

Abstract
Rationale:
X-linked dominant hypophosphatemia rickets (XLH, OMIM 307800) is the most common hereditary hypophosphatemic rickets and characterized by growth retardation, skeletal malformations, dental dysplasia, spontaneous fractures and osteomalacia. PHEX gene was identified for XLH and novel mutations were consistent with loss of function.
Patient concerns:
Case1: the proband 1 III3 in family 1 was a fourteen-year-old boy with bowing of bilateral legs, obviously enlarged joints, tooth absence and difficulty in walking; X-rays showed bilateral femoral multiple fractures with sclerosis at the fracture edge. Case 2: the proband 2 III2, a five-year-old boy in family 2, showed growth retardation, dental dysplasia, gingiva abscess and bilateral legs malformations; X-rays showed low bone density, delayed bone age, bowing of legs, frayed and widened metaphyses of the distal femurs and proximal tibias. Both of their mothers suffered from skeletal malformations, tooth absence and were performed with osteotomy due to fractures of lower limb. Their biochemical parameters showed hypophosphatemia, elevated alkaline phosphatase.
Diagnoses:
X-linked dominant hypophosphatemia rickets (XLH).
Interventions and outcomes:
Treatment with high doses of phosphate and 1,25-dihydroxyvitamin D3, the height of proband 2 increased 10 cm and femoral Multiple fracture of proband1 almost healed after treatment for 6 months and the patients's PHEX gene was investigated.
Lessons:
Two novel pathogenic PHEX mutations were found: c.497delG in family 1 and c.388G> T in family 2, both of which caused early termination of translation and produced truncated protein. Serum FGF23 concentration in our XLH patients were obviously higher than the normal and may be related to age to some extent. Early initiation of treatment produces better effect.
Keywords: FGF23, PHEX, X-linked dominant hypophosphatemia rickets
1. Introduction
Hypophosphatemic rickets is a group of skeletal mineralization disorders characterized by hypophosphatemia caused by renal reabsorption dysfunction of phosphorus, including X-linked dominant hypophosphatemia rickets (XLH, OMIM 307800), autosomal dominant hypophosphatemic rickets, autosomal recessive hypophosphatemic rickets, tumor-induced osteomalacia, primary or secondary Fanconi syndrome, and so on. XLH is the most common hereditary hypophosphatemic rickets and clinical manifestations included growth retardation, skeletal malformations, dental dysplasia in childhood and spontaneous fractures, and osteomalacia in adult.[1] PHEX gene was identified for XLH, which located in Xp22.1 to 22. 2, contained 22 exons, spanned approximately 243 kb, and encoded 749-amino-acid protein.[2] PHEX protein was a kind of membrane-bound protein, including a short N-terminal cytoplasmic domain, a single transmembrane hydrophobic region, and a large C-terminal extracellular domain containing several conserved cysteine residues and zinc binding sites, and is highly homologous to the M13 family of neutral endopeptidases, which could activate or degrade peptides.[3] PHEX protein chiefly expressed in osteoblast and osteoclast of bone and teeth, but not in the kidney.[4]
Fibroblastic growth factor 23 (FGF23) was candidate for circulating phosphaturic factor. Circulatory FGF23 concentration was significantly higher in the patients with inactive mutation of PHEX gene compared with healthy people at a low level.[5] Inactivating mutations of PHEX protein resulted in overexpression of FGF23 transcripts in the bone and cultured osteoblasts of the Hyp mouse, which was highly homolog of XLH. Nevertheless, some research also pointed that FGF23 could not be degradated by PHEX protein in vitro[6] and some unidentified maybe correlate with PHEX and FGF23 and further controls systemic phosphate homeostasis and mineralization.
Two novel mutations of PHEX gene: c.497delG and c.388G>T were found in our study by Sanger sequencing in 2 families manifesting growth retardation, skeletal malformations, dental dysplasia, reported as follows.
