the soft tips used. Two approach–withdraw cycles were performed per cell. Data analysis of FD curves and calculation of Young’s modulus were performed using XEI software (Park Systems) and SPIP software (Image Metrology, Hørsholm, Denmark).
Visualization and comparison of distributions
Distributions were visualized using boxplots with the features indicated below. The bold black line parallel to the x-axis indicates the median (second quartile) of the distribution and the boxplot’s notched section shows the 95% confidence interval around the median (Wilcoxon–Mann–Whitney nonparametric test). Regular black lines extended vertically below and above the medians represent the first and third quartiles of the distribution. Lines extending vertically from the boxplot (whiskers) represent the lowest datum within 1.5 IQR (interquartile range) of the lower quartile and the highest datum within 1.5 IQR of the upper quartile. Data points beyond the whiskers are considered outliers and displayed as circles. To assess whether the differences between the medians of two distributions are significant, we run a nonparametric Wilcoxon rank sum test, since the distributions are not normal. One or two asterisks denote statistically significant differences between the medians of two distributions (<0.05 or <0.01, respectively).
Supplementary Material
Acknowledgments
Our thanks to M. Footer, R. Lamason, M. Rengaranjan, G. Skariah, and members of the Theriot Lab for discussions and experimental support. This work was supported by National Institutes of Health R01AI036929 (J.A.T.), the Howard Hughes Medical Institute (J.A.T.), and the American Heart Association (E.E.B. and Y.Y.). Flow cytometry was performed at the Stanford Shared FACS Facility, and RT-qPCR was performed at the Stanford PAN Facility.
Abbreviations used:
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- HMEC-1
human microvascular endothelial cell
- Li
Listeria innocua
- Lm
Listeria monocytogenes
- MOI
multiplicity of infection
- TC
tissue culture
- VEC
vascular endothelial cell.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-04-0228) on May 2, 2018.
REFERENCES
- Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. (2015). Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) , 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed EM. (2015). Hydrogel: preparation, characterization, and applications: a review. J Adv Res , 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird WC. (2007). Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res , 158–173. [DOI] [PubMed] [Google Scholar]
- Aird WC. (2012). Endothelial cell heterogeneity. Cold Spring Harb Perspect Med , a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P, Deokule SP, Thompson K, Wizeman J, Srinivasan C, Vooturi S, Kompella UB, Mohan R. (2013). Withaferin A effectively targets soluble vimentin in the glaucoma filtration surgical model of fibrosis. PLoS One , e63881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P, Hamza A, Kim Y-E, Khuan Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan C-G, et al (2007) The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol , 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E, Meili R, Alonso-Latorre B, del Álamo J, Lasheras J, Firtel R. (2011). The SCAR/WAVE complex is necessary for proper regulation of traction stresses during amoeboid motility. Mol Biol Cell , 3995–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E, Meili R, Álvarez-González B, Francois J, del Álamo JC, Firtel RA, Lasheras JC. (2014). Both contractile axial and lateral traction force dynamics drive amoeboid cell motility. J Cell Biol , 1045–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Gonzalez AM, DeBiase PJ, Trejo HE, Goldman RD, Flitney FW, Jones JCR. (2009). Recruitment of vimentin to the cell surface by β3 integrin and plectin mediates adhesion strength. J Cell Sci , 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Cossart P. (2002). InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J Cell Sci , 3357. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Tian X, Cokic I, Beckham Y, Gardel M, Birukov KG. (2013). Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res , 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacher J, Asmar R, Djane S, London GM, Safar ME. (1999). Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension , 1111–1117. [DOI] [PubMed] [Google Scholar]
- Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, Laurent S. (2002). Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension , 10–15. [DOI] [PubMed] [Google Scholar]
- Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. (1993). Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci USA , 11890–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan SG, Hendle J, Lee PS, Smith CR, Bounaud PY, Jessen KA, Tang CM, Huser NH, Felce JD, Froning KJ, et al (2009). SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther , 3181–3190. [DOI] [PubMed] [Google Scholar]
- Burgess-Cassler A, Johansen JJ, Santek DA, Ide JR, Kendrick NC. (1989). Computerized quantitative analysis of coomassie-blue-stained serum proteins separated by two-dimensional electrophoresis. Clin Chem , 2297–2304. [PubMed] [Google Scholar]
- Chi F, Jong TD, Wang L, Ouyang Y, Wu C, Li W, Huang SH. (2010). Vimentin-mediated signalling is required for IbeA+E. coli K1 invasion of human brain microvascular endothelial cells. Biochem J , 79–90. [DOI] [PubMed] [Google Scholar]
- Chien S, Li S, Shiu YT, Li YS. (2005). Molecular basis of mechanical modulation of endothelial cell migration. Front Biosci , 1985–2000. [DOI] [PubMed] [Google Scholar]
- Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, Missiakas D, Schneewind O, Hoylaerts MF, Heying R, Verhamme P. (2014). Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood , 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke EJ, Allan V. (2002). Intermediate filaments: vimentin moves in. Curr Biol , R596–R598. [DOI] [PubMed] [Google Scholar]
- Collins C, Osborne LD, Guilluy C, Chen Z, O’Brien ET, Reader JS, Burridge K, Superfine R, Tzima E. (2014). Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat Commun , 3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig LE, Spelman JP, Strandberg JD, Zink MC. (1998). Endothelial cells from diverse tissues exhibit differences in growth and morphology. Microvasc Res , 65–76. [DOI] [PubMed] [Google Scholar]
- Darie CC, Deinhardt K, Zhang G, Cardasis HS, Chao MV, Neubert TA. (2011). Identifying transient protein–protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry. Proteomics , 4514–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey PA, Wang YL. (2005). Tissue cells feel and respond to the stiffness of their substrate. Science , 1139–1143. [DOI] [PubMed] [Google Scholar]
- Drevets DA, Sawyer RT, Potter TA, Campbell PA. (1995). Listeria monocytogenes infects human endothelial cells by two distinct mechanisms. Infect Immun , 4268–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zu Y, Li J, Du S, Xu Y, Zhang L, Jiang L, Wang Z, Chien S, Yang C. (2016). Extracellular matrix stiffness dictates Wnt expression through integrin pathway. Sci Rep , 20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du N, Cong H, Tian H, Zhang H, Zhang W, Song L, Tien P. (2014). Cell surface vimentin is an attachment receptor for enterovirus 71. J Virol , 5816–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussurget O, Pizarro-Cerda J, Cossart P. (2004). Molecular determinants of Listeria monocytogenes virulence. Annu Rev Microbiol , 587–610. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. (2014). Advanced methods of microscope control using μManager software. J Biol Methods , e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia G. (2012). Cell surface protein biotinylation for SDS–PAGE analysis. Methods Mol Biol , 361–372. [DOI] [PubMed] [Google Scholar]
- Fiedler F. (1988). Biochemistry of the cell surface of Listeria strains: a locating general view. Infection (Suppl 2), S92–S97. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Weber K. (1994). Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem , 345–382. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. (1992). Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J Cell Biol , 1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Barnes PF, Porgador A, Roy S, Wu S, Nanda JS, Griffith DE, Girard WM, Rawal N, Shetty S, Vankayalapati R. (2006). Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. J Immunol , 6192–6198. [DOI] [PubMed] [Google Scholar]
- Gattazzo F, Urciuolo A, Bonaldo P. (2014). Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta , 2506–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. (2009). Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol , 21–33. [DOI] [PubMed] [Google Scholar]
- Georges P, Miller W, Meaney D, Sawyer E, Janmey PA. (2006). Matrices with compliance comparable to that of brain tissue select neuronal over glial growth in mixed cortical cultures. Biophys J , 3012–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Halvorsen EM, Ammendolia DA, Mor-Vaknin N, O’Riordan MXD, Brumell JH, Markovitz DM, Higgins DE. (2018). Invasion of the brain by Listeria monocytogenes is mediated by InlF and host cell vimentin. MBio , e00160–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, et al (2001). Comparative genomics of Listeria species. Science , 849–852. [DOI] [PubMed] [Google Scholar]
- Gopal S, Bober A, Whiteford JR, Multhaupt HA, Yoneda A, Couchman JR. (2010). Heparan sulfate chain valency controls syndecan-4 function in cell adhesion. J Biol Chem , 14247–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco S, Elia MG, Muscella A, Storelli C, Marsigliante S. (2002). AT1 angiotensin II receptor mediates intracellular calcium mobilization in normal and cancerous breast cells in primary culture. Cell Calcium , 1–10. [DOI] [PubMed] [Google Scholar]
- Greiffenberg L, Goebel W, Kim KS, Weiglein I, Bubert A, Engelbrecht F, Stins M, Kuhn M. (1998). Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun , 5260–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiffenberg L, Sokolovic Z, Schnittler H-J, Spory A, Böckmann R, Goebel W, Kuhn M. (1997). Listeria monocytogenes-infected human umbilical vein endothelial cells: internalin-independent invasion, intracellular growth, movement, and host cell responses. FEMS Microbiol Lett , 163–170. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol , R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry-Stanley MJ, Hess DJ, Erickson EA, Garni RM, Wells CL. (2003). Role of heparan sulfate in interactions of Listeria monocytogenes with enterocytes. Med Microbiol Immunol , 107–115. [DOI] [PubMed] [Google Scholar]
- Higuita-Castro N, Mihai C, Hansford DJ, Ghadiali SN. (2014). Influence of airway wall compliance on epithelial cell injury and adhesion during interfacial flows. J Appl Physiol , 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Butler PJ, Tong S, Muddana HS, Bao G, Zhang S. (2013). Substrate stiffness regulates cellular uptake of nanoparticles. Nano Lett , 1611–1615. [DOI] [PubMed] [Google Scholar]
- Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. (2011). Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med , 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC. (2004). Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood , 2752–2760. [DOI] [PubMed] [Google Scholar]
- Ise H, Kobayashi S, Goto M, Sato T, Kawakubo M, Takahashi M, Ikeda U, Akaike T. (2010). Vimentin and desmin possess GlcNAc-binding lectin-like properties on cell surfaces. Glycobiology , 843–864. [DOI] [PubMed] [Google Scholar]
- Jackson KA, Iwamoto M, Swerdlow D. (2010). Pregnancy-associated listeriosis. Epidemiol Infect , 1503–1509. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Miller RT. (2011). Mechanisms of mechanical signaling in development and disease. J Cell Sci , 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhemo P-R, Ravela S, Laakso M, Ritamo I, Tatti O, Mäkinen S, Goodison S, Stenman U-H, Hölttä E, Hautaniemi S, et al (2012). An optimized isolation of biotinylated cell surface proteins reveals novel players in cancer metastasis. J Proteomics , 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatiwala CB, Peyton SR, Putnam AJ. (2006). Intrinsic mechanical properties of the extracellular matrix affect the behavior of pre-osteoblastic MC3T3-E1 cells. Am J Physiol Cell Physiol , C1640. [DOI] [PubMed] [Google Scholar]
- Kirchner M, Higgins DE. (2008). Inhibition of ROCK activity allows InlF-mediated invasion and increased virulence of Listeria monocytogenes. Mol Microbiol , 749–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, Levental I, Hawthorne E, Janmey PA, Assoian RK. (2009). Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol , 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. (1992). L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell , 521–531. [DOI] [PubMed] [Google Scholar]
