

Abstract
The dynein-2 complex mediates trafficking of ciliary proteins by powering the intraflagellar transport (IFT) machinery containing IFT-A and IFT-B complexes. Although 11 subunits are known to constitute the dynein-2 complex, with several light-chain subunits shared by the dynein-1 complex, the overall architecture of the dynein-2 complex has not been fully clarified. Utilizing the visible immunoprecipitation assay, we demonstrated the interaction modes among the dynein-2 subunits, including previously undefined interactions, such as that between WDR60 and the TCTEX1D2–DYNLT1/DYNLT3 dimer. The dynein-2 complex can be divided into three subcomplexes, namely DYNC2H1–DYNC2LI1, WDR34–DYNLL1/DYNLL2–DYNLRB1/DYNLRB2, and WDR60–TCTEX1D2–DYNLT1/DYNLT3. We established cell lines lacking WDR60 or TCTEX1D2, both of which are dynein-2–specific subunits encoded by ciliopathy-causing genes, and found that both WDR60-knockout (KO) and TCTEX1D2-KO cells show defects in retrograde ciliary protein trafficking, with WDR60-KO cells demonstrating more severe defects probably due to failed assembly of the dynein-2 complex. The exogenous expression of a WDR60 mutant lacking TCTEX1D2 binding partially restored retrograde trafficking to a level comparable to that of TCTEX1D2-KO cells. Thus, our results demonstrated that WDR60 plays a major role and TCTEX1D2 plays an auxiliary role in the dynein-2 complex to mediate retrograde ciliary protein trafficking.
INTRODUCTION
The appropriate movement of proteins and the positioning of intracellular organelles are fundamental to normal cellular functions. Microtubule motor proteins play crucial roles in a variety of cellular processes, such as the trafficking of proteins and carrier vesicles, positioning and dynamics of intracellular organelles, and organization of microtubules during cell division. Generally, plus-end–directed movement of proteins and organelles along microtubules is driven by kinesins, whereas minus-end–directed movement is driven by dyneins (Hirokawa et al., 2010).
Cilia are cell-surface appendages observed in various eukaryotic cells and consist of a microtubule-based axoneme extending from the basal body and the ciliary membrane surrounding the axoneme (Ishikawa and Marshall, 2011). Although the ciliary membrane is continuous with the plasma membrane, the composition of proteins and lipids within cilia and on the ciliary membrane are considerably different from those of the cytoplasm and the plasma membrane, because the transition zone located at the ciliary base functions as a diffusion/permeability barrier (Garcia-Gonzalo and Reiter, 2017; Gonçalves and Pelletier, 2017). Cilia play key roles in the perception of extracellular stimuli and the coordination of various signaling pathways, such as those involving Hedgehog (Hh). To fulfill these functions, cilia contain a number of unique proteins, including ion channels, G protein–coupled receptors (GPCRs), and adenylyl cyclases (Mukhopadhyay and Rohatgi, 2014). The roles of cilia as cellular sensory organelles have been underscored by the fact that defects in ciliary assembly and functions result in phenotypically diverse hereditary diseases, which are collectively called the ciliopathies.
Bidirectional protein trafficking along the ciliary axoneme is mediated by intraflagellar transport (IFT) particles, which contain the two large multisubunit complexes IFT-A and IFT-B (Taschner and Lorentzen, 2016; Nakayama and Katoh, 2018). We as well as others recently demonstrated the architecture of these complexes; IFT-A is composed of six subunits with associated TULP3 (Hirano et al., 2017), and the IFT-B complex is composed of 16 subunits and is divided into the core (B1) subcomplex composed of 10 subunits and the peripheral (B2) subcomplex composed of six subunits, and these two subcomplexes are connected by composite interactions involving the IFT38, IFT52, IFT57, and IFT88 subunits (Boldt et al., 2016; Katoh et al., 2016; Taschner et al., 2016). Anterograde protein trafficking from the base to the tip of cilia is mediated by the IFT-B complex assisted by kinesin-2 motor proteins, whereas retrograde trafficking is mediated by the IFT-A complex powered by the dynein-2 complex (Ishikawa and Marshall, 2011; He et al., 2016).
There are two cytoplasmic dynein complexes in most ciliated eukaryotic cells (Roberts et al., 2013). The dynein-1 complex is involved in membrane trafficking, organelle dynamics, and chromosome segregation during cell division (Kardon and Vale, 2009). On the other hand, the dynein-2 complex mediates retrograde ciliary protein trafficking in concert with the IFT-A complex. The dynein-2 complex, at least in humans, is composed of a heavy chain (DYNC2H1), a light intermediate chain (DYNC2LI1), two intermediate chains (WDR34 and WDR60), and a light chain (TCTEX1D2), all of which are unique to dynein-2. In addition, some light-chain subunits (DYNLL1, DYNLL2, DYNLT1, DYNLT3, DYNLRB1, and DYNLRB2) are shared by dynein-1 and dynein-2 complexes (Asante et al., 2014; Hou and Witman, 2015; He et al., 2016). Mutations in all of the dynein-2–specific subunits, as well as those in some of the IFT-A and IFT-B subunits are known to cause skeletal ciliopathies, including short-rib thoracic dysplasia (SRTD) (Schmidts, 2014; McInerney-Leo et al., 2015; Reiter and Leroux, 2017).
The molecular composition of the mammalian dynein-2 complex has not been well defined to date. Recently, Asante et al. (2014) biochemically analyzed the composition of the dynein-2 subunit in human telomerase reverse transcriptase-immortalized retinal pigment epithelial 1 (hTERT-RPE1) cells and found that WDR34/SRTD11, WDR60/SRTD8, and TCTEX1D2/SRTD17 are bona fide dynein-2–specific subunits, in addition to DYNC2H1/SRTD3 and DYNC2LI1/SRTD15. On the basis of the interaction data, they proposed a rough architectural model of the dynein-2 complex (Asante et al., 2014). However, specific functions of individual subunits remain largely unclear (for a review, see Prevo et al. [2017]). Therefore, to understand the roles of individual subunits in the context of the holocomplex, a finer architectural map of the dynein-2 complex is required.
We recently established a convenient and flexible strategy to visually detect protein–protein interactions, named the visible immunoprecipitation (VIP) assay (Katoh et al., 2015), which can visually detect not only binary protein interactions but also one-to-many and many-to-many protein interactions without the need of SDS–PAGE and immunoblotting. Taking advantage of this strategy, we determined the intricate architecture of the IFT-A, IFT-B, and BBSome complexes (Katoh et al., 2015, 2016; Hirano et al., 2017). Therefore, in the present study, we determined the architecture of the dynein-2 complex. In our predicted architectural model, WDR34 and WDR60 interact with distinct sets of light chains and in turn interact with DYNC2H1–DYNC2LI1.
On the other hand, we also recently established a practical method for the targeted disruption of genes using the CRISPR/Cas9-mediated homology-independent knock-in system (Katoh et al., 2017). As this method can be used effectively in hTERT-RPE1 cells, which have been used in a number of studies analyzing ciliary assembly and function, we recently clarified the functions of various ciliary proteins by disrupting the genes encoding these proteins (Funabashi et al., 2017; Hirano et al., 2017; Katoh et al., 2017; Nishijima et al., 2017; Nozaki et al., 2017; Takahara et al., 2018). In this study, we therefore established WDR60-knockout (KO) and TCTEX1D2-KO hTERT-RPE1 cell lines and analyzed the roles of these dynein-2–specific subunits in the dynein-2 complex.