2. Study subjects and ethics statement
The clinical manifestations of the 2 family members affected with XLH showed skeletal malformations, bowing of legs, and dental dysplasia. The biochemical parameters showed hypophosphatemia, elevated alkaline phosphatase, procollagen type I amino-terminal propeptide (PINP), and FGF23 (Table 1). Case 1: in family 1, the proband 1 III3, a 14-year-old boy, was found with bowing of bilateral legs at 3 years old and had difficulty in walking for 3 months. Physical examination showed shoulder joints, elbow joints, wrist joints obviously enlarged, tooth absence, lower extremities showed “O” type bending and “penguin” gait (Fig. 1A and B). X-rays displayed bilateral femoral multiple fractures with sclerosis at the fracture edge, low bone density, and bowing of bilateral distal tibias (Fig. 1C and D). Case 2: the proband 2 III2 in family 2 was a 5-year-old boy with growth retardation, tooth absence, gingival abscess, and bowing of legs (Fig. 1E and F). X-rays showed low bone density, delayed bone age, frayed and widened metaphases of the distal femurs and proximal tibias (Fig. 1G and H). Both of the proband's mothers suffered from skeletal malformations and tooth absence and had an osteotomy operation due to fractures when they were teenagers. They were diagnosed with XLH based on clinical manifestations and treated with phosphate and 1,25-dihydroxyvitamin D3. To investigate gene mutations in 2 families, their blood sample was collected. Patient informed consent was given, and the approval of First Affiliated Hospital of Zhengzhou University Clinical and Scientific Research Ethics Committee was obtained.
Table 1.
Clinical manifestations and biochemical parameters of the patients.
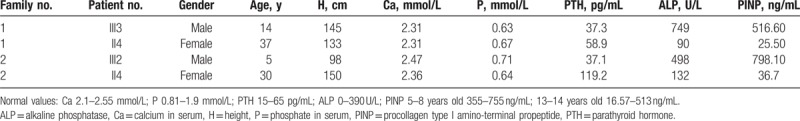
Figure 1.
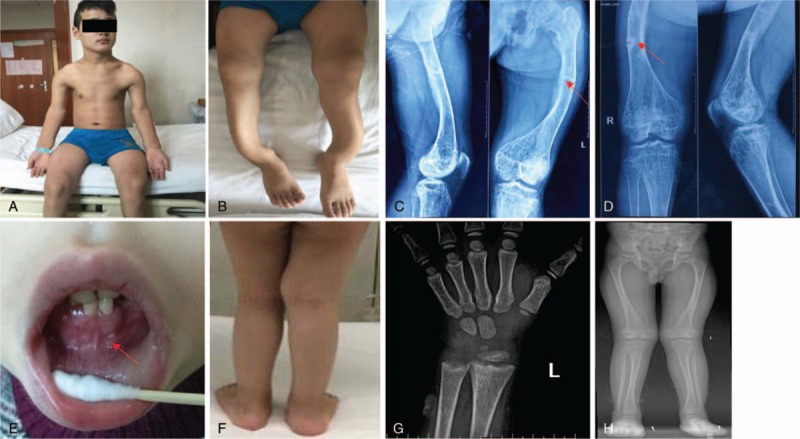
Proband 1: (A) shoulder joints, elbow joints, wrist joints obviously enlarged; (B) lower extremities showed “O” type bending; (C, D) bilateral femoral multiple fractures and low bone density (marked by red arrow). Proband 2: (E) tooth absence, gingival abscess (marked by red arrow); (F) bowing of legs; (G) delayed bone age and widened metaphases of the distal radial; (H) widened metaphases of the distal femurs and proximal tibias.
3. Mutation analysis
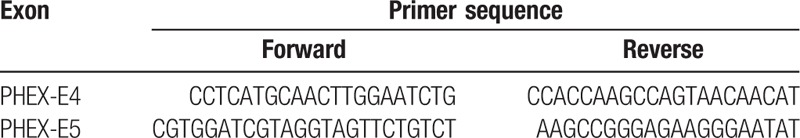
Genomic DNA of the probands including their parents and sisters was extracted from peripheral blood by Omega DNA extraction kit and subjected to DNA analysis. All 22 exons and their exon–intron boundaries of the PHEX gene were amplified by polymerase chain reaction (PCR) with 22 pairs of primers designed by GeneTool (Table 2 just showed primers of exon 4 and 5). PCR amplification product was directly sequenced by using BIBigDye3.1 on the ABI3130XI sequencer and analyzed in BeiJing MyGenostics company. Serum concentration of FGF23 was measured with KAINOS FGF23 ELISA Kit.
Table 2.
Primer sequence of exon 4 and 5 in the PHEX gene.
4. Results
In family 1, hemizygous novel mutation c.497delG in exon 5 was found in III3 (proband 1) and heterozygous c.497delG was found in II4 (Fig. 2, family 1), which changed codon CGC to CCT, resulted in a subsequent frameshift change of the codon and was predicted to terminate translation at 220 codon. In family 2, hemizygous nonsense mutation c.388G>T in exon 4 was found in III2 (proband 2) and II4 was heterozygous (Fig. 2, family 2), which was predicted to terminate translation at 129 codon. The proband's fathers and sisters were normal. We sequenced exon 4 and 5 of the PHEX gene from 100 unrelated healthy people to identify nucleotide polymorphism, they did not carry the 2 variations. The function of mutant proteins was predicted to be pathological and had not been reported by searching in the HGMD and PHEX MUTATION database (http://www.phexdb.mcgill.ca/).
Figure 2.
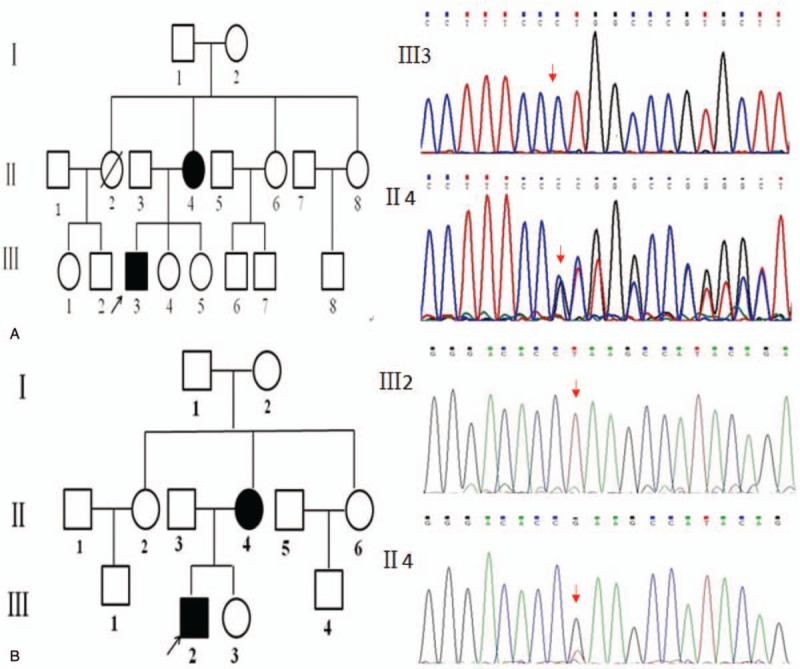
Genetic analysis in the 2 pedigrees of X-linked hypophosphatemic rickets. Family 1: Deletion mutation: c.497delG of PHEX gene was found in proband 1 III3 and his mother II4 affected with XLH in family 1 (marked by red arrow). The proband 1 was hemizygous and his mother was heterozygous. Family 2: Missense mutation: c.388G > T of PHEX gene was found in proband 2 III2 and his mother II4 suffering from XLH in family 2 (marked by red arrow). The proband 1 was hemizygous and his mother was heterozygous.
Serum FGF23 concentration of proband 1 was 149.9 pg/mL and his mother was 58.4 pg/mL; proband 2 was 308.2 pg/mL and his mother was 83.5 pg/mL (the health control: 21 ± 5.0 pg/mL).
5. Discussion
Three hundred fifty mutations of PHEX have been found from the rickets families and sporadic cases, including missense, nonsense, deletions, insertions, frameshift and splice site mutations as well as polymorphisms, most of which are consistent with the loss-of-function of PHEX by searching HGMD. Sabbagh et al demonstrated that defects in protein trafficking, endopeptidase activity, and protein conformation accounted for loss-of-function of protein in the study of structure and function of disease-causing missense mutations in the PHEX gene.[7] There was no clearly correlation between genotype and phenotype in previous studies, and also no correlation between the severity and the genotypes.[8] In our study, 2 novel mutations of PHEX gene: c.497delG and c.388G>T were found in 2 rickets families with characteristics of defective bone mineralization and hypophosphatemia, and were predicted to produce truncated protein and cause loss-of-function of PHEX protein. In family 1, hemizygous novel mutation c.497delG causing P.R166P was found in proband 1 and heterozygous c.497delG was found in his mother, which changed codon CGC to CCT and resulted in a subsequent frameshift change of the codon. In family 2, hemizygous nonsense mutation c.388G>T causing P.E130X was found in proband 2 and his mother was heterozygous, which stopped translation at amino acid 130. Both of the 2 novel mutations were predicted to terminate translation and lead to losing several conserved cysteine residues and zinc binding sites, which was likely to affect the catalytic activity of the PHEX protein.