- Kohn JC, Dennis Zhou W, Bordeleau F, Allen Zhou L, Brooke Mason N, Michael Mitchell J, Michael King R, Reinhart-King CA. (2015). Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophys J , 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolewe KW, Peyton SR, Schiffman JD. (2015). Fewer bacteria adhere to softer hydrogels. ACS Appl Mater Interfaces , 19562–19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka JB. (2012). N-acetylglucosamine (GlcNAc) functions in cell signaling. Scientifica . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothapalli D, Liu S-L, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, et al (2012). Cardiovascular protection by apoE and apoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep , 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VWM, Carman CV, Brain JD, Fredberg JJ, et al (2011). Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol , C146–C154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. (2002). Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol , 4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun M, Mengaud J, Ohayon H, Nato F, Cossart P. (1996). Internalin must be on the bacterial surface to mediate entry of Listeria monocytogenes into epithelial cells. Mol Microbiol , 579–592. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Hurme R, Pizarro-Cerda J, Ohayon H, Geiger B, Cossart P. (2000). A role for alpha-and beta-catenins in bacterial uptake. Proc Natl Acad Sci USA , 10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemichez E, Lecuit M, Nassif X, Bourdoulous S. (2010). Breaking the wall: targeting of the endothelium by pathogenic bacteria. Nat Rev Microbiol , 93–104. [DOI] [PubMed] [Google Scholar]
- Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY. (1997). Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J Biol Chem , 30455–30462. [DOI] [PubMed] [Google Scholar]
- Liu D, Shriver Z, Venkataraman G, El Shabrawi Y, Sasisekharan R. (2002). Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis. Proc Natl Acad Sci , 568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis H, Kakou A, Regnault V, Labat C, Bressenot A, Gao-Li J, Gardner H, Thornton SN, Challande P, Li Z, Lacolley P. (2007). Role of alpha1beta1-integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am J Physiol Heart Circ Physiol , H2597–H2604. [DOI] [PubMed] [Google Scholar]
- Mak TN, Brüggemann H. (2016). Vimentin in bacterial infections. Cells , 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu SV, Aragão KS, Imberty A, Varrot A. (2010). Discoidin I from Dictyostelium discoideum and interactions with oligosaccharides: specificity, affinity, crystal structures and comparison with Discoidin II. J Mol Biol , 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Ohayon H, Gounon P, Mege RM, Cossart P. (1996). E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell , 923–932. [DOI] [PubMed] [Google Scholar]
- Mih JD, Sharif AS, Liu F, Marinkovic A, Symer MM, Tschumperlin DJ. (2011). A multiwell platform for studying stiffness-dependent cell biology. PLoS One , e19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. (2015). Cell-surface vimentin: A mislocalized protein for isolating csVimentin(+) CD133(-) novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer , 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel M, de la Blanca EP, Jimenez E. (2005). Angiotensin II induces focal adhesion kinase/paxillin phosphorylation and cell migration in human umbilical vein endothelial cells. Biochem Biophys Res Commun , 971–978. [DOI] [PubMed] [Google Scholar]
- Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. (2003). Vimentin is secreted by activated macrophages. Nat Cell Biol , 59–63. [DOI] [PubMed] [Google Scholar]
- Niddam AF, Ebady R, Bansal A, Koehler A, Hinz B, Moriarty TJ. (2017). Plasma fibronectin stabilizes Borrelia burgdorferi–endothelial interactions under vascular shear stress by a catch-bond mechanism. Proc Natl Acad Sci USA , E3490–E3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura K, Nagano K, Itagaki C, Taoka M, Okamura N, Yamauchi Y, Sugano S, Takahashi N, Izumi T, Isobe T. (2005). Cell surface labeling and mass spectrometry reveal diversity of cell surface markers and signaling molecules expressed in undifferentiated mouse embryonic stem cells. Mol Cell Proteomics , 1968–1976. [DOI] [PubMed] [Google Scholar]
- Oakley BR, Kirsch DR, Morris NR. (1980). A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem , 361–363. [DOI] [PubMed] [Google Scholar]
