

Abstract
Background
The clinical phenotype associated with germline SMARCB1 mutations has as yet not been fully documented. It is known that germline SMARCB1 mutations may cause rhabdoid tumor predisposition syndrome (RTPS1) or schwannomatosis. However, the co‐occurrence of rhabdoid tumor and schwannomas in the same patient has not so far been reported.
Methods
We investigated a family with members harboring a germline SMARCB1 deletion by means of whole‐body MRI as well as high‐resolution microstructural magnetic resonance neurography (MRN). Breakpoint‐spanning PCRs were performed to characterize the SMARCB1 deletion and its segregation in the family.
Results
The index patient of this family was in complete continuous remission for an atypical teratoid/rhabdoid tumor (AT/RT) treated at the age of 2 years. However, at the age of 21 years, she exhibited paraparesis of her legs and MRI investigations revealed multiple intrathoracic and spinal schwannomas. Breakpoint‐spanning PCRs indicated that the germline deletion segregating in the family encompasses 6.4‐kb and includes parts of SMARCB1 intron 7, exons 8–9 and 3.3‐kb located telomeric to exon 9 including the SMARCB1 3′ UTR. The analysis of sequences at the deletion breakpoints showed that the deletion has been caused by replication errors including template‐switching. The patient had inherited the deletion from her 56‐year‐old healthy mother who did not exhibit schwannomas or other tumors as determined by whole‐body MRI. However, MRN of the peripheral nerves of the mother's extremities revealed multiple fascicular microlesions which have been previously identified as indicative of schwannomatosis‐associated subclinical peripheral nerve pathology.
Conclusion
The occurrence of schwannomatosis‐associated clinical symptoms independent of the AT/RT as the primary disease should be considered in long‐term survivors of AT/RT. Furthermore, our investigations indicate that germline SMARCB1 mutation carriers not presenting RTs or schwannomatosis‐associated clinical symptoms may nevertheless exhibit peripheral nerve pathology as revealed by MRN.
Keywords: atypical teratoid/rhabdoid tumor, deletion breakpoint, RTPS1, schwannomatosis, SMARCB1 gene
1. INTRODUCTION
Rhabdoid tumors (RTs) are aggressive malignancies termed atypical teratoid (AT)/RTs when they arise in the central nervous system, malignant rhabdoid tumor (MRT) when located in extra‐renal soft tissues or RT of the kidney (RTK). The majority of RTs are caused by biallelic loss of function of the SMARCB1 gene located at 22q11.23 (Biegel et al., 1999; Versteege et al., 1998). Approximately 30% of patients with RTs exhibit germline SMARCB1 mutations causing RT predisposition syndrome 1 (RTPS1, OMIM #609322) (Bourdeaut et al., 2011; Eaton, Tooke, Wainwright, Judkins, & Biegel, 2011; Janson et al., 2006; Kordes et al., 2010; Sévenet et al., 1999). So far, six families have been reported with siblings affected by RTs due to the transmission of a SMARCB1 mutation from a healthy parent, with gonadal mosaicism for the respective SMARCB1 mutation (Bourdeaut et al., 2011; Bruggers et al., 2011; Eaton et al., 2011; Gigante et al., 2016; Sévenet et al., 1999).
Germline mutations in SMARCB1 also cause schwannomatosis (OMIM #162091). It has been shown that 48% of familial and 9.8% of sporadic patients with schwannomatosis exhibit germline SMARCB1 mutations (Boyd et al., 2008; Hadfield et al., 2008; Hulsebos et al., 2007; Rousseau, Noguchi, Bourdon, Sobol, & Olschwang, 2011; Sestini, Bacci, Provenzano, Genuardi, & Papi, 2008; Smith, Wallace, Bowers, Eaton, & Evans, 2014; Smith et al., 2012). Genetic heterogeneity is observed in schwannomatosis since germline mutations in LZTR1 have also been identified in patients with the disease (Hutter et al., 2014; Louvrier et al., 2018; Paganini et al., 2015; Piotrowski et al., 2012; Smith et al., 2015). Furthermore schwannomatosis predisposition genes are likely to exist since germline mutations in SMARCB1 or LZTR1 are not detectable in at least 50% of sporadic patients with schwannomatosis (Kehrer‐Sawatzki, Farschtschi, Mautner, & Cooper, 2017). However, the frequency of somatic mosaicism of SMARCB1 or LZTR1 mutations in patients with schwannomatosis is as yet unclear. Schwannomatosis is characterized by the occurrence of multiple schwannomas most commonly affecting the peripheral nerves (89%) and the spine (74%) (Merker, Esparza, Smith, Stemmer‐Rachamimov, & Plotkin, 2015). The incidence of schwannomatosis is still unclear, but most sources suggest an incidence comparable to that of NF2 (Antinheimo et al., 2000; Koontz et al., 2013). Schwannomatosis is most commonly diagnosed during adulthood. Merker et al. (2015) analyzed 87 patients with schwannomatosis and reported a median age at initial symptom presentation of 30 years (with a range between 8 and 59 years) and a median age at diagnosis of 40 years (with a range between 16 and 70 years). Schwannomas are benign nerve sheath tumors that generally do not transform toward malignancy (Carter et al., 2012). In contrast, the vast majority of AT/RTs are tumors of infancy and early childhood with a poor prognosis.
At least 30% of patients with RTs have germline SMARCB1 mutations which are mostly de novo (Bourdeaut et al., 2011; Eaton et al., 2011; Kordes et al., 2010). Approximately one‐third of patients with RTs and germline SMARCB1 mutations inherited the mutation from a parent who also harbors the mutation in his or her blood cells (Eaton et al., 2011). So far, only four families have been reported, with germline SMARCB1 mutation carriers being either affected by schwannomatosis or RT (Table 1) (Carter et al., 2012; Eaton et al., 2011; Sredni & Tomita, 2015; Swensen et al., 2009). The co‐occurrence of RT and schwannomatosis in the same patient has however not been reported as yet. AT/RT is often associated with a poor prognosis and early death but long‐term survival is possible by means of intensive multimodal treatment (Ammerlaan et al., 2008; Bartelheim et al., 2016; Kordes et al., 2014; Squire, Chan, & Marcus, 2007; Tekautz et al., 2005). Favorable prognostic factors are inheritance of the SMARCB1 germline mutation from a healthy carrier of the mutation, absence of synchronous tumors and older age at presentation (Bartelheim et al., 2016; Kordes et al., 2014).
Table 1.
Familial SMARCB1 mutations and family members affected by schwannomatosis or rhabdoid tumor (RT) as previously published
| Number of SMARCB1 mutation carriers and clinical details | SMARCB1 germline mutation | Reference |
|---|---|---|
| Four generations of the family were affected. Six family members were mutation carriers; three of them had schwannomatosis, two died from RT as infants and one mutation carrier was clinically unaffected. However, MRI investigations were not performed | Direct duplication of 2,631‐bp including parts of intron 5 and 6, as well as complete exon 6 causing a frameshift and protein truncation (p.Leu266fs) | Swensen et al. (2009) |
| The female mutation carrier was affected by RT. Her father and paternal grandmother were also mutation carriers and had schwannomatosis but not RT | c.472C>T, exon 4 | Eaton et al. (2011) |
| The mother carried the mutation and had schwannomatosis. One of her schwannomas transformed to an epithelioid malignant peripheral nerve sheath tumor. All three of her children harbored the mutation, two of whom had RT, whereas one was asymptomatic | Frameshift mutation c.245_246insAT, exon 3 | Carter et al. (2012) |
| Two sisters exhibited RT, while their father and paternal grandmother had schwannomatosis | Not investigated | Sredni & Tomita (2015) |
In this study, we report the follow‐up of a patient (II.4, Figure 1) with a germline SMARCB1 deletion of exons 8–9 who survived an AT/RT treated at the age of 2 years without tumor recurrence as determined by analysis of the patient at the age of 17 years (Kordes et al., 2014). She had inherited the germline SMARCB1 deletion from her healthy mother, proband I.5 (Figure 1). In this study, we analyzed the SMARCB1 deletion segregating in the family of patient II.4 in greater detail and report on the subsequent course of the disease in patient II.4 who was diagnosed with schwannomatosis at the age of 21 years. We used whole‐body and cranial magnetic resonance imaging (MRI) to investigate the tumor load as well as high‐resolution microstructural magnetic resonance neurography (MRN) to quantify peripheral nerve pathology at the fascicular level in patient II.4 as well as in her clinically unaffected mother I.5.
Figure 1.

Pedigree of the family investigated in this study. Individuals II.2 and II.4 had atypical teratoid/rhabdoid tumor (AT/RT). Patient II.4 underwent surgery at the age of two years to resect an AT/RT located in the posterior fossa. Patient II.2 died at the age of one year owing to complications at primary surgery of a SMARCB1‐negative AT/RT. The germline SMARCB1 deletion was detected in patient II.4, her mother and was also in the AT/RT of patient II.2. Consequently, proband I.2 also carries the germline SMARCB1 deletion of exons 8–9
2. PATIENTS AND METHODS
The patients provided written informed consent and the study was approved by the institutional review boards.
2.1. Whole‐body MRI examination
Patient II.4 and her mother I.5 were investigated by means of whole‐body MRI using a Magnetic Resonance scanner (Magnetom SKYRA; Magnetom VERIO and Magnetom TIM TRIO, Siemens Healthineers) at 3 Tesla with T1‐ and T2‐weighted sequences according to standard protocols in order to determine the occurrence of tumorous lesions >1 cm in diameter.
2.2. High‐resolution, microstructural peripheral nerve MRI
Large coverage, high‐resolution, microstructural peripheral nerve MRI also termed microstructural magnetic resonance neurography (MRN) was performed at 3‐Tesla in order to investigate the presence of small fascicular nerve lesions in the extremities of patient II.4 and her clinically unaffected mother I.5. Contiguous coverage of upper and lower extremities was achieved by applying an array of fat‐saturated T2‐weighted high‐resolution sequences as described previously (Bäumer et al., 2013). Quantitative analysis was performed by the evaluation of cross‐sectional images. The fascicular nerve lesions were categorized as non‐compressive fascicular microlesions (<2 mm in diameter), intermediate lesions (2–5 mm in diameter) and compressive macrolesions (≥5 mm in diameter).
2.3. Microarray analysis
CytoScan HD arrays (Affymetrix) were used to investigate blood‐derived genomic DNAs from patient II.4 and her mother I.5 following the manufacturer's protocols and instructions. The array consists of 2,696,550 probes that include 743,304 SNPs and 1,953,246 non‐polymorphic probes. The Affymetrix Chromosome Analysis Suite (ChAS) software was used to detect and evaluate copy‐number variants (CNVs).
2.4. Breakpoint‐spanning PCRs
Long‐range breakpoint‐spanning PCRs were performed using the Expand™ Long Range dNTPack (Merck) which contains a mixture of Taq polymerase and a thermostable DNA polymerase with proof reading activity optimized to efficiently amplify long PCR products. The primers used to amplify breakpoint‐spanning PCR products are listed in Supporting Information Table S1 (SMARCB1 GenBank accession number: NG_009303.1). In addition, we performed short breakpoint‐spanning PCRs with primers listed in Supporting Information Table S2 using the AmpliTaq Gold™ polymerase (Thermo Fisher Scientific) and DNA isolated from paraffin‐embedded AT/RT tissue from patient II.2 or DNA isolated from fresh‐frozen schwannoma tissue (T2763) of patient II.4. PCR products were sequenced using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and an ABI 3130 genetic analyzer (Applied Biosystems).
2.5. Mutation analysis
Blood‐derived genomic DNA of patient II.4 as well as genomic DNA isolated from fresh‐frozen schwannoma tissue of patient II.4 were analyzed by PCR and Sanger sequencing of the respective PCR products in order to identify mutations within the NF2 and LZTR1 genes (GenBank accession numbers: NG_009057.1; NG_034193.1). The primers used for these analyses are listed in Supporting Information Tables S3 and S4. The DNA samples were also investigated by MLPA in order to identify intragenic copy‐number variants within NF2 and LZTR1 by means of the SALSA MLPA probemix P455 LZTR1 (version A1) and the SALSA MLPA P044 NF2 probemix (version B3; MRC Holland, The Netherlands).
2.6. Analysis of the sequences flanking the SMARCB1 deletion breakpoints
Genomic regions immediately flanking the germline SMARCB1 deletion were investigated for the presence of sequences that may adopt alternative conformations such as non‐B DNA using the non‐B DB database (https://nonb-abcc.ncifcrf.gov/apps/nBMST/default/) (Cer et al., 2013).
3. RESULTS
3.1. Clinical findings
3.1.1. Patient II.4
Female patient II.4 underwent surgery at the age of 2 years to resect an AT/RT located in the posterior fossa. The tumor was diagnosed by immunohistochemistry as a SMARCB1/INI negative AT/RT (Kordes et al., 2014). After high‐dose chemotherapy and autologous stem cell rescue she survived and was without evidence of malignancy recurrence at the age of 17 years as confirmed by MRI (Kordes et al., 2014). In the present study, we report on the clinical follow‐up of patient II.4. At the age of 21 years, the patient exhibited mild aconuresis, paraparesis of both legs and progressive gait disturbances present since 4 months. MRI analysis of the patient revealed a large intraspinal schwannoma (T2763) with paraspinal extension located in the region of the thoracic vertebral bodies 8 and 9. The tumor encompassed 5.3 × 3.6 cm and caused profound compression of the spinal cord. It was removed by surgery when the patient was 21 years of age and her clinical symptoms, including aconuresis and paraparesis, improved considerably afterward. The tumor was classified by means of histopathological examination as a schwannoma (WHO grade I) with rhabdoid features (Figure 2).
Figure 2.
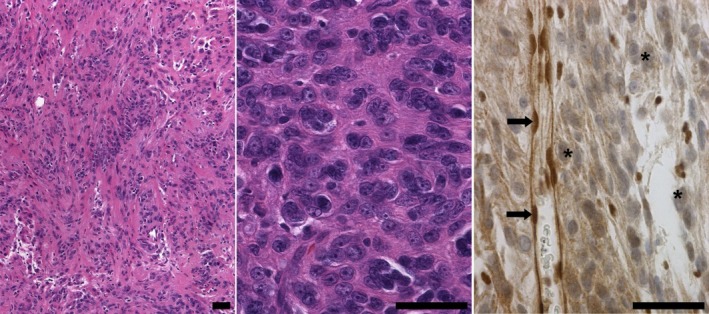
Histopathological investigation of the schwannoma (T2763) of patient II.4 revealed palisading of nuclei typical for schwannomas (left, H&E) and areas of high cellularity and rhabdoid features comprising round, sharply demarcated cells with eccentric nuclei and large nucleoli (middle, H&E). Immunohistochemical staining with the INI1/BAF47 antibody (BD Transduction Laboratories) indicated loss of nuclear SMARCB1 expression in most tumor cells (asterisk) but not in endothelial cells (arrow). Scale bars: 50 μm
Whole‐body MRI of patient II.4 exhibited additional intraabdominal, intrathoracic and spinal T2‐hyperintensive nodules of 2–3 cm in diameter suggestive of schwannomas. The nodules were located paravertebrally along the thoracic and the lumbar spine, within the intercostal and pelvic regions, the left pectoral girdle and left upper arm. Multiple T2‐hyperintense neurogenic nodules up to 3 cm in diameter were observed in both lower legs located intramuscular and along the major nerve trunks.
By means of high‐resolution, microstructural peripheral nerve MRI of the extremities, multiple fascicular microlesions (<2 mm), intermediate lesions (2–5 mm) and macrolesions (≥5 mm) were detected (Table 2, Figure 3). Cranial MRI performed at the age of 21 years did not indicate RT relapse or other brain tumors.
Table 2.
Number of fascicular peripheral nerve lesions observed by high‐resolution microstructural nerve MRI in patient II.4 and her mother I.5
| Individual investigated | Extremity | Affected nerve | Number of fascicular | ||
|---|---|---|---|---|---|
| IMicrolesions (2–5 mm) | Intermediate lesions (2–5 mm) | Macrolesions (≥5 mm) | |||
| Patient II.4 | Proximal left thigh | Sciatic | 6 | 5 | 6 |
| Distal left thigh | Tibial | 2 | – | 5 | |
| Distal left thigh | Peroneal | 3 | 1 | – | |
| Left lower leg | Tibial | 5 | 5 | 1 | |
| Proximal right thigh | Sciatic | 5 | 1 | 1 | |
| Distal right thigh | Tibial | 4 | – | 1 | |
| Distal right thigh | Peroneal | 1 | – | – | |
| Right lower leg | Tibial | 4 | 3 | 1 | |
| Left upper arm | Median | 3 | – | – | |
| Left upper arm | Ulnar | 1 | – | – | |
| Left upper arm | Radial | 4 | 1 | 1 | |
| Right upper arm | Median | 7 | – | – | |
| Right upper arm | Ulnar | 5 | – | – | |
| Right upper arm | Radial | – | – | ||
| Proband I.5 | Proximal left thigh | Sciatic | 2 | – | – |
| Distal left thigh | Tibial | 2 | – | – | |
| Distal left thigh | Peroneal | 2 | – | – | |
| Left lower leg | Tibial | 3 | 2 | – | |
| Proximal right thigh | Sciatic | 1 | – | – | |
| Distal right thigh | Tibial | 1 | – | – | |
| Distal right thigh | Peroneal | 1 | – | – | |
| Right lower leg | Tibial | 2 | – | – | |
| Left upper arm | Median | – | – | – | |
| Left upper arm | Ulnar | 2 | – | – | |
| Left upper arm | Radial | 1 | – | – | |
| Right upper arm | Median | 1 | – | – | |
| Right upper arm | Ulnar | 4 | – | – | |
| Right upper arm | Radial | – | – | – | |
–, none detected.
Figure 3.
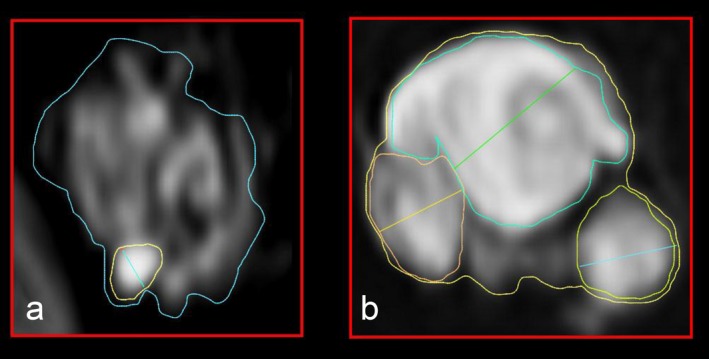
Microstructural images of fascicular T2 lesions of the sciatic nerve. (a) In the clincially unaffected mother (proband I.5), microstructural magnetic resonance neurography (MRN) revealed a non‐compressive fascicular microlesion (<2 mm in diameter). (b) Intermediate fascicular lesions (2–5 mm) and macrolesions (>5 mm) were detected in patient II.4
3.1.2. Mother of patient II.4
The mother of patient II.4 (proband I.5, Figure 1) did not exhibit any clinical symptoms of schwannomatosis at the age of 56 years. Whole‐body and cranial MRI investigation did not indicate any tumorous T2‐hyperintense lesions. We also performed high‐resolution, microstructural peripheral nerve MRI to detect small fascicular abnormalities in the nerves which are not detectable by conventional MRI. These investigations did not indicate the presence of compressive macrolesions (≥5 mm) along the peripheral nerves of her extremities. However, we detected non‐compressive fascicular microlesions (<2 mm) of the peripheral nerves of all four extremities and two intermediate lesions (2–5 mm) in the left lower leg indicative of peripheral nerve pathology in proband I.5 (Table 2). Taken together, 11 microlesions were detected in her left leg and five microlesions in her right leg.
In contrast, microlesions are very rare in healthy control individuals. Previous analyses indicated microlesions in only three of 25 healthy control individuals investigated (Farschtschi et al., 2016). Furthermore, the three control individuals exhibited microlesions only in the lower extremities. On average, 3.5 microlesions were detected per extremity in these three controls (Farschtschi et al., 2016). This number of microlesions is considerably lower than the number of microlesions detected in proband I.5. Remarkably, the 25 healthy controls analyzed previously by means of MRN did not exhibit microlesions in the upper extremities (Farschtschi et al., 2016). In contrast to this, proband I.5 exhibited three microlesions in the left upper arm and five microlesions in the right upper arm.
3.2. Molecular findings
3.2.1. Identification of the SMARCB1 germline deletion breakpoints
The SMARCB1 deletion detected in patient II.4 and her mother I.5 includes exons 8 and 9 as previously determined by MLPA (Kordes et al., 2014). In the present study, we characterized the deletion in greater detail. We first performed CytoScan HD array analysis using blood‐derived genomic DNA of patient II.4 in order to narrow down the deletion breakpoint regions (Figure 4). Subsequently, we performed breakpoint‐spanning PCRs with primers US1_for10 and rev8 (Figure 4c, Supporting Information Figure S1). Sequence analysis of the 1,138‐bp breakpoint‐spanning PCR product identified the deletion junction indicating that the deletion encompasses 6,388‐bp. The deletion includes parts of SMARCB1 intron 7, exons 8–9 and 3,302‐bp located telomeric to exon 9 including the 3′ UTR of SMARCB1 as well as the 3′‐end of the DERL3 gene which flanks SMARCB1 in a telomeric direction. A breakpoint‐spanning PCR product was also obtained using primers for9 and rev8 (Supporting Information Figure S2). Both breakpoint‐spanning products (for9/rev8 and US1_for10/rev8) were also amplified from genomic DNA of the mother of patient II.4, proband I.5. Sequence analyses of these breakpoint‐spanning PCR products did not indicate any sequence differences between patient II.4 and her mother within 3,756‐bp flanking the deletion breakpoints.
Figure 4.

Schema indicating the results of the MLPA and the CytoScan HD array analyses performed to narrow down the breakpoints of the SMARCB1 germline deletion identified in the family studied here. According to these results, breakpoint‐spanning PCRs were performed to identify the breakpoints at highest resolution. (a) The genomic positions of the MLPA probes are indicated as well as the relative locations of the SMARCB1 exons. (b) The relative positions of the array probes as well as their genomic locations are presented in this part of the schema. MLPA and array probes indicating the loss of one copy in the patient are labeled as “del” whereas those indicating diploid copy numbers are labeled as “2n”. (c) Breakpoint‐spanning PCRs performed with primers represented as green arrows revealed that the deletion region encompasses 6,388‐bp and involves parts of SMARCB1 intron 7, complete exon 8, intron 8, exon 9 and 3,302‐bp located telomeric to exon 9. The genomic positions indicated are according to the human genome reference sequence (hg19)
3.2.2. Mechanism causing the germline SMARCB1 deletion
Neither the proximal deletion breakpoint in SMARCB1 intron 7 nor the distal deletion breakpoint located 3′ to the SMARCB1 gene were situated within high‐copy repeat sequences such as ALU‐ or L1‐elements. Furthermore, extended sequence homology between proximal and distal breakpoint regions was not observed. We conclude that the deletion has not been mediated by a mechanism dependent upon extended sequence similarity at the breakpoints. A 13‐bp insertion was identified at the deletion junction, which may have been templated by a sequence located 5.5‐kb telomeric to the distal deletion breakpoint (Figure 5). Hence, a template‐switching mechanism during replication such as fork stalling and template switching (FoSTeS), microhomology‐mediated break‐induced replication (MMBIR) or serial replication slippage has most likely caused the SMARCB1 deletion. Indeed, DNA replication‐based mechanisms are frequently causing disease‐associated multi‐exon copy number variants as well as polymorphic structural variants in the human genome (Abyzov et al., 2015; Ankala et al., 2012; Hsiao et al., 2015; Zhang et al., 2009, 2010). We identified a short inverted repeat located close to the proximal deletion breakpoint which may have templated the formation of a cruciform structure responsible for replication stalling (Figure 5). Short inverted repeats are known to induce DNA instability mediated by replication‐related mechanisms (Lu et al., 2015).
Figure 5.
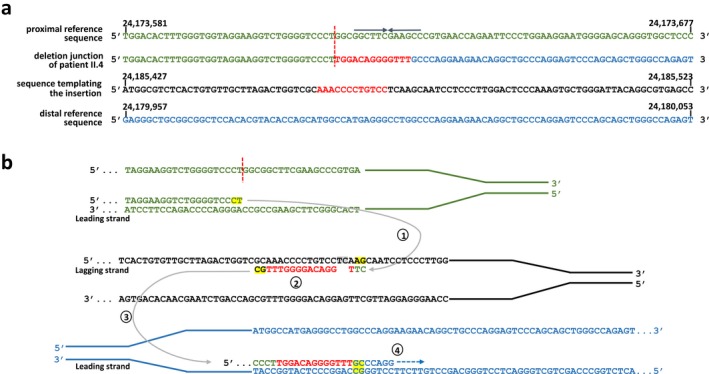
The germline SMARCB1 deletion investigated in this study, as well as the insertion of 13‐bp (red) at the deletion breakpoints, most likely resulted from replication‐associated template switching. (a) Alignment of the deletion breakpoint‐flanking sequences of patient II.4 against the reference sequence of the human genome (hg19). Sequences located at the proximal (centromeric) deletion breakpoint are indicated in green, while sequences at the distal (telomeric) breakpoint are given in blue. The vertical red line highlights the position of the proximal deletion breakpoint. The 13‐bp insertion (red) identified at the deletion junction exhibits homology to a sequence located 5.5‐kb telomeric to the distal breakpoint region. (b) Model proposed to explain the origin of the deletion‐associated insertion. In the proximal breakpoint‐flanking region, DNA synthesis at the leading strand is interrupted but appears to have resumed, after an interstrand template switch, to a replication fork located 5.5‐kb telomeric to the distal breakpoint‐flanking region (step 1). Subsequently, the 13‐bp indicated in red are newly synthesized and included in the nascent DNA strand at the replication fork (step 2). Single‐nucleotide changes due to DNA polymerase errors are highlighted in gray. Subsequently, another template switch occurs on the leading strand (step 3) upon which replication is continued (step 4). The nucleotides exhibiting microhomology at sites of template switching are marked in yellow. An inverted repeat of 6‐bp marked by arrows was identified close to the proximal deletion breakpoint which may have caused a cruciform structure responsible for replication stalling
3.2.3. Segregation of the germline SMARCB1 deletion in other family members
A maternal niece (patient II.2, Figure 1) had died at the age of one year due to complications at primary surgery of a SMARCB1‐negative AT/RT, but genetic analysis was not performed (Kordes et al., 2014). In the present study, we analyzed DNA isolated from paraffin‐embedded AT/RT tissue of patient II.2 by means of breakpoint‐spanning PCR and confirmed the presence of the germline SMARCB1 deletion (Supporting Information Figure S3). This finding indicates the segregation of the SMARCB1 germline deletion in the family and that proband I.2 must be an obligate carrier of the deletion (Figure 1). Analysis of proband I.2 was however not possible since she declined clinical or genetic investigations.
3.2.4. Genetic analysis of a schwannoma from patient II.4
Germline mutations within LZTR1 or NF2 were neither detected in blood‐derived DNA of patient II.4 nor in genomic DNA isolated from schwannoma T2763 of patient II.4. MLPA analysis of schwannoma‐derived DNA indicated the somatic loss of one copy of LZTR1 and NF2 which was not detected in the patient's blood. Our findings imply that a large tumor‐specific deletion had occurred on the chromosome 22 not harboring the germline SMARCB1 deletion of exons 8–9. This somatic large deletion includes one allele of LZTR1 and NF2 as well as the wild‐type allele of SMARCB1 (Supporting Information Figure S5). We were able to conclude this from the observation that the SMARCB1 exon 8–9 deletion breakpoint‐spanning PCR fragment was strongly amplified from DNA of schwannoma T2763 but not SMARCB1 exons 8 and 9. Hence, most of the tumor cells exhibit biallelic loss of SMARCB1 exons 8 and 9.
4. DISCUSSION
Atypical teratoid/RTs are brain tumors most commonly affecting children younger than 3 years of age (Hilden et al., 2004; Rorke, Packer, & Biegel, 1996). AT/RTs are predominantly infratentorial malignancies, most of them located within the posterior fossa. However, AT/RTs may also be located in supratentorial brain regions (Dho et al., 2015). AT/RT caused by the complete loss of function of SMARCB1 is often associated with a poor prognosis and early death but long‐term survival is possible in some cases (Ammerlaan et al., 2008; Bartelheim et al., 2016; Kordes et al., 2014; Squire et al., 2007; Tekautz et al., 2005). In this study, we report the follow‐up of female patient II.4 with a germline SMARCB1 deletion who survived an AT/RT surgically removed at the age of 2 years (Kordes et al., 2014). Patient II.4 is the first case documented to be in complete continuous remission for a AT/RT but diagnosed with schwannomatosis during adulthood. Our findings indicate that schwannomatosis‐associated tumorigenesis and related clinical symptoms need to be taken into account in patients surviving RTs and harboring germline SMARCB1 mutations.
Reinvestigation of the SMARCB1 deletion previously identified in patient II.4 indicated that the deletion encompasses 6.4‐kb and parts of SMARCB1 intron 7, exons 8–9. Analysis of the breakpoint sequences implies that the deletion has been mediated by a mutational mechanism involving replication‐associated template‐switching (Figure 5). The mother of patient II.4 also carries the SMARCB1 deletion. Sequence analysis of regions flanking the deletion breakpoints did not indicate any sequence differences between the mother and her daughter. By means of deletion junction‐spanning PCR, we were able to demonstrate that the niece of proband I.5 and the sister of proband I.5 (individuals II.2 and I.2, Figure 1) are both carriers of the germline SMARCB1 deletion.
Probands I.2 and I.5 were not affected by AT/RT as children even although they are carriers of the germline SMARCB1 deletion. The risk of RT development seems to be time‐dependent in the sense that a specific developmental time window exists during which the tumor progenitor cell is vulnerable to complete SMARCB1 protein loss initiating RT growth. If this sensitive period is completed without biallelic SMARCB1 inactivation, the growth of an AT/RT is not initiated. This hypothesis has been supported by tissue and developmental stage‐specific conditional knockout mouse models which showed that biallelic Smarcb1 loss in early neural crest cells (embryonic day 9.5) initiates RT growth in the cranial nerves and meninges. After embryonic day 12.5, inactivation of Smarcb1 in peripheral Schwann cells or in brain glial cells does not initiate RTs (Vitte, Gao, Coppola, Judkins, & Giovannini, 2017). Hence, an early spatio‐temporal window must exist during which Smarcb1 loss results in malignant transformation of specific neural crest cells. The absence of AT/RT in SMARCB1 mutation carriers is thus explained by retention of the SMARCB1 wild‐type allele and its activity in neural crest cells during early stages of embryonic development.
It is however surprising that the mother of patient II.4 (proband I.5) does not show clinical symptoms of schwannomatosis even though she also carries the germline SMARCB1 deletion. Whole‐body MRI and cranial MRI investigations of proband I.2 did not indicate any tumorous T2‐hyperintense lesions. However, high‐resolution, microstructural peripheral nerve MRI, also termed microstructural magnetic resonance neurography (MRN) revealed non‐compressive fascicular microlesions (<2 mm) and intermediate lesions (2–5 mm) along the peripheral nerves of the extremities in proband I.5 (Table 2; Figure 3). Intrafascicular microlesions have previously been shown to represent frequent features of peripheral nerve pathology in patients with schwannomatosis (Farschtschi et al., 2016). It is however unclear as yet whether these microlesions are predictive of future symptomatic tumors. Our results nevertheless imply that microstructural MRN is a method well suited to detecting subclinical peripheral nerve pathology in clinically unaffected SMARCB1 mutation carriers such as proband I.5. According to the recommended surveillance protocols for patients with AT/RT and truncating germline SMARCB1 mutations, whole‐body MRI should be considered up to the age of 5 years (Foulkes et al., 2017). Our findings suggest that long‐term survivors of AT/RT and truncating germline SMARCB1 mutations should receive regular surveillance for schwannomatosis‐associated clinical symptoms.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGMENT
US is supported by the Fördergemeinschaft Kinderkrebs‐Zentrum Hamburg.
Kehrer‐Sawatzki H, Kordes U, Seiffert S, et al. Co‐occurrence of schwannomatosis and rhabdoid tumor predisposition syndrome 1. Mol Genet Genomic Med. 2018;6:627–637. 10.1002/mgg3.412
REFERENCES
- Abyzov, A. , Li, S. , Kim, D. R. , Mohiyuddin, M. , Stütz, A. M. , Parrish, N. F. , … Gerstein, M. B. (2015). Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nature Communications, 6, 7256 10.1038/ncomms8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerlaan, A. C. , Ararou, A. , Houben, M. P. , Baas, F. , Tijssen, C. C. , Teepen, J. L. , … Hulsebos, T. J. (2008). Long‐term survival and transmission of INI1‐mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. British Journal of Cancer, 98, 474–479. 10.1038/sj.bjc.6604156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankala, A. , Kohn, J. N. , Hegde, A. , Meka, A. , Ephrem, C. L. , Askree, S. H. , … Hegde, M. R. (2012). Aberrant firing of replication origins potentially explains intragenic nonrecurrent rearrangements within genes, including the human DMD gene. Genome Research, 22, 25–34. 10.1101/gr.123463.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinheimo, J. , Sankila, R. , Carpén, O. , Pukkala, E. , Sainio, M. , & Jääskeläinen, J. (2000). Population‐based analysis of sporadic and type 2 neurofibromatosis‐associated meningiomas and schwannomas. Neurology, 54, 71–76. 10.1212/WNL.54.1.71 [DOI] [PubMed] [Google Scholar]
- Bartelheim, K. , Nemes, K. