

Abstract
CK1 protein kinases contribute to multiple biological processes, but how they are tailored to function in compartmentalized signaling events is largely unknown. Hhp1 and Hhp2 (Hhp1/2) are the soluble CK1 family members in Schizosaccharomyces pombe. One of their functions is to inhibit the septation initiation network (SIN) during a mitotic checkpoint arrest. The SIN is assembled by Sid4 at spindle pole bodies (SPBs), and though Hhp1/2 colocalize there, it is not known how they are targeted there or whether their SPB localization is required for SIN inhibition. Here, we establish that Hhp1/2 localize throughout the cell cycle to SPBs, as well as to the nucleus, cell tips, and division site. We find that their catalytic domains but not their enzymatic function are used for SPB targeting and that this targeting strategy is conserved in human CK1δ/ε localization to centrosomes. Further, we pinpoint amino acids in the Hhp1 catalytic domain required for SPB interaction; mutation of these residues disrupts Hhp1 association with the core SPB protein Ppc89, and the inhibition of cytokinesis in the setting of spindle stress. Taken together, these data have enabled us to define a molecular mechanism used by CK1 enzymes to target a specific cellular locale for compartmentalized signaling.
INTRODUCTION
Casein kinases are among the most abundant serine/threonine protein kinases found in eukaryotic cells (Desjardins et al., 1972; Matsumura and Takeda, 1972; Hathaway and Traugh, 1979; Tuazon and Traugh, 1991; Carpy et al., 2014) and generally recognize substrate motifs consisting of acidic or phosphorylated amino acid residues (Agostinis et al., 1989; Flotow et al., 1990; Flotow and Roach, 1991; Meggio et al., 1991, 1992; Graves et al., 1993). Members of the casein kinase 1 (CK1) family have been implicated in multiple cellular processes, including regulation of autophagy, DNA repair, circadian rhythm, ribosome assembly, intracellular trafficking, meiotic progression, and Wnt signaling (Knippschild et al., 2005; Schittek and Sinnberg, 2014; Ghalei et al., 2015; Nakatogawa, 2015). CK1 kinases share highly related catalytic domains (53–98% sequence identity) that consist of a bilobed structure with a smaller N-terminal lobe consisting primarily of β-sheets and a larger, primarily α-helical C-terminal lobe (Carmel et al., 1994; Xu et al., 1995; Longenecker et al., 1996). However, CK1 family members have divergent C-terminal noncatalytic domains that are thought to dictate intracellular localization and govern catalytic activity (Graves and Roach, 1995; Longenecker et al., 1996, 1998; Cegielska et al., 1998; Gietzen and Virshup, 1999; Babu et al., 2002; Dahlberg et al., 2009; Greer and Rubin, 2011; Ianes et al., 2015; Meng et al., 2016). Indeed, some CK1 isoforms are anchored to membranes via C-terminal palmitoylation (Wang et al., 1992; Vancura et al., 1994; Babu et al., 2004; Sun et al., 2004).
Hhp1 and Hhp2 (hereafter referred to as Hhp1/2) are the sole soluble CK1s in Schizosaccharomyces pombe and are orthologues of Saccharomyces cerevisiae Hrr25p and human CK1δ and CK1ε (hereafter referred to as CK1δ/ε) (Dhillon and Hoekstra, 1994; Hoekstra et al., 1994). In addition to functioning redundantly in meiosis, DNA damage repair, and mitotic commitment (Dhillon and Hoekstra, 1994; Sakuno and Watanabe, 2015; Chan et al., 2017), Hhp1/2 are essential for preventing cytokinesis during a mitotic stress imposed by microtubule depolymerization (Johnson et al., 2013). In this checkpoint pathway, Hhp1/2 function redundantly upstream of the ubiquitin ligase Dma1 to inhibit the septation initiation network (SIN) (Murone and Simanis, 1996; Guertin et al., 2002b), a hippo-related signaling pathway localized at the spindle pole body (SPB) that triggers cytokinesis (Johnson and Gould, 2011; Johnson et al., 2012; Simanis, 2015). Specifically, Hhp1/2 phosphorylate the SIN scaffold Sid4 at T275 and S278 to create a docking site for Dma1’s FHA domain (Johnson et al., 2013). Dma1 then concentrates at SPBs and ubiquitinates Sid4, also a scaffold for Polo-like kinase Plo1, to inhibit Plo1’s SPB localization and ability to activate the SIN (Guertin et al., 2002; Johnson and Gould, 2011).
In addition to other subcellular localizations, Hhp1/2, as well as Hrr25p and human CK1δ/ε, localize to the SPB or its equivalent in vertebrates, the centrosome (Hutchins et al., 2010; Greer and Rubin, 2011; Johnson et al., 2013; Peng et al., 2015b). Interaction with AKAP450 and the γ-tubulin complex is reported to be important for anchoring CK1δ and Hrr25p to the centrosome and SPB, respectively (Sillibourne et al., 2002; Peng et al., 2015b). However, the residues within CK1 enzymes required for SPB/centrosome recruitment are unknown, as is the SPB tether in S. pombe.
In this work, we establish that Hhp1/2 localize throughout the cell cycle to the SPB and also to the nucleus and cell division site, with Hhp2 showing additional localization to cell tips. We report that the catalytic domains of Hhp1/2 are sufficient to support this localization pattern independently of enzymatic function. Further, we define conserved residues at the base of the Hhp1 catalytic domain necessary for SPB localization. Mutation of these residues eliminates Hhp1’s SPB localization and mitotic checkpoint function by disrupting the interaction with the core SPB protein Ppc89 (Rosenberg et al., 2006) but does not affect other critical Hhp1 functions. We also find that the centrosomal targeting information in human CK1δ/ε is analogously contained within their respective kinase domains, indicating conservation of this localization mechanism from yeast to humans. This study provides novel insight into how specific docking residues within the catalytic domain of protein kinases can direct them to subcellular locations to control substrate phosphorylation and downstream signaling.
RESULTS
Hhp1/2 localization during a normal and perturbed cell cycle
Both Hhp1 and Hhp2 localize to SPBs, and either is sufficient to promote the Dma1-mediated mitotic checkpoint that delays cytokinesis (Johnson et al., 2013). For determination of their intracellular distribution throughout the cell cycle and during a mitotic arrest, each was tagged at its endogenous C-terminus with mNeonGreen (mNG) (Shaner et al., 2013; Willet et al., 2015) and coimaged with the SPB protein Sid4-RFP (Chang and Gould, 2000). Both Hhp1 and Hhp2 localize to the nucleus and the division site in addition to SPBs; Hhp2 was also detected at cell tips (Figure 1A). By immunoblotting whole-cell lysates from a strain producing both enzymes tagged with green fluorescent protein (GFP), we determined that Hhp1 is more abundant than Hhp2 (Supplemental Figure S1A), consistent with the relative protein levels determined by quantitative mass spectrometry (Marguerat et al., 2012; Carpy et al., 2014) and the more severe growth defects and phenotypes associated with the deletion of hhp1+ relative to hhp2+ (Supplemental Figure S1, B–D) (Dhillon and Hoekstra, 1994; Hoekstra et al., 1994; Bimbo et al., 2005; Carpy et al., 2014; Chen et al., 2015). To better define the localization of Hhp1/2 during the mitotic checkpoint signaling window, we performed time-lapse imaging of cells progressing through mitosis. Hhp1-mNG and Hhp2-mNG localized to the SPB and nucleus throughout mitosis, and also to the division site following the completion of spindle elongation with comparable timing (Supplemental Figure S1, E and F). These data indicate that the spatial and temporal distributions of Hhp1 and Hhp2 are similar but not identical.