- O’Farrell PH. (1975). High resolution two-dimensional electrophoresis of proteins. J Biol Chem , 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Onken MD, Mooren OL, Mukherjee S, Shahan ST, Li J, Cooper JA. (2014). Endothelial monolayers and transendothelial migration depend on mechanical properties of the substrate. Cytoskeleton (Hoboken) , 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski Maggie A, Huang NF, Walker TW, Verwijlen T, Poplawski C, Khoo AS, Cooke JP, Fuller GG, Dunn AR. (2014). Microvascular endothelial cells migrate upstream and align against the shear stress field created by impinging flow. Biophys J , 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Päll T, Pink A, Kasak L, Turkina M, Anderson W, Valkna A, Kogerman P. (2011). Soluble CD44 interacts with intermediate filament protein vimentin on endothelial cell surface. PLoS One , e29305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida SK, Domann E, Rohde M, Muller S, Darji A, Hain T, Wehland J, Chakraborty T. (1998). Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol Microbiol , 81–93. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Wang YL. (1997). Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA , 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerdá J, Cossart P. (2006). Bacterial adhesion and entry into host cells. Cell , 715–727. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. (2009). Matrix density-induced mechanoregulation of breast cell phenotype, signaling, and gene expression through a FAK-ERK linkage. Oncogene , 4326–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SC, Lamason RL, Risca VI, Abernathy E, Welch MD. (2014). Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr Biol , 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart-King CA. (2008). Endothelial cell adhesion and migration. Methods Enzymol , 45–64. [DOI] [PubMed] [Google Scholar]
- Rengarajan M, Hayer A, Theriot JA. (2016). Endothelial cells use a formin-dependent phagocytosis-like process to internalize the bacterium Listeria monocytogenes. PLoS Pathog , e1005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesli C, Mumprecht D, Fau-Neri V, Neri M, Fau-Detmar D, Detmar M. (2008). Identification of the surface-accessible, lineage-specific vascular proteome by two-dimensional peptide mapping. FASEB J , 1933–1944. [DOI] [PubMed] [Google Scholar]
- Rohrbeck A, Schroder A, Hagemann S, Pich A, Holtje M, Ahnert-Hilger G, Just I. (2014). Vimentin mediates uptake of C3 exoenzyme. PLoS One , e101071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA. (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol , a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinummi J, Seppala J, Yli-Harja O, Puhakka JA. (2005). Software for quantification of labeled bacteria from digital microscope images by automated image analysis. Biotechniques , 859–863. [DOI] [PubMed] [Google Scholar]
- Senger DR, Perruzzi CA, Streit M, Koteliansky VE, de Fougerolles AR, Detmar M. (2002). The α(1)β(1) and α(2)β(1) integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am J Pathol , 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Naujokas M, Park M, Ireton K. (2000). InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell , 501–510. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem , 850–858. [DOI] [PubMed] [Google Scholar]
- Shi Z-D, Tarbell JM. (2011). Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann Biomed Eng , 1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigyo M, Kuboyama T, Sawai Y, Tada-Umezaki M, Tohda C. (2015). Extracellular vimentin interacts with insulin-like growth factor 1 receptor to promote axonal growth. Sci Rep , 12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowska I, Dorobantu C, Woods AG, Macovei A, Branza-Nichita N, Darie CC. (2012a). Proteomic analysis of plasma membranes isolated from undifferentiated and differentiated HepaRG cells. Proteome Sci , 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowska I, Gawinowicz MA, Ngounou Wetie AG, Darie CC. (2012b). Disulfide proteomics for identification of extracellular or secreted proteins. Electrophoresis , 2527–2536. [DOI] [PubMed] [Google Scholar]
- Sokolowska I, Woods AG, Gawinowicz MA, Roy U, Darie CC. (2012c). Identification of potential tumor differentiation factor (TDF) receptor from steroid-responsive and steroid-resistant breast cancer cells. J Biol Chem , 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. (2008). Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics , 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling LH, Friedman DW. (1969). Synthesis and mechanical behavior of interpenetrating polymer networks: poly(ethyl acrylate) and polystyrene. J Polym Sci A-2: Polymer Physics , 425–427. [Google Scholar]
