

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a rare autosomal dominant disorder characterized by vascular dysplasia, including typically systemic telangiectases and arteriovenous malformations (AVMs). Due to its variable clinical manifestations, HHT patients often seek medical care from different medical subspecialties and thus experience delays in diagnosis and treatment.
This study is designed to analyze the clinical features and treatment options for patients with HHT.
Hospitalized patients with a definitive diagnosis of HHT from November 1973 to July 2016 in Peking Union Medical College Hospital were identified after reviewing medical records and electronic databases. Further follow-up data of these patients were collected from outpatient clinical visits and/or telephone interviews.
We identified a total of 20 patients, 7 males and 13 females. The mean age was 42.4 ± 20.3 years. Epistaxis (18/20) was the most common presentation, followed by telangiectases of the oral buccal mucosa, tongue and/or lips (14/20), pulmonary AVMs (12/19), hepatic AVMs (9/17), gastrointestinal telangiectases (9/9), and encephalic AVMs (1/12). The correct diagnosis of HHT was delayed on average by about 26.4 ± 17.0 years from the onset of HHT-related clinical signs and symptoms. Although epistaxis is usually presented in childhood (mean age 11 ± 7.1 years), gastrointestinal telangiectasia was often encountered in late middle age (mean age 55.4 ± 12.8 years). Bleeding and anemia were the most common complications. Molecular analysis was conducted in 4 patients. Only 1 patient was found to have a single-base deletion in ENG gene. The mean duration of follow-up of the patients was 41.8 months. The efficacy of locoregional therapy was of limited value and short-lived. Two patients were treated systemically with thalidomide, and their symptoms of epistaxis, melena, and anemia were notably improved.
Patients with HHT have variable clinical characteristics, and their diagnoses were delayed on average by about 26 years. An experienced multidisciplinary team is needed for the early diagnosis and optimal management of patients with HHT. Thalidomide may be an effective choice to alleviate the bleeding symptoms of patients with HHT.
Keywords: arteriovenous malformation, complications, features, hereditary hemorrhagic telangiectasia, thalidomide, treatment
1. Introduction
Hereditary hemorrhagic telangiectasia (HHT), also called Osler–Weber–Rendu syndrome, is a rare autosomal dominant disorder characterized by vascular malformations in multiple organ systems. The clinical manifestations of HHT vary from epistaxis, mucocutaneous telangiectases to arteriovenous malformations (AVM) in lungs, brain, and liver.[1] As a result, patients with HHT usually visit different departments. However, due to the disease's rarity and physicians’ lack of relevant knowledge, patients often experience long diagnostic delays.[2] In this study, we retrospectively analyzed the clinical features and treatment of patients with HHT diagnosed in our hospital.
2. Methods
This study was a single-center retrospective analysis. By reviewing medical records and electronic databases, patients with diagnosis of HHT from November 1973 to July 2016 at Peking Union Medical College Hospital were enrolled. To be included in the study, all of the patients should meet at least 3 of the 4 Curacao criteria for HHT diagnosis: spontaneous, recurrent nose bleeding; multiple telangiectases, especially in the lips, oral mucosa, fingers and nose; visceral lesions such as gastrointestinal (GI) telangiectases and hepatic and cerebral AVMs; and a first-degree relative with HHT.[3] The data collected from patients’ medical records included the age of disease onset and the initial clinical HHT manifestation, the age of first medical visit for HHT-related symptoms, and age of first definite HHT diagnosis, clinical features, screening for AVMs, complications from HHT, family history, gene analysis results, and treatment. The current study was approved by the Institutional Review Board of Peking Union Medical College Hospital (No. S-K234).
A total of 20 eligible patients were included in this study. Mucocutaneous telangiectases were identified by visual inspection with or without nasopharyngoscopy. Endoscopic examinations were performed only in patients with melena or bright red blood per rectum. AVMs were analyzed with ultrasonography, computed tomography (CT) and CT angiography, and/or magnetic resonance angiography. GI bleeding and hematuria were confirmed by medical history, presence of bleeding telangiectases on endoscopy, or ongoing symptoms (bright red or dark stools, bloody urine). Liver bleeding was suspected if the patient complained of dull pain in the right upper abdomen and was confirmed by contrast-enhanced CT or magnetic resonance imaging (MRI). Anemia severity was defined as mild if hemoglobin ≥9 g/dL, moderate if 6 to 9 g/dL, and severe if <6 g/dL. Iron-deficiency anemia was diagnosed if serum ferritin and transferrin saturation were low and total iron binding capacity was high. Cerebral and coronary embolisms were confirmed by cerebral MRI and coronary angiography, respectively. Pulmonary hypertension and portal hypertension were diagnosed by echocardiogram and abdominal ultrasound, respectively. High-output heart failure was defined by clinical signs and symptoms of heart failure with preserved ejection fraction of >50% in echocardiogram.
Follow-up was conducted by clinical visits, phone contact with patients, and reviewing records of patients’ subsequent consultations. The following clinical data were collected: age, sex, medical, and surgical interventions, and their efficacies. “Effective” was defined as patients experienced improvement or remission after procedure or medical treatment, and vice versa. “Recurrence” was defined as the clinical features occurred or aggravated after a period of improvement.
Continuous variables were expressed as means with standard deviations. Categorical variables were expressed as frequencies and percentages. The diagnosis time lag (DTL) was defined as the time interval from the onset of signs and symptoms to definitive diagnosis of HHT. The visit time lag (VTL) was defined as the time interval from the onset of the signs and symptoms of HHT to the first clinic visit to a physician for HHT-associated clinical presentations. In addition, using Chi-square test, we conducted comparative analyses among the onset age of epistaxis and the diagnostic age of GI telangiectasia, pulmonary, and liver AVM in patients with HHT. All tests were 2-tailed, and a P value < .05 was considered significant. The software package SPSS 23.0 (SPSS Inc., Chicago, IL) was used for statistical analysis.
3. Results
3.1. Clinical manifestations
Twenty eligible patients, 7 males and 13 females, were included in this study, with age ranging from 3 to 80 years and a mean age of 42.4 ± 20.3 years (Table 1). Epistaxis was the earliest and most common manifestation (18/20 cases), followed by telangiectasia in oral buccal mucosa, tongue, and lips (14/20 cases). Lungs were the most common involved sites of AVM (12/19 cases), followed by liver (9/17 cases) and brain (1/12 cases) (Fig. 1).
Table 1.
Clinical features in 20 patients with HHT.

Figure 1.
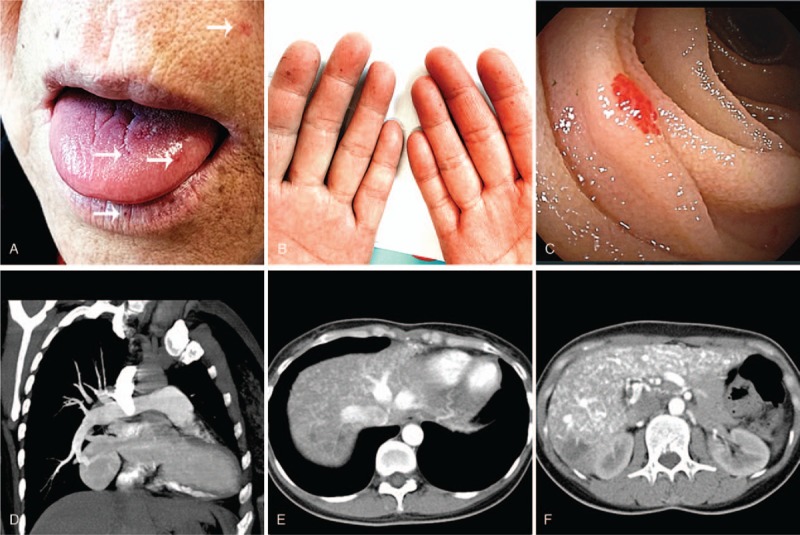
Clinical manifestations in patients with HHT. (A, B) were collected from a 62-year-old woman with HHT. Multiple maroon-colored telangiectases (arrows) were seen on her lips, tongue, face (A), and fingers (B). Enteroscopy shows scattered telangiectases (C). CT pulmonary angiogram shows an AVM in the lower lobe of the right lung, associated with the right inferior pulmonary vein (D). CTs with contrast of another 35-year-old woman with HHT who had hepatic AVMs showed that the hepatic vein, portal vein, and their branches enhanced earlier during the arterial phase, indicating the existence of hepatic arteriovenous shunt (E) and arterioportal shunt (F). AVM = arteriovenous malformation, CT = computed tomography, HHT = hereditary hemorrhagic telangiectasia.
Our hospital is a tertiary referral hospital that is specialized in the diagnosis and management of rare diseases and complicated patients. Patients in this study all had visited other hospitals, but none of them received the correct diagnosis of HHT before they were referred to us. It was also noteworthy that there were long delays among the age of HHT onset, the age of first medical visit to physicians for HHT-related features, and the age of first definite HHT diagnosis (mean age of 13.2 ± 10.1, 32.3 ± 18.0, and 42.4 ± 20.3 years, respectively) (Fig. 2). Correspondingly, the VTL and DTL were 1 decade (10.2 ± 10.7 years) and nearly 3 decades (26.4 ± 17.0 years), respectively.
Figure 2.
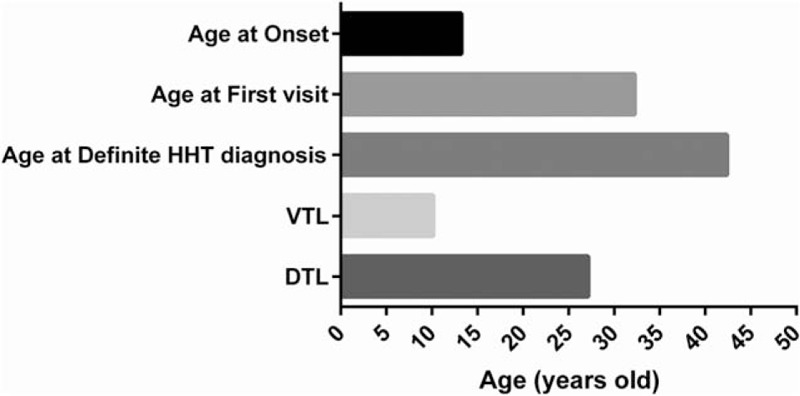
Time delays in diagnosis of HHT. DTL = diagnosis time lag, VTL = visiting time lag.
With a mean age of 11.0 ± 7.1 years, epistaxis often occurred in childhood, which was significantly earlier than the diagnoses of visceral involvements, including GI telangiectasia, pulmonary, and hepatic AVM (mean diagnostic age of 55.4 ± 12.8, 35.7 ± 20.5, and 42.9 ± 15.55 years, respectively) (Fig. 3). The diagnostic age of pulmonary AVM was significantly earlier than that of GI telangiectasia (P < .05), whereas no significant difference was found between other visceral involvements.
Figure 3.