FIGURE 1:
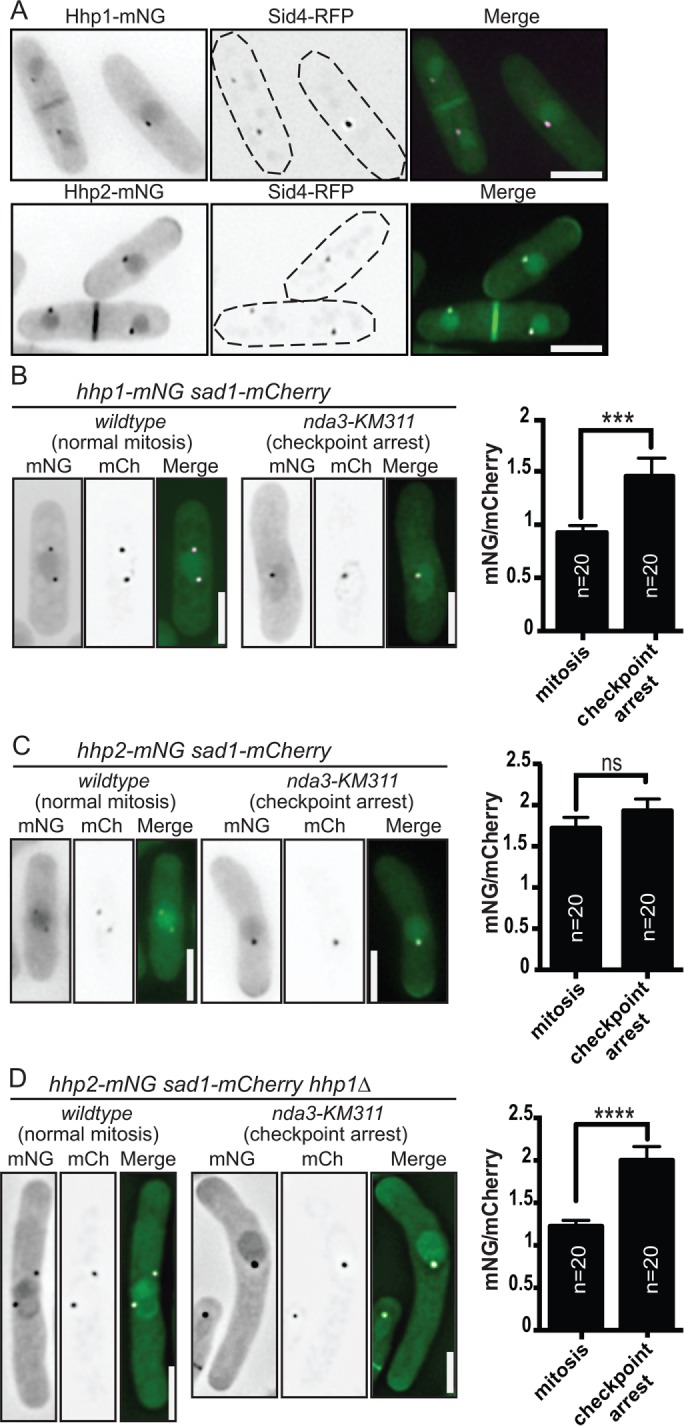
Intracellular localization patterns of Hhp1/2. (A) Live-cell imaging of Hhp1-mNG and Hhp2-mNG with Sid4-RFP. (B–D) Hhp1-mNG (B) and Hhp2-mNG (C, D) were imaged in prometaphase-arrested and wild-type mitotic sad1-mCherry or sad1-mCherry hhp1∆ cells. Representative inverted grayscale images are shown in the left panels with quantitation to the right. mCh, mCherry. Scale bars: 5 µm. Values are represented as mNG/mCherry intensity ratios. ***, p < 0.005, ****, p < 0.001 determined using Student’s t test. ns, not significant. Error bars represent SEM.
When the mitotic checkpoint is activated by preventing microtubule polymerization in the nda3-KM311β-tubulin mutant (Toda et al., 1983), Sid4 is phosphorylated by Hhp1/2, and Hhp1 increases in intensity at SPBs (Johnson et al., 2013) (Figure 1B). By coimaging with the SPB marker Sad1-mCherry in checkpoint-activated and unperturbed mitotic cells, we found that Hhp2 intensity at duplicated but unseparated SPBs was also increased in checkpoint-activated cells, but only if they lacked Hhp1 (Figure 1, C and D). These results indicate that both Hhp1/2 can accumulate at SPBs during the checkpoint.
Hhp1/2 catalytic domains direct SPB localization and checkpoint function
To determine the mechanism of Hhp1/2 SPB targeting, we first defined the regions of the enzymes necessary for this localization. Hhp1/2 contain conserved N-terminal catalytic domains and kinase domain extensions (KDEs) that are conserved among mammalian but not S. cerevisiae CK1 family members (reviewed in Knippschild et al., 2014) (Figure 2A). Given that both Hhp1/2 localize to SPBs and participate in checkpoint signaling, we anticipated that homologous sequences dictated these functions and that their unrelated C-termini were not involved. Accordingly, Hhp1/2 C-terminal truncations, constructed by inserting sequences encoding GFP in the endogenous hhp1/2 loci to produce Hhp1-(1-296)-GFP and Hhp2-(1-295)-GFP (Figure 2B), both colocalized with Sid4-RFP at SPBs (Figure 2C). Moreover, Hhp1-(1-296)-GFP and Hhp2-(1-295)-GFP recapitulated all other subcellular localizations of the full-length proteins (Figure 2C). The C-terminus of Hhp1 fused to GFP was not targeted to any particular subcellular location, although it was produced in cells (Supplemental Figure S2, A and B), indicating that the C-terminus is neither necessary nor sufficient for SPB localization of Hhp1/2.
FIGURE 2:

The C-termini of Hhp1/2 are dispensable for their functions. (A) Schematic diagrams of Hhp1/2 with relative positions of N-terminal extensions (N, green), kinase domains (blue), kinase domain extensions (KDE, yellow), and unrelated C-termini in red or purple indicated, drawn to scale. (B) Anti-GFP immunoblots of whole-cell extracts prepared from untagged and the indicated GFP-tagged strains. Anti-PSTAIRE (Cdc2) immunoblots served as protein loading controls. (C) Live-cell imaging of endogenously tagged Hhp1-(1-296)-GFP and Hhp2-(1-295)-GFP with Sid4-RFP. Scale bars: 5 µm. (D) Serial 10-fold dilutions of the indicated strains were spotted on YE plates and incubated at the indicated temperatures. (E) In vitro kinase assays of recombinant MBP-Hhp1, MBP-Hhp1-(1-296), MBP-Hhp2, and MBP-Hhp2-(1-295) detected by Coomassie blue (CB) staining of SDS–PAGE gels, with casein as substrate. Phosphorylated casein was detected by autoradiography (32P). (F) Sid4 from the indicated strains was immunoprecipitated from denatured cell lysates, treated with phosphatase, and visualized by immunoblotting.
To ascertain the functionality of the C-terminal truncation mutants of Hhp1/2, we performed growth, in vitro kinase, and mitotic checkpoint assays. First, we found that each of the hhp1/2 C-terminal truncation mutants integrated as the sole hhp1/2 allele in cells rescued the severe growth defect of the double-deletion mutant in vivo (Figure 2D). Consistent with this finding and previous reports (Graves and Roach, 1995; Cegielska et al., 1998; Gietzen and Virshup, 1999), recombinant Hhp1/2 C-terminal truncation mutants phosphorylated the exogenous substrate casein in vitro even more robustly than the full-length proteins (Figure 2E). Importantly, we found that the truncation mutants were sufficient to inhibit the SIN during the mitotic checkpoint. GFP-tagged Hhp1 and Hhp2 C-terminal truncations accumulated at SPBs during a mitotic checkpoint arrest (Supplemental Figure S2, C and D), and Sid4 was appropriately ubiquitinated (Figure 2F). Taken together, these data indicate that the C-termini of Hhp1/2 are dispensable for their SPB localization and function during the Dma1-mediated mitotic checkpoint.
Because the KDE of CK1δ was reported to mediate its centrosomal localization (Greer and Rubin, 2011), we tested whether Hhp1/2 KDEs influence SPB targeting. Further C-terminal truncations of Hhp1/2 were made such that only the predicted catalytic domains (Hhp1-(1-280)-GFP and Hhp2-(1-278)-GFP) were produced from endogenous loci (Supplemental Figure S3A). While these Hhp1/2 truncation mutants were still SPB targeted, as evidenced by colocalization with Sid4-RFP (Supplemental Figure S3B), functional tests revealed that they only partially rescued the severe growth defect of the double-deletion mutant in vivo (Supplemental Figure S3C), and they did not support mitotic checkpoint signaling as assayed by Sid4 ubiquitination (Supplemental Figure S3D). This loss of in vivo function is likely due to significantly reduced kinase activity (Supplemental Figure S3, E and F). We conclude that, while the KDEs of Hhp1/2 are required for their full enzymatic function, SPB targeting information is contained within the Hhp1/2 catalytic domains.