RESULTS
Architecture of the dynein-2 complex determined by the VIP assay
To delineate the architecture of the dynein-2 complex, we first analyzed binary interactions between the 11 known constituents of the human dynein-2 complex (Asante et al., 2014) (see Figure 1A) using the VIP assay; we have recently reported a detailed protocol for the VIP assay (Katoh et al., 2018). In short, lysates prepared from HEK293T cells cotransfected with any of the 121 possible combinations of expression vectors for the dynein-2 subunits fused to enhanced green fluorescent protein (EGFP) and mCherry (mChe) were subjected to immunoprecipitation with glutathione S-transferase (GST)-tagged anti-GFP nanobody (Nb) prebound to glutathione–Sepharose 4B beads. The EGFP and mChe signals on the precipitated beads were in turn detected by fluorescence microscopy under constant conditions in the same sets of experiments. Routinely, we evaluate binary interactions as “positive” only when red signals can be detected on the surface of the precipitated beads in reciprocal combinations of EGFP- and mChe-fused proteins under fixed conditions.
FIGURE 1:

Comprehensive VIP assay for dynein-2 subunits and interaction of WDR60 with light chains. (A) Schematic representation of structures and domain organizations of dynein-2 subunits. (B) Comprehensive VIP assay. Lysates prepared from HEK293T cells transfected with a combination of expression vectors for EGFP-fused and mChe-fused dynein-2 subunits, as indicated, were processed for immunoprecipitation with GST-tagged anti-GFP Nb prebound to glutathione–Sepharose beads. The red fluorescence signals on the precipitated beads were observed, and images of the beads were acquired using a BZ-8000 microscope. (C, D) Identification of light chains interacting with WDR60. Lysates prepared from cells cotransfected with expression vectors for mChe-WDR60 and EGFP-fused light chains, as indicated, were subjected to the VIP assay using GST-tagged anti-GFP Nb. (E) WDR60 interacts with the TCTEX1D2–DYNLT1/DYNLT3 dimer. Lysates prepared from cells cotransfected with expression vectors for mChe-WDR60 and EGFP-fused light chains, as indicated, were immunoprecipitated with GST-tagged anti-GFP Nb prebound to glutathione–Sepharose beads and subjected to the VIP assay or immunoblotting analysis using anti-RFP or anti-GFP antibodies, as indicated. WDR60 was found to interact with the dimers TCTEX1D2–DYNLT1 and TCTEX1D2–DYNLT3. IP, immunoprecipitation; IB, immunoblotting. (F) Schematic representation of the H1, WDR34, and WDR60 subcomplexes demonstrated by the experiments shown in this figure.
Figure 1B shows mChe signals on the immunoprecipitated beads in comprehensive assays of the dynein-2 subunits; note that for DYNC2H1, the N-terminal 1650-amino-acid tail region (H1 tail), which lacks the AAA+ motor domains, was used for the interaction assay. It is also noteworthy that most of the interactions detected with a combination of EGFP-fused and mChe-fused proteins were also detected with a reciprocal combination of fluorescent fusion proteins, highlighting the reliability of the VIP assay. For example, the DYNC2H1 tail showed a weak but substantial interaction with DYNC2LI1. The intermediate-chain WDR34 interacted with a limited set of light chains, namely DYNLL1/DYNLL2 and DYNLRB1/DYNLRB2. Groups of light chains (DYNLL1/DYNLL2, DYNLT1/DYNLT3/TCTEX1D2, and DYNLRB1/DYNLRB2) demonstrated homophilic and heterophilic interactions within individual groups, except that TCTEX1D2 did not show a homophilic interaction; the light-chain interaction data are in line with a previous interaction study using a yeast two-hybrid assay (Lo et al., 2007). In contrast to these dynein-2 subunits, we did not detect an interaction of the other intermediate chain, WDR60, with any other dynein-2 subunits, although a previous study indicated that WDR60 is a key component of the dynein-2 complex (Asante et al., 2014).
To identify potential interacting partners of WDR60, we utilized an advantage of the VIP assay; namely that this assay can easily detect one-to-many and many-to-many protein interactions. When cells were cotransfected with expression vectors for mChe-WDR60 and each light-chain subunit fused to EGFP, and then processed for the VIP assay with GST–anti-GFP Nb, red signals were detected on the precipitated beads (Figure 1C, column 1), indicating that WDR60 was indeed coimmunoprecipitated with some of the light chains. We then performed the subtractive VIP assay to determine which light chains are required for the interaction with WDR60. When limited sets of light chains fused to EGFP were omitted from the VIP assay for mChe-WDR60, red signals were abolished when DYNLT1/DYNLT3 (column 3) or TCTEX1D2 (column 4) was omitted but not when DYNLL1/DYNLL2 (column 2) or DYNLRB1/DYNLRB2 (column 5) was omitted. mChe-WDR60 was coimmunoprecipitated when EGFP-fused TCTEX1D2, DYNLT1, and DYNLT3 were simultaneously expressed (column 9), indicating involvement of the three light chains in the interaction with WDR60. Furthermore, interactions with WDR60 were detected when a combination of TCTEX1D2 and either DYNLT1 or DYNLT3 were coexpressed (Figure 1D, columns 5 and 6). The VIP data were confirmed by conventional immunoblotting analysis (Figure 1E, lanes 6–8). From these results, we conclude that WDR60 interacts with a dimer of TCTEX1D2–DYNLT1 or TCTEX1D2–DYNLT3. Overall, the dynein-2 complex is likely to consist of the following three subcomplexes (Figure 1F): the H1 subcomplex containing DYNC2H1 and DYNC2LI1; the WDR34 subcomplex containing WDR34, DYNLL1/DYNLL2, and DYNLRB1/DYNLRB2; and the WDR60 subcomplex containing WDR60, TCTEX1D2, and DYNLT1/DYNLT3. Our data, however, cannot exclude the possibility that DYNLL1/DYNLL2, DYNLRB1/DYNLRB2, and DYNLT1/DYNLT3 are included in the respective subcomplexes as homodimers and/or heterodimers.
We then attempted to detect a potential interaction between the WDR34 and WDR60 subcomplexes. To this end, we cotransfected HEK293T cells with an expression vector for EGFP-fused WDR60 alone or in combination with those for light chains (TCTEX1D2 and DYNLT1), along with an expression vector for mChe-fused WDR34 alone or in combination with those for DYNLL2 and DYNLRB1, and processed the lysates prepared from the transfected cells for immunoprecipitation with GST–anti-GFP Nb followed by the VIP assay (Figure 2A) or immunoblotting analysis (Figure 2B). When EGFP-WDR60 and mChe-WDR34 were coexpressed, red signals were hardly detectable in the VIP assay (Figure 2A, column 2), although a weak but substantial band for mChe-WDR34 was detected by immunoblotting (Figure 2B, lane 2). Moderate levels of the interaction were detected when EGFP-fused WDR60+DYNLT1+TCTEX1D2 and mChe-WDR34 (column 3 and lane 3) or EGFP-WDR60 and mChe-fused WDR34+DYNLL2+DYNLRB1 (column 4 and lane 4) were coexpressed. Most prominent signals/bands were detected in both the VIP assay and immunoblotting when EGFP-fused WDR60+TCTEX1D2+DYNLT1 and mChe-fused WDR34+DYNLL2+DYNLRB1 were simultaneously expressed (column 5 and lane 5). On the basis of the data shown in Figure 2, A and B, we conclude that the intermediate chains WDR34 and WDR60 robustly interact with each other when they form subcomplexes with the appropriate combinations of light chains, although they can interact weakly in the absence of light chains.
FIGURE 2:

Interactions between the WDR34, WDR60, and H1 subcomplexes. (A, B) Interaction between the WDR34 and WDR60 subcomplexes. Lysates prepared from cells cotransfected with expression vectors for EGFP-fused subunits of the WDR60 subcomplex and mChe-fused subunits of the WDR34 subcomplex, as indicated, were immunoprecipitated with GST-tagged anti-GFP Nb prebound to glutathione–Sepharose beads and subjected to the VIP assay (A) or immunoblotting analysis using anti-RFP or anti-GFP antibodies, as indicated (B). The asterisk indicates the position of a putative degradation product of the EGFP-WDR60 protein. (C, D) Interactions of the WDR34 and WDR60 subcomplexes with the H1 subcomplex. Lysates prepared from cells cotransfected with expression vectors for EGFP-fused subunits of the H1 subcomplex and mChe-fused subunits of the WDR34 and WDR60 subcomplexes, as indicated, were immunoprecipitated with GST-tagged anti-GFP Nb prebound to glutathione–Sepharose beads and subjected to the VIP assay (C) or immunoblotting analysis (D). (E) Schematic representation of interactions involving the H1, WDR34, and WDR60 subcomplexes, demonstrated by the experiments shown in this figure.
We then addressed the interaction mode of the H1 subcomplex (DYNC2H1+DYNC2LI1) with other dynein-2 subunits (Figure 2, C and D). EGFP-DYNC2H1 or EGFP-DYNC2LI1 alone demonstrated a weak interaction with WDR34 but not with WDR60 (columns 2 and 3 and lanes 2 and 3); note that, for an unknown reason, the expression level of mChe-WDR60 was substantially lower than that of mChe-WDR34 in this set of experiments (see lanes 10–18 in Figure 2D). When EGFP-fused DYNC2H1 and DYNC2LI1 were simultaneously expressed, a substantial interaction was observed with mChe-WDR34 (column 4 and lane 4) or mChe-WDR60 (column 5 and lane 5) alone, as well as in the presence of light chains (columns 7–9 and lanes 7–9). Thus, interactions of the H1 subcomplex with the intermediate chains appear not necessarily to require the presence of light chains.
Collectively considering the data shown in Figures 1, B–E, and 2, A–D, we propose the following model for the efficient assembly of the dynein-2 complex (Figure 2E): 1) the intermediate chain WDR34 forms a subcomplex with DYNLL1/DYNLL2 and DYNLRB1/DYNLRB2 (the WDR34 subcomplex); 2) the other intermediate-chain WDR60 forms a subcomplex with a light-chain dimer composed of TCTEX1D2 and DYNLT1/DYNLT3 (the WDR60 subcomplex); 3) DYNC2H1 and DYNC2LI1 form another subcomplex (the H1 subcomplex); 4) WDR34 and WDR60 interact robustly with each other as their corresponding subcomplexes; and 5) the H1 subcomplex interacts with WDR34 and WDR60 to form the dynein-2 holocomplex.
Interaction of WDR60 with the light-chain dimer via a conserved region upstream of the WD40 domain
We then focused on WDR60, as there are virtually no reports on the characterization of this intermediate chain at the molecular or cellular level, except for a study on a Chlamydomonas WDR60 paralogue (Patel-King et al., 2013). We initially set out to determine the interaction mode of WDR60 with the light chains. When the WDR60 protein was divided into the C-terminal WD40 domain (residues 627–1066) and the remaining N-terminal region (residues 1–626), the N-terminal region (Figure 3A, left) was found to interact with a mixture of DYNLT1, DYNLT3, and TCTEX1D2. We therefore constructed various deletion mutants of WDR60 (Figure 3B). As shown in Figure 3C, a construct covering residues 473–1066 (Figure 3C, column 4), but not that covering residues 523–1066 (column 5), retained the ability to interact with the light chains TCTEX1D2+DYNLT1. Finally, the Δ473–522 construct (column 6) failed to interact with the light chains. The VIP data were confirmed by immunoblotting analysis. As shown in Figure 3D, the 574–1066 and Δ473–522 constructs lacked the ability to interact with the light chains (lanes 5 and 6). Thus, the 50-amino-acid region (residues 473–522), which is highly conserved among vertebrate species (see Figure 3E), is responsible for the interaction of WDR60 with the dimer composed of TCTEX1D2 and DYNLT1/DYNLT3.
FIGURE 3:
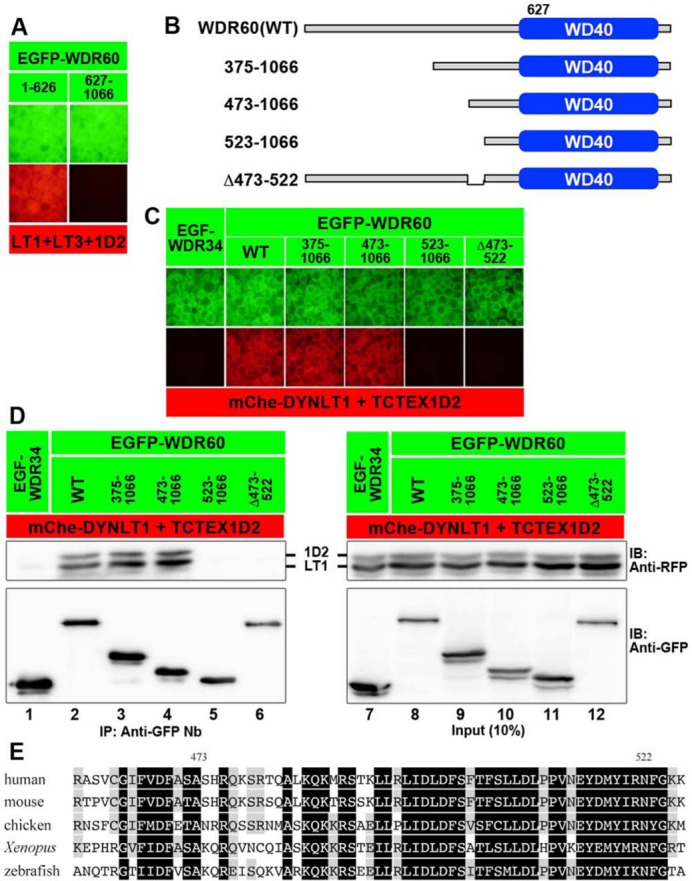
Region of WDR60 involved in its interaction with the TCTEX1D2–DYNLT1 dimer. (A) The N-terminal half of the WDR60 protein participates in its interaction with TCTEX1D2–DYNLT. Lysates prepared from cells cotransfected with expression vectors for EGFP-fused WDR60(1–626) or WDR60(627–1066) and mChe-fused DYNLT1+DYNLT3+TCTEX1D2 were subjected to the VIP assay. (B–D) Determination of the region of WDR60 involved in its interaction with the TCTEX1D2–DYNLT1 dimer. Lysates prepared from cells cotransfected with expression vectors for an EGFP-fused WDR60 construct, as schematically shown in B, and mChe-fused TCTEX1D2+DYNLT1 were immunoprecipitated with GST-tagged anti-GFP Nb prebound to glutathione–Sepharose beads and subjected to the VIP assay (C) or immunoblotting analysis (D). (E) Sequence alignment of the vertebrate WDR60 region involved in binding to the TCTEX1D2–DYNLT dimer. Residues conserved in all species are shown in black boxes, and those conserved in at least three species are shaded.
Similar, but slightly different, defects in retrograde trafficking in WDR60-KO and TCTEX1D2-KO cells
Given that both WDR60 and TCTEX1D2 are dynein-2–specific subunits interacting with each other and are encoded by the causative genes of SRTD (SRTD8 and SRDT17, respectively) (McInerney-Leo et al., 2013; Gholkar et al., 2015; Schmidts et al., 2015; Cossu et al., 2016; Kakar et al., 2017), we then investigated the roles of these dynein-2 subunits in ciliary protein trafficking by establishing KO hTERT-RPE1 cell lines using the CRISPR/Cas9 system; although we analyzed two independent WDR60-KO cell lines established using distinct target sequences (#W60-1-8 and #W60-2-2) and two independent TCTEX1D2-KO cell lines established using distinct target sequences (#1D2-1-1 and #1D2-2-9) (for characterization of these cell lines, see Supplemental Figure S1), typical data are shown for one of the two established cell lines.
When control RPE1, WDR60-KO, and TCTEX1D2-KO cells were immunostained for ARL13B (a marker of the ciliary membrane; Figure 4, A′–C′) and FOP (also known as FGFR1OP; a marker of the basal body (Nishijima et al., 2017; Figure 4, A′′–C′′), we noticed that some population of WDR60-KO cells had very short cilia (<1.5 µm; for example, see those indicated by arrows in Figure 4, B–B′′); note that cells positive for ARL13B around the basal body indeed have cilia even though they are very short, since ARL13B is specifically localized on the ciliary membrane. Quantitative analysis revealed that in addition to #W60-1-8 and #W60-2-2 cell lines, the #1D2-1-1 and #1D2-2-9 cell lines also demonstrated a tendency to possess very short cilia (Figure 4G). However, ciliary lengths of WDR60-KO and TCTEX1D2-KO cells, except for those with very short cilia, were comparable to the length of cilia of control RPE1 cells (Figure 4H). These observations are compatible with those in previous studies on WDR60 and TCTEX1D2 mutant fibroblasts from SRTD patients that the percentage of ciliated cells was lower compared with controls, although there was no obvious difference in ciliary length in mutant fibroblasts with detectable cilia, compared with control fibroblasts (McInerney-Leo et al., 2013; Schmidts et al., 2015).
FIGURE 4:

Accumulation of IFT88 and IFT140 within cilia in WDR60-KO and TCTEX1D2-KO cells. Control RPE1 cells (A, D), the WDR60-KO cell line #W60-2-2 (B, E), and the TCTEX1D2-KO cell line #1D2-2-9 (C, F) were serum starved for 24 h and triple immunostained for either IFT88 (A–C) or IFT140 (D–F), ARL13B (A′–F′), and FOP (A′′–F′′). Merged, enlarged images of the boxed regions of A–F and A′–F′ are shown in A′′′–F′′′. Scale bars, 10 µm. (G) Cells with very short cilia (<1.5 µm; for example, see those indicated by arrows in B–B′′) were counted, and the percentages of these cells are expressed as bar graphs. Values are the means ± SD (error bars) of three independent experiments. In each set of experiments, 31–53 cells were observed, and the total numbers of observed ciliated cells (n) are shown. p, one-way analysis of variance (ANOVA) followed by Tukey post-hoc analysis. (H) Ciliary lengths of individual cells, except for those with very short cilia, were measured and expressed as bar graphs. Values are means ± SD (error bars) of three independent experiments. In each experiment, 28–41 ciliated cells were observed, and the total numbers of ciliated cells observed (n) are shown. (I) The staining intensity for IFT88 within cilia in control RPE1 cells, and in the WDR60-KO and TCTEX1D2-KO cell lines were measured and expressed as bar graphs. Values are means ± SD (error bars) of three independent experiments. In each experiment, 28–87 ciliated cells were observed, and the total numbers of ciliated cells observed (n) are shown. p, one-way ANOVA followed by Tukey post-hoc analysis. (J) Localization of IFT140 in control RPE1 cells, and in the WDR60-KO and TCTEX1D2-KO cell lines were classified as “ciliary base,” “ciliary base and tip,” “even distribution within cilia,” and “no ciliary localization,” and the cells with each category of IFT140 localization were counted. The percentages of these populations are expressed as stacked bar graphs. Values are the means of three independent experiments. In each set of experiments, 31–67 ciliated cells were observed, and the total numbers of ciliated cells observed (n) are shown. p, the Pearson χ2 test.
When control RPE1 cells were immunostained for IFT88 (a subunit of the IFT-B complex), the staining was found mainly around the ciliary base and faintly along the axoneme (Figure 4A). In the TCTEX1D2-KO cells, the IFT88 staining within cilia (Figure 4C) was slightly but significantly more intense than that in control cells (Figure 4I). In considerable contrast, in WDR60-KO cells, IFT88 was observed throughout cilia and often found around the distal tips (Figure 4B; also see Figure 4I); this phenotype is reminiscent of that of IFT139-KO and IFT121-KO cells (Hirano et al., 2017; Takahara et al., 2018); both IFT139 and IFT121 are peripheral subunits of the IFT-A complex and are encoded by the causative genes of SRTD (SRTD4 and SRTD7, respectively).
We then stained control, WDR60-KO, and TCTEX1D2-KO cells with an antibody against IFT140 (a subunit of the IFT-A complex). In control and TCTEX1D2-KO cells, the majority of IFT140 staining was detected at the ciliary base (Figure 4, D and F). Note that, as described previously (Hirano et al., 2017; Takahara et al., 2018), the commercially available anti-IFT140 antibody also stained undetermined nuclear structures in RPE1 cells (see the manufacturer’s website, www.ptglab.com/Products/IFT140-Antibody-17460-1-AP.htm). Owing to the high background staining for IFT140, we classified the localization of IFT140 by visual observation (Figure 4J), instead of mechanically measuring the signal intensity within cilia. In WDR60-KO cells, similarly to IFT88 staining, IFT140 staining was observed within cilia, occasionally at the ciliary tips (Figure 4E). These observations indicate that retrograde trafficking of the IFT machinery is severely impaired in the absence of WDR60.
To address whether retrograde protein trafficking within cilia is indeed impaired in WDR60-KO cells and whether it is unaffected in TCTEX1D2-KO cells, we next analyzed the localization of Smoothened (SMO) and GPR161, both of which are GPCRs that participate in the Hh signaling pathway (Mukhopadhyay and Rohatgi, 2014). This was because we previously showed that, due to a block in retrograde trafficking, both SMO and GPR161 are accumulated within cilia in IFT139-KO cells (Hirano et al., 2017) and that GPR161 is accumulated within cilia in IFT121-KO cells (Takahara et al., 2018).
Under basal conditions in normal cells, SMO is excluded from cilia (Figure 5A), whereas GPR161 is found on the ciliary membrane (Figure 5G), where it negatively regulates Hh signaling (Mukhopadhyay and Rohatgi, 2014). On the other hand, when the Hh pathway is activated by treating cells with a small molecule activator, Smoothened Agonist (SAG), SMO is cued to enter cilia (Figure 5D), whereas GPR161 is exported from cilia (Figure 5J), resulting in cancelation of the negative regulation (Mukhopadhyay and Rohatgi, 2014; Schou et al., 2015). In WDR60-KO cell lines, however, the situation of these GPCRs was completely different. Even in the absence of SAG, a substantial amount of SMO was found along cilia (Figure 5B). When WDR60-KO cells were treated with SAG, the proportion of cells with ciliary SMO was further increased (Figure 5E; also see Figure 5M). By contrast, a substantial amount of GPR161 was retained within cilia even when cells were treated with SAG (Figure 5K; also see Figure 5N). Overall, these data indicate that retrograde trafficking of ciliary GPCRs is impaired in the absence of WDR60.