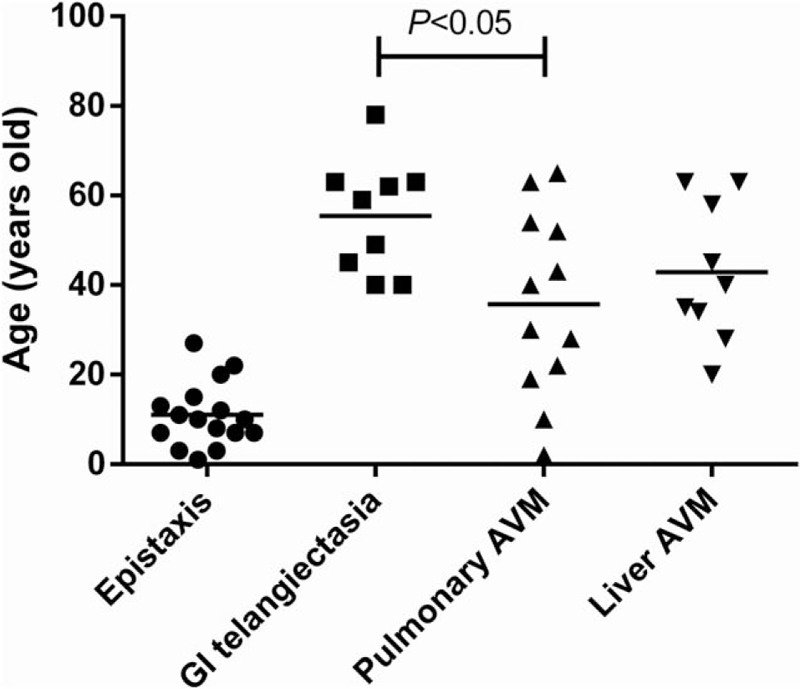
The age of patients with HHT at the time epistaxis and the age when diagnosis of GI telangiectasia, pulmonary, or liver AVM was confirmed. The onset of epistaxis was significantly earlier than the diagnosis of the visceral lesions (P < .01). There was no significant difference between the ages of diagnosis of visceral lesions, except that pulmonary AVM was diagnosed significantly earlier than GI telangiectasia (P < .05). AVM = arteriovenous malformation, GI = gastrointestinal, HHT = hereditary hemorrhagic telangiectasia.
Fourteen patients had positive family histories. Four of them conducted gene analysis, among which 3 cases were negative in ENG and ALK1 mutations, and the other one had a single-base deletion mutation in ENG gene. The patient with ENG gene mutation presented with epistaxis, mucocutaneous and GI telangiectases, and AVMs in lungs and liver.
3.2. Complications
The major complications are bleeding and anemia, followed by embolism and pulmonary hypertension. As summarized in Table 2, 8 patients (40%) had documented anemia, of whom 2 were mild, 4 were moderate, and 2 were severe. All anemia patients presented with both epistaxis and GI bleeding except 1 patient with moderate anemia who presented with epistaxis alone. Four cases (20%) had embolism, of which all presented with cerebrovascular embolism and stroke, while 1 patient with coronary embolism and acute myocardial infarction. It was noted that 3 of the 4 patients with embolism had pulmonary AVM. Pulmonary hypertension was diagnosed in 4 cases (20%), of which 1 patient had both pulmonary and hepatic AVMs, while the other 3 patients had pulmonary AVM, pulmonary interstitial fibrosis, and hepatic AVM, respectively. Two patients with hepatic AVM were complicated with portal hypertension. One patient had high-output heart failure secondary to severe anemia and hepatic AVM.
Table 2.
Complications of patients with HHT.

3.3. Treatment and follow-up
Clinical follow-ups were available in 12 of the 20 patients. The mean follow-up time was 41.8 months (range from 6 months to 12.5 years). According to previous treatment histories, main clinical manifestations, and patients’ willingness, managements were given as summarized in Table 3.
Table 3.
Treatments and outcomes in 12 follow-up patients with HHT.
