

Abstract
Tetraploid cells, which are most commonly generated by errors in cell division, are genomically unstable and have been shown to promote tumorigenesis. Recent genomic studies have estimated that ∼40% of all solid tumors have undergone a genome-doubling event during their evolution, suggesting a significant role for tetraploidy in driving the development of human cancers. To safeguard against the deleterious effects of tetraploidy, nontransformed cells that fail mitosis and become tetraploid activate both the Hippo and p53 tumor suppressor pathways to restrain further proliferation. Tetraploid cells must therefore overcome these antiproliferative barriers to ultimately drive tumor development. However, the genetic routes through which spontaneously arising tetraploid cells adapt to regain proliferative capacity remain poorly characterized. Here, we conducted a comprehensive gain-of-function genome-wide screen to identify microRNAs (miRNAs) that are sufficient to promote the proliferation of tetraploid cells. Our screen identified 23 miRNAs whose overexpression significantly promotes tetraploid proliferation. The vast majority of these miRNAs facilitate tetraploid growth by enhancing mitogenic signaling pathways (e.g., miR-191-3p); however, we also identified several miRNAs that impair the p53/p21 pathway (e.g., miR-523-3p), and a single miRNA (miR-24-3p) that potently inactivates the Hippo pathway via down-regulation of the tumor suppressor gene NF2. Collectively, our data reveal several avenues through which tetraploid cells may regain the proliferative capacity necessary to drive tumorigenesis.
INTRODUCTION
Cytokinesis, the final stage of cell division, is a highly complex process that cleaves a single cell into two daughter cells following segregation of replicated chromosomes. Failures in cytokinesis give rise to tetraploid cells, which harbor twice the normal chromosomal content. Proliferating tetraploid cells are highly genomically unstable and rapidly accumulate both numerical and structural chromosomal abnormalities (Ganem and Pellman, 2007; Storchova and Kuffer, 2008; Davoli and de Lange, 2011). Moreover, tetraploid cells, by virtue of their doubled genomes, are able both to buffer deleterious mutations as they arise and to rapidly acquire beneficial mutations that promote adaptation to stressful environments (Fujiwara, Bandi, et al., 2005; Storchova et al., 2006; Storchova and Kuffer, 2008; Dewhurst, McGranahan, et al., 2014; Selmecki et al., 2015). Consequently, multiple models have validated the conclusion that genomically unstable tetraploid cells can promote tumorigenesis, and current estimates indicate that ∼40% of human solid tumors pass through a tetraploid intermediate at some stage during their evolution (Duelli et al., 2005; Fujiwara, Bandi, et al., 2005; Mazumdar et al., 2006; Nguyen et al., 2009; Davoli and de Lange, 2012; Zack, Schumacher, et al., 2013).
To combat the potentially oncogenic effects of tetraploidy, nontransformed cells have evolved mechanisms to limit the proliferation of tetraploid cells (Carter, 1967; Andreassen, Lohez, et al., 2001; Ganem et al., 2007; Senovilla et al., 2012; Kuffer et al., 2013). First, it is now known that tetraploid cells activate two core kinases of the Hippo tumor suppressor pathway, LATS1 and LATS2 (Ganem, Cornils, et al., 2014; McKinley and Cheeseman, 2017). Activated LATS1/2 kinases phosphorylate the pro-growth transcriptional co-activators YAP and TAZ, ultimately leading to their cytoplasmic retention and subsequent degradation (Pan, 2007; Ganem, Cornils, et al., 2014; Moroishi, Hansen, and Guan, 2015a). Second, tetraploid cells promote the stabilization of p53 through caspase-2- and LATS2-dependent mechanisms that inhibit the activity of the p53 E3 ubiquitin ligase MDM2 (Aylon et al., 2006; Ganem, Cornils, et al., 2014; Fava et al., 2017). Stabilization of p53 leads to a corresponding increase in the expression of one of its core target genes, CDKN1A, which encodes the CDK inhibitor p21. Consequently, tetraploid cells arrest in the G1 phase of the cell cycle, after which they commonly senesce (Andreassen, Lohez, et al., 2001; Ganem et al., 2014; Panopoulos et al., 2014).
To facilitate tumorigenesis, tetraploid cells must overcome these barriers to cell proliferation (Ganem and Pellman, 2007). There are three broad routes through which tetraploid cells are known to regain proliferative capacity. First, they can proliferate if they have a nonfunctional or defective p53/p21 pathway (Carter, 1967; Andreassen, Lohez, et al., 2001; Krzywicka-Racka and Sluder, 2011; Ganem, Cornils, et al., 2014; Panopoulos et al., 2014). Indeed, analysis of near-tetraploid tumors in the Cancer Genome Atlas (TCGA) reveals a significant enrichment in TP53-inactivating mutations, demonstrating the importance of nonfunctional p53 in the tolerance of tetraploidy (Crockford, Zalmas, Gronroos, et al., 2017). Second, they can regain proliferative capacity through functional impairment of the Hippo tumor suppressor pathway, which leads to a corresponding restoration of YAP activity (Ganem, Cornils, et al., 2014). Analysis of the Cancer Cell Line Encyclopedia (CCLE) reveals that both YAP overexpression and LATS1/2 deletion are highly enriched in near-tetraploid compared with near-diploid cancer cell lines (Ganem, Cornils, et al., 2014). Importantly, active YAP is sufficient to promote tetraploid cell proliferation even in the presence of a functional p53 pathway (Ganem, Cornils, et al., 2014; Fava et al., 2017). Finally, they can regain proliferative capability by promoting cyclin D accumulation through hyperactivation of mitogenic signaling, or by amplification of the gene encoding for cyclin D itself (Ganem, Cornils, et al., 2014; Potapova et al., 2016; Crockford, Zalmas, Gronroos, et al., 2017). When in excess, cyclin D1 has been shown to inhibit p21 through noncatalytic sequestration, resulting in the formation of cyclin D1/p21 complexes that negate p21-mediated growth inhibition and permit tetraploid proliferation (Crockford, Zalmas, Gronroos, et al., 2017).