Hhp1/2 localize to SPBs independent of catalytic activity
It was previously reported that the kinase activity of CK1δ and of Hrr25 is necessary for their proper centrosomal and SPB localization, respectively (Milne et al., 2001; Peng et al., 2015a,b). Because the Hhp1-(1-280)-GFP and Hhp2-(1-278)-GFP mutants had severely reduced activity but were still able to localize, we hypothesized that, in contrast, Hhp1/2 kinase activity may not impact their SPB targeting. We generated catalytically inactive mutants of each enzyme (Hhp1-K40R and Hhp2-K41R) and verified their inactivity in vitro (Figure 3A). When produced at the endogenous loci as sole gene copies tagged with mNG in cells, these mutants mimicked null alleles in growth and Sid4 ubiquitination assays (Supplemental Figure S4, A and B), although they were produced at wild-type levels as determined by immunoblotting (Figure 3B) and by measuring whole-cell fluorescence intensities (Supplemental Figure S4, C and D). Coimaging of Sid4-RFP and Hhp1-(K40R)-mNG or Hhp2-(K41R)-mNG showed that both catalytically inactive mutants localized to SPBs (Figure 3C), indicating that Hhp1/2 kinase activity is not necessary for SPB localization. Furthermore, Hhp1-(K40R)-mNG colocalized with Sid4-RFP in a hhp2∆ background, and Hhp2-(K41R)-mNG colocalized with Sid4-RFP in a hhp1∆ background. Taken together, these data indicate that hhp2 is not required for the SPB localization of Hhp1 and vice versa.
FIGURE 3:
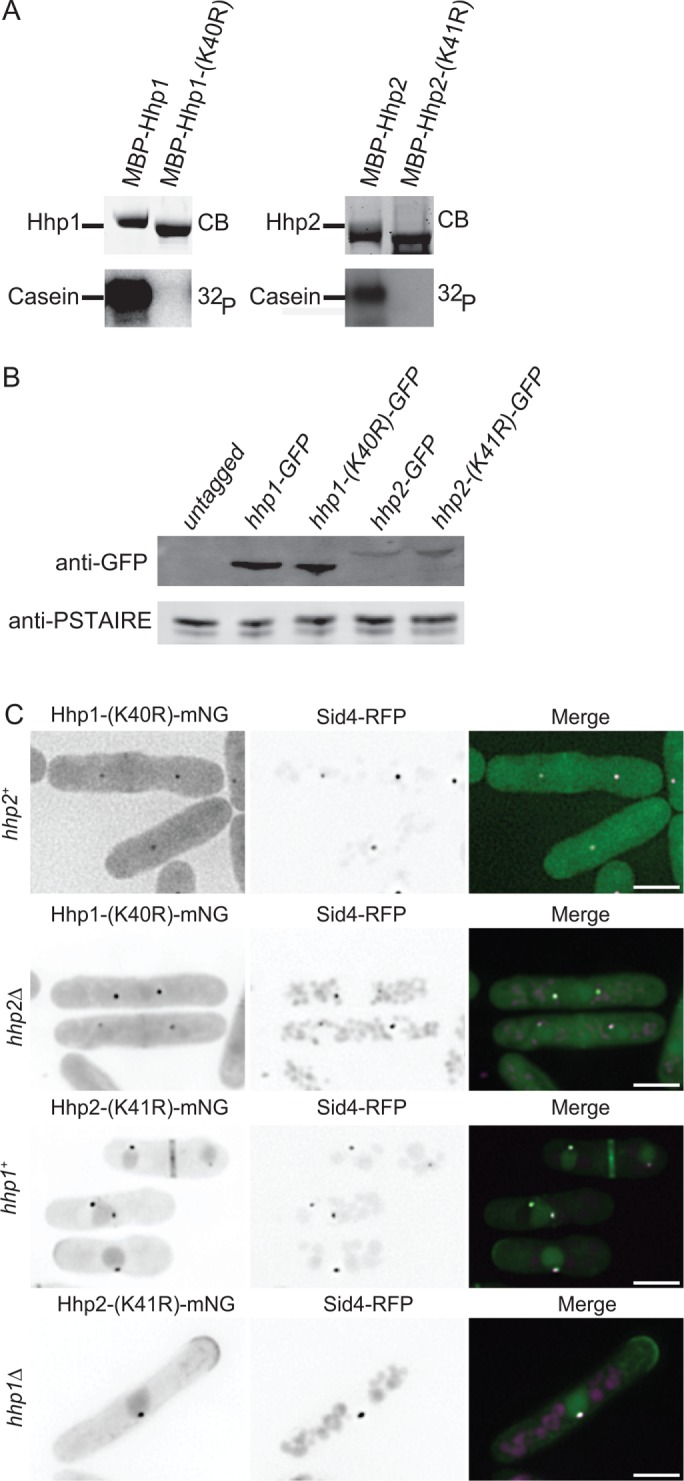
Kinase activity of Hhp1/2 is not required for SPB localization. (A) In vitro kinase assays of recombinant MBP-Hhp1-(K40R) and MBP-Hhp2-(K41R) detected by CB staining of SDS–PAGE gels, with casein as substrate. Phosphorylated casein was detected by autoradiography. (B) Anti-GFP immunoblot of whole-cell extracts prepared from untagged and the indicated GFP-tagged strains. Anti-PSTAIRE antibody served as loading control for lysates. (C) Live-cell imaging of endogenously tagged Hhp1-(K40R)-mNG with Sid4-RFP in wildtype and hhp2∆ cells along with Hhp2-(K41R)-mNG with Sid4-RFP in wildtype and hhp1∆ cells. Scale bars: 5 µm.
The kinase domains of CK1δ/ε dictate centrosomal localization
CK1δ/ε, the vertebrate orthologues of Hhp1/2, localize to the centrosome (Milne et al., 2001; Hutchins et al., 2010) (Figure 4A). To determine whether the intrinsic cues necessary for Hhp1/2 SPB localization are conserved in CK1δ/ε, we made C-terminal truncations of CK1δ/ε analogous to those of Hhp1/2 that did or did not contain the KDE (Supplemental Figure S5A). These truncation mutants or wild-type versions of the proteins were then expressed in RPE-1 cells as N-terminal GFP fusions (Figure 4A and Supplemental Figure S5B). In the majority of transfected cells, all three versions of the proteins colocalized with the centrosomal marker γ-tubulin, although truncation of the KDE from either protein resulted in a lower percentage of centrosomal localization compared with wild type or truncations including the KDEs (Figure 4, A and B). These data support the idea that, as in Hhp1/2, the centrosome-targeting information is contained within the CK1δ/ε kinase domains, and we hypothesize that the KDE may impart stability to their association with centrosomes.
FIGURE 4:

The centrosomal targeting information of CK1δ/ε is located within the kinase domain. RPE-1 cells were transiently transfected with GFP N-terminal fusions to full length or truncations of CK1δ and CK1ε. Cells were fixed with 100% methanol and stained with γ-tubulin (magenta) and DAPI (blue). (A) Localization of full-length protein and truncation mutants in RPE-1 cells. (B) Quantification of the colocalization between γ-tubulin and full-length proteins or truncation mutants. (C) Localization of full-length GFP-CK1δ and GFP-CK1ε catalytically inactive mutants. (D, E) Quantification of the colocalization between γ-tubulin and GFP-CK1δ (D) or GFP-CK1ε (E) wild type or catalytically inactive mutants. For B, D, and E, 100 cells per experiment. n = 3. *, p < 0.05, **, p < 0.01, ***, p < 0.005, ****, p < 0.001, p values determined using ANOVA; ns, not significant. Error bars represent SEM. Scale bars: 15 μm.
We next tested whether kinase activity was important for CK1δ/ε centrosomal localization. K38R mutations in both enzymes render them inactive (Gietzen and Virshup, 1999) (Supplemental Figure S5C). When these mutations were expressed in RPE-1 cells as GFP fusions (Supplemental Figure S5D), both colocalized at centrosomes with γ-tubulin to the same extent as wild type (Figure 4, C–E), indicating that protein kinase activity is dispensable for their centrosomal targeting. Taken together, these data indicate that soluble CK1 family members use their catalytic domains to associate with the major microtubule organizing centers.
Residues in the C-terminal lobe of the Hhp1 kinase domain are required for SPB localization
To probe the mechanism by which one of these enzymes targets spindle poles, we identified residues in the Hhp1 catalytic domain that were necessary. To do this, we performed a comparative analysis of Hhp1 and its paralogue, Cki2. Cki2 does not localize to SPBs; Cki2 C-terminally tagged with GFP localized to vacuolar membranes (Supplemental Figure S6, A and B, top), as previously reported (Matsuyama et al., 2006). Furthermore, a C-terminally tagged mutant of Cki2 lacking the predicted palmitoylation sites (Cki2-(1-429)-GFP) (Sun et al., 2004) localized to the nucleus, cytoplasm, and division site, but it still did not localize to SPBs (Supplemental Figure S6, A and B, bottom), indicating that Cki2 lacks SPB-targeting information.
To detect differences between Hhp1 and Cki2, we generated structural models of the kinase domains of Hhp1 and Cki2 (Figure 5A and Supplemental Figure S6C) using the Protein Homology/analogY Recognition Engine v. 2.0 (Phyre2) (Kelley et al., 2015). A comparison revealed 19 surface residues of Cki2 that differed in charge potential from analogous residues on Hhp1 (Supplemental Figure S6, C and D). To determine whether any of these residues mediate SPB localization, the corresponding residues in Hhp1 were mutated to either mimic the Cki2 residue or reverse the charge of the Hhp1 residue, and the resultant mutants were integrated at the hhp1 endogenous locus and tagged at their C-termini with mNG. Of the 14 mutants tested, only Hhp1-(R261E)-mNG and Hhp1-(R272E K273E)-mNG failed to localize to SPBs (Figure 5B and Supplemental Figure S6D). Hhp1-(R261E)-mNG and Hhp1-(R272E K273E)-mNG were still recruited to other subcellular locations, including the nucleus and division site. Both mutant proteins were produced at levels similar to those of wild type (Figure 5C). Interestingly, these mutants retained function, as measured by their ability to support wild-type S. pombe growth and phosphorylate casein in vitro (Supplemental Figure S6, E and F).
FIGURE 5:

Residues at the base of the Hhp1 catalytic domain are critical for SPB localization. (A) Homology model of Hhp1 catalytic domain generated from Phyre2 software and visualized with MacPymol. Residues critical for Hhp1 SPB localization are in blue. (B) Live-cell imaging of endogenously tagged Hhp1-(R261E)-mNG and Hhp1-(R272E K273E)-mNG with Sid4-RFP. Scale bars: 5 μm. (C) Anti-GFP immunoblot of whole-cell extracts prepared from the indicated strains. Anti-PSTAIRE immunoblots served as protein loading controls.
Interaction with Ppc89 mediates Hhp1/2 SPB localization
Though Sid4 is an SPB-localized Hhp1/2 target (Johnson et al., 2013), Sid4 is not required for Hhp1 (Johnson et al., 2013) or Hhp2 (Supplemental Figure S7A) SPB localization, and it is not known which protein(s) tethers Hhp1/2 to SPBs. However, Hhp1/2 copurify with multiple SPB proteins, including a central component of the SPB, Ppc89, which also secures Sid4 at the SPB (Rosenberg et al., 2006; Johnson et al., 2013). Ppc89 is required for Hhp1 (Johnson et al., 2013) and Hhp2 (Supplemental Figure S7B) SPB localization, and ppc89 interacts with hhp1 (Vo et al., 2016) and hhp2 in a yeast two-hybrid assay (Figure 6, A–C). We further determined that a C-terminal truncation of Ppc89 supports two-hybrid interactions with hhp1/2 (Figure 6A), indicating that the interaction site is distinct from that of Sid4, which interacts with the Ppc89 C-terminus (Rosenberg et al., 2006).
FIGURE 6:

Basic residues within the kinase domain of Hhp1 are critical for interaction with the SPB protein Ppc89. (A, B) S. cerevisiae strain PJ69-4A was cotransformed with plasmids expressing hhp1/2 or their variants, hhp1-(1-296), or hhp2-(1-295) and indicated sections of ppc89. KD = kinase domain. Black boxes in A indicate regions of predicted coiled-coil. Transformants were scored for growth on −Trp −Leu −His plates supplemented with 5 mM 3-AT. Pluses indicate strong growth and minuses indicate no growth. (C) Bar graph shows β-galactosidase activity of the indicated bait and prey plasmids tested for growth in B (represented in relative light units). Each assay was performed in triplicate. ****, p < 0.001 determined using ANOVA. Error bars represent SEM.
We next tested whether the residues that were critical for Hhp1 SPB localization affected interaction with Ppc89. We found that neither hhp1-R261E nor hhp1-R272E K273E interacted with ppc89 in the two-hybrid assay (Figure 6, B and C). Thus, these residues likely mediate an interaction with Ppc89, which may position Hhp1/2 proximal to their mitotic checkpoint substrate, Sid4.
Hhp1/2 SPB localization is required for checkpoint signaling
To test the hypothesis that CK1 must be present at SPBs to execute its checkpoint function, we examined the ability of Hhp1-R261E and Hhp1-R272E K273E to prevent septation when a mitotic checkpoint is imposed. Because either Hhp1 or Hhp2 can redundantly activate the Dma1-mediated mitotic checkpoint, we combined hhp1-R261E and hhp1-R272E K273E with an hhp2∆ mutant to analyze mitotic checkpoint function. We found that the tubulin mutant nda3-KM311, nda3-KM311 hhp1-(R272E K273E) and nda3-KM311 hhp1-(R261E) delayed septation, whereas nda3-KM311 hhp1-(R272E K273E) hhp2∆ and nda3-KM311 hhp1-(R261E) hhp2∆ could not hold the arrest (Figure 7, A and B). Because of its ability to inhibit the SIN via Sid4 ubiquitination, overexpression of dma1 is lethal (Guertin et al., 2002); however, hhp1-(R261E) hhp2∆ and hhp1-(R272E K273E) hhp2∆ mutants were refractory to dma1 overexpression–induced lethality (Figure 7C). Also consistent with checkpoint failure, Sid4 ubiquitination was not detected in checkpoint-activated hhp1-(R261E) hhp2∆ or hhp1-(R272E K273E) hhp2∆ cells (Figure 7D). Taken together, these data indicate that Hhp1/2 localization to SPBs is critical for their role in mitotic checkpoint signaling.
FIGURE 7:

Hhp1 SPB localization is required for mitotic checkpoint function. (A, B) The indicated strains were synchronized in S phase with hydroxyurea and shifted to 19°C to activate the spindle checkpoint, and septation indices were measured periodically for 9 h. (C) Overexpression of dma1 from the nmt41 promoter in wild type, hhp1-(R261E) hhp2∆, or hhp1-(R272E K273E) hhp2∆ mutant cells. Growth of the transformants was observed on agar plates in the presence (repression) or absence (derepression) of thiamine. (D) Sid4 from the indicated strains was immunoprecipitated from denatured cell lysates, treated with phosphatase, and visualized by immunoblotting.
DISCUSSION
The mechanisms governing CK1 enzyme targeting to specific intracellular locales are not well defined. The S. pombe CK1 enzymes Hhp1/2 are the most upstream components yet identified in the Dma1-mediated mitotic checkpoint that stalls cytokinesis when the mitotic spindle is disrupted. To define how they access their SPB substrate Sid4 in this pathway, we determined how they localize to SPBs and whether this was an essential feature of the checkpoint pathway. We found that Hhp1/2 catalytic domains but not enzymatic activities provide the localization cue and that this targeting strategy is conserved in human CK1δ/ε localization to centrosomes. In further probing the specific requirements for spindle pole localization in one of these enzymes, we identified positively charged residues at the base of the Hhp1 catalytic domain that support interaction with a key SPB scaffold, Ppc89. This interaction is necessary for Hhp1 SPB association, Sid4 phosphorylation, and a mitotic checkpoint response, but not for other functions carried out by this enzyme to promote cell proliferation. Our findings reveal a conserved mechanism by which CK1 enzymes can be tailored to perform a specific function at a discrete subcellular location and time.