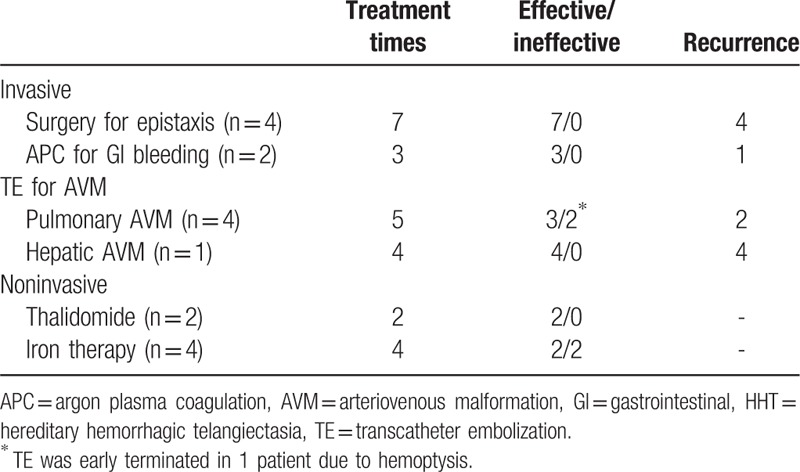
Four patients with epistaxis were given electrochemical cautery or septal dermoplasty for 7 times, with recurrence rate of more than 50%. Active bleeding was observed by endoscope in 2 cases with GI telangiectasia. And they accepted argon plasma coagulation (APC) 3 times with 1 relapsed. Four patients with pulmonary AVM received 5 times of transcatheter embolization (TE) with 3 times effective and 2 times relapsed. One patient with hepatic AVM has undergone her fourth TE to date, but only temporary efficacy of 2 months was achieved.
Two patients received oral thalidomide for 100 to 125 mg per night. Their epistaxis, melena, and anemia were markedly improved. One patient who had failed iron treatment became transfusion independent, and the changes of hemoglobin and ferritin are shown in Fig. 4. Four patients with iron-deficiency anemia accepted long-term oral and/or intermittent intravenous iron treatment, and 2 patients’ anemia improved (including the 1 simultaneously taking thalidomide above-mentioned). By the end of follow-up, 2 cases have deceased, of which 1 died from gastric cancer and GI bleeding, while the other 1 died from pulmonary infection.
Figure 4.
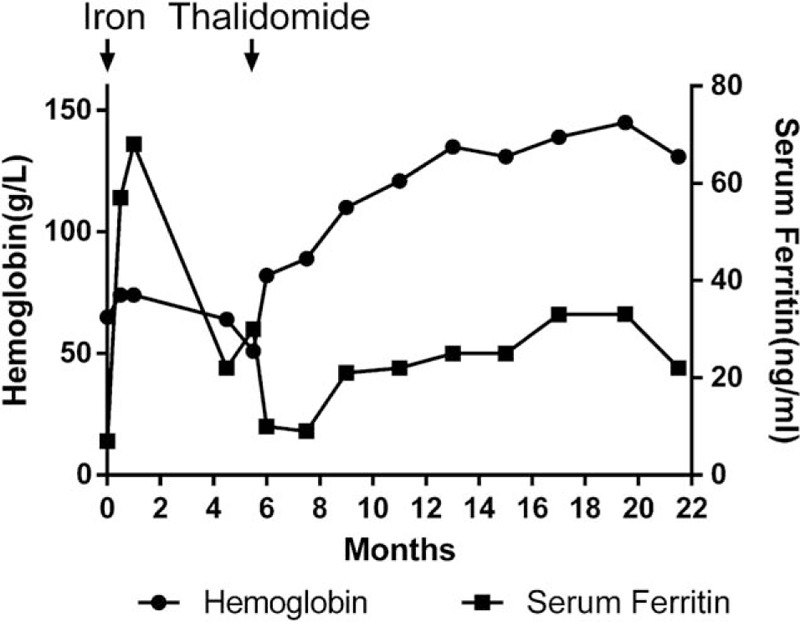
Efficacy of iron treatment and thalidomide in one patient with HHT. HHT = hereditary hemorrhagic telangiectasia.
4. Discussion
HHT is a rare autosomal dominant disorder, with a prevalence of 1/5000 to 1/8000.[4,5] Patients with HHT present with various manifestations and multiple involvements that often appear gradually with age. By the age of 16, about 70% of individuals have some features of HHT, and 90% by the age of 40.[6] Epistaxis was the earliest and most common symptom in our study, with a mean age of onset of 11.0 ± 7.1 years. Visceral lesions were usually diagnosed in adults, while pulmonary AVM was more commonly diagnosed in middle age (35.7 ± 20.5 years). GI telangiectasia was often diagnosed in middle to older aged patients (55.4 ± 12.8 years). The clinical manifestations and the age of diagnosis of our patients were generally in agreement with published data. However, our patients experienced a relatively late diagnosis for pulmonary AVM.[7]
In our study, we observed a long diagnostic delay of nearly 3 decades in patients with HHT from disease onset. Pierucci et al[2] in Italy had reported a similar diagnostic delay of 25.7 ± 17.4 years. The main reason accounting for such a long diagnostic time was most likely due to the rarity of the disease, heterogeneity of clinical features, and the insufficient knowledge of clinical practitioners. Moreover, HHT tends to remain relatively silent for decades before presenting as sudden, acute life-threating events. In our study, 14 of 20 cases (70%) first presented with isolated epistaxis, and none of them sought medical attention. They usually regarded the nosebleed as normal annoyance for many years rather than perceived it as the symptom of an underlying disease.[2]
Pierucci et al[2] and we also observed a prolonged delay from the age of first visit to a physician until the age when definitive HHT diagnosis was made (14.9 ± 14 and 10.2 ± 10.7 years, respectively). These results highlight the insufficient knowledge of HHT among medical community. The time needed to accomplish a definite correct diagnosis depends on the complexity of essential diagnostic steps and the cognitive biases of the physician.[2] Part of the value and aims of our study is to raise physicians’ awareness of the clinical characteristics of HHT so that diagnosis earlier can be made.
There are 5 genes associated with HHT. Endoglin (ENG) and ALK1 (aka activin-like receptor kinase 1 or ACVRL1) genes are the major ones, whose mutations cause HHT1 and HHT2, respectively. Mutations in a third gene known as SMAD4 may present a combined syndrome of juvenile polyposis and HHT. The roles of other 2 genes have not been identified yet.[8] The above 3 known genes are components of the transforming growth factor-beta (TGF-β) signaling pathway, which regulates cell differentiation and proliferation. Thus, the TGF-β signaling pathway has been considered as a major role in pathogenetic mechanism of HHT, and eventually leads to vascular dysplasia and malformation. McDonald et al[8] found that 96% of patients who fulfilled 3 or more Curacao criteria had either an ENG or ALK1 mutation. A retrospective analysis by Arthur et al[9] showed that 11% of patients with HHT did not develop symptoms until age 50, indicating the importance of gene analysis. In our series, 4 patients and some of their first-degree relatives underwent genetic analysis. One patient was found to have ENG mutation. Further studies are needed to clarify the demographic genotype in Chinese patients with HHT.
Bleeding and anemia are the most common complications of HHT. All 20 cases in our study had more than 1 bleeding site, with the most common being epistaxis and GI bleeding. Infrequent bleeding occurred in the liver and urinary tract. Eight cases (40%) developed secondary anemia, with moderate anemia as the most common (50%). Almost all anemic patients presented with both epistaxis and GI bleeding except 1 who presented with epistaxis only. The 2 patients with severe anemia all had active GI bleeding. Pahl et al retrospectively analyzed 168 patients with HHT and found that 50% had documented anemia. Mild anemia was most common (52%), followed by moderate (37%) and severe anemia (11%). Epistaxis was the most common cause for anemia in the mild and moderate groups (75% and 59%, respectively), while both GI bleeding and epistaxis were present in the majority of patients with severe anemia (44%). The mean severity score of epistaxis in patients with moderate anemia was higher than those with mild anemia.[10] Stroke is a recognized complication of HHT and occurs in 10% to 19% of affected individuals.[11] In young patients with HHT and pulmonary AVM, stroke was most often due to paradoxical embolism. A cross-sectional study also showed that 20% patients with HHT and pulmonary AVM had embolic complications.[12] In our study, 20% cases (4/20) with HHT experienced embolism, and 25% (3/12) in patients with HHT and pulmonary AVM. All 4 patients with embolism had documented cerebrovascular embolism, and 1 had a coronary embolism. Stroke should be closely screened and monitored in patients with HHT and pulmonary AVM. Conversely, HHT should be considered and screened in young patients with stroke.[13]
There is no permanent effective cure for HHT available to date. The major treatment in clinical practice is supportive and symptom relieving. Bleeding of patients with HHT is often intractable. Physicians generally treat the bleeding with various types of hemostasis, such as electrical cauterization, photocoagulation, astringent, intravascular embolization, and surgery, but the efficacy is often limited, and has a high recurrence rate, as suggested in our study.