While functional impairment of the p53/p21 pathway, reactivation of YAP, or increased mitogenic signaling is sufficient to initiate tetraploid proliferation, it remains unclear how cells initially achieve these growth-promoting adaptations. We hypothesized that overexpression of individual microRNAs (miRNAs) may be one route through which tetraploid cells subtly disrupt these pathways to initially escape G1 cell-cycle arrest. miRNAs, which are small noncoding RNAs, function as critical regulators of gene expression by repressing the translation of complementary mRNAs. The complementarity region of miRNAs, known as the seed sequence, is ∼6–8 nucleotides in length and may target multiple sites within the same mRNA and/or simultaneously modulate the translation of hundreds of mRNAs (Bartel, 2009). In this manner, miRNAs can control the regulation of multiple signaling pathways at once. Indeed, deregulation of miRNA expression is a common feature of human cancers, with overexpression observed in a variety of tumors (Jansson and Lund, 2012).
In this study, we conducted a genome-wide gain-of-function screen to comprehensively identify individual miRNAs that are sufficient to drive tetraploid proliferation when overexpressed. The results of this screen, detailed herein, establish a list of putative oncogenic miRNAs (oncomiRs), many of which have been implicated in driving tumorigenesis. Importantly, we mechanistically define the pathways these miRNAs modulate to overcome tetraploidy-induced G1 arrest.
RESULTS AND DISCUSSION
miRNA screen to identify regulators of tetraploid cell proliferation
We performed a genome-wide gain-of-function screen to identify individual miRNAs that promote the proliferation of G1-arrested tetraploid cells. For this screen, we used diploid, nontransformed retinal pigmented epithelial cells (RPE-1), as they contain an intact p53 pathway and undergo durable G1 arrest following cytokinesis failure (Ganem, Cornils, et al., 2014). RPE-1 cells were engineered to stably express the fluorescence ubiquitination cell-cycle indicator (FUCCI) system. The FUCCI reporter system distinguishes cell-cycle position based on a fluorescence readout, where G1 cells exhibit red fluorescence (due to expression of a fragment of Cdt1 fused to m-Cherry) and S/G2/M cells exhibit green fluorescence (due to expression of a fragment of Geminin fused to Azami-Green; Sakaue-Sawano et al., 2008).
To generate G1-arrested tetraploid cells for the screen, diploid RPE-1 FUCCI cells were treated with 4 μM dihydrocytochalasin B (DCB), an inhibitor of actin polymerization, for 16 h to inhibit cytokinesis (Ganem, Cornils, et al., 2014). This treatment induces tetraploidy in ∼60% of cells. Fluorescence-activated cell sorting (FACS) was then used to purify tetraploid cells from diploid cells, as previously described (Shenk and Ganem, 2016). In brief, cells exhibiting both 4N DNA content and red fluorescence, indicative of G1 tetraploids, were collected (Figure 1A). This methodology results in a highly enriched (>95% pure) tetraploid population (Ganem, Cornils, et al., 2014). Consistent with previous studies, we found that the isolated tetraploid cells exhibited strong G1 cell-cycle arrest. Live-cell imaging revealed that >80% of all binucleated tetraploid cells failed to enter S-phase over 72 h, as indicated by persistent red fluorescence (Figure 1, D and E). In contrast, the vast majority of isolated diploid cells, which were exposed to the same culture conditions, drug treatments, and FACS as tetraploids, showed continued proliferation, as evidenced by a change from red to green fluorescence indicative of the G1–S transition (Figure 1, D and E; Ganem, Cornils, et al., 2014). As a positive control, small interfering RNA (siRNA)–-mediated depletion of p53 was observed to restore proliferation to tetraploid cells (Figure 1D).
FIGURE 1:

Genome-wide screen to identify miRNAs that promote tetraploid cell proliferation. (A) Protocol for a genome-wide miRNA screen to identify overexpressed miRNAs that promote tetraploid cell proliferation. Adapted from Ganem, Cornils, et al. (2014). (B) Average Z-scores for the ∼800 screened individual miRNAs in the primary screen. The primary screen yielded ∼80 miRNAs hits (green) that were subsequently rescreened. (C) The 23 miRNA hits following secondary screening ranked by average Z-score. (D) Still images from a live-cell imaging experiment with RPE-FUCCI cells. Sorted diploids and tetraploids transfected with the indicated siRNA/miRNA are shown at 24, 48, and 72 h. Representative images of mitotic cells for each condition are shown on the right. Microtubules (red), chromosomes (white), centrosomes (green). (E) Quantitation of the percentage of cells that enter S/G2 for each condition shown in D over 72 h. Error bars represent mean ± SEM from three separate experiments (n = 50 cells counted per experiment), *** p ≤ .001, two-tailed t test and one-way analysis of variance (ANOVA).
To perform the screen, freshly purified G1-arrested binucleated tetraploids were seeded in 96-well plates in triplicate and reverse transfected with a library of ∼880 precursor miRNA mimics to emulate overexpression of endogenous miRNA (Figure 1A). Nontargeting miRNA sequences were used as negative controls, while siRNAs targeting p53 were used as positive controls. At 96 h post–miRNA transfection, all plates were fixed and automated image analysis was used to determine the fraction of tetraploid cells per well that had escaped G1 arrest and entered S-phase (as judged by a transition from red to green fluorescence; Figure 1A). For the primary screen, miRNA hits were identified as having a Z-score ≥6.0, which was determined from multiple intraplate negative controls. Using this strict criterion, the primary screen identified 81 miRNAs for which overexpression enabled tetraploid cells to escape G1 arrest (Figure 1B). To validate and reproduce these hits, each of the 81 miRNAs was then rescreened under the same protocol and manually analyzed. This secondary screen resulted in the identification of 23 miRNAs whose overexpression gave an average Z-score value ≥5.0 after both biological replicates, indicating robust escape from G1 arrest (Figure 1C). To complement the Z-score ranking, we further determined how well each miRNA promoted S-phase entry in tetraploids relative to RNA interference (RNAi) depletion of p53. Remarkably, multiple miRNAs enhanced S-phase entry of tetraploid cells nearly as well as p53 loss, including the top two hits, miR-523-3p and miR191-3p, which essentially mirrored the ability of p53 knockdown to overcome G1 arrest in tetraploid cells.