FIGURE 5:

Accumulation of SMO and GPR161 within cilia in WDR60-KO and TCTEX1D2-KO cells. (A–L) Control RPE1 cells (A, D, G, and J), the #W60-2-2 cell line (B, E, H, and K), and the #1D2-2-9 cell line (C, F, I, and L) were serum-starved for 24 h and further cultured for 24 h in the absence (–SAG) or presence (+SAG) of 200 nM SAG. The cells were triple immunostained for either SMO (A–F) or GPR161 (G–L), ARL13B (A′–L′), and FOP (A′′–L′′). Scale bars, 10 µm. (M, N) Fluorescence staining intensities of SMO (M) and GPR161 (N) in control, WDR60-KO, and TCTEX1D2-KO cells were measured, and relative intensities of the cells are expressed as bar graphs. Values are means ± SD of three independent experiments. In each set of experiments, 31–63 (M) and 31–53 (N) ciliated cells were analyzed and the total numbers of ciliated cells analyzed (n) are shown. p, one-way ANOVA followed by Tukey post-hoc analysis for comparison among the cell lines, and the Student t test for comparison between the cells with and without SAG treatment.
TCTEX1D2-KO cells showed mild defects with respect to GPCR localization. However, like in control RPE1 cells, SMO was absent from cilia under basal conditions (Figure 5C) and entered cilia on SAG treatment (Figure 5F), GPR161 demonstrated a tendency to remain within cilia under SAG-treated conditions, although this was not statistically significant (Figure 5L; also see Figure 5N).
WDR60–TCTEX1D2 interaction is required for dynein-2 function
To exclude the possibility that the phenotypes of WDR60-KO and TCTEX1D2-KO cells resulted from off-target effects of the CRISPR/Cas9 system, we performed rescue experiments by stably expressing an mChe-fused WDR60 or TCTEX1D2 construct. When mChe-WDR60(WT) was expressed in WDR60-KO cells, the accumulation of IFT88 within cilia was significantly rescued (Figure 6B′), compared with those expressing mChe as a negative control (Figure 6A′; also see Figure 6P). Similarly, stable expression of mChe-TCTEX1D2(WT), but not mChe, in TCTEX1D2-KO cells resulted in the normal localization of IFT88 (Figure 6, compare D′ and E′; also see Figure 6P).
FIGURE 6:

Rescue of IFT88 and GPR161 localization in WDR60-KO and TCTEX1D2-KO cells on the expression of a WDR60 and TCTEX1D2 construct, respectively. The #W60-2-2 cell line stably expressing mChe (A, F, K), mChe-fused WDR60(WT) (B, G, L), or WDR60(Δ473–522) (C, H, M), or the #1D2-2-9 cell line stably expressing mChe (D, I, N,) or mChe-TCTEX1D2(WT) (E, J, O) were cultured under serum-starved conditions to induce ciliogenesis, as described in the legend to Figure 4. For examining the localization of GPR161, cells were incubated in the presence or absence of 200 nM SAG, as described in the legend to Figure 5. The cells were quadruple immunostained for RFP (A–O), either IFT88 (A′–E′) or GPR161 (F′–O′), and ARL13B+FOP (A′′–O′′). Scale bars, 10 μm. The staining intensity for IFT88 within cilia in the cells were measured and expressed as bar graphs. Values are means ± SD of three independent experiments. In each experiment, 28–87 ciliated cells were observed, and the total numbers of ciliated cells observed (n) are shown. p, one-way ANOVA followed by Tukey post-hoc analysis. Relative staining intensities for IFT88 (P) or GPR161 (Q) within cilia were estimated and expressed as described in the legends to Figures 4 and 5. Values are means ± SD of three independent experiments. In each set of experiments, 31–49 (A–E) or 30–56 (F–O) ciliated cells were analyzed, and the total numbers of ciliated cells analyzed (n) are shown. p, one-way ANOVA followed by Tukey post-hoc analysis for comparison among the cell lines, and the Student t test for comparison between the cells with and without SAG treatment.
We also expressed mChe-WDR60(Δ473–522), which lacks the ability to interact with the TCTEX1D2–DYNLT1 dimer (Figure 3, C and D), in WDR60-KO cells. As shown in Figure 6C′ (also see Figure 6P), expression of mChe-WDR60(Δ473–522) moderately restored the localization of IFT88; a larger amount of IFT88 was retained within cilia in mChe-WDR60(Δ473–522)-expressing cells than in mChe-WDR60(WT)-expressing cells.
We also analyzed the ciliary levels of SMO and GPR161 under basal conditions, and their changes on SAG treatment in WDR60-KO and TCTEX1D2-KO cells expressing a WDR60 and TCTEX1D2 construct, respectively. In WDR60-KO cells, the accumulation of SMO within cilia under basal conditions was canceled by the stable expression of mChe-fused WDR60(WT) or WDR60(Δ473–522) (Supplemental Figure S2, compare B′ and C′ with A′; also see Supplemental Figure S2K). The accumulation of GPR161 under both basal and SAG-stimulated conditions was canceled by mChe-WDR60(WT) expression (Figure 6, compare G′ with F′ and L′ with K′; also see Figure 6Q). On the other hand, stable expression of mChe-WDR60(Δ473–522) rescued the basal ciliary GPR161 level (Figure 6H′) but not SAG-stimulated exit of GRP161 from cilia (Figure 6M′; also see Figure 6Q); this phenotype resembled that of TCTEX1D2-KO cells expressing mChe (Figure 6, I′ and N′; also see Figure 6Q). The exogenous expression of mChe-TCTEX1D2(WT) in TCTEX1D2-KO cells restored the SAG-stimulated exit of GPR161 (Figure 6O′; also see Figure 6Q).
Thus, loss of the interaction of WDR60 with the TCTEX1D2–DYNLT1/DYNLT3 dimer or lack of TCTEX1D2 appears to moderately impair ciliary retrograde trafficking.
Dynein-2 localization is impaired in WDR60-KO and TCTEX1D2-KO cells
The above data using WDR60-KO and TCTEX1D2-KO cells suggested that in the absence of WDR60 or TCTEX1D2, impaired dynein-2 function leads to defects in retrograde protein trafficking. To confirm this, we next aimed to determine the localization of the dynein-2 subunits in WDR60-KO and TCTEX1D2-KO cells. However, none of the commercially available antibodies against dynein-2 subunits that we examined worked well in immunofluorescence experiments. Eventually, we used total internal reflection fluorescence (TIRF) microscopy and found that EGFP-DYNC2LI1 stably expressed in RPE1 cells undergoes intraciliary trafficking. Supplemental Video S1 and kymographic analysis of the consecutive images (Figure 7A) revealed that EGFP-positive particles underwent both anterograde and retrograde movement along cilia. As shown in Figure 7C (also see Figure 7F), EGFP-DYNC2LI1 was found within cilia and at the base of cilia in fixed RPE1 cells. In fixed TCTEX1D2-KO cells, EGFP-DYNC2LI1 signals were detectable at the ciliary base but were below the detection level within cilia in a high level of cytoplasmic background signals (Figure 7E; also see Figure 7F). When TCTEX1D2-KO cells expressing EGFP-DYNC2LI1 were examined by TIRF microscopy followed by kymographic analysis, however, we could detect weak signals of shuttling particles (Supplemental Video S2 and Figure 7B). These observations suggested that, in the absence of TCTEX1D2, the dynein-2 complex containing DYNC2LI1 assembles around the ciliary base, although less efficiently than in its presence. In contrast to control and TCTEX1D2-KO cells, we could not detect EGFP-DYNC2LI1 signals within nor at the base of cilia in WDR60-KO cells (Figure 7D; also see Figure 7F). Taking into account the dynein-2 architectural model (Figure 2E), it is thus likely that the functional dynein-2 complex containing DYNC2LI1 does not assemble in the absence of WDR60.