- Stroka KM, Aranda-Espinoza H. (2011). Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood , 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Wen Q, Kuhlenschmidt TB, Kuhlenschmidt MS, Janmey PA, Saif TA. (2012). Attenuation of cell mechanosensitivity in colon cancer cells during in vitro metastasis. PLoS One , e50443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsoni AS, Marin TM, Velloso LA, Franchini KG. (2005). RhoA/ROCK signaling is critical to FAK activation by cyclic stretch in cardiac myocytes. Am J Physiol Heart Circ Physiol , H1488–H1496. [DOI] [PubMed] [Google Scholar]
- Trepat X, Lenormand G, Fredberg JJ. (2008). Universality in cell mechanics. Soft Matter , 1750–1759. [Google Scholar]
- Tse JR, Engler AJ. (2010). Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol Chapter 10:Unit 10.16. [DOI] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. (2001). Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev , 584–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent LG, Choi YS, Alonso-Latorre B, del Alamo JC, Engler AJ. (2013). Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol J , 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman BF, Prehoda KE, Scott JA, Peterson FC, Lim WA. (2002). Structure of the N-WASP EVH1 domain-WIP complex: insight into the molecular basis of Wiskott–Aldrich syndrome. Cell , 565–576. [DOI] [PubMed] [Google Scholar]
- Wells RG. (2008). The role of matrix stiffness in regulating cell behavior. Hepatology , 1394–1400. [DOI] [PubMed] [Google Scholar]
- Weng Y-I, Shukla SD. (2002). Angiotensin II activation of focal adhesion kinase and pp60c-Src in relation to mitogen-activated protein kinases in hepatocytes. Biochim Biophys Acta , 285–297. [DOI] [PubMed] [Google Scholar]
- Wilson SL, Drevets DA. (1998). Listeria monocytogenes infection and activation of human brain microvascular endothelial cells. J Infect Dis , 1658–1666. [DOI] [PubMed] [Google Scholar]
- Wood JA, Liliensiek SJ, Russell P, Nealey PF, Murphy CJ. (2010). Biophysical cueing and vascular endothelial cell behavior. Materials , 1620–1639. [Google Scholar]
- Yam PT, Theriot JA. (2004). Repeated cycles of rapid actin assembly and disassembly on epithelial cell phagosomes. Mol Biol Cell , 5647–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zou L, Yang Y, Yuan J, Hu Z, Liu H, Peng H, Shang W, Zhang X, Zhu J, Rao X. (2016). Superficial vimentin mediates DENV-2 infection of vascular endothelial cells. Sci Rep , 38372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Sanchez HM, Hultz M, Yang Z, Bogorad M, Wong AD, Searson PC. (2014). Brain microvascular endothelial cells resist elongation due to curvature and shear stress. Sci Rep , 4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh Y-T, Hur SS, Chang J, Wang K-C, Chiu J-J, Li Y-S, Chien S. (2012). Matrix stiffness regulates endothelial cell proliferation through septin 9. PLoS One , e46889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. (2005). Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton , 24–34. [DOI] [PubMed] [Google Scholar]
- Yu YT-C, Chien S-C, Chen IY, Lai C-T, Tsay Y-G, Chang SC, Chang M-F. (2016). Surface vimentin is critical for the cell entry of SARS-CoV. J Biomed Sci , 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebda N, Dubrovskyi O, Birukov KG. (2012). Focal adhesion kinase regulation of mechanotransduction and its impact on endothelial cell functions. Microvasc Res , 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldovich VB, Robbins JR, Kapidzic M, Lauer P, Bakardjiev AI. (2011). Invasive extravillous trophoblasts restrict intracellular growth and spread of Listeria monocytogenes. PLoS Pathog , e1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieman SJ, Melenovsky V, Kass DA. (2005). Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol , 932–943. [DOI] [PubMed] [Google Scholar]
- Zou Y, He L, Huang SH. (2006). Identification of a surface protein on human brain microvascular endothelial cells as vimentin interacting with Escherichia coli invasion protein IbeA. Biochem Biophys Res Commun , 625–630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.