We performed live-cell imaging to confirm that tetraploid cells overcame G1 arrest following transfection with several of our strongest miRNA hits (Figure 1, D and E). We also assessed the fraction of tetraploid cells that progressed through the cell cycle and ultimately underwent mitosis (Supplemental Figure S1A). Interestingly, even though all miRNA hits were sufficient to promote G1/S transition in tetraploids, not all miRNAs enabled progression to mitosis (Supplemental Figure S1A). As expected, those tetraploid cells that did enter mitosis displayed abnormal multipolar spindles as a consequence of containing supernumerary centrosomes (Figure 1D). Despite these mitotic defects, expression of the top two miRNA hits, miR-523-3p and miR-191-3p, was sufficient to induce multiple rounds of tetraploid cell division (Supplemental Figure S1B). Thus, our data demonstrate that overexpression of individual miRNAs is sufficient to promote tetraploid cell proliferation and to promote the abnormal mitotic spindle assembly that leads to subsequent genome instability (Ganem et al., 2009; Silkworth et al., 2009).
The majority of miRNA hits hyperactivate mitogenic signaling or impair the p53/p21 pathway
As an initial assessment, we performed cluster analysis to identify whether any of the miRNA hits share seed sequence homology. This analysis revealed that over half of the miRNAs share a recently identified oncomotif seed sequence, AAGUGC (Supplemental Figure S1C). Previous work has demonstrated that miRNAs with an AAGUGC seed motif act as oncomiRs by targeting multiple tumor suppressors to promote the proliferation of cancer cells (Zhou et al., 2017). Thus, this result validated our screening approach and revealed that many clinically relevant oncomiRs may act, at least in part, by promoting the proliferation of tetraploid cells.
We then sought to identify the specific pathways that are disrupted by overexpression of the strongest miRNA hits. Previous studies have demonstrated that hyperactivation of mitogenic signaling is sufficient to promote tetraploid cell proliferation; therefore, we investigated whether any of our miRNA hits enhanced MAPK or PI3K signaling. To do this, RPE-1 cells transfected with scrambled or endogenous miRNA mimics were serum-starved for 24 h to turn off mitogenic pathways and then restimulated with 5% fetal bovine serum (FBS). Following restimulation, both MAPK and PI3K pathway activation were assessed by quantitating phosphorylated ERK as a fraction of total ERK and phosphorylated AKT as a fraction of total AKT (Supplemental Figure S1, D and E). Results from this assay revealed that overexpression of miR-191-3p, the second strongest hit, significantly stimulates both the MAPK and PI3K pathways and leads to a corresponding increase in cyclin D and cyclin E protein levels in both diploid and tetraploid cells (Figure 2, A and B, and Supplemental Figures S1, D and E, and S2D). Furthermore, we found that miR-191-3p overexpression not only hyperactivates the initial mitogenic response to serum, but also maintains a prolonged activation of MAPK and PI3K signaling over multiple hours (Figure 2A). Owing to its overexpression in 16 different cancer subtypes, miR-191-3p is largely classified as an oncogenic miRNA (Di Leva, Piovan, et al., 2013; Nagpal and Kulshreshtha, 2014). Our data reveal that the strong and enduring increase in mitogenic signaling induced by miR-191 overexpression may underpin its capacity to promote tetraploid cell cycle progression, and offer a mechanistic explanation for its frequent overexpression in human cancers.
FIGURE 2:

miRNA hits increase mitogenic signaling and/or disable the p53/p21 pathway. (A) Representative Western blot of RPE-1 cells, transfected with either a control siRNA (si-C) or miR-191-3p, and serum starved for 24 h prior to restimulation for the indicated time with 5% FBS. (B) Representative Western blot of RPE-1 cells, transfected with either a nontargeting miRNA control (miR-NC) or miR-191-3p, and serum starved for 24 h prior to restimulation with 5% FBS for the indicated duration. (C) Representative Western blot of RPE-1 cells, transfected with the indicated siRNA/miRNA, and treated with or without 100 ng/ml doxorubicin for 4 h. (D) Representative fixed images of RPE-1 cells, transfected with the indicated siRNA/miRNA, and treated with or without 100 ng/ml doxorubicin for 4 h prior to fixation. (E) Normalized fluorescence intensity quantification of images in D. Error bars represent mean ± SEM (n > 650 cells/condition from one experiment), ****p ≤ 0.0001 and ns = nonsignificant, two-tailed t test and one-way ANOVA.
In addition to hyperactive mitogenic signaling, abrogation of the p53/p21 signaling axis is also known to restore proliferation to tetraploid cells (Figure 1E; Andreassen, Lohez, et al., 2001; Fujiwara, Bandi, et al., 2005; Ganem, Cornils, et al., 2014). Thus, we investigated whether overexpression of any of the strongest miRNA hits disrupts p53-induced increases in p21 protein (Supplemental Figure S2, A and B). In normal RPE-1 cells, endogenous levels of p53 and p21 are quite low, making detection of decreases in their protein levels difficult. To address this, control cells or cells transfected with miRNA hits were first treated with 100 ng/ml of the DNA-damaging agent doxorubicin for 4 h to induce the p53/p21 pathway. In control cells, this treatment strongly activated p53 and elicited a threefold increase in p21 protein levels (Figure 2, C–E). Using this approach, we identified that our strongest overall hit, miR-523-3p, significantly reduces p21 protein levels in both diploid (Figure 2, C–E, and Supplemental Figure S2, A and B) and tetraploid (Supplemental Figure S2C) cells. This effect is likely to be indirect, as bioinformatics approaches do not strongly predict miR-523-3p to target the 3′ untranslated region of CDKN1A mRNA, which we then confirmed with luciferase assays (unpublished data). Thus, our data reveal that disruption of normal p53/p21 signaling by overexpression of individual miRNAs is another route through which tetraploid cells can escape cell-cycle arrest.