FIGURE 7:

Localization of EGFP-DYNC2LI1 in WDR60-KO and TCTEX1D2-KO cells. (A, B) Kymograms showing the movement of EGFP-DYNC2LI1-positive particles in control RPE1 cells (A) and #1D2-2-9 cells (B). See Supplemental Videos S1 and S2, respectively. (C–E) Control RPE1 cells (C), the #W60-2-2 cell line (D), or the #1D2-2-9 cell line (E) stably expressing EGFP-DYNC2LI1 were double immunostained for ARL13B (C′–E′) and FOP (C′′–E′′). Merged, enlarged images of the boxed regions of C–E and C′–E′ are shown in C′′′–E′′′. Scale bars, 10 µm. (F) Localization of EGFP-DYNC2LI1 in control RPE1 cells, and in the #W60-2-2 and #1D2-2-9 cell lines was classified as “even distribution within cilia,” “ciliary base,” and “no ciliary localization,” and the cells in each category were counted. The percentages of these populations are expressed as stacked bar graphs. Values are the means of three independent experiments. In each set of experiments, 33–60 ciliated cells were observed, and the total numbers of ciliated cells observed (n) are shown. p, the Pearson χ2 test.
Video S1.
TIRF microscopy of RPE1 cells expressing EGFP-DYNC2LI1. Scale bar: 5 μm
Video S2.
TIRF microscopy of the #1D2-2-9 cells expressing EGFP-DYNC2LI1. Scale bar: 5 μm
DISCUSSION
While a substantial amount of insights into the structures and functions of the dynein-1 complex and the dynactin complex, which is an essential cofactor for dynein-1, have been obtained over the past decade (Roberts et al., 2013; Carter et al., 2016), information about dynein-2 has remained limited, although a rough architectural model of the dynein-2 complex has been proposed (Asante et al., 2014). We here demonstrated the overall architecture of the dynein-2 complex. The holocomplex can be divided into the following three subcomplexes: 1) the H1 subcomplex (the heavy chain DYNC2H1 directly interacting with the light intermediate chain DYNC2LI1); 2) the WDR34 subcomplex (the intermediate chain WDR34 interacting with the light chains DYNLL1/DYNLL2 and DYNLRB1/DYNLRB2); and 3) the WDR60 subcomplex (the other intermediate chain WDR60 interacting with the light-chain dimer TCTEX1D2–DYNLT1/DYNLT3) (Figure 1F). Thus, the two intermediate chains interact with specific sets of light chains. The WDR34 and WDR60 subcomplexes interact with each other, and in turn interact with the H1 subcomplex (Figure 2E); thus, the dynein-2 complex demonstrates structural asymmetry with respect to the intermediate chains. This is in striking contrast to the dynein-1 intermediate chains; in the dynein-1 complex, two copies of the same intermediate chain (DYNC1I1 or DYNC1I2) are included (Kardon and Vale, 2009). Although WDR60 did not demonstrate a binary interaction with any other subunit (Figure 1B), we found that it interacts with the TCTEX1D2–DYNLT1/DYNLT3 dimer via its conserved region upstream of the WD40 domain (Figure 3). Although a previous study suggested that a conserved motif, (K/R)XTQT, in DYNC1I1 is required for its binding to DYNLL1 (Lo et al., 2001), such a motif sequence was not found in the conserved region of WDR60. Thus, the recognition of intermediate chains by different light chains appears to be based on distinct mechanisms.
We then established cell lines lacking WDR60 and TCTEX1D2 and compared their phenotypes, as their protein products interact with each other and mutations of these genes cause SRTDs (McInerney-Leo et al., 2013; Gholkar et al., 2015; Schmidts et al., 2015; Cossu et al., 2016; Kakar et al., 2017). In both WDR60-KO and TCTEX1D2-KO cells, IFT88 (an IFT-B subunit) was significantly accumulated within cilia, with WDR60-KO cells demonstrating a severer phenotype (Figure 4, A–C and I). In the case of IFT140 (an IFT-A subunit), WDR60-KO cells demonstrated its accumulation within cilia, whereas TCTEX1D2-KO cells did not show significant IFT140 accumulation (Figure 4, D–F and J).
In WDR60-KO cells, SMO was found within cilia even under basal conditions and GPR161 was not exported from cilia on SAG stimulation (Figure 5), indicating that the retrograde trafficking of SMO and GPR161 was significantly impaired. The trafficking defect in WDR60-KO cells probably resulted from the lack of functional dynein-2 assembly in the absence of WDR60 (Figure 7). The phenotype of WDR60-KO cells resembled those of IFT139-KO and IFT121-KO cells (Hirano et al., 2017; Takahara et al., 2018), which are peripheral subunits of the IFT-A complex that mediate retrograde ciliary protein trafficking (Hirano et al., 2017). On the other hand, TCTEX1D2-KO cells, as well as WDR60-KO cells exogenously expressing a WDR60 mutant defective in binding to the TCTEX1D2–DYNLT dimer, demonstrated mild defects in retrograde trafficking (Figure 5). This mild defect is probably a result of the dynein-2 complex not being able to efficiently enter cilia in the absence of TCTEX1D2 (Figure 7 and Supplemental Video S2).
The present study on dynein-2 subunits, as well as our previous studies on IFT-A subunits (Hirano et al., 2017; Takahara et al., 2018), unequivocally show that both the IFT-A and dynein-2 complexes are involved in retrograde ciliary protein trafficking in mammalian cells. However, there has been no direct evidence for the interaction between the IFT-A and dynein-2 complexes, even though such an interaction has been schematically illustrated in many review articles. In addition, our analysis using TIRF microscopy (Supplemental Video S1; also see Figure 7A) showed that dynein-2 undergoes not only retrograde but also anterograde trafficking. This is consistent with previous dynein-2 studies in Caenorhabditis elegans neuronal cilia (Mijalkovic et al., 2017) and in mouse olfactory cilia (Williams et al., 2014). The bidirectional dynein-2 movement indirectly suggests that the dynein-2 complex also associates with the IFT machinery, probably as a cargo, while it is trafficked in an anterograde direction. Therefore, our strategy utilizing the VIP assay will pave the way to clarifying the IFT-A–dynein-2 interaction and the potential IFT-B–dynein-2 interaction and understanding the physiological relevance of the interactions. Indeed, we recently demonstrated the mode of interaction between the IFT-B complex and heterotrimeric kinesin-II, wherein four IFT-B subunits and all the kinesin-II subunits participate in the interaction and demonstrated the importance of the interaction in ciliogenesis (Funabashi et al., 2018).
MATERIALS AND METHODS
Plasmids, antibodies, and reagents
cDNAs for the dynein-2 subunits listed in Supplemental Table S1 were cloned into fluorescent protein vectors as shown in Supplemental Table S2. Expression vectors for dynein-2 subunits and their deletion constructs used in this study are also listed in Supplemental Table S2. Antibodies used in this study are listed in Supplemental Table S3. GST-tagged anti-GFP Nb prebound to glutathione–Sepharose 4B beads were prepared as described previously (Katoh et al., 2015).
VIP assay and immunoblotting analysis
The VIP assay was performed as described previously (Katoh et al., 2015, 2016); we have recently reported a detailed protocol for the VIP assay (Katoh et al., 2018). Expression vectors for EGFP- and mChe-fused proteins were transfected into HEK293T cells grown on a six-well plate (∼1.6 × 106 cells) using Polyethylenimine Max (20 µg; Polysciences) and cultured for 24 h. Lysates were prepared from the transfected cells in 250 µl of the HNTG cell lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol) containing protease inhibitor cocktail (Nacalai Tesque) for 15 min on ice. The lysates were centrifuged at 16,100 × g for 15 min at 4°C, and the supernatant (200 µl) was transferred to a 0.2 ml 8-tube strip, which contained GST-tagged anti-GFP Nb bound to glutathione–Sepharose 4B beads (∼5 µl bed volume) and incubated for 1 h at 4°C with constant rotation of the tube. After centrifugation at 2000 × g for 30 s, the precipitated beads were washed three times with the lysis buffer (180 µl), and transferred to a 96-well plate. The beads bearing fluorescent fusion proteins were observed under an all-in-one type microscope (BZ-8000; Keyence) with a 20 ×/0.75 NA objective lens under fixed conditions (sensitivity ISO 400, exposure 1/30 s for green fluorescence; and sensitivity ISO 800, exposure 1/10 s for red fluorescence). Image acquisition was performed under fixed conditions in each set of experiments.
Establishment of KO cell lines using the CRISPR/Cas9 system
The strategy for the KO of genes in hTERT-RPE1 cells (American Type Culture Collection; CRL-4000) by the CRISPR/Cas9 system using homology-independent repair (the version 2 method) was described previously in detail (Katoh et al., 2017). The single guide RNA (sgRNA) sequences targeting the human WDR60 and TCTEX1D2 genes (see Supplemental Table S4) were designed using CRISPOR (http://crispor.tefor.net/). Double-stranded oligonucleotides for the sequences were inserted into an all-in-one sgRNA expression vector, peSpCAS9(1.1)-2 × sgRNA (Addgene 80768). hTERT-RPE1 cells were grown on a 12-well plate to ∼3.0 × 105 cells and transfected with 1 µg of the sgRNA vector and 0.25 µg of the donor knock-in vector, pDonor-tBFP-NLS-Neo(universal) (Addgene 80767), using X-tremeGENE9 Reagent (Roche Applied Science). After selection in the presence of G418 (600 µg/ml), the cells with blue nuclear fluorescence were isolated. To confirm disruption of the WDR60 and TCTEX1D2 genes, genomic DNA was extracted from the isolated cells and subjected to PCR using KOD FX Neo DNA polymerase (TOYOBO). Three sets of primers (Supplemental Table S4) were used for PCR to distinguish the following three states of integration of the donor vector: forward integration, reverse integration, and no integration with a small insertion or deletion (Supplemental Figure S1A). Direct sequencing of the PCR products was performed to confirm disruption of both alleles of the WDR60 and TCTEX1D2 genes; a small deletion resulting in a frameshift in one allele and a forward integration of the donor vector in the other allele (Supplemental Figure S1, B and C).
Preparation of cells stably expressing mChe-WDR60, mChe-TCTEX1D2, or EGFP-DYNC2LI1
Lentiviral vectors for WDR60, TCTEX1D2, and DYNC2LI1 constructs were prepared as described previously (Takahashi et al., 2012). Briefly, pRRLsinPPT-mChe-WDR60 or its deletion construct pRRLsinPPT-mChe-TCTEX1D2, or pRRLsinPPT-EGFP-DYNC2LI1 was transfected into HEK293T cells with packaging plasmids (pRSV-REV, pMD2.g, and pMDL/pRRE; kind gifts from Peter McPherson, McGill University [Thomas et al., 2009]). The culture medium was replaced 8 h after transfection. Culture media containing lentiviral particles were collected at 24, 36, and 48 h after transfection, passed through a 0.45-µm filter (Sartorius), and centrifuged at 32,000 × g at 4°C for 4 h using an R15A rotor and Himac CR22G centrifuge (Hitachi Koki, Japan). Precipitated viral particles were resuspended in Opti-MEM (Invitrogen) and stored at –80°C until use. Cells expressing mChe-WDR60, mChe-TCTEX1D2, or EGFP-DYNC2LI1 were prepared by addition of the lentiviral suspension to the culture medium and processed for immunofluorescence analysis or TIRF microscopy.
Immunofluorescence analysis and TIRF microscopy
hTERT-RPE1 cells were cultured in DMEM/F-12 (Nacalai Tesque) supplemented with 10% fetal bovine serum (FBS) and 0.348% sodium bicarbonate. To induce ciliogenesis, cells were grown to 100% confluence on coverslips and starved for 24 h in Opti-MEM containing 0.2% bovine serum albumin.
For immunofluorescence analysis, cells were fixed and permeabilized with 3% paraformaldehyde at 37°C for 5 min, and subsequently in 100% methanol for 5 min at −20°C, and washed three times with phosphate-buffered saline. The fixed/permeabilized cells were blocked with 10% FBS and stained with antibodies diluted in 5% FBS. The stained cells were observed using an Axiovert 200M microscope (Carl Zeiss). Statistical analyses were performed using JMP Pro 13 software (SAS Institute).
TIRF microscopy was performed as described previously (Takahashi et al., 2012; Kubo et al., 2015). In short, RPE1 cells expressing EGFP-DYNC2LI1 were serum starved for 24 h on a glass-bottom culture dish, placed on a microscope stage prewarmed to 37°C, and observed using a TIRFM ECLIPSE Ti microscope (NIKON) at a video rate using NIS-Elements imaging software. Kymograms were generated with NIS-Elements imaging software.
Supplementary Material
Acknowledgments
We thank Peter McPherson for kindly providing plasmids for recombinant lentivirus production, Helena Akiko Popiel for critical reading of the manuscript, and Kentaro Nakamura for technical assistance. The cDNA clones of DYNC2LI1, WDR34, and WDR60 were provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT/AMED, Japan, and the clone of DYNC2H1 was provided by the Kazusa DNA Research Institute. This work was supported in part by Grants-in-Aid for Scientific Research on Innovative Areas “Cilia and Centrosome” from the Ministry of Education, Culture, Sports, Science and Technology, Japan (grant number 15H01211 to K.N.); grants from the Japan Society for the Promotion of Science (JSPS) (grant numbers 15H04370 and 15K14456 to K.N., 15K07929 and 18H02403 to Y.K., and 16J03865 to S.N.); and grants from the Uehara Memorial Foundation (to K.N. and Y.K.) and from the Takeda Science Foundation (to Y.K.). S.N. was supported by a JSPS Research Fellowship.
Abbreviations used:
- EGFP
enhanced green fluorescent protein
- GPCR
G protein–coupled receptor
- GST
glutathione S-transferase
- Hh
Hedgehog
- hTERT-RPE1
human telomerase reverse transcriptase-immortalized retinal pigment epithelial 1
- IFT
intraflagellar transport
- KO
knockout
- mChe
mCherry
- Nb
nanobody
- SAG
Smoothened agonist
- sgRNA
single guide RNA
- SMO
Smoothened
- SRTD
short-rib thoracic dysplasia
- TIRF
total internal reflection fluorescence
- VIP
visible immunoprecipitation.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-03-0173) on May 9, 2018.
REFERENCES
- Asante D, Stevenson NL, Stephens DJ. (2014). Subunit composition of the human cytoplasmic dynein-2 complex. J Cell Sci , 4774–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen TM, Texier Y, van Beersum SEC, Horn N, Willer JR, Mans D, et al (2016). An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun , 11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Diamant AG, Urnavicius L. (2016). How dynein and dynactin transport cargos: a structural perspective. Curr Opin Struct Biol , 62–70. [DOI] [PubMed] [Google Scholar]
- Cossu C, Incani F, Serra ML, Coiana A, Crisponi G, Boccone L, Rosatelli MC. (2016). New mutations in DYNC2H1 and WDR60 genes revealed by whole-exome sequencing in two unrelated Sardinian families with Jeune asphyxiating thoracic dystrophy. Clin Chim Acta , 172–180. [DOI] [PubMed] [Google Scholar]
- Funabashi T, Katoh Y, Michisaka S, Terada M, Sugawa M, Nakayama K. (2017). Ciliary entry of KIF17 is dependent on its binding to the IFT-B complex via IFT46-IFT56 as well as on its nuclear localization signal. Mol Biol Cell , 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabashi T, Katoh Y, Okazaki M, Sugawa M, Nakayama K. (2018). Interaction of heterotrimeric kinesin-II with IFT-B–connecting tetramer is crucial for ciliogenesis. J Cell Biol , DOI: 10.1083/jcb.201801039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Reiter JF. (2017). Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol , a028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholkar AA, Senese S, Lo YC, Capri J, Deardorff WJ, Dharmarajan H, Contreras E, Hodara E, Whitelegge JP, Jackson PK, Torres JZ. (2015). Tctex1d2 associates with short-rib polydactyly syndrome proteins and is required for ciliogenesis. Cell Cycle , 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J, Pelletier L. (2017). The ciliary transition zone: finding the pieces and assembling the gate. Mol Cells , 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Agbu S, Anderson KV. (2016). Microtubule-motors drive Hedgehog signaling in primary cilia. Trends Cell Biol , 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Katoh Y, Nakayama K. (2017). Intraflagellar transport-A complex mediates ciliary entry and retrograde trafficking of ciliary G protein-coupled receptors. Mol Biol Cell , 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron , 610–638. [DOI] [PubMed] [Google Scholar]
- Hou Y, Witman GB. (2015). Dynein and intraflagellar transport. Exp Cell Res , 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. (2011). Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol , 222–234. [DOI] [PubMed] [Google Scholar]
- Kakar M, Horn D, Decker E, Sowada N, Kubisch C, Ahmad J, Borck G, Bergmann C. (2017). Expanding the phenotype associated with biallelic WDR60 mutations: siblings with retinal degeneration and polydactyly lacking other features of short rib thoracic dystrophies. Am J Med Genet , 438–442. [DOI] [PubMed] [Google Scholar]
- Kardon JR, Vale RD. (2009). Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol , 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Michisaka S, Nozaki S, Funabashi T, Hirano T, Takei R, Nakayama K. (2017). Practical method for targeted disruption of cilia-related genes by using CRISPR/Cas9-mediated homology-independent knock-in system. Mol Biol Cell , 898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Nakamura K, Nakayama K. (2018). Visible immunoprecipitation (VIP) assay: a simple and versatile method for visual detection of protein-protein interactions. Bio-protocol , e2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Nozaki S, Hartanto D, Miyano R, Nakayama K. (2015). Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J Cell Sci , 2351–2362. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Terada M, Nishijima Y, Takei R, Nozaki S, Hamada H, Nakayama K. (2016). Overall architecture of the intraflagellar transport (IFT)-B complex containing Cluap1/IFT38 as an essential component of the IFT-B peripheral subcomplex. J Biol Chem , 10962–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K, Kobayashi M, Nozaki S, Yagi C, Hatsuzawa K, Katoh Y, Shin H-W, Takahashi S, Nakayama K. (2015). SNAP23/25 and VAMP2 mediate exocytic event of transferrin receptor-containing recycling vesicles. Biol Open , 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KW-H, Kogoy JM, Rasoul BA, King SM, Pfister KK. (2007). Interaction of the DYNLT (TCTEX1/RP3) light chains and the intermediate chains reveals novel intersubunit regulation during assembly of the dynein complex. J Biol Chem , 36871–36878. [DOI] [PubMed] [Google Scholar]
- Lo KW-H, Naisbitt S, Fan J-S, Sheng M, Zhang M. (2001). The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J Biol Chem , 14059–14066. [DOI] [PubMed] [Google Scholar]
- McInerney-Leo AM, Harris JE, Marshall MS, Gardiner B, Kinning E, Leong HY, McKenzie F, Ong WP, Vodopiutz J, Wicking C, et al (2015). Whole exome sequencing is an efficient, sensitive and specific method for determining the genetic cause of short-rib thoracic dystrophies. Clin Genet , 550–557. [DOI] [PubMed] [Google Scholar]
- McInerney-Leo AM, Schmidts M, Cortés CR, Leo PJ, Gener B, Courtney AD, Gardiner B, Harris JA, Lu Y, Marshall M, et al (2013). Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet , 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijalkovic J, Prevo B, Oswald F, Mangeol P, Peterman EJG. (2017). Ensemble and single-molecule dynamics of IFT dynein in Caenorhabditis elegans cilia. Nat Commun , 14591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Rohatgi R. (2014). G-protein-coupled receptors, Hedgehog signaling and primary cilia. Sem Cell Dev Biol , 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Katoh Y. (2018). Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin-2 and dynein-2 motors. J Biochem , 155–164. [DOI] [PubMed] [Google Scholar]
- Nishijima Y, Hagiya Y, Kubo T, Takei R, Katoh Y, Nakayama K. (2017). RABL2 interacts with the intraflagellar transport B complex and CEP19 and participates in ciliary assembly. Mol Biol Cell , 1652–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki S, Katoh Y, Terada M, Michisaka S, Funabashi T, Takahashi S, Kontani K, Nakayama K. (2017). Regulation of ciliary retrograde protein trafficking by the Joubert syndrome proteins ARL13B and INPP5E. J Cell Sci , 563–576. [DOI] [PubMed] [Google Scholar]
- Patel-King RS, Gilberti RM, Hom EFY, King SM. (2013). WD60/FAP163 is a dynein intermediate chain required for retrograde intraflagellar transport in cilia. Mol Biol Cell , 2668–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo B, Scholey JM, Peterman EJG. (2017). Intraflagellar transport: mechanisms of motor action, cooperation, and cargo delivery. FEBS J , 2905–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR. (2017). Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol , 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. (2013). Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol , 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M. (2014). Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet , 49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Hou Y, Cortés C, Mans DA, Huber C, Boldt K, Patel M, van Reeuwijk J, Plaza JM, van Beersum SEC, et al (2015). TCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport. Nat Commun , 7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schou KB, Pedersen LB, Christensen ST. (2015). Ins and outs of GPCR signaling in primary cilia. EMBO Rep , 1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara M, Katoh Y, Nakamura K, Hirano T, Sugawa M, Tsurumi Y, Nakayama K. (2018). Ciliopathy-associated mutations of IFT122 impair ciliary protein trafficking but not ciliogenesis. Hum Mol Genet , 516–528. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kubo K, Waguri S, Yabashi A, Shin H-W, Katoh Y, Nakayama K. (2012). Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J Cell Sci , 4049–4057. [DOI] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E. (2016). The intraflagellar transport machinery. Cold Spring Harb Perspect Biol , a028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Weber K, Mourão A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, Lorentzen E. (2016). Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. EMBO J , 773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Ritter B, Verbich D, Sanson C, Bourbonnière L, McKinney RA, McPherson PS. (2009). Intersectin regulates dendritic spine development and somatodendritic endocytosis but not synaptic vesicle recycling in hippocampal neurons. J Biol Chem , 12410–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, McIntyre JC, Norris SR, Jenkins PM, Zhang L, Pei Q, Verhey K, Martens JR. (2014). Direct evidence for BBSome-associated intraflagellar transport reveals distinct properties of native mammalian cilia. Nat Commun , 5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.