

Abstract
Aim:
PDE5A is a leading factor contributing to cGMP signaling and cardiac hypertrophy. However, microRNA-mediated posttranscriptional regulation of PDE5A has not been reported. The aim of this study is to screen the microRNAs that are able to regulate PDE5A and explore the function of the microRNAs in cardiac hypertrophy and remodeling.
Methods and Results:
Although miR-19a/b-3p (microRNA-19a-3p and microRNA-19b-3p) have been reported to be differentially expressed during cardiac hypertrophy, the direct targets and the functions of this microRNA family for regulation of cardiac hypertrophy have not yet been investigated. The present study identified some direct targets and the underlying functions of miR-19a/b-3p by using bioinformatics tools and gene manipulations within mouse neonatal cardiomyocytes. Transfection of miR-19a/b-3p down-regulated endogenous expressions of PDE5A at both mRNA and protein levels with real-time PCR and western blot. Luciferase reporter assays showed that PDE5A was a direct target of miR-19a/b-3p. In mouse models of cardiac hypertrophy, we found that miR-19a/b-3p was expressed in cardiomyocytes and that its expression was reduced in pressure overload-induced hypertrophic hearts. miR-19a/b-3p transgenic mice prevented the progress of cardiac hypertrophy and cardiac remodeling in response to angiotensin II infusion with echocardiographic assessment and pressure–volume relation analysis.
Conclusion:
Our study elucidates that PDE5A is a novel direct target of miR-19a/b-3p, and demonstrates that antihypertrophic roles of the miR-19a/b-3p family in Ang II-induced hypertrophy and cardiac remodeling, suggests that endogenous miR-19a/b-3p might have clinical potential to suppress cardiac hypertrophy and heart failure.
Keywords: angiotensin II, cardiac hypertrophy, microRNA-19a-3p and microRNA-19b-3p, PDE5A
INTRODUCTION
Progressive cardiac hypertrophy owing to pathological stimuli, such as pressure overload, is frequently associated with the development of heart failure, a major cause of morbidity and mortality worldwide [1]. Growing evidence has shown that microRNAs (miRNAs) are extensively involved in the pathogenesis of cardiac hypertrophy [2]. miRNAs are evolutionarily conserved noncoding RNAs, approximately 22 nucleotides in length, which negatively regulate gene expression by translational repression or mRNA destabilization [3,4]. miRNAs have been implicated in numerous diseases [5,6], including heart diseases [7]. Among them, miRNAs associated with cardiac hypertrophy have been identified. For example, miR-23a, miR-199, miR-208, miR-350 and miR-499 have a pro-hypertrophic function [8–11], whereas miR-1, miR-9, miR-19a/b, miR-98 and miR-133 are antihypertrophic miRNAs [2,12–15]. miR-19a/b (miR-19a-3p and miR-19b-3p) are located in the miR-17–92 cluster, which also encodes four other mature miRNAs, miR-17, miR-18a, miR-20a and miR-92a [16]. Although miR-19a/b-3p have been reported to be differentially expressed during cardiac hypertrophy [17–19], specific roles of miR-19a/b, among the miR-17–92 components, in cardiac hypertrophy and detailed underlying molecular mechanisms have not been studied. PDE5A is one of the most widely studied members of the phosphodiesterase superfamily, which catabolizes cyclic guanosine monophosphate (cGMP) [20,21]. PDE5A is expressed and active in the myocardium [22,23], however, its role in the heart has been questioned as its inhibition has minimal effects on resting heart function [24]. It has potent effects on vascular tone in the corpus cavernosum and pulmonary vasculature [25–27]. Sildenafil, a phosphodiesterase 5 (PDE5) inhibitor, elevates cardiac cGMP at high doses, increases cGKI activity, and has been reported to reverse TAC-induced cardiac hypertrophy [28,29]. However, miRNA-mediated posttranscriptional regulation of PDE5A has not been reported. Here, we reveal the role of miR-19a/b-3p in hearts subjected to sustained pressure load and show that miR-19a/b-3p prevents angiotensin II (Ang II)-induced molecular, cellular and cardiac chamber remodeling through PDE5A degradation.
MATERIALS AND METHODS
In this study, we established a transgenic mouse harboring both microRNA-19a-3p and microRNA-19b-3p in rosa26 locus, which is used for Cre-mediated gene expression in mice. An expanded Materials and Methods section is available in the supplemental data, which includes detailed information on the following aspects: Ang II administration, histological analysis, culture of neonatal mouse ventricular myocytes, lenti-virus infection [30], immunofluorescence analysis, immunoprecipitation [31], western blot, quantitative real-time PCR, echocardiographic measurement and left ventricular function analysis. These methods were performed according to routine protocols.
Statistical analysis
Results are expressed as mean ± SEM. Comparisons between two groups were performed using a two-tailed Student's t-test. Statistical comparison among different groups was performed by one-way analysis of variance (ANOVA) with an appropriate post hoc test for multiple-sample comparisons, and two-way ANOVA for comparisons of the means of populations that were classified in two different ways or the mean responses in an experiment with two factors. Values of P < 0.05 were considered statistically significant.
RESULTS
To screen specific miRNAs that could modulate PDE5A expression. 11 miRNAs were overlappingly predicted to bind to PDE5A-3′-untranslated region (UTR) based on three different algorithms, ‘microrna.org’ (http://www.microrna.org/) [32], TargetScan (http://www.targetscan.org/vert_71/) [33] and Exiqon (https://www.exiqon.com/miRSearch) [34,35]. Eleven mimics (Agomir) of these predicted miRNA were transfected into neonatal mouse cardiomyocytes (NMCMs). Indeed, quantitative real-time PCR (Fig. 1b) and western blot analysis (Fig. 1a) showed the decrease of PDE5A expression in cardiomyocytes transfected with miR-19a-3p mimic (ago-19a-3p) compared with other miRNA mimics (Fig. S1). MicroRNAs expression efficiency was shown in Fig. S1. We tested whether miR-19a-3p is able to regulate PDE5A levels. As shown in Fig. 1b, transfection with Ago-19a-3p reduced PDE5A mRNA and inhibitor of miR-19a-3p (anta-19a-3p) increased PDE5A mRNA level. Consistently, protein of PDE5A expression is decreased by miR-19a/b-3p and increased by anta-19a/b-3p (Fig. 1c). However, its underlying molecular mechanisms, including a possible role for miRNAs, remain to be elucidated fully. Enforced expression of miR-19a-3p alleviated Ang II-induced increase on PDE5A protein level and inhibition of miR-19a-3p exacerbated the PDE5A protein expression (Fig. 1e and f). To further identify for direct and specific regulation of the presumed mRNA targets by miR-19a/b-3p, we generated luciferase reporter constructs in which firefly luciferase cDNA was linked to the 3′ UTR of PDE5A. NMCMs were transfected with each of the reporter constructs, together with ago-19a/b-3p or anta-19a/b-3p, and the luciferase signal was quantified after cell lysis. As an internal control for transfection, the reporter constructs also constituted renilla luciferase cDNA under an autonomous promoter, and data were assessed as the ratio of both luciferase signals. Indeed, ago-19a-3p efficiently reduced firefly luciferase luminescence, whereas anta-19a-3p conferred the opposite (Fig. 1h). Mutation of seed binding sites for miR-19a-3p made the reporter insensitive to ago-19a-3p or anta-19a-3p transfection (Fig. 1h), indicating the specific dependence of PDE5A 3′ UTRs on miR-19a-3p. Ang II has been well documented to induce cardiac hypertrophy. To delve the underlying mechanism about miR-19a-3p binding with PDE5A mRNA, we performed RNA co-immunoprecipitation with Ago2, which is required for RNA-mediated gene silencing (RNAi) by the RNA-induced silencing complex (RISC) [36]. We found that miR-19a-3p showed decreased association with the PDE5A mRNA after Ang II treatment (Fig. 1i), consistent with miR-19a-3p expression in whole cell lysate (Fig. 1g). However, PDE5A expression was also decreased by Ang II in the pulldown complex, which is exactly opposite of Fig. 1g and expectation. The possible explanation is that the pulldown PDE5A mRNA is binding with miR-99a-3p through ago2, it may be consistent with miR-99a-3p amount, less miR-99a-3p by Ang II, more total PDE5A mRNA. miR-19a/b-3p also resulted in a reduction of hypertrophic responses because of Ang II stimulation, including hypertrophic marker atrial natriuretic factor (ANF), brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC; Fig. S2). These data suggest that miR-19a-3p regulates PDE5A and participates in antagonizing hypertrophy in cardiomyocytes.
FIGURE 1.
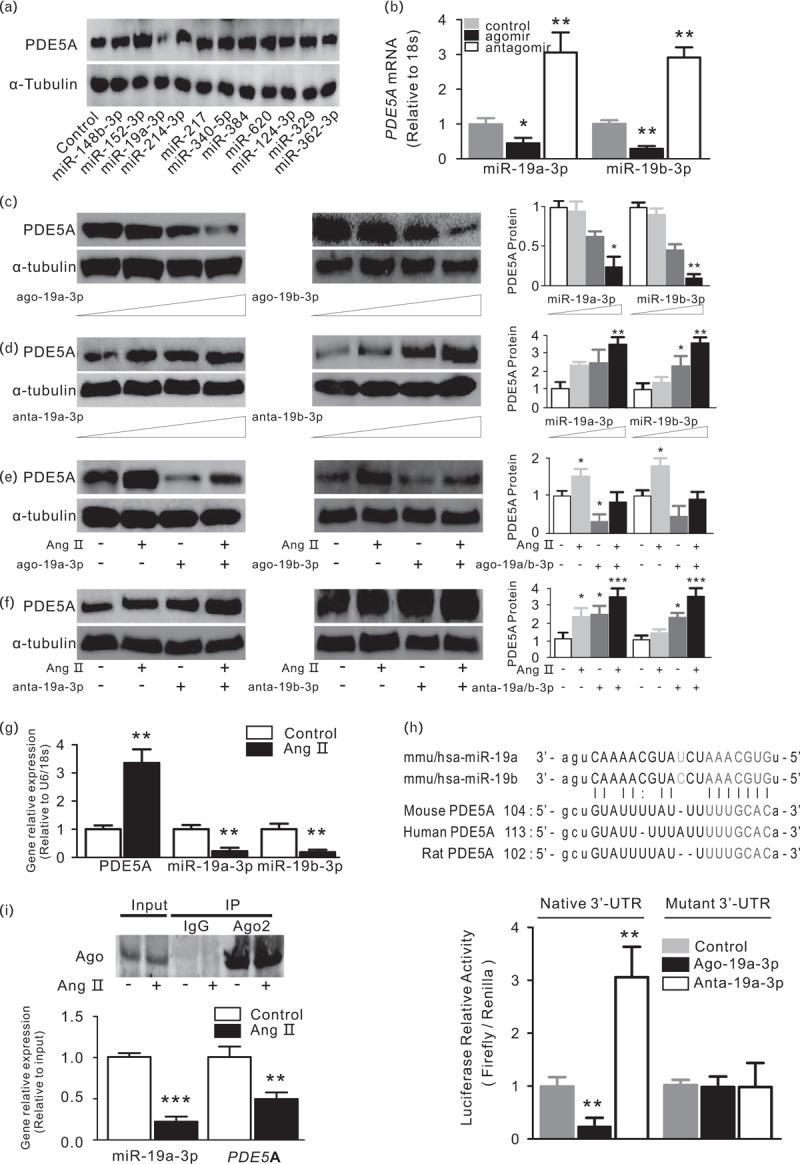
Mmu-miR-19a-3p represses PDE5A in neonatal mouse cardiomyocytes (NMCMs). (a) Mmu-miR-19a-3p downregulated PDE5A protein level in NMCMs. NMCMs were transfected with 11 miRNA mimics predicted to bind PDE5A mRNA 3′ UTR. Cells were harvested and analyzed by western blotting for the PDE5A expression, α-tubulin was used as internal control. (b) Overexpression/inhibition of miR-19a/b-3p induces an downregulation/upregulation in PDE5A mRNA levels. NMCMs were transfected with miR-19a/b-3p mimic (agomir, ago-19a-3p) or inhibitor (antagomir, anta-19a-3p). Cells were harvested for the analysis of PDE5A expression by real-time PCR (b) and immunoblot (c and d), 18S and α-tubulin were used as internal control, respectively. ∗P < 0.05; ∗∗P < 0.01, compared with control group, n = 3 in each group. (e and f) Overexpression of miR-19a/b-3p abolished an Ang II-induced PDE5A protein increase. Inhibition of miR-19a/b-3p aggravate Ang II-induced PDE5A protein upregulation. Cardiomyocytes transfected with ago-19a/b-3p or anta-19a/b-3p and then were treated with Ang II at 100 nmol/l for 24 h. Cells were harvested and analyzed for PDE5A protein expression by immunoblot. α-tubulin were used as internal control. ∗P < 0.05; ∗∗∗P < 0.005, compared with control group, n = 3 in each group. (g) Ang II decreased miR-19a/b-3p and increased PDE5A expression. NMCMs were treated with Ang II at 100 nmol/l for 24 h. Cardiomyocytes were collected and analyzed for RNA expression by real-time PCR, 18S and U6 were used for internal control for mRNA and microRNA, respectively. Data are from three independent experiments performed in triplicate. ∗∗P < 0.01, compared with control group, n = 3 in each group. (h) Luciferase activity of reporter constructs carrying cDNA of luciferase and the 3′ untranslated region (3′ UTR) of PDE5A mRNA. Top, localization of binding sites for mmu-miR-19a-3p in the 3′ UTR of PDE5A mRNA and their evolutionary conservation. mmu-miR-19a-3p seed pairing in the target regions is marked in red. Bottom, quantitative analysis of luciferase activities. NMCMs were transfected with miR-19a-3p or anta-19a-3p in parallel to a control along with reporter constructs with native 3′ UTR or mutated 3′ UTR. Data are from three independent experiments performed in triplicate. ∗∗P < 0.01, compared with control group, n = 3 in each group. (i) The interaction between the PDE5A mRNA and miR-19a-3p involved with Ago2 was weakened by Ang II stimulus. NMCMs stimulated with Ang II were lysated and immunoprecipitated by Ago2 antibody, the pulldowned complex was reverse transcribed for RNA analysis. Data are from three independent experiments performed in triplicate. ∗∗P < 0.01; ∗∗∗P < 0.005, compared with control group, n = 3 in each group. Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
MicroRNA-19a-3p and microRNA-19b-3p were expressed in cardiomyocytes and its expression was reduced in hypertrophic hearts
We examined the expression of miR-19a/b-3p in the hearts of fetal, postnatal and adult mice. qPCR analyses showed that the expression of miR-19a/b-3p were relatively low in embryonic and neonatal hearts. However, miR-19a/b-3p expression was increased in postnatal day 7 and adult hearts (Fig. 2a). These data suggest that miR-19a/b-3p may play an important role in the heart embryonic development and adult heart. Next, we examined the distribution of miR-19a/b-3p expression in cardiomyocyte and noncardiomyocyte fractions of the adult heart. We found that miR-19a/b-3p expression were enriched in cardiomyocytes of adult hearts (Fig. 2b). For positive controls, we showed that expression of the cardiomyocyte-specific genes cardiac troponin T and miR-1 were enriched in the cardiomyocyte fraction. In contrast, expression of periostin, which marked cardiac epithelial cells, was restricted to the noncardiomyocyte fraction. The miR-19a/b-3p expression was reduced in cardiac hypertrophy induced by pressure overload via Ang II at 5, 10 and 15 days (Fig. 2c). To determine the role of miR-19a/b-3p on cardiac hypertrophy in vivo, we subjected miR-19a/b-3p cardiac-specific overexpression transgenic and wild type mice to pressure overload (Fig. 3a). Six-week-old miR-19a/b-3p transgenic and control littermate mice were subjected to Ang II or PBS infusion. No differences in basal SBP were observed between PBS-infused groups. In Ang II-infused miR-19a/b-3p mice, SBP rised progressively at the first 5 days at which time SBP became significantly lower than Ang II-treated wild type mice (Fig. 3b). The PDE5A mRNA was up-regulated by Ang II and this increase was abolished by miR-19a/b-3p (Fig. 3d). PDE5A protein level (Fig. 3c) is consistent with mRNA level. Quantification of miR-9a/b-3p in mouse hearts showed that miR-19a/b-3p effectively compensated for the Ang II-associated loss of miR-19a/b-3p (Fig. 3e). Together, these data demonstrated that miR-19a/b-3p were expressed in cardiomyocytes of adult hearts and that its expression were regulated in hypertrophic hearts.
FIGURE 2.
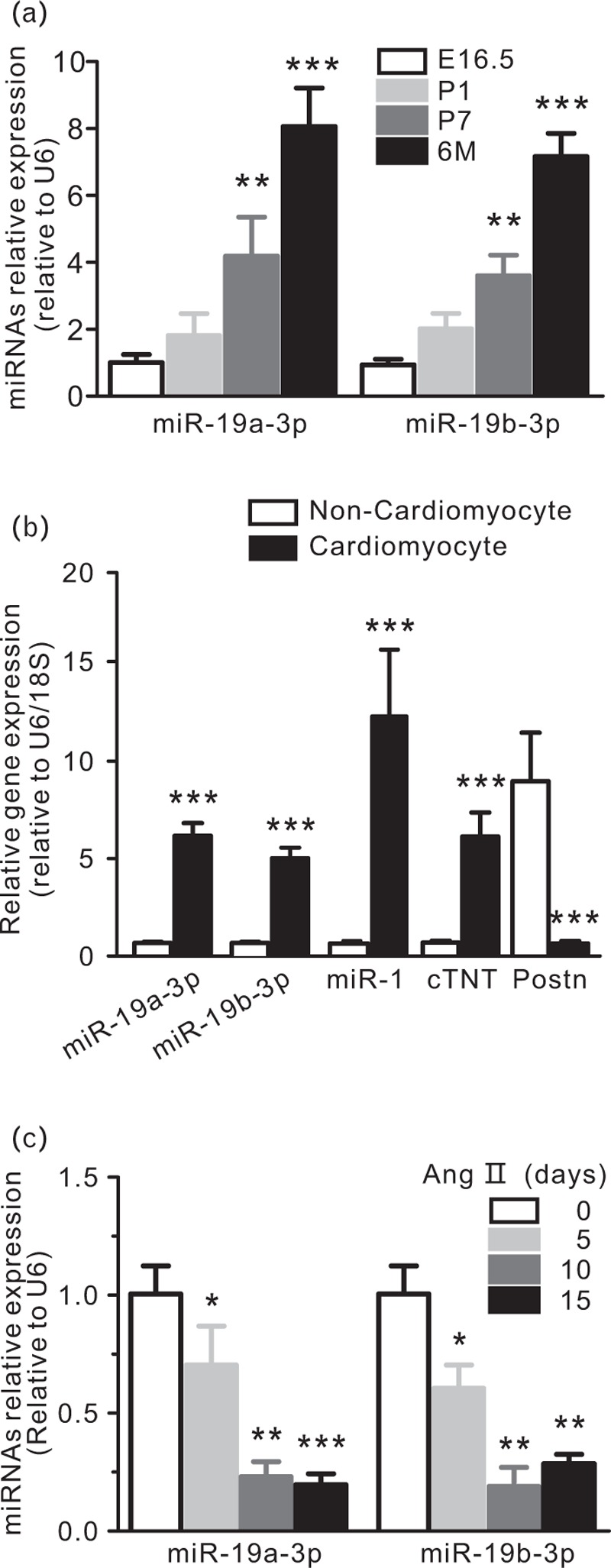
miR-19a/b-3p expression in hypertrophic mouse hearts involves in heart development. (a) Expression of miR-19a/b-3p in embryonic (E16.5), postnatal (P1, P7), and adult (6 months) hearts as determined by quantitative real-time PCR assays, n = 3 in each group. ∗∗P < 0.01; ∗∗∗P < 0.005, compared with E16.5 group. (b) Expression of miR-19a/b-3p in cardiomyocytes or noncardiomyocytes of adult mouse hearts. miR-1 and cardiac troponin T (cTNT) serve as positive controls for cardiomyocytes. Periostin (Postn) marks cardiac fibroblasts, ∗∗∗P < 0.005. (c) Expression of miR-19a/b-3p in Ang II-induced hypertrophic hearts at 5, 10 and 15 days, ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.005. Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
FIGURE 3.
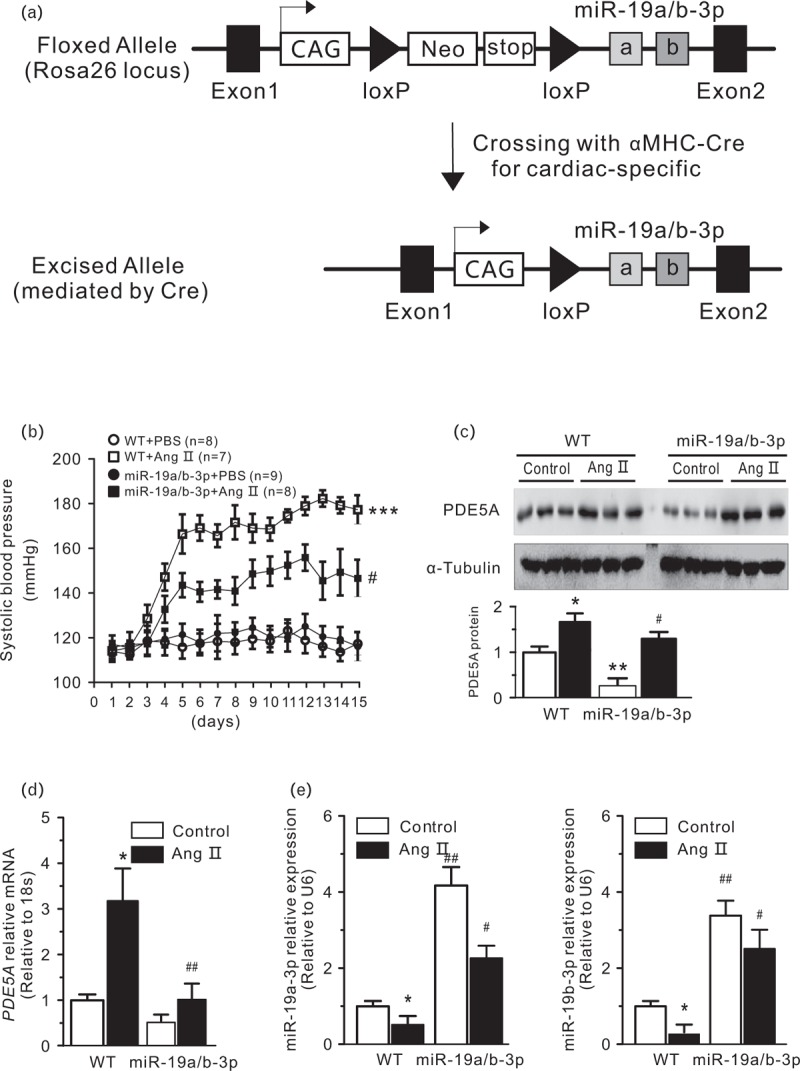
Generation of transgenic mice to overexpress miR-19a/b-3p in the heart. (a) Strategy of cardiac-specific overexpression of miR-19a/b-3p cluster in vivo. (b) SBP in miR-19a/b-3p transgenic and wild type mice infused with Ang II or PBS (control) for 2 weeks. SBP was measured daily for 15 days by tail-cuff plethysmography, ∗∗∗P < 0.005, compared with wild type and PBS groups. #P < 0.05, compared with wild type and Ang II group, n = 7–9 in each group. (c) The protein and mRNA level (d) of PDE5A were negatively correlated with the miR-19a/b-3p overexpression. ∗P < 0.05, ∗∗P < 0.01, compared with control; ##P < 0.01, compared with wild type and Ang II group, n = 3 in each group. (e) qPCR analysis showed miR-19a/b-3p was effectively expressed in miR-19a/b-3p transgenic mice group. Ang II inhibited the increase of miR-19a/b-3p in transgenic mice. ∗P < 0.05, compared with control. #P < 0.05; ##P < 0.01, compared with wild type, n = 3 in each group. Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
Cardiomyocyte expression of microRNA-19a-3p and microRNA-19b-3p in vivo partially prevents cardiac hypertrophy
Two weeks after mini-pump implantation, Ang II-treated wild type mice developed severe cardiac hypertrophy and fibrosis with left ventricular dilatation and reduced fractional shortening. miR-19a/b-3p transgenic mice developed much less disease on heart and lung weight index, remarkably alleviated Ang II-induced cardiomyocyte enlargement and cardiac dysfunction (Fig. 4a–e). Consistently, echocardiographic measurements confirmed that enforced expression of miR-19a/b-3p dramatically assuaged Ang II-induced ventricular dilation and contractile dysfunction (Fig. 4f–h). Fractional shortening and ejection fraction were slightly decreased by Ang II treatment in wild type mice in this study and some others [37–39]; however, significant increasing of these value were also widely reported [40–42], the possible explanation may be individuals differences, or operation by different instruments and people may have errors, I think the effect of Ang II on fractional shortening and ejection fraction is still controversial. Collectively, these function studies suggested that miR-19a/b-3p contributed to the improvement of pathological cardiac hypertrophy induced by chronic pressure overload.
FIGURE 4.
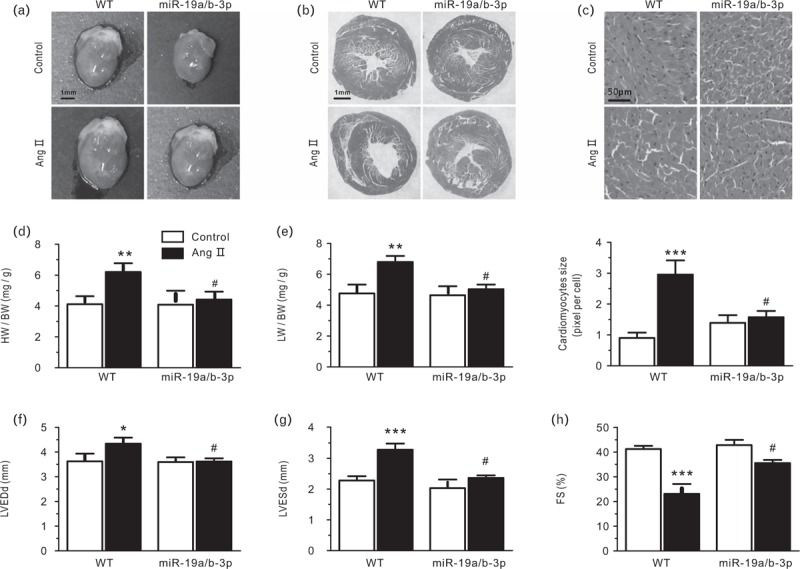
Cardiotropic expression of miR-19a/b-3p prevents cardiac hypertrophy remodeling in response to Ang II-induced pressure overload. (a) Heart size (scale bar, 1 mm). Heart slices (b and c) were subjected to hematoxylin–eosin (H&E) staining for the assessment of cardiomyocyte sectional area and fibrosis 2 weeks after Ang II mini-pump implantation (scale bar, 1 mm for b; scale bar, 50 μm for c). (d) Heart weight/body weight (HW/BW) and lung weight/body weight (LW/BW, e) ratio of hearts were assessed in indicated groups. ∗∗P < 0.01, compared with control; #P < 0.05, compared with wild type, n = 7–9 in each group. (f) Echocardiographic assessment of left ventricular end-diastolic diameter (LVEDd), left ventricular end-systolic diameter (LVESd, g), and fractional shortening (FS; h) in different groups. ∗P < 0.05, compared with control; #P < 0.05, compared with wild type, n = 7–9 in each group.
Detailed examination of heart function was carried out by invasive pressure–volume analysis. Concurrent transgene with miR-19a/b-3p resulted in preservation of cardiac volumes in the normal range and an increase in systolic function (for example, slope of end-systolic pressure– volume relation). PBS treatment for 2 weeks showed no change. Ventricular afterload (indexed by arterial elastance; Fig. 5a) was identically elevated by Ang II regardless of miR-19a/b-3p, yet only Ang II-exposed mice showed a decline in ejection fraction (Fig. 5b). Contractile function assessed through load-independent parameters (dP/dt max-EDV and preload recruitable stroke work) was restored or improved by miR-19a/b-3p as compared with the hearts of Ang II or control mice (Fig. 5c and d). Similar results were observed for diastolic function (Tau and dP/dtmin; Fig. 5e and f). Pressure–volume loops were measured before and during transient reduction of chamber preload to generate specific systolic and diastolic function indexes (Fig. 5g; Table S1). As shown, pressure–volume loops and corresponding systolic and diastolic boundary relations shifted rightward with Ang II, consistent with cardiac dysfunction.
FIGURE 5.
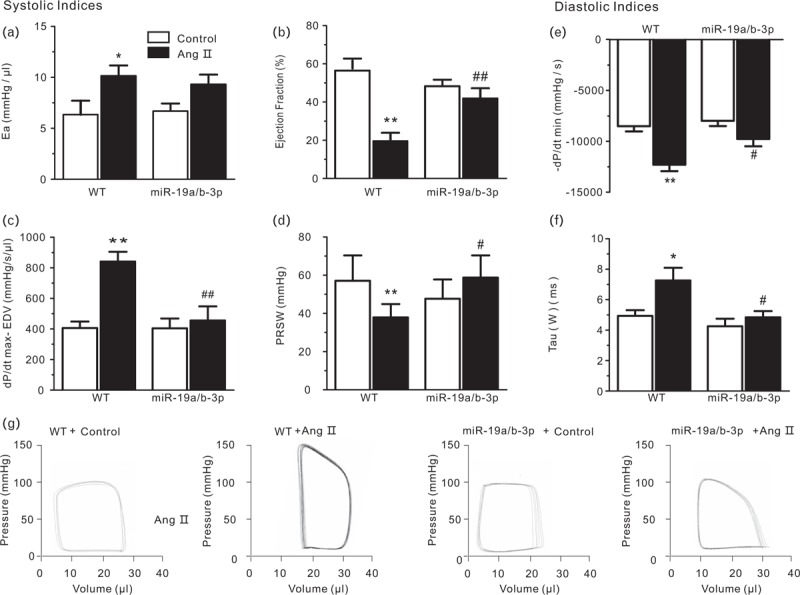
In-vivo heart function as shown by pressure–volume relations in wild-type and miR-19a/b-3p mice infused with PBS or Ang II. (a–d) Systolic indices. Ea, arterial elastance. (measure of ventricular afterload) EF, ejection fraction; dP/dt max, peak rate of pressure rise; dP/dt max-EDV, slope of dP/dt max-end diastolic volume relationship; PRSW, preload recruitable stroke work (contractility indexes), n = 7–9 in each group. (e and f) Diastolic indices. −dP/dt min, peak rate of pressure decline; Tau (W/G), relaxation time constant calculated by Weiss method. (g) Left-ventricular pressure–volume loops. Ang II-treated hearts showed rightward shift of the loops and end-systolic pressure–volume relation consistent with cardiac dysfunction. miR-19a/b-3p transgenic mice hearts showed less remodeling and improved contractile function in Ang II-treated mice. miR-19a/b-3p itself did not affect cardiac function. ∗P < 0.05; ∗∗P < 0.01 versus control; #P < 0.05; ##P < 0.01 versus wild type (n = 7–9 in each group). Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
miR-19a-3p decreases fibrotic gene markers. Next, quantitative real-time PCR was performed on total heart RNA samples to measure transcription of several genes associated with hypertrophy and fibrosis (primers are shown in supplemental Table S3). ANP mRNA levels, a classic marker of maladaptive cardiac hypertrophy, was increased in both groups by Ang II, and this increase was slightly abated in the presence of miR-19a/b-3p (Fig. 6a). Similarly, although the β-myosin heavy-chain gene transcript was elevated by Ang II in control and miR-19a/b-3p mice, in miR-19a/b-3p group, the increase was statistically significant only compared with the control group, the increase was alleviated versus wild type group (Fig. 6b). Analysis of the transcription of four genes involved in the fibrotic response, two pro-fibrotic hormones, namely connective tissue growth factor, and transforming growth factor β, and two extracellular matrix proteins, fibronectin1 and collagen 1, showed a significant increase in Ang II groups both in wild type and miR-19a/b-3p transgenic mice. miR-19a/b-3p lowered this increase in Ang II, but not in wild type mice (Fig. 6c–e). These findings indicate that miR-19a/b-3p inhibits the expression of pro-fibrotic genes induced by Ang II.
FIGURE 6.