With a better understanding of the pathogenetic mechanism of vascular malformation in HHT, several drugs have been investigated, such as bevacizumab (an antivascular endothelial growth factor monoclonal antibody), and thalidomide (an antiangiogenic and immunomodulatory agent).[14] Thalidomide may modulate the activation of mural cells and enhance both their proliferation and ability to embrace blood vessels, which means that thalidomide can make HHT vessels more firm and less prone to breaking.[15,16] A recent prospective phase II clinical trial analyzed the efficacy of thalidomide for severe recurrent epistaxis in HHT and showed that thalidomide could effectively reduce epistaxis and transfusion requirement, ameliorate anemia, and allow for a rapid, often durable clinical improvement.[16] Another prospective study also indicated that thalidomide was effective in improving epistaxis severity and anemia, but not in GI and hepatic bleeding.[17] In our study, 2 cases underwent thalidomide treatment and achieved significant improvements in epistaxis, melena, and anemia. Nonetheless, more clinical trials are needed to clarify the timing and dosage of thalidomide and its efficacy for GI bleeding and AVMs. On the contrary, side effects of thalidomide such as neuropathy, constipation, and thromboembolism may sometimes limit its use in clinical practice.[17]
To our best knowledge, there is a lack of data proving any significant benefit of prophylactic anticoagulation for patients with HHT and pulmonary AVM. However, as for patients who have a strong indication for antiplatelet or anticoagulant use, they should not have these agents withheld.[18] Many HHT patients can tolerate anticoagulant or antithrombotic therapy without further bleeding complications.[18,19] Treatments with a low risk of bleeding complications, such as low-dose warfarin, low-dose aspirin, or apixaban, are preferred.[18] Only 1 patient in our study was given anticoagulant therapy after TE. She received low molecular weight heparin for 3 days without apparent hemorrhagic events.
Several limitations of this study should be noted. First, this is a retrospective study conducted at a single center. Therefore, our results cannot be extrapolated to a wider range of population. Second, due to the small sample size of only 20 cases in our study, it would be difficult to draw any general conclusions about clinical features and treatment efficacies. For example, only 2 cases were treated with thalidomide. Despite both experienced solid improvements, studies with a larger population of patients would be needed to further confirm the efficacy. We also showed that there are significant delays in correct diagnosis of HHT as well as lack of effective treatment options in clinical practice. Our data provided some useful information. However, prospective studies with large sample and long-term follow-up are required.
5. Conclusion
HHT is a rare systemic disease, with diverse clinical features affecting different organs. Close collaboration among different disciplines is essential for timely diagnosis and adequate management. Thalidomide may be an effective option for alleviating bleeding symptoms of patients with HHT.
Acknowledgment
We would like to thank Dr Wang Junfeng for his assistance of polishing the language.
Author contributions
Conceptualization: Shujie Wang, Yongqiang Zhao.
Data curation: Sen Li.
Formal analysis: Sen Li.
Methodology: Sen Li.
Resources: Sen Li.
Software: Sen Li.
Supervision: Shujie Wang.
Validation: Shujie Wang.
Visualization: Sen Li.
Writing – original draft: Sen Li.