Overexpression of miR-24-3p strongly promotes YAP activation and tetraploid cell proliferation
Functional inactivation of the Hippo tumor suppressor pathway, which leads to YAP activation, is also known to confer proliferative ability on tetraploid cells (Ganem, Cornils, et al., 2014). Therefore, we explored whether overexpression of any miRNA hits is sufficient to increase YAP activity. To do this, we quantitated YAP localization in cells transfected with miRNA hits as an indirect readout of its activity, as it is known that YAP is predominantly cytoplasmic (inactive) upon Hippo pathway activation, but predominantly nuclear (active) upon Hippo pathway inactivation (Pan, 2007; Yu et al., 2015). Utilizing this assay, we identified two miRNAs (miR-24-3p and miR-553) that significantly increase nuclear YAP localization relative to controls, indicative of Hippo pathway attenuation (Figure 3, A and B, and Supplemental Figure S3A). Interestingly, miR-24-3p overexpression results in more nuclear-localized YAP than even RNAi-mediated depletion of the kinases LATS1/2, which served as the positive control in this assay. This finding was reproduced in tetraploid RPE-1 cells as well as in HEK293A cells, a cell line frequently employed to study Hippo signaling (Supplemental Figures S2E and S3, B and C; Meng et al., 2015; Moroishi et al., 2015b). In contrast to overexpression of miR-523-3p, overexpression of miR-24-3p did not abrogate the p53/p21 pathway, nor did it activate mitogenic signaling like miR-191-3p (Figure 2C and Supplemental Figure S1D).
FIGURE 3:

miR-24 overexpression promotes YAP activation. (A) Relative localization of YAP in RPE-1 cells following transfection with the indicated siRNA/miRNA. Plot depicts the normalized ratio of YAP immunofluorescence intensity in the nucleus:cytoplasm (N/C) performed in triplicate (n = 3). Error bars represent mean ± SEM (n >200 cells/condition, ****p ≤ 0.0001, one-way ANOVA). (B) Representative fixed images of YAP localization in RPE-1 cells 48 h posttransfection with miR-24-3p or negative control (n > 3). (C) Gene-set enrichment analysis of RPE-1 cells transfected with miR-24-3p compared with RPE-1 cells expressing YAP-5SA and two YAP dependent gene-sets. For reference: Park et al. (2016), Zanconato et al. (2015).
To examine whether the observed increase in nuclear YAP from miR-24-3p overexpression led to a corresponding increase in the transcription of YAP-regulated target genes, we performed gene expression analysis. We compared the gene expression profiles of three cell lines: control RPE-1 cells, RPE-1 cells overexpressing miR-24-3p, and RPE-1 cells expressing a constitutively active version of YAP in which five serines phosphorylated by LATS are mutated to alanines (YAP-5SA; Zhao et al., 2007). Gene set enrichment analysis (GSEA) of the resultant expression data confirmed that YAP-target genes, as defined by the RPE-1 YAP-5SA cells as well as previously published YAP-dependent gene sets, are significantly up-regulated in RPE-1 cells expressing miR-24-3p (Figure 3C; Zanconato et al., 2015; Park et al., 2016). Thus, our data reveal that overexpression of the miRNA miR-24-3p leads to YAP activation.
miR-24-3p overexpression results in down-regulation of the tumor suppressor NF2
We aimed to identify potential targets of miR-24-3p to elucidate how it impairs the Hippo pathway to activate YAP. First, we assessed the effect of miR-24-3p overexpression on the levels of the best-characterized Hippo pathway components (e.g., LATS1, LATS2, MST1, MST2; Supplemental Figure S3D). However, none of the core Hippo kinases was down-regulated, due to miR-24-3p overexpression. We then focused our attention on gene expression data, where we found that an upstream activator of the Hippo pathway, the tumor suppressor gene Neurofibromin 2 (NF2), was one of the 50 most down-regulated genes upon miR-24-3p overexpression (Supplemental Figure S3E). Known as Merlin in Drosophila, NF2 promotes activation of the LATS1/2 kinases, resulting in the phosphorylation and subsequent cytoplasmic sequestration of YAP (Mohseni et al., 2014). Loss of NF2 reduces Hippo pathway activity and promotes active, nuclear YAP (Yin et al., 2013; Moroishi et al., 2015b; Meng et al., 2016). This led us to test whether miR-24-3p overexpression activates YAP through down-regulation of NF2.
Consistent with our expression data, we found that miR-24-3p overexpression results in a greater than 50% decrease in NF2 protein levels (Figure 4, A and B). To compare relative YAP activation between miR-24-3p overexpression and depletion of NF2, we performed immunofluorescence imaging of YAP localization following treatment with siRNAs targeting NF2. Indeed, RNAi knockdown of NF2 significantly increases nuclear YAP localization; however, the observed effect does not recapitulate the magnitude of increased nuclear YAP localization achieved by miR-24-3p (Figure 4, C and D). These data suggest that miR-24-3p likely targets multiple mRNAs to inactivate the Hippo pathway and promote YAP activity.
FIGURE 4:

miR-24 activates YAP through down-regulation of NF2. (A) Representative Western blot of NF2 protein levels in RPE-1 cells transfected with the indicated siRNA/miRNA after 48 h. (B) Quantification of Western blots in A. Error bars represent mean ± SEM (n > 3, ***p ≤ .001, ****p ≤ .0001, ns = nonsignificant, one-way ANOVA). (C) Representative fixed images of YAP localization in RPE-1 cells transfected with the indicated siRNA/miRNA after 48 h (n > 3). (D) Plot depicts the normalized ratio of YAP immunofluorescence intensity in the nucleus:cytoplasm (N/C) from C (n > 450 cells/condition, ****p ≤ .0001, one-way ANOVA).