Hhp1/2 have overlapping but not identical subcellular localizations
Hhp1/2 have redundant roles in the Dma1-dependent mitotic checkpoint (Johnson et al., 2013), and deletion of either enzyme also renders cells sensitive to multiple types of DNA-damaging agents (Dhillon and Hoekstra, 1994; Bimbo et al., 2005; Chen et al., 2015), observations that are congruent with these enzymes having similar functions. However, in addition to detecting Hhp1/2 in the nucleus, in accord with their known roles in DNA damage repair, and at SPBs and the cell division site, we found that Hhp2 but not Hhp1 is present at cell tips. This observation raises the possibility that these enzymes could have, in addition to common functions, variable, specialized roles that remain to be determined.
Kinase domains mediate spindle pole recruitment of Hhp1/2 and CK1δ/ε
Because neither hhp1 nor hhp2 are essential genes (the double-deletion mutant can also be maintained; e.g., Figure 2D), we were able to clearly define the requirements for their SPB localization without the potentially confounding effects of overexpression and/or the presence of wild-type enzymes. By making gene-replacement strains producing C-terminal truncations, we pinpointed the SPB-targeting information of Hhp1/2 to within their kinase domains. In contrast, the noncatalytic C-terminus of S. cerevisiae Hrr25 was reported to be necessary for SPB localization (Peng et al., 2015b). This difference may be explained by a “central domain” in the Hrr25 kinase that is not conserved in the S. pombe or mammalian CK1 enzymes and that has been found to confer functions specific to Saccharomyces species (Ye et al., 2016). Interestingly, we found that the cell tip localization of Hhp2 is also supported by its kinase domain. Because of the high degree of sequence similarity between the Hhp1/2 kinase domains (Dhillon and Hoekstra, 1994), it seems likely that a subtle difference in the enzymes’ structures imparts this distinction in localization pattern.
Conflicting evidence has been reported regarding the requirements for CK1δ/ε centrosomal targeting. While one report indicated that the kinase domain of human CK1δ is necessary and sufficient for proper subcellular localization (Milne et al., 2001), another suggested that a C-terminal centrosomal localization sequence (equivalent to what we have termed the KDE) in mouse CK1δ was necessary and sufficient for centrosomal localization (Greer and Rubin, 2011). Here, we found that the catalytic domains of CK1δ/ε (lacking the KDEs) are sufficient for centrosome targeting, although constructs containing the KDEs are more stably localized.
In the course of characterizing the hhp1/2 C-terminal truncation mutants, we found that removal of the C-terminal tails, but not the KDEs, led to increased catalytic activity toward an exogenous substrate in vitro. These findings agree with previous results demonstrating that phosphorylation of the C-terminal noncatalytic domain by intramolecular autophosphorylation inhibits CK1 catalytic activity (Graves and Roach, 1995; Longenecker et al., 1996, 1998; Cegielska et al., 1998; Gietzen and Virshup, 1999). Interestingly, hhp1-(1-296) and hhp2-(1-295) did not display any growth defects, signifying that the autoinhibition observed in vitro may not be essential in vivo, or that hypermorphic Hhp1/2 activity is not detrimental to cells. Removal of the KDEs of Hhp1/2, however, led to decreased function in vitro and in vivo. It is possible that the KDEs interact with the C-terminal lobe of the kinase domain, as does the central domain of Hrr25p (Ye et al., 2016). Such an interaction could influence the structure and function of Hhp1/2 and CK1δ/ε, thereby explaining defects in both localization and activity of mutants lacking KDEs.
Hhp1/2 and CK1δ/ε localize to spindle poles independently of kinase activity
We found that Hhp1/2 and CK1δ/ε do not require kinase activity for SPB or centrosomal localization. This differs from what has been reported for Hrr25 (Peng et al., 2015a,b) and one report on CK1δ (Milne et al., 2001). However, our results agree with another report (Qi et al., 2015) indicating that CK1δ/ε kinase activity is dispensable for centrosomal targeting. Because Hhp1/2 are the only protein kinases that function upstream of Dma1 in the Dma1-mediated mitotic checkpoint (Johnson et al., 2013), our results indicate that Hhp1/2 do not need to phosphoprime their tether(s) at spindle poles. Thus, to accumulate at SPBs/centrosomes, these enzymes may use scaffolds whose accessibility or abundance is modulated.
Notably, only one scaffolding protein, AKAP450, has been implicated in mediating CK1 localization to centrosomes (Sillibourne et al., 2002). In other signaling pathways, scaffolds are essential organizing platforms that recruit a kinase and its substrate to the same intracellular location. For example, mitogen-activated protein kinase cascades and protein kinase A pathways depend on scaffolding proteins to maintain kinase specificity (Schwartz and Madhani, 2004). In the context of Hhp1/2, which are involved in multiple cellular processes and have potentially constitutive activity, binding the scaffold Pcp89 may compartmentalize a subset of Hhp1/2 from a wider cellular population to direct CK1 enzymes to a specialized signaling complex. Therefore, interaction with Ppc89 may 1) provide an additional level of specificity by localizing Hhp1/2 and its substrate Sid4 to the appropriate site of action, 2) generate a stronger signal by concentrating Hhp1/2 and Sid4, and/or 3) allow for coordination of the Hhp1/2-Dma1-dependent mitotic checkpoint with other signaling pathways that are centered at the SPB.
Defining the CK1 SPB-binding interface
Our results, along with previous work, indicate that CK1 catalytic domains moonlight as protein interaction domains for kinase localization. Hhp1 requires conserved basic residues on its C-terminal lobe to interact with the SPB. In only one prior case have residues in the catalytic domain of CK1 enzymes been implicated in docking interactions. Specifically, residues in the N-terminal lobe of the Hrr25p catalytic domain, together with its central domain, bind the monopolin subunit Mam1 and promote phosphorylation of monopolin’s kinetochore receptor Dsn1 (Petronczki et al., 2006; Ye et al., 2016). Our findings therefore represent an important advance for the understanding of CK1 kinase domain–mediated subcellular targeting. Though new for CK1, several other catalytic domains of broadly acting protein kinases are known to associate with scaffolds. As one example, the MAPK Fus3 is recruited and also allosterically regulated by the scaffold protein Ste5 (Choi et al., 1994; Bhattacharyya et al., 2006). Thus, Ppc89 may not only scaffold Hhp1/2 at the SPB but also modulate their catalytic activity.
Our work demonstrates that CK1 interaction with SPBs is essential for mitotic checkpoint function in S. pombe. Similarly, CK1δ centrosomal localization has been reported to be required for Wnt-3a–dependent neuritogenesis and proper ciliogenesis (Greer and Rubin, 2011; Greer et al., 2014); thus, specific subcellular localization of CK1 enzymes is essential for mediating proper cellular signaling. Future work on the role of these enzymes at the SPB/centrosome is expected to reveal additional functions that are coordinated from this important cellular signaling nexus.
MATERIALS AND METHODS
Yeast strains, media, and genetic methods
Schizosaccharomyces pombe strains used in this study (Supplemental Table S1) were grown in yeast extract (YE) media (Moreno et al., 1991). Crosses were performed in glutamate medium (Moreno et al., 1991), and strains were constructed by tetrad analysis. hhp1, hhp1-K40R, hhp1-R261E, hhp1-R272E K273E, hhp2, and cki2 were tagged endogenously at the 3′ end of their open reading frames (ORFs) with GFP:kanR or mNG:kanR using pFA6 cassettes as previously described (Bahler et al., 1998). G418 (100 μg/ml; Sigma-Aldrich, St. Louis, MO) in YE media was used for selecting kanR cells. mNG, a recently reported GFP derived from the lancelet Branchiostoma lanceolatum, was chosen for imaging experiments because of its superior brightness (Shaner et al., 2013; Willet et al., 2015). A lithium acetate transformation method (Keeney and Boeke, 1994) was used for introducing sequences encoding tags, and integration of tags was verified using whole-cell PCR and/or microscopy. Tagged hhp1/2 alleles were fully functional as determined by growth assays and their ability to support Sid4 ubiquitination (Figure 2, D and F, and Supplemental Figures S3, C and D, and S5E). Introduction of tagged loci into other genetic backgrounds was accomplished using standard S. pombe mating, sporulation, and tetrad-dissection techniques. Fusion proteins were expressed from their native promoters at their normal chromosomal locus unless otherwise indicated. For hhp1 and hhp2 gene replacements, haploid hhp1::ura4+ and hhp2::ura4+ strains were transformed with linear hhp1 and hhp2 mutant gene fragments (digested with BamHI and PstI from pIRT2-hhp1 and pIRT2-hhp2 plasmids) using standard lithium acetate transformations. Integrants were selected based on resistance to 1.5 mg/ml 5-fluoroorotic acid (Fisher Scientific) and validated by colony PCR using primers homologous to endogenous sequences that flank the genomic clone within pIRT2 in combination with those within the ORF. All constructs and integrants were sequenced to ensure their accuracy.