Writing – review & editing: Shujie Wang, Yongqiang Zhao.
Author name: orcid number
Footnotes
Abbreviations: ALK1 = aka activin-like receptor kinase 1, APC = argon plasma coagulation, AVM = arteriovenous malformations, CT = computed tomography, DTL = diagnosis time lag, ENG = Endoglin, GI = gastrointestinal, HHT = hereditary hemorrhagic telangiectasia, MRI = magnetic resonance imaging, TE = transcatheter embolization, TGF-β = transforming growth factor-beta, VTL = visit time lag.
The authors of this work have nothing to disclose.
References
- [1].Lesca G, Olivieri C, Burnichon N, et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network. Genet Med 2007;9:14–22. [DOI] [PubMed] [Google Scholar]
- [2].Pierucci P, Lenato GM, Suppressa P, et al. A long diagnostic delay in patients with hereditary haemorrhagic telangiectasia: a questionnaire-based retrospective study. Orphanet J Rare Dis 2012;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66–7. [DOI] [PubMed] [Google Scholar]
- [4].Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002;19:140–8. [DOI] [PubMed] [Google Scholar]
- [5].Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J 2003;79:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Plauchu H, de Chadarevian JP, Bideau A, et al. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989;32:291–7. [DOI] [PubMed] [Google Scholar]
- [7].Salaria M, Taylor J, Bogwitz M, et al. Hereditary haemorrhagic telangiectasia, an Australian cohort: clinical and investigative features. Intern Med J 2014;44:639–44. [DOI] [PubMed] [Google Scholar]
- [8].McDonald J, Damjanovich K, Millson A, et al. Molecular diagnosis in hereditary hemorrhagic telangiectasia: findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin Genet 2011;79:335–44. [DOI] [PubMed] [Google Scholar]
- [9].Arthur H, Geisthoff U, Gossage JR, et al. Executive summary of the 11th HHT international scientific conference. Angiogenesis 2015;18:511–24. [DOI] [PubMed] [Google Scholar]
- [10].Kristy P, Arkopal C, Raj SK. Causes and severity of anemia in hereditary hemorrhagic telangiectasia. Blood 2016;128:3776. [Google Scholar]
- [11].Cottin V, Dupuis-Girod S, Lesca G, et al. Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (rendu-osler disease). Respiration 2007;74:361–78. [DOI] [PubMed] [Google Scholar]
- [12].Angriman F, Ferreyro BL, Wainstein EJ, et al. Pulmonary arteriovenous malformations and embolic complications in patients with hereditary hemorrhagic telangiectasia. Arch Bronconeumol 2014;50:301–4. [DOI] [PubMed] [Google Scholar]
- [13].Ribeiro E, Cogez J, Babin E, et al. Stroke in hereditary hemorrhagic telangiectasia patients. New evidence for repeated screening and early treatment of pulmonary vascular malformations: two case reports. BMC Neurol 2011;11:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 2012;307:948–55. [DOI] [PubMed] [Google Scholar]
- [15].Lebrin F, Srun S, Raymond K, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 2010;16:420–8. [DOI] [PubMed] [Google Scholar]
- [16].Invernizzi R, Quaglia F, Klersy C, et al. Efficacy and safety of thalidomide for the treatment of severe recurrent epistaxis in hereditary haemorrhagic telangiectasia: results of a non-randomised, single-centre, phase 2 study. Lancet Haematol 2015;2:e465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hosman A, Westermann CJ, Snijder R, et al. Follow-up of thalidomide treatment in patients with hereditary haemorrhagic telangiectasia. Rhinology 2015;53:340–4. [DOI] [PubMed] [Google Scholar]
- [18].Dittus C, Streiff M, Ansell J. Bleeding and clotting in hereditary hemorrhagic telangiectasia. World J Clin Cases 2015;3:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Devlin HL, Hosman AE, Shovlin CL. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. N Engl J Med 2013;368:876–8. [DOI] [PubMed] [Google Scholar]