Previous work has identified multiple roles for miR-24-3p in carcinogenesis. miR-24 is overexpressed in many cancer subtypes (e.g., breast, hepatic, and Hodgkin lymphoma), and its up-regulation promotes cell proliferation through disruption of the cyclin-dependent kinase inhibitors p27Kip1 and p16INK4a (Hatziapostolou et al., 2011; Giglio et al., 2013; Kerimis et al., 2017; Liu, Wang, et al., 2017; Yuan et al., 2017). Interestingly, small-RNA sequencing has revealed several miRNAs that are highly overexpressed in patient hepatocellular carcinoma (HCC) samples relative to cirrhotic nodules, including miR-24-3p with a more than sevenfold increase in expression (Sulas, Di Tommaso, Novello, et al., 2018). This is relevant since hepatocytes from adult animals are predominantly tetraploid, and YAP activation has been shown to promote their proliferation in vivo and encourage the development of HCC (Zhang, Bai, et al., 2010; Zhang, Chen, Liu, et al., 2017; Ganem, Cornils, et al., 2014; Patel et al., 2017). As such, our data suggest that miR-24-3p overexpression in tetraploid hepatocytes may promote cell-cycle progression through activation of YAP, thus providing one potential explanation for this clinical observation.
Summary
We have identified 23 miRNAs whose expression enables tetraploid cells to escape G1 arrest and reenter the cell cycle. We have demonstrated that these miRNAs promote tetraploid cell proliferation by either hyperactivating mitogenic signaling, disrupting the p53/p21 pathway, or functionally impairing the Hippo tumor suppressor pathway to activate YAP. As miRNAs are known to have multiple targets, it is likely that simultaneous modulation of several genes by each miRNA combines to induce the strong phenotypic effects observed. Our data suggest that subtle modulation of miRNA expression levels, even transiently, provides routes through which tetraploid cells can overcome cell-cycle arrest and regain proliferative capacity. As proliferating tetraploids are highly genomically unstable, this initial proliferative push may set in motion a subsequent series of mutational events that ultimately lead to tumorigenesis.
MATERIALS AND METHODS
Cell culture
Telomerase-immortalized RPE-1 cells (American Type Culture Collection) and all derivative cell lines generated in this study were grown in phenol red–free DMEM:F12 containing 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained at 37°C under 5% CO2 atmosphere.
Viral infections and siRNA transfections
RPE-1 cells were infected for 12–16 h with viruses carrying genes of interest in the presence of 10 μg/ml polybrene, washed, and allowed to recover for 24 h before selection. All siRNA transfections were performed using 50 nM siRNA with Lipofectamine RNAi MAX according to the manufacturer’s instructions.
Tetraploid miRNA screen
Day 1: Dishes (15 cm) were seeded with 6 million exponentially growing RPE-FUCCI cells, so that they were ∼65% confluent the following day. Day 2: DCB (4 μM) was added to each 15-cm dish for 16 h. Day 3: DCB-treated cells were washed 5 × 5 min, incubated in medium containing 2.5 μg/ml Hoechst dye for 1 h, trypsinized in 0.05% trypsin, pelleted, resuspended in fresh medium, and FACS sorted. G1 diploids (2C DNA content; mCherry+, GFP–) and G1 tetraploids (4C DNA content; mCherry+, GFP–) were isolated by FACS. Sorted cells were pelleted, resuspended in fresh medium without antibiotics, and replated at a density of 5000 cells per well of a 96-well plate. Cells were then reverse transfected using Lipofectamine RNAi Max (according to the manufacturer’s specifications). The final concentration of miRNAs per well was 50 nM. Each plate was screened in triplicate and was internally controlled with multiple p53 siRNA-positive controls and scrambled miRNA negative controls. Day 4: All transfected wells were fed with fresh medium containing penicillin/streptomycin. Day 6: Monastrol (100 μM) was added to each well for 12 h to arrest proliferating GFP+ cells in mitosis. Day 7: At 96 h following transfection, cells were fixed with 4% paraformaldehyde. Fluorescence images from each well were acquired using a Nikon TE2000-E2 inverted microscope equipped with a cooled CCD camera (Orca ER, Hamamatsu), and Nikon Perfect Focus. An encoded precision stage was used to capture nine fields of view from each well of the 96-well dish. Subsequently, the total number of proliferating S/G2/M cells (based on the GFP positiveness of the FUCCI system) was calculated as a fraction of the total number of cells (based on nuclear counts using Hoechst) for each well. The Z-score was calculated using the formula 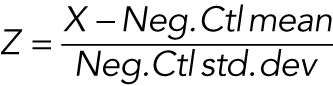 . All miRNAs that had a median Z-score of 6.0 were retested in a secondary screen under the same protocol. For the secondary screen, all miRNAs that had a median Z-score above 3.0 were manually examined, and any miRNAs that caused cell death or yellow binucleates (indicative of a failure of the FUCCI degradation system) were removed. The Z-scores from the two screens were then averaged for each miRNA, resulting in the list of hits.
. All miRNAs that had a median Z-score of 6.0 were retested in a secondary screen under the same protocol. For the secondary screen, all miRNAs that had a median Z-score above 3.0 were manually examined, and any miRNAs that caused cell death or yellow binucleates (indicative of a failure of the FUCCI degradation system) were removed. The Z-scores from the two screens were then averaged for each miRNA, resulting in the list of hits.
Immunofluorescence microscopy
All cells were washed in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 10 min. Cells were extracted in PBS-0.5% Triton X-100 for 5 min., blocked for 30 min in TBS-BSA (10 mM Tris, pH 7.5, 150 mM NaCl, 5% bovine serum albumin [BSA], 0.2% sodium azide), and incubated with primary antibodies diluted in TBS-BSA for 30–60 min in a humidified chamber. Primary antibodies were visualized using species-specific fluorescent secondary antibodies (Molecular Probes), and DNA was detected with 2.5 µg/ml Hoechst. Immunofluorescence images were collected on a Nikon Ti-E inverted microscope (Nikon Instruments). Immunofluorescence images of RPE-FUCCI cells, including all data collected from the RNAi screen, were captured on a Nikon TE2000-E2 inverted microscope. Confocal immunofluorescence images were collected on a Nikon Ti-E inverted microscope equipped with a C2+ laser scanning confocal head with 405-, 488-, 561-, and 640-nm laser lines. Images were analyzed using NIS-Elements software and ImageJ. To assess YAP localization, two small boxes were drawn in individual cells: one in the nucleus and one in the cytoplasm. The mean fluorescence intensity of YAP was measured in these regions of interest, and a nuclear:cytoplasmic ratio was determined. All quantifications of YAP fluorescence localization were completed in a blinded manner.
Live-cell imaging
RPE-FUCCI cells were grown on glass-bottom 12-well tissue culture dishes (Mattek) and imaged on a Nikon TE2000-E2 inverted microscope equipped with the Nikon Perfect Focus system. The microscope was enclosed within a temperature- and CO2-controlled environment that maintained an atmosphere of 37°C and 5% humidified CO2. Fluorescence and phase contrast images were captured every 10–20 min with a 10 × 0.5 NA Plan Fluor objective, at multiple points for 2–4 d. All captured images were analyzed using NIS-Elements software.
Protein extraction, immunoprecipitation, and immunoblotting
Cells were washed twice in ice-cold PBS and lysed using ice-cold Laemmli lysis buffer containing HALT (dual phosphatase and protease inhibitor, Thermo Fisher). Lysates were sonicated at 20% amplitude for 20 s, diluted in 4X Sample Buffer (Boston BioProducts), and resolved using SDS gel electrophoresis. Proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes, blocked for 1 h with TBS–0.5% Tween containing 5% skim milk powder, and then probed overnight at 4°C with primary antibodies. Bound antibodies were detected by horseradish peroxidase–linked secondary antibodies and processed with ECL (Amersham) or Clarity ECL (Bio-Rad). Chemiluminescence acquisition was carried out using the Bio-Rad ChemiDox XRS+ system and analyzed using the Bio-Rad Image Lab.
Growth factor restimulation
Control RPE-1 cells, or RPE-1 cells transfected with miRNAs/siRNAs for 48 h, were serum starved overnight; they were then stimulated with medium containing 5% serum for the indicated time points and collected immediately for quantitative Western blot analysis.
Microarray analysis and gene set enrichment analysis
Total RNA was extracted from exponentially growing control RPE-1 cells (transfected with scrambled siRNA), RPE-1 cells expressing YAP-5SA, and RPE-1 cells transfected with miR-24-3p for 48 h with the RNeasy kit (Qiagen) and hybridized onto Affymetrix HG-U133_Plus_2 arrays per the manufacturer’s instructions. GSEA was performed using the GSEA application from the Broad Institute. Analysis was performed using preranked gene lists with phenotype as permutation type, 1000 permutations, and log2 ratio of classes as metric for ranking genes (Mootha, Lindgren, et al., 2003; Subramanian, Tamayo, et al., 2005). YAP/TAZ dependent gene sets were curated from the following expression data: GSE66083 and GSE54617 (Zanconato et al., 2015; Park et al., 2016).
Reagents and antibodies
The following antibodies were from Cell Signaling Technologies: AKT (#9272), Phospho-AKT (#4051), ERK (#9107), Phospho-ERK (#9101), LATS1 (#3477), LATS2 (#5888 and #13646), p21 (#2947), YAP (#4912), MST1 (#3682), MST2 (#3952), NF2 (#12896), and GAPDH (#2118). Antibodies against p53 (DO-1), p21 (F-5), Cyclin D1 (A-12), Cyclin E (M-20), MARK2 (H-86) and YAP (63.7) were from Santa Cruz. Antibody against beta-actin (AC-74) was from Sigma. Antibodies against vinculin (ab18058) and AMOTL1 (ab84049) were from Abcam. Doxorubicin and dihydrocytochalasin B were from Sigma.
Plasmids
Plasmids encoding cDNA for YAP-5SA (#33093) were obtained from Addgene (Zhao et al., 2007). YAP-5SA was cloned into the pBABE backbone, making use of PCR-based cloning (In-Fusion HD; Clontech).
Statistics
Prism 7 was used for all statistical analyses and for the creation of all graphs.
miRNA and siRNA Sequences
The miRNA library was from Ambion. Accession numbers and sequences for hits are listed in Table 1. siRNAs used in this study are listed in Table 2.
Table 1:
Accession numbers and sequences for hits.
| Ambion ID | Human ID | Accession number | Sequence |
|---|---|---|---|
| hsa-miR-523 | hsa-miR-523-3p | MIMAT0002840 | GAACGCGCUUCCCUAUAGAGGGU |
| hsa-miR-191* | hsa-miR-191-3p | MIMAT0001618 | GCUGCGCUUGGAUUUCGUCCCC |
| hsa-miR-372 | hsa-miR-372-3p | MIMAT0000724 | AAAGUGCUGCGACAUUUGAGCGU |
| mmu-miR-302a | hsa-miR-302a-3p | MIMAT0000684 | UAAGUGCUUCCAUGUUUUGGUGA |
| hsa-miR-767-5p | hsa-miR-767-5p | MIMAT0003882 | UGCACCAUGGUUGUCUGAGCAUG |
| hsa-miR-520c-3p | hsa-miR-520c-3p | MIMAT0002846 | AAAGUGCUUCCUUUUAGAGGGU |
| hsa-miR-302b | hsa-miR-302b-3p | MIMAT0000715 | UAAGUGCUUCCAUGUUUUAGUAG |
| hsa-miR-302e | hsa-miR-302e | MIMAT0005931 | UAAGUGCUUCCAUGCUU |
| xtr-miR-148b | hsa-miR-148b-3p | MIMAT0000759 | UCAGUGCAUCACAGAACUUUGU |
| bta-miR-24 | hsa-miR-24-3p | MIMAT0000080 | UGGCUCAGUUCAGCAGGAACAG |
| hsa-miR-520b | hsa-miR-520b | MIMAT0002843 | AAAGUGCUUCCUUUUAGAGGG |
| hsa-miR-520a-3p | hsa-miR-520a-3p | MIMAT0002834 | AAAGUGCUUCCCUUUGGACUGU |
| hsa-miR-302d | hsa-miR-302d-3p | MIMAT0000718 | UAAGUGCUUCCAUGUUUGAGUGU |
| hsa-miR-520e | hsa-miR-520e | MIMAT0002825 | AAAGUGCUUCCUUUUUGAGGG |
| hsa-miR-520d-3p | hsa-miR-520d-3p | MIMAT0002856 | AAAGUGCUUCUCUUUGGUGGGU |
| gga-miR-302c | hsa-miR-302c-3p | MIMAT0000717 | UAAGUGCUUCCAUGUUUCAGUGG |
| hsa-miR-32 | hsa-miR-32-5p | MIMAT0000090 | UAUUGCACAUUACUAAGUUGCA |
| hsa-miR-298 | hsa-miR-298 | MIMAT0004901 | AGCAGAAGCAGGGAGGUUCUCCCA |
| rno-miR-874 | hsa-miR-874-3p | MIMAT0004911 | CUGCCCUGGCCCGAGGGACCGA |
| mmu-miR-200a* | hsa-miR-200a-5p | MIMAT0001620 | CAUCUUACCGGACAGUGCUGGA |
| hsa-miR-520g | hsa-miR-520g-3p | MIMAT0002858 | ACAAAGUGCUUCCCUUUAGAGUGU |
| hsa-miR-553 | hsa-miR-553 | MIMAT0003216 | AAAACGGUGAGAUUUUGUUUU |
| hsa-miR-517b | hsa-miR-517b | MIMAT0002857 | UCGUGCAUCCCUUUAGAGUGUU |
Table 2:
The siRNAs used in this study.
| Company | siRNA | Sequence |
|---|---|---|
| Dharmacon OnTARGETplus | LATS1-1 | GGUGAAGUCUGUCUAGCAA |
| LATS1-2 | UAGCAUGGAUUUCAGUAAU | |
| LATS1-3 | GGUAGUUCGUCUAUAUUAU | |
| LATS1-4 | GAAUGGUACUGGACAAACU | |
| LATS2-1 | GCACGCAUUUUACGAAUUC | |
| LATS2-2 | ACACUCACCUCGCCCAAUA | |
| LATS2-3 | AAUCAGAUAUUCCUUGUUG | |
| LATS2-4 | GAAGUGAACCGGCAAAUGC | |
| TP53-1 | GAAAUUUGCGUGUGGAGUA | |
| TP53-2 | GUGCAGCUGUGGGUUGAUU | |
| TP53-3 | GCAGUCAGAUCCUAGCGUC | |
| TP53-4 | GGAGAAUAUUUCACCCUUC | |
| SPINT2-1 | GAAGACCACUCCAGCGAUA | |
| SPINT2-2 | GCAAUAACUUCAUCUAUGG | |
| SPINT2-3 | CCUGCCAGCUGUUUGUGUA | |
| SPINT2-4 | CAGCAGGAAUGCAGCGGAU | |
| Ambion Silencer Select | NF2 (s224112) | AGAUGGAGUUCAAUUGCGATT |
Supplementary Material
Acknowledgments
We thank members of the Ganem lab for comments on the manuscript, members of the Institute of Chemistry and Cell Biology (ICCB) screening facility at Harvard Medical School for their expertise in high-throughput screening, and David Pellman for supporting the initial stages of this work. M.V., E.S., and A.B. were supported by a training grant from the National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) (5T32GM008541-20). M.V. is supported by an F30 Award from the National Cancer Institute (NCI) (1F30CA228388-01). R.Q. is supported by a Canadian Institutes of Health Research Doctoral Foreign Study Award. K.O. is supported by an F30 Award from the NCI (1CA200110-01A1). N.G. is a member of the Shamim and Ashraf Dahod Breast Cancer Research Laboratories and is supported by NIH Grants CA154531 and GM117150, the Karin Grunebaum Foundation, the Smith Family Awards Program, the Melanoma Research Alliance, and the Searle Scholars Program.
Abbreviations used:
- AKT
also known as protein kinase B (PKB)
- CDKN1A
cyclin-dependent kinase inhibitor 1A (p21, Cip1)
- ECL
enhanced chemiluminescence
- ERK
extracellular signal-regulated kinase
- LATS1
large tumor suppressor kinase 1
- LATS2
large tumor suppressor kinase 2
- MAPK
mitogen-activated protein kinase
- MDM2
mouse double minute 2
- MST1
mammalian STE20-like protein kinase 1 (STK4)
- MST2
mammalian STE20-like protein kinase 2 (STK3)
- PI3K
phosphoinositide 3-kinase
- YAP
Yes-associated protein 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-02-0141) on May 23, 2018.
REFERENCES
Boldface names denote co–first authors.
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. (2001). Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell , 1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. (2006). A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev , 2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. (2009). MicroRNAs: target recognition and regulatory functions. Cell , 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SB. (1967). Effects of cytochalasins on mammalian cells. Nature , 261–264. [DOI] [PubMed] [Google Scholar]
- Crockford A, Zalmas LP, Gronroos E, Dewhurst SM, McGranahan N, Cuomo ME, Encheva V, Snijders AP, Begum J, Purewal S, et al (2017). Cyclin D mediates tolerance of genome-doubling in cancers with functional p53. Ann Oncol , 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, de Lange T. (2011). The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol , 585–610. [DOI] [PubMed] [Google Scholar]
- Davoli T, de Lange T. (2012). Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell , 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al (2014). Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov , 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva G, Piovan C, Gasparini P, Ngankeu A, Taccioli C, Briskin D, Cheung DG, Bolon B, Anderlucci L, Alder H, et al (2013). Estrogen mediated-activation of miR-191/425 cluster modulates tumorigenicity of breast cancer cells depending on estrogen receptor status. PLoS Genet , e1003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli DM, Hearn S, Myers MP, Lazebnik Y. (2005). A primate virus generates transformed human cells by fusion. J Cell Biol , 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev , 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. (2005). Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature , 1043–1047. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. (2014). Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell , 833–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature , 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. (2007). Limiting the proliferation of polyploid cells. Cell , 437–440. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Storchova Z, Pellman D. (2007). Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev , 157–162. [DOI] [PubMed] [Google Scholar]
- Giglio S, Cirombella R, Amodeo R, Portaro L, Lavra L, Vecchione A. (2013). MicroRNA miR-24 promotes cell proliferation by targeting the CDKs inhibitors p27Kip1 and p16INK4a. J Cell Physiol , 2015–2023. [DOI] [PubMed] [Google Scholar]
- Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, et al (2011). An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell , 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson MD, Lund AH. (2012). MicroRNA and cancer. Mol Oncol , 590–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimis D, Kontos CK, Christodoulou S, Papadopoulos IN, Scorilas A. (2017). Elevated expression of miR-24–3p is a potentially adverse prognostic factor in colorectal adenocarcinoma. Clin Biochem , 285–292. [DOI] [PubMed] [Google Scholar]
- Krzywicka-Racka A, Sluder G. (2011). Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol , 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffer C, Kuznetsova AY, Storchova Z. (2013). Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma , 305–318. [DOI] [PubMed] [Google Scholar]
- Liu W, Wang S, Zhou S, Yang F, Jiang W, Zhang Q, Wang L. (2017). A systems biology approach to identify microRNAs contributing to cisplatin resistance in human ovarian cancer cells. Mol Biosyst , 2268–2276. [DOI] [PubMed] [Google Scholar]
- Mazumdar M, Lee JH, Sengupta K, Ried T, Rane S, Misteli T. (2006). Tumor formation via loss of a molecular motor protein. Curr Biol , 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM. (2017). Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev Cell , 405–420 e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Guan KL. (2016). Mechanisms of Hippo pathway regulation. Genes Dev , 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo JS, Lu W, Lu S, et al (2015). MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun , 8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, et al (2014). A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol , 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet (3), 267–273. [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL. (2015a). The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer , 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, et al (2015b). A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev , 1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal N, Kulshreshtha R. (2014). miR-191: an emerging player in disease biology. Front Genet , 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HG, Makitalo M, Yang D, Chinnappan D, St Hilaire C, Ravid K. (2009). Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J , 2741–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. (2007). Hippo signaling in organ size control. Genes Dev , 886–897. [DOI] [PubMed] [Google Scholar]
- Panopoulos A, Pacios-Bras C, Choi J, Yenjerla M, Sussman MA, Fotedar R, Margolis RL. (2014). Failure of cell cleavage induces senescence in tetraploid primary cells. Mol Biol Cell , 3105–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Sohn BH, Johnson RL, Kang MH, Kim SB, Shim JJ, Mangala LS, Kim JH, Yoo JE, Rodriguez-Aguayo C, et al (2016). Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology , 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SH, Camargo FD, Yimlamai D. (2017). Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology , 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Seidel CW, Box AC, Rancati G, Li R. (2016). Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol Biol Cell , 3065–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell , 487–498. [DOI] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al (2015). Polyploidy can drive rapid adaptation in yeast. Nature , 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al (2012). An immunosurveillance mechanism controls cancer cell ploidy. Science , 1678–1684. [DOI] [PubMed] [Google Scholar]
- Shenk EM, Ganem NJ. (2016). Generation and purification of tetraploid cells. Methods Mol Biol , 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D. (2009). Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One , e6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E, Pellman D. (2006). Genome-wide genetic analysis of polyploidy in yeast. Nature , 541–547. [DOI] [PubMed] [Google Scholar]
- Storchova Z, Kuffer C. (2008). The consequences of tetraploidy and aneuploidy. J Cell Sci (Pt 23), 3859–3866. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA , 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulas P, Di Tommaso L, Novello C, Rizzo F, Rinaldi A, Weisz A, Columbano A, Roncalli M. (2018). A large set of miRNAs is dysregulated since the earliest steps of human hepatocellular carcinoma development. Am J Pathol , 785–794. [DOI] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. (2013). Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell , 1342–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell , 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Kluiver J, Koerts J, de Jong D, Rutgers B, Abdul Razak FR, Terpstra M, Plaat BE, Nolte IM, Diepstra A, et al (2017). miR-24–3p is overexpressed in Hodgkin lymphoma and protects Hodgkin and Reed–Sternberg cells from apoptosis. Am J Pathol , 1343–1355. [DOI] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al (2013). Pan-cancer patterns of somatic copy number alteration. Nat Genet , 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol , 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. (2010). The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell , 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen Q, Liu Q, Li Y, Sun X, Hong L, Ji S, Liu C, Geng J, Zhang W, et al (2017). Hippo signaling suppresses cell ploidy and tumorigenesis through Skp2. Cancer Cell , 669–684 e667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev , 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Frings O, Branca RM, Boekel J, le Sage C, Fredlund E, Agami R, Orre LM. (2017). microRNAs with AAGUGC seed motif constitute an integral part of an oncogenic signaling network. Oncogene , 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.