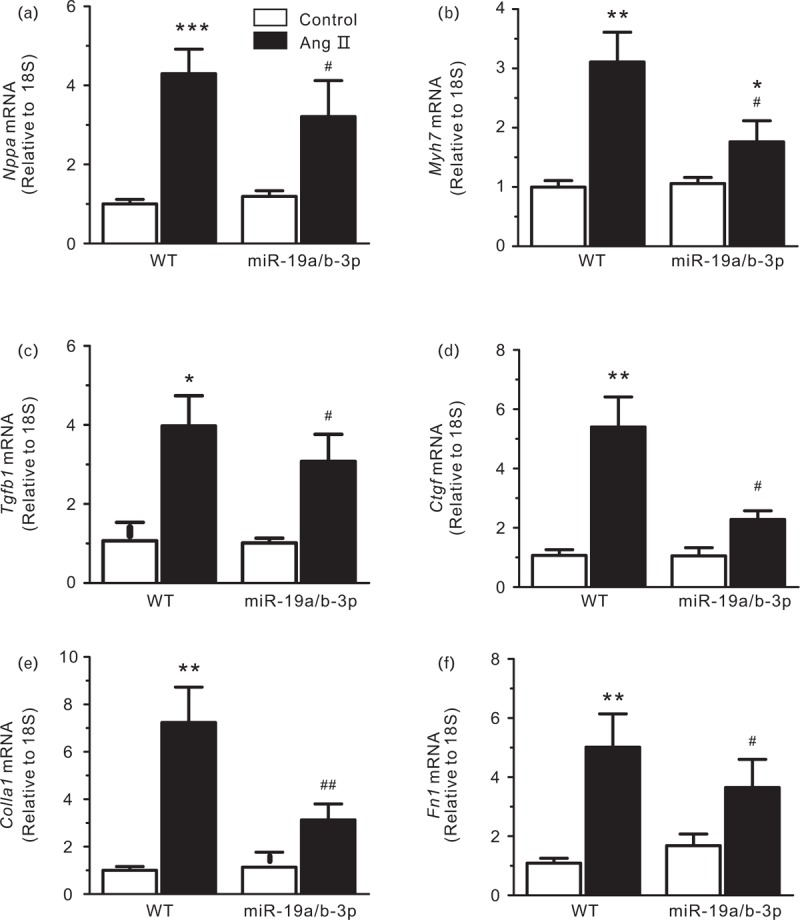
mRNA expression levels of Nppa (ANP) (a), Myh7 (ßMHC) (b), Tgfb1 (TGF-ß) (c), Ctgf (CTGF) (d), Colla1 (collagen 1) (e) and Fn1 (fibronectin) (f) in wild type and miR-19a/b-3p mice infused with PBS or Ang II. mRNA expression level was assessed by real-time PCR and normalized to 18S rRNA. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.005 versus control group; #P < 0.05; ##P < 0.01 versus wild type and Ang II group, n = 3 in each group. Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
miR-19a/b-3p expression was also decreased in human heart biopsies of end-stage heart failure patients, both those with dilated cardiomyopathy and those with hypertrophic cardiomyopathy (Fig. 7a), PDE5A was increased in the same samples (Fig. 7b and c). These results suggest that the up-regulation of PDE5A and down-regulation of miR-19a/b-3p are associated with cardiac hypertrophy and heart failure in humans.
FIGURE 7.
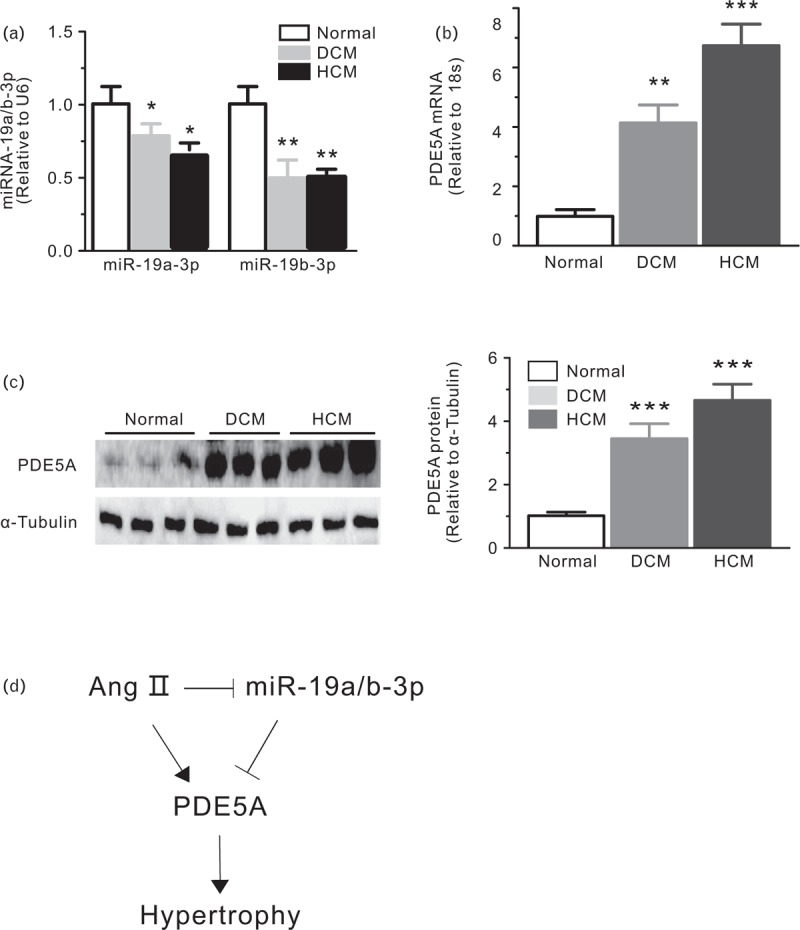
Increased PDE5A expression during cardiac hypertrophy and heart failure. (a) Real-time PCR analysis of miR-19a/b-3p and PDE5A mRNA (b) in human heart biopsies from normal and end-stage heart failure patients, n = 6–8 in each group. (c) Western blot analysis of PDE5A in human heart biopsies from normal and end-stage heart failure patients (DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy). Densitometry results are expressed as PDE5A: α-tubulin ratio, n = 3 in each group. (d) A schematic model showing the essential role of miR-19a/b-3p and PDE5A in Ang II-induced hypertrophy. Ang II, angiotensin II; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p.
DISCUSSION
Cardiac hypertrophy is a growth of the heart in response to an increased workload. Although hypertrophic growth appears to be beneficial for the maintenance of heart function at an early stage, excessive and prolonged hypertrophic stimuli could lead to fatal heart failure [43]. In hearts exposed to sustained pressure overload, cellular, molecular and morphologic changes are activated that often become maladaptive and contribute to progressive cardiac dysfunction and heart failure. The heart also has an intrinsic signaling system coupled to cGMP pathway. cGMP synthesis is often increased by chronic exposure to such stresses, however, this seems to be insufficient to effectively impede hypertrophy and remodeling progression. One possibility is that cGMP catabolism is also increased. If so, reducing the catabolism by PDE5A, serving as a negative feedback loop to modulate cGMP levels, may augment cGMP-dependent antihypertrophic effects. As revealed in models with enhanced cGMP synthesis resulting from genetic upregulation of natriuretic peptide receptor signaling [19,44], or activation of PKG-1 [45], hyperstimulation of this pathway can blunt hypertrophy in vitro and in vivo despite sustained pressure load or neurohormonal stress, whereas inhibition of this signaling worsens hypertrophy [45,46]. Therefore, modulation of PDE5A expression appears to be a leading factor contributing to cGMP signaling and cardiac hypertrophy.
A number of studies have suggested that major hypertrophic changes are associated with increased cardiomyocyte size, up-regulation of fetal gene expression and enhanced protein synthesis owing to involvement of key signaling molecules. The pro-hypertrophic signaling cascades are initially triggered by the activation of membrane receptors, such as G-protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs), eventually leading to changes in the cardiac gene program through regulation of essential transcription factors [47]. A number of miRNAs have been shown to affect cardiac remodeling and diseases. This study demonstrates a previously unknown, potent effect of miR-19a/b-3p in reversing chamber, cellular and molecular remodeling while improving cardiac function in hearts exposed to sustained pressure overload. microRNA pathways that prevents hypertrophy observed here are uncommon, suggesting that the underlying mechanisms linked to modulation of PDE5A, cGMP and/or PKG-1 are potent and are likely to interfere with several pathways. The simplicity of the therapy, the wide clinical experience and safety record of PDE5A inhibitors suggest that this may be an important therapeutic target for human hypertrophic heart disease [48]. Preclinical and clinical studies suggest that PDE-5 inhibitor therapy improved exercise tolerance and clinical status in patients with idiopathic pulmonary arterial hypertension and in patients with heart failure and reduced ejection fraction [49–52]. However, another randomized clinical trial reported that among patients with heart failure with preserved ejection fraction (HFPEF), PDE-5 inhibition with administration of sildenafil for 24 weeks, compared with placebo, did not result in significant improvement in exercise capacity or clinical status [53]. PDE5A inhibitor, sildenafil's role in treating heart failure is still controversial. Our study in animal model shows that decrease of PDE5A has a negligible effect on the normal heart, but this situation changes in hearts under loading stress.
In the present study, we examined the hypothesis that the miR-19a/b-3p family acts as a key regulator of Ang II-induced cardiac hypertrophy. miR-19a/b-3p are ubiquitously expressed and have been shown to be sufficient for promoting the miR-17/92 cluster's oncogenic properties [54,55]. Many of the experimentally validated targets (e.g. PTEN, STAT2, TSP-1 and CTGF) of these two miRNAs are involved in such processes as cell death, proliferation, immune response and angiogenesis. However, to date only a few miRNAs have been shown to target genes in the cGMP pathway, and in particular, PDE5A. The ubiquitous expression of PDE5A and its long 3′ UTR (820 nts) make the study of its miRNA posttranscriptional regulation an uniquely attractive and challenging task. In the above, we examined the interaction between these two competing molecular classes. Specifically, we studied the miRNA-mediated posttranscriptional regulation of PDE5A and determined that miR-19a-3p and miR-19b-3p directly interact with an MRE (microRNA response element) in PDE5A 3′ UTR. The predicted heteroduplexes with miR-19a/b-3p constitute classical Watson–Crick base pairing in the seed region of the respective miRNA. The identified MRE is conserved across species, for example, it is also present in mouse and rat. Several lines of evidence showed that these interactions result in a decrease of PDE5A mRNA and protein abundance. First, over-expression of each of the two miRNAs resulted in a decrease of PDE5A mRNA and protein levels in a cell-dependent manner. Second, by using a luciferase construct containing the native or mutant PDE5A 3′ UTR, we showed a decrease in luciferase activity and proved miR-19a/b-3p and PDE5A MRE direct physical interactions. According to the results of in-vivo study, Ang II-induced hypertrophy was greatly attenuated by miR-19a/b-3p, consistent with previous in-vitro results. miR-19a/b-3p appear to act as a powerful antihypertrophic regulator and the miR-19 a/b-3p-mediated dysregulation of PDE5A may contribute to pathogenesis of cardiac hypertrophy.
miR-19a/b-3p have been identified as the most important oncogenic miRNAs of the miR-17–92 cluster and such an oncogenic role was attributed to their ability to induce cell proliferation and survival by targeting the tumor suppressor PTEN (phosphatase and tensin homologue) on chromosome 10 [55]. Recently, two interesting reports have demonstrated similar roles for the miR-19a/b-3p family in the cardiovascular system [56–58]. In particular, overexpression of miR-19b-3p increased proliferation, survival and differentiation of the multipotent P19 cells, which can differentiate into cardiomyocytes [59]. In these studies, miR-19b-3p or miR-17–92 cluster were thought to promote the potential progenitor cardiomyocytes (P19 cells) proliferation, which was not contradictory with our result. The potential therapeutic function of miR-19a/b-3p in cardiac repair and regeneration has been also found in a study examining the in-vivo overexpression of the miR-17–92 cluster [56]. The main finding of this work is the discovery of a link between miR-19a/b-3p and the posttranscriptional regulation of PDE5A, one of the regulators of the cGMP pathway. The functional implications of this link are not known and need to be investigated. The results would improve our understanding of PDE5A's posttranscriptional regulation by miRNAs and could prove helpful in improving clinical management of the disease and designing improved therapies that target the cGMP metabolism pathway.
Our findings are of clinical interest, given the high prevalence of hypertensive heart disease and hypertrophy, which has a prominent role in many forms of heart failure. PDE5A expression level decrease by miR-19a/b-3p may play different roles compared with sildenafil. The expanding use of PDE5A inhibition/knockdown to treat disorders such as pulmonary hypertension supports their use as a chronic therapy. This should facilitate future clarification of the utility of PDE5A inhibition/knockdown for hypertrophic heart disease.
In conclusion, the present study suggests that the miR-19a/b-3p family regulates cardiac hypertrophy through repression of the novel target gene PDE5A. We suggest that knowledge of such miRNA functions will improve our understanding of pathogenesis during cardiac remodeling under stress conditions.
ACKNOWLEDGEMENTS
Contract grant sponsor: This work was supported in part by National Natural Science Foundation of China (81570326) and Accelerating Excellence in Translational Science Pilot Grants G0812D05, NIH/NCI SC1CA200517 to Y.W.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
Kun Liu, Qiongyu Hao and Jie Wei contributed equally to this work.
Abbreviations: 3′ UTR, 3′ untranslated region; ANF, atrial natriuretic factor; BNP, brain natriuretic peptide; Colla1, collagen 1; CTGF, connective tissue growth factor; Ctgf, CTGF; cTNT, cardiac troponin T; Fn1, fibronectin1; GPCRs, G-protein-coupled receptors; HFPEF, heart failure with preserved ejection fraction; HW/BW, heart weight/body weight; LVEDd, left ventricular end-diastolic diameter; LVESd, left ventricular end-systolic diameter; LW/BW, lung weight/body weight; miR-19a/b-3p, microRNA-19a-3p and microRNA-19b-3p; Myh7, myosin heavy chain 7; NMCMs, neonatal mouse cardiomyocytes; Nppa, natriuretic peptide A; PDE5, phosphodiesterase 5; Postn, periostin; RTKs, receptor tyrosine kinases; Tgfb1, TGF-β; TGF-β, transforming growth factor β1; β-MHC, β-myosin heavy chain
REFERENCES
- 1.Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, et al. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis 2011; 2:e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song DW, Ryu JY, Kim JO, Kwon EJ, Kim DH. The miR-19a/b family positively regulates cardiomyocyte hypertrophy by targeting atrogin-1 and MuRF-1. Biochem J 2014; 457:151–162. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol 2010; 17:1169–1174. [DOI] [PubMed] [Google Scholar]
- 5.Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011; 474:649–653. [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, et al. A microRNA feedback circuit in midbrain dopamine neurons. Science 2007; 317:1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingwell K. Cardiovascular disease: microRNA protects the heart. Nat Rev Drug Discov 2011; 10:98. [DOI] [PubMed] [Google Scholar]
- 8.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A 2009; 106:12103–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 2009; 119:2772–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge Y, Pan S, Guan D, Yin H, Fan Y, Liu J, et al. MicroRNA-350 induces pathological heart hypertrophy by repressing both p38 and JNK pathways. Biochim Biophys Acta 2013; 1832:1–10. [DOI] [PubMed] [Google Scholar]
- 11.Shieh JT, Huang Y, Gilmore J, Srivastava D. Elevated miR-499 levels blunt the cardiac stress response. PLoS One 2011; 6:e19481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol 2009; 29:2193–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K, Long B, Zhou J, Li PF. miR-9 and NFATc3 regulate myocardin in cardiac hypertrophy. J Biol Chem 2010; 285:11903–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res 2011; 108:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong DL, Chen C, Huo R, Wang N, Li Z, Tu YJ, et al. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: a novel mechanism for progressive cardiac hypertrophy. Hypertension 2010; 55:946–952. [DOI] [PubMed] [Google Scholar]
- 16.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008; 133:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A 2006; 103:18255–18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 2007; 42:1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem 2003; 278:47694–47699. [DOI] [PubMed] [Google Scholar]
- 20.Francis SH, Turko IV, Corbin JD. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog Nucleic Acid Res Mol Biol 2001; 65:1–52. [DOI] [PubMed] [Google Scholar]
- 21.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res 2003; 93:280–291. [DOI] [PubMed] [Google Scholar]
- 22.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, et al. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J 2001; 15:1718–1726. [DOI] [PubMed] [Google Scholar]
- 23.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res 2005; 96:100–109. [DOI] [PubMed] [Google Scholar]
- 24.Corbin J, Rannels S, Neal D, Chang P, Grimes K, Beasley A, Francis S. Sildenafil citrate does not affect cardiac contractility in human or dog heart. Curr Med Res Opin 2003; 19:747–752. [DOI] [PubMed] [Google Scholar]
- 25.Reffelmann T, Kloner RA. Therapeutic potential of phosphodiesterase 5 inhibition for cardiovascular disease. Circulation 2003; 108:239–244. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein I, Lue TF, Padma-Nathan H, Rosen RC, Steers WD, Wicker PA. Oral sildenafil in the treatment of erectile dysfunction. Sildenafil Study Group. N Engl J Med 1998; 338:1397–1404. [DOI] [PubMed] [Google Scholar]
- 27.Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover study. J Am Coll Cardiol 2004; 43:1149–1153. [DOI] [PubMed] [Google Scholar]
- 28.Patrucco E, Domes K, Sbroggió M, Blaich A, Schlossmann J, Desch M, et al. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A 2014; 111:12925–12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wegener JW, Nawrath H, Wolfsgruber W, Kühbandner S, Werner C, Hofmann F, Feil R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ Res 2002; 90:18–20. [DOI] [PubMed] [Google Scholar]
- 30.Song T, Hao Q, Zheng YM, Liu QH, Wang YX. Inositol 1,4,5-trisphosphate activates TRPC3 channels to cause extracellular Ca2+ influx in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2015; 309:L1455–L1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Hao Q, Luo J, Xiong J, Zhang S, Wang T, et al. USP4 inhibits p53 and NF-kappaB through deubiquitinating and stabilizing HDAC2. Oncogene 2016; 35:2902–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res 2008; 36 (Database issue):D149–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15–20. [DOI] [PubMed] [Google Scholar]
- 35.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol 2011; 18:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 2004; 15:185–197. [DOI] [PubMed] [Google Scholar]
- 37.Alghamri MS, Weir NM, Anstadt MP, Elased KM, Gurley SB, Morris M. Enhanced angiotensin II-induced cardiac and aortic remodeling in ACE2 knockout mice. J Cardiovasc Pharmacol Ther 2013; 18:138–151. [DOI] [PubMed] [Google Scholar]
- 38.Misaka T, Suzuki S, Miyata M, Kobayashi A, Ishigami A, Shishido T, et al. Senescence marker protein 30 inhibits angiotensin II-induced cardiac hypertrophy and diastolic dysfunction. Biochem Biophys Res Commun 2013; 439:142–147. [DOI] [PubMed] [Google Scholar]
- 39.Wang AW, Song L, Miao J, Wang HX, Tian C, Jiang X, et al. Baicalein attenuates angiotensin II-induced cardiac remodeling via inhibition of AKT/mTOR, ERK1/2, NF-kappaB, and calcineurin signaling pathways in mice. Am J Hypertens 2015; 28:518–526. [DOI] [PubMed] [Google Scholar]
- 40.Guan XH, Hong X, Zhao N, Liu XH, Xiao YF, Chen TT, et al. CD38 promotes angiotensin II-induced cardiac hypertrophy. J Cell Mol Med 2017; 21:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galan M, Varona S, Guadall A, Orriols M, Navas M, Aguiló S, et al. Lysyl oxidase overexpression accelerates cardiac remodeling and aggravates angiotensin II-induced hypertrophy. FASEB J 2017; 31:3787–3799. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto E, Sasaki S, Kinoshita H, Kito T, Ohta H, Konishi M, et al. Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells 2013; 18:544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 2003; 65:45–79. [DOI] [PubMed] [Google Scholar]
- 44.Kishimoto I, Rossi K, Garbers DL. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci U S A 2001; 98:2703–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wollert KC, Fiedler B, Gambaryan S, Smolenski A, Heineke J, Butt E, et al. Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension 2002; 39:87–92. [DOI] [PubMed] [Google Scholar]
- 46.Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest 2001; 107:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohli S, Ahuja S, Rani V. Transcription factors in heart: promising therapeutic targets in cardiac hypertrophy. Curr Cardiol Rev 2011; 7:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 2005; 11:214–222. [DOI] [PubMed] [Google Scholar]
- 49.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353:2148–2157. [DOI] [PubMed] [Google Scholar]
- 50.Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation 2007; 116:1555–1562. [DOI] [PubMed] [Google Scholar]
- 51.Guazzi M, Vicenzi M, Arena R. Phosphodiesterase 5 inhibition with sildenafil reverses exercise oscillatory breathing in chronic heart failure: a long-term cardiopulmonary exercise testing placebo-controlled study. Eur J Heart Fail 2012; 14:82–90. [DOI] [PubMed] [Google Scholar]
- 52.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail 2011; 4:8–17. [DOI] [PubMed] [Google Scholar]
- 53.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 2009; 23:2806–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev 2009; 23:2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 2013; 112:1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han F, Zhang S, Zhang L, Hao Q. The overexpression and predictive significance of MMP-12 in esophageal squamous cell carcinoma. Pathol Res Pract 2017; 213:1519–1522. [DOI] [PubMed] [Google Scholar]
- 58.Liu X, Yang L, Wang H, Xu G, Zhu S, Li M, et al. Effects of miR-19b knockdown on the cardiac differentiation of P19 mouse embryonic carcinoma cells. Mol Med Rep 2015; 11:2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin DN, Qian L, Hu DL, Yu ZB, Han SP, Zhu C, et al. Effects of miR-19b overexpression on proliferation, differentiation, apoptosis and Wnt/beta-catenin signaling pathway in P19 cell model of cardiac differentiation in vitro. Cell Biochem Biophys 2013; 66:709–722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.