For serial-dilution growth assays, cells were cultured in liquid YE at 25°C, three serial 10-fold dilutions starting at 4 × 106 cells/ml were made, 4 μl of each dilution was spotted on YE plates, and cells were grown at the indicated temperatures for 3–4 d. The GFP-Hhp1 (aa 280–365) fragment was cloned into a pREP81-GFP vector using NdeI and BamHI restriction sites. This construct and a construct with GFP alone were expressed in a wild-type strain under the control of the nmt81 promoter. Cells were grown in media containing thiamine and then washed into media without thiamine to induce protein production, then grown for 22 h at 32°C before imaging. For imaging experiments, temperature-sensitive mutants were grown at 25°C and then shifted to 36°C before imaging. Cold-sensitive nda3-km311 strains were grown at 32°C and shifted to 19°C for 6 h, and images were acquired immediately at room temperature. All other strains were grown and imaged at 25°C.
Cell culture, transfection, fixation, and antibody staining
RPE-1 cells were cultured in DMEM/F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were seeded 24 h before transfection onto 25-mm coverslips that were contained in a six-well plate. Six hours before transfection, cells were incubated in serum-free media. Plasmid DNA (1.5 μg) was transfected into cells using Lipofectamine 3000 (Life Technologies). Cells were allowed to grow for an additional 24 h before fixation. Cells were washed once with cold 1X PBS and once with 100% cold methanol, followed by fixation with 100% cold methanol for 15 min at −20°C. Cells were then washed three times with 0.1% TBST (1× Tris-buffered saline, 0.1% Tween 20), followed by blocking with AbDil (0.1% TBST + 2% bovine serum albumin) for 10 min. Cells were stained with anti-γ-tubulin antibody (Sigma-Aldrich, GTU88; 1:500) in AbDil overnight at 4°C, washed three times with 0.1% TBST, followed by 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/μl) and secondary antibody staining (Alexa Fluor 594 goat-anti mouse immunoglobulin G [IgG] [H+L], ThermoFisher; 1:500) for 45 min at 22°C. Coverslips were mounted on slides using ProLong Gold antifade mounting media (Invitrogen by ThermoFisher Scientific). Each experiment was performed in triplicate.
Molecular biology methods
All plasmids were generated by standard molecular biology techniques. hhp1 and hhp2 genes including 500 base pairs upstream and downstream of the ORFs were amplified by PCR and ligated into a PCR-Blunt vector (Life Technologies) and then subcloned into a pIRT2 vector (Hindley et al., 1987). hhp1 and hhp2 mutants were created by mutagenizing pIRT2-plasmids containing hhp1+ and hhp2+ using a QuikChange site-directed mutagenesis kit (Agilent Technologies). Plasmids were validated by DNA sequencing.
The ORF of CK1δ was amplified by PCR from a plasmid (CK1δ pGEX-6p-2) kindly provided by Fanni Gergely (University of Cambridge). V405 4HA-CK1ε, a gift from David Virshup (Duke University Medical School) (Addgene plasmid #13724), was used as a template for PCR amplification of the CK1ε ORF. Each PCR product was cloned into pEGFP-C1, and the correct sequence was validated by DNA sequencing. Mutant CK1δ/ε variants were made using the QuikChange site-directed mutagenesis kit (Agilent Technologies) and confirmed by DNA sequencing.
Schizosaccharomyces pombe protein methods
Cell pellets were frozen in a dry ice/ethanol bath and lysed by bead disruption in NP-40 lysis buffer under denaturing SDS lysis conditions as previously described (Gould et al., 1991), except with the addition of a complete protease inhibitor mixture (Calbiochem). Cell pellets for Hhp1 and Hhp2 immunoblots were lysed by bead disruption using a FastPrep cell homogenizer (MP Biomedicals). For analysis of Sid4 ubiquitination, Sid4 was immunoprecipitated under denaturing SDS conditions using Sid4 antiserum (Johnson et al., 2013). Proteins were separated on a 4–12% Bis-Tris gel (Life Technologies), transferred to Immobilon-P polyvinylidene fluoride (Millipore) membrane, and immunoblotted with anti-GFP (Roche, 1:1000), anti-Sid4 (1:2000), and fluorescent anti-mouse and anti-rabbit secondary antibodies (Li-Cor Biosciences) according to the manufacturers’ instructions.
Microscopy methods
Live-cell images of S. pombe cells were acquired using a Personal DeltaVision microscope system (Applied Precision) that includes an Olympus IX71 microscope, 60×/1.42 NA Plan-Apo and 100×/1.40 NA UPlanSApo objectives, a Photometrics CoolSnap HQ2 camera, and softWoRx imaging software. Images in figures are maximum-intensity projections of z-sections spaced at 0.2–0.5 µm. Images used for quantification were not deconvolved and were sum projected. Other images were deconvolved with 10 iterations. Time-lapse imaging was performed on cells in log phase using a microfluidics perfusion system (CellASIC ONIX; EMD Millipore). Cells were loaded into Y04C plates for 5 s at 8 psi, and YE liquid medium was flowed into the chamber at 5 psi throughout imaging. Quantitative analysis of microscopy data was performed using Fiji (a version of ImageJ software available at https://fiji.sc). For comparison of populations of cells for all genotypes, cells were imaged on the same day with the same microscope parameters. The average mNG fluorescence intensity of each protein at SPBs was measured in at least 20 cells. For all intensity measurements, the background was subtracted by creating a region of interest (ROI) in the same image where there were no cells. The raw intensity of the background was divided by the area of the background, which was multiplied by the area of the ROI. This number was subtracted from the raw integrated intensity of that ROI. For SPB intensity quantification, an ROI was drawn around the SPB and measured for raw integrated density; for whole-cell intensity quantification, an ROI was drawn around the entire cell. The average mCherry fluorescence intensities were measured similarly, and final values for each cell are expressed as mNG/mCherry ratios. Measurements for the 20 cells in each group were averaged for statistical analysis using a two-tailed Student’s t test or analysis of variance (ANOVA) implemented in Prism 6 (GraphPad Software). All statistical ANOVAs used Tukey’s post hoc analysis.
In addition, for comparison of two populations of cells within the same field of view, one population was incubated with fluorescently conjugated lectin (Sigma-Aldrich), which labels cell walls. Specifically, 1 µl of a 5 mg/ml stock of tetramethylrhodamine-lectin in water was added to 1 ml of cells for a final concentration of 5 µg/ml. Cells were then incubated for 10 min at room temperature, washed three times, and resuspended in media. The lectin-labeled cell population and unlabeled cell population were mixed 1:1 immediately before imaging. The reciprocal labeling of populations was also performed to account for any signal bleed-through. The fluorescence intensity of the SPB was quantified, and background fluorescence was subtracted (Willet et al., 2015).
In vitro kinase assays
MBP-Hhp1 and MBP-Hhp2 fusion proteins were purified on amylose beads (New England Biolabs) in column buffer (20 mM Tris, pH 7.0, 150 mM NaCl, 2 mM EDTA, and 0.1% NP40) and eluted with maltose (10 mM). Kinase reactions were performed with 500 ng kinase, 500 ng casein, 10 μM ATP plus 1 µCi γ-[32P]ATP in kinase buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, and 5 mM dithiothreitol) in 20 µl at 30°C for 30 min. Reactions were quenched by adding SDS–PAGE sample buffer, and proteins were separated by SDS–PAGE. Phosphorylated proteins were visualized by autoradiography, and relative protein quantities were assessed by Coomassie blue staining relative to known standards using Odyssey software (Li-Cor Biosciences). Kinetic assays were performed using recombinant MBP-Hhp1 and MBP-Hhp2 fusion proteins treated for 2 h with lambda phosphatase (New England Biolabs). These kinase reactions were performed with 0.25 μM of kinase, 25 μM casein, 5 μM cold ATP, 2 μCi [γ32P]-ATP in CK1 kinase buffer. Reactions were quenched at different time points by adding SDS sample buffer, and proteins were separated by SDS–PAGE. Phosphorylated proteins were visualized and quantitated using an FLA7000IP Typhoon Storage Phosphorimager (GE Healthcare Life Sciences). Kinetic measurements were calculated using Prism 6 software. Relative protein quantities were assessed by Coomassie blue staining.
Two-hybrid analyses
Two-hybrid experiments were performed as described previously (Vo et al., 2016). hhp1 and ppc89, cloned into pDEST DB and pDEST AD vectors, respectively, were generously provided by Haiyuan Yu (Cornell University). These or fragments thereof were cotransformed into S. cerevisiae strain PJ69-4A. Leu+ and Trp+ transformants were selected and then scored for positive interactions by streaking onto synthetic dextrose plates containing 5 mM 3-amino-1,2,4-triazole and lacking tryptophan, leucine, and histidine. β-Galactosidase reporter enzyme activity in the two-hybrid strains was measured using the Galacto-Star chemiluminescent reporter assay system according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA), except that cells were lysed by glass bead disruption. Each experiment was performed in triplicate. Reporter assays were recorded on a Multi-Detection Microplate Reader (Bio-TEK Instruments).
Checkpoint assay
Schizosaccharomyces pombe cells were synchronized in S phase using hydroxyurea (HU; Sigma) at a final concentration of 12 mM for 3–3.5 h at 32°C. Cells were then filtered into HU-free media and immediately incubated at 19°C to activate the spindle checkpoint. Cells were fixed in 70% ethanol, and septation indices were measured periodically for 10 h by methyl blue staining of the septa.
Protein modeling
Structural models of Hhp1 and Cki2 were generated using the Protein Homology/analogY Recognition Engine v. 2.0 (Phyre2) (Kelley et al., 2015). Homology models were generated from crystal structures of the kinase domains of Hhp1 and Cki2 homologues.
Supplementary Material
Acknowledgments
We thank Sierra Cullati, Jun-Song Chen, Alaina Willet, Christine Jones, and Janel Beckley for critical comments on the article. Liping Ren and Jun-Song Chen are thanked for outstanding technical assistance. pDEST two-hybrid vectors were a gracious gift from the Yu lab (Weill Institute for Cell and Molecular Biology, Cornell University). Z.C.E. was supported by the Integrated Biological Systems Training in Oncology Program (2T32CA119925-06). R.X.G. was supported by the Vanderbilt Initiative for Maximizing Student Diversity program (R25 5R25GM062459-11). This work was funded by National Institutes of Health grant R01GM112989 to K.L.G.
Abbreviations used:
- ANOVA
analysis of variance
- CK1
casein kinase 1
- CK1δ/ε
human CK1δ and CK1ε
- GFP
green fluorescent protein
- Hhp1/2
Hhp1 and Hhp2
- HU
hydroxyurea
- IgG
immunoglobulin G
- KDE
kinase domain extension
- mNG
mNeonGreen
- ORF
open reading frame
- ROI
region of interest
- SIN
septation initiation network
- SPB
spindle pole body
- YE
yeast extract.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-02-0129) on May 9, 2018.
REFERENCES
- Agostinis P, Pinna LA, Meggio F, Marin O, Goris J, Vandenheede JR, Merlevede W. (1989). A synthetic peptide substrate specific for casein kinase I. FEBS Lett , 75–78. [DOI] [PubMed] [Google Scholar]
- Babu P, Bryan JD, Panek HR, Jordan SL, Forbrich BM, Kelley SC, Colvin RT, Robinson LC. (2002). Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J Cell Sci , 4957–4968. [DOI] [PubMed] [Google Scholar]
- Babu P, Deschenes RJ, Robinson LC. (2004). Akr1p-dependent palmitoylation of Yck2p yeast casein kinase 1 is necessary and sufficient for plasma membrane targeting. J Biol Chem , 27138–27147. [DOI] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast , 943–951. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. (2006). The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science , 822–826. [DOI] [PubMed] [Google Scholar]
- Bimbo A, Jia Y, Poh SL, Karuturi RK, den Elzen N, Peng X, Zheng L, O’Connell M, Liu ET, Balasubramanian MK, Liu J. (2005). Systematic deletion analysis of fission yeast protein kinases. Eukaryot Cell , 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel G, Leichus B, Cheng X, Patterson SD, Mirza U, Chait BT, Kuret J. (1994). Expression, purification, crystallization, and preliminary x-ray analysis of casein kinase-1 from Schizosaccharomyces pombe. J Biol Chem , 7304–7309. [PubMed] [Google Scholar]
- Carpy A, Krug K, Graf S, Koch A, Popic S, Hauf S, Macek B. (2014). Absolute proteome and phosphoproteome dynamics during the cell cycle of Schizosaccharomyces pombe (fission yeast). Mol Cell Proteomics , 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A, Gietzen KF, Rivers A, Virshup DM. (1998). Autoinhibition of casein kinase I epsilon (CKI epsilon) is relieved by protein phosphatases and limited proteolysis. J Biol Chem , 1357–1364. [DOI] [PubMed] [Google Scholar]
- Chan KY, Alonso-Nunez M, Grallert A, Tanaka K, Connolly Y, Smith DL, Hagan IM. (2017). Dialogue between centrosomal entrance and exit scaffold pathways regulates mitotic commitment. J Cell Biol , 2795–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Gould KL. (2000). Sid4p is required to localize components of the septation initiation pathway to the spindle pole body in fission yeast. Proc Natl Acad Sci USA , 5249–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Beckley JR, McDonald NA, Ren L, Mangione M, Jang SJ, Elmore ZC, Rachfall N, Feoktistova A, Jones CM, et al (2015). Identification of new players in cell division, DNA damage response, and morphogenesis through construction of Schizosaccharomyces pombe deletion strains. G3 (Bethesda) , 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KY, Satterberg B, Lyons DM, Elion EA. (1994). Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell , 499–512. [DOI] [PubMed] [Google Scholar]
- Dahlberg CL, Nguyen EZ, Goodlett D, Kimelman D. (2009). Interactions between Casein kinase Iε (CKIε) and two substrates from disparate signaling pathways reveal mechanisms for substrate-kinase specificity. PLoS One , e4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins PR, Lue PF, Liew CC, Gornall AG. (1972). Purification and properties of rat liver nuclear protein kinases. Can J Biochem , 1249–1259. [DOI] [PubMed] [Google Scholar]
- Dhillon N, Hoekstra MF. (1994). Characterization of two protein kinases from Schizosaccharomyces pombe involved in the regulation of DNA repair. EMBO J , 2777–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. (1990). Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem , 14264–14269. [PubMed] [Google Scholar]
- Flotow H, Roach PJ. (1991). Role of acidic residues as substrate determinants for casein kinase I. J Biol Chem , 3724–3727. [PubMed] [Google Scholar]
- Ghalei H, Schaub FX, Doherty JR, Noguchi Y, Roush WR, Cleveland JL, Stroupe ME, Karbstein K. (2015). Hrr25/CK1δ-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J Cell Biol , 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietzen KF, Virshup DM. (1999). Identification of inhibitory autophosphorylation sites in casein kinase I epsilon. J Biol Chem , 32063–32070. [DOI] [PubMed] [Google Scholar]
- Gould KL, Moreno S, Owen DJ, Sazer S, Nurse P. (1991). Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J , 3297–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves PR, Haas DW, Hagedorn CH, DePaoli-Roach AA, Roach PJ. (1993). Molecular cloning, expression, and characterization of a 49-kilodalton casein kinase I isoform from rat testis. J Biol Chem , 6394–6401. [PubMed] [Google Scholar]
- Graves PR, Roach PJ. (1995). Role of COOH-terminal phosphorylation in the regulation of casein kinase I delta. J Biol Chem , 21689–21694. [DOI] [PubMed] [Google Scholar]
- Greer YE, Rubin JS. (2011). Casein kinase 1 delta functions at the centrosome to mediate Wnt-3a-dependent neurite outgrowth. J Cell Biol , 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer YE, Westlake CJ, Gao B, Bharti K, Shiba Y, Xavier CP, Pazour GJ, Yang Y, Rubin JS. (2014). Casein kinase 1δ functions at the centrosome and Golgi to promote ciliogenesis. Mol Biol Cell , 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Venkatram S, Gould KL, McCollum D. (2002). Dma1 prevents mitotic exit and cytokinesis by inhibiting the septation initiation network (SIN). Dev Cell , 779–790. [DOI] [PubMed] [Google Scholar]
- Hathaway GM, Traugh JA. (1979). Cyclic nucleotide-independent protein kinases from rabbit reticulocytes. Purification of casein kinases. J Biol Chem , 762–768. [PubMed] [Google Scholar]
- Hindley J, Phear G, Stein M, Beach D. (1987). Sucl+ encodes a predicted 13-kilodalton protein that is essential for cell viability and is directly involved in the division cycle of Schizosaccharomyces pombe. Mol Cell Biol , 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra MF, Dhillon N, Carmel G, DeMaggio AJ, Lindberg RA, Hunter T, Kuret J. (1994). Budding and fission yeast casein kinase I isoforms have dual-specificity protein kinase activity. Mol Biol Cell , 877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, et al (2010). Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science , 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianes C, Xu P, Werz N, Meng Z, Henne-Bruns D, Bischof J, Knippschild U. (2015). CK1δ activity is modulated by CDK2/E- and CDK5/p35-mediated phosphorylation. Amino Acids , 579–592. [DOI] [PubMed] [Google Scholar]
- Johnson AE, Chen JS, Gould KL. (2013). CK1 is required for a mitotic checkpoint that delays cytokinesis. Curr Biol , 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AE, Gould KL. (2011). Dma1 ubiquitinates the SIN scaffold, Sid4, to impede the mitotic localization of Plo1 kinase. EMBO J , 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AE, McCollum D, Gould KL. (2012). Polar opposites: fine-tuning cytokinesis through SIN asymmetry. Cytoskeleton (Hoboken) , 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney JB, Boeke JD. (1994). Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics , 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc , 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. (2005). The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal , 675–689. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Kruger M, Richter J, Xu P, Garcia-Reyes B, Peifer C, Halekotte J, Bakulev V, Bischof J. (2014). The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front Oncol , 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker KL, Roach PJ, Hurley TD. (1996). Three-dimensional structure of mammalian casein kinase I: molecular basis for phosphate recognition. J Mol Biol , 618–631. [DOI] [PubMed] [Google Scholar]
- Longenecker KL, Roach PJ, Hurley TD. (1998). Crystallographic studies of casein kinase I delta toward a structural understanding of auto-inhibition. Acta Crystallogr D Biol Crystallogr , 473–475. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J. (2012). Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell , 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Takeda M. (1972). Phosphoprotein kinases from rat liver cytosol. Biochim Biophys Acta , 237–241. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, et al (2006). ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol , 841–847. [DOI] [PubMed] [Google Scholar]
- Meggio F, Perich JW, Marin O, Pinna LA. (1992). The comparative efficiencies of the Ser(P)-, Thr(P)- and Tyr(P)-residues as specificity determinants for casein kinase-1. Biochem Biophys Res Commun , 1460–1465. [DOI] [PubMed] [Google Scholar]
- Meggio F, Perich JW, Reynolds EC, Pinna LA. (1991). A synthetic beta-casein phosphopeptide and analogues as model substrates for casein kinase-1, a ubiquitous, phosphate directed protein kinase. FEBS Lett , 303–306. [DOI] [PubMed] [Google Scholar]
- Meng Z, Bischof J, Ianes C, Henne-Bruns D, Xu P, Knippschild U. (2016). CK1δ kinase activity is modulated by protein kinase C alpha (PKCα)-mediated site-specific phosphorylation. Amino Acids , 1185–1197. [DOI] [PubMed] [Google Scholar]
- Milne DM, Looby P, Meek DW. (2001). Catalytic activity of protein kinase CK1δ (casein kinase 1δ) is essential for its normal subcellular localization. Exp Cell Res , 43–54. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. (1991). Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol , 795–823. [DOI] [PubMed] [Google Scholar]
- Murone M, Simanis V. (1996). The fission yeast dma1 gene is a component of the spindle assembly checkpoint, required to prevent septum formation and premature exit from mitosis if spindle function is compromised. EMBO J , 6605–6616. [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H. (2015). Hrr25: an emerging major player in selective autophagy regulation in Saccharomyces cerevisiae. Autophagy , 432–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Grassart A, Lu R, Wong CC, 3rd Yates J, Barnes G, Drubin DG. (2015a). Casein kinase 1 promotes initiation of clathrin-mediated endocytosis. Dev Cell , 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Moritz M, Han X, Giddings TH, Lyon A, Kollman J, Winey M, Yates J, 3rd, Agard DA, Drubin DG, Barnes G. (2015b). Interaction of CK1δ with γTuSC ensures proper microtubule assembly and spindle positioning. Mol Biol Cell , 2505–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Matos J, Mori S, Gregan J, Bogdanova A, Schwickart M, Mechtler K, Shirahige K, Zachariae W, Nasmyth K. (2006). Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell , 1049–1064. [DOI] [PubMed] [Google Scholar]
- Qi ST, Wang ZB, Huang L, Liang LF, Xian YX, Ouyang YC, Hou Y, Sun QY, Wang WH. (2015). Casein kinase 1 (α, δ and ε) localize at the spindle poles, but may not be essential for mammalian oocyte meiotic progression. Cell Cycle , 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JA, Tomlin GC, McDonald WH, Snydsman BE, Muller EG, Yates JR, 3rd, Gould KL. (2006). Ppc89 links multiple proteins, including the septation initiation network, to the core of the fission yeast spindle-pole body. Mol Biol Cell , 3793–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuno T, Watanabe Y. (2015). Phosphorylation of cohesin Rec11/SA3 by casein kinase 1 promotes homologous recombination by assembling the meiotic chromosome axis. Dev Cell , 220–230. [DOI] [PubMed] [Google Scholar]
- Schittek B, Sinnberg T. (2014). Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer , 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Madhani HD. (2004). Principles of MAP kinase signaling specificity in Saccharomyces cerevisiae. Annu Rev Genet , 725–748. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods , 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillibourne JE, Milne DM, Takahashi M, Ono Y, Meek DW. (2002). Centrosomal anchoring of the protein kinase CK1δ mediated by attachment to the large, coiled-coil scaffolding protein CG-NAP/AKAP450. J Mol Biol , 785–797. [DOI] [PubMed] [Google Scholar]
- Simanis V. (2015). Pombe’s thirteen—control of fission yeast cell division by the septation initiation network. J Cell Sci , 1465–1474. [DOI] [PubMed] [Google Scholar]
- Sun B, Chen L, Cao W, Roth AF, Davis NG. (2004). The yeast casein kinase Yck3p is palmitoylated, then sorted to the vacuolar membrane with AP-3-dependent recognition of a YXXφ adaptin sorting signal. Mol Biol Cell , 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Umesono K, Hirata A, Yanagida M. (1983). Cold-sensitive nuclear division arrest mutants of the fission yeast Schizosaccharomyces pombe. J Mol Biol , 251–270. [DOI] [PubMed] [Google Scholar]
- Tuazon PT, Traugh JA. (1991). Casein kinase I and II—multipotential serine protein kinases: structure, function, and regulation. Adv Second Messenger Phosphoprotein Res , 123–164. [PubMed] [Google Scholar]
- Vancura A, Sessler A, Leichus B, Kuret J. (1994). A prenylation motif is required for plasma membrane localization and biochemical function of casein kinase I in budding yeast. J Biol Chem , 19271–19278. [PubMed] [Google Scholar]
- Vo TV, Das J, Meyer MJ, Cordero NA, Akturk N, Wei X, Fair BJ, Degatano AG, Fragoza R, Liu LG, et al (2016). A proteome-wide fission yeast interactome reveals network evolution principles from yeasts to human. Cell , 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PC, Vancura A, Mitcheson TG, Kuret J. (1992). Two genes in Saccharomyces cerevisiae encode a membrane-bound form of casein kinase-1. Mol Biol Cell , 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet AH, McDonald NA, Bohnert KA, Baird MA, Allen JR, Davidson MW, Gould KL. (2015). The F-BAR Cdc15 promotes contractile ring formation through the direct recruitment of the formin Cdc12. J Cell Biol , 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RM, Carmel G, Sweet RM, Kuret J, Cheng X. (1995). Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J , 1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Ur SN, Su TY, Corbett KD. (2016). Structure of the Saccharomyces cerevisiae Hrr25:Mam1 monopolin subcomplex reveals a novel kinase regulator. EMBO J , 2139–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.