The circulatory FGF23 concentration of our XLH patients was obviously higher than the normal. FGF23 was a bone-derived hormone with protease recognition motif and could form a ternary complex with a cognate fibroblastic growth factor receptor and the klotho protein, which downregulated the Na-phosphate II cotransporters activities at the brush border membrane and the vitamin D hydroxylases and leaded to hypophosphatemia.[9] A study showed that increased FGF23 expression in Hyp bone resulted from a local effect of PHEX deficiency by researching the change of FGF23 level by cross-transplantations between wild-type mice and the Hyp mouse model of XLH. Alternatively, the study also displayed that FGF23 was inhibited in juvenile Hyp bone explanted into adult Hyp mice, indicating that FGF23 was age-dependent to some extent.[10] Our study also observed that FGF23 concentration of the probands was obviously higher than their mother. In our study, the biochemical parameters alkaline phosphatase and PINP were higher in the probands, which indicated the high bone turnover state, but their mother was normal. It was speculated that the symptoms of rickets could improve by themselves in adults because of the decreasing phosphate requirement as described before and maybe also related to FGF23.
Treatment with high doses of phosphate and 1,25-dihydroxyvitamin D3 was the basic measures of XLH at present, but mineralization disorder just could be improved partially.[11] In the follow-up process, the height of proband 2 increased 10 cm and femoral multiple fracture of proband 1 almost healed after treatment for 6 months. Early initiation of treatment appeared to improve height outcomes in adults. The proband 2 showed catchup growth, but the height of proband 1 had no change. Early initiation of treatment appeared to improve height outcomes in adults. Quinlan found that those who started treatment before 1 year old got higher median height standard deviation score and even normalize growth of XLH patients, regardless of gender and genotype.[12]
In conclusion, 2 novel pathogenic mutations of PHEX gene were found in the 2 families with hypophosphatemic rickets: c.497delG and c.388G>T, both of which caused early termination of translation and produced truncated protein. Serum FGF23 concentration in our XLH patients was obviously higher than the normal and may be related with age to some extent. Further research about genetic molecular basis and acquired disorders of phosphate homeostasis is necessary, which will provide new insights and approaches to XLH diagnosis and treatment.
Author contributions
Investigation: Zhi-Min Wang.
Project administration: Xia-Lian Li.
Writing – original draft: Yue Gao.
Footnotes
Abbreviations: XLH= X-linked dominant hypophosphatemia rickets, FGF23 = fibroblastic growth factor 23.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Fuente R, Gil-Pena H, Claramunt-Taberner D, et al. X-linked hypophosphatemia and growth. Rev Endocr Metab Disord 2017;18:107–15. [DOI] [PubMed] [Google Scholar]
- [2].Rowe PS, Goulding JN, Francis F, et al. The gene for X-linked hypophosphataemic rickets maps to a 200–300 kb region in Xp22.1, and is located on a single YAC containing a putative vitamin D response element (VDRE). Hum Genet 1996;97:345–52. [DOI] [PubMed] [Google Scholar]
- [3].Turner AJ, Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J 1997;11:355–64. [DOI] [PubMed] [Google Scholar]
- [4].Ruchon AF, Tenenhouse HS, Marcinkiewicz M, et al. Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. Bone Miner Res 2000;15:1440–50. [DOI] [PubMed] [Google Scholar]
- [5].Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. Clin Endocrinol Metab 2002;87:4957–60. [DOI] [PubMed] [Google Scholar]
- [6].Liu S, Guo R, Simpson LG, et al. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. Biol Chem 2003;278:37419–26. [DOI] [PubMed] [Google Scholar]
- [7].Sabbagh Y, Boileau G, Campos M, et al. Structure and function of disease-causing missense mutations in the PHEX gene. Clin Endocrinol Metab 2003;88:2213–22. [DOI] [PubMed] [Google Scholar]
- [8].Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. Clin Endocrinol Metab 2001;86:3889–99. [DOI] [PubMed] [Google Scholar]
- [9].Kinoshita Y, Fukumoto S. X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases-prospect for new treatment. Endocr Rev 2018;39:274–91. [DOI] [PubMed] [Google Scholar]
- [10].Liu S, Tang W, Zhou J, et al. Distinct roles for intrinsic osteocyte abnormalities and systemic factors in regulation of FGF23 and bone mineralization in Hyp mice. Am J Physiol Endocrinol Metab 2007;293:E1636–44. [DOI] [PubMed] [Google Scholar]
- [11].Carpenter TO, Imel EA, Holm IA, et al. A clinician's guide to X-linked hypophosphatemia. Bone Miner Res 2011;26:1381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Quinlan C, Guegan K, Offiah A, et al. Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment. Pediatr Nephrol 2012;27:581–8. [DOI] [PubMed] [Google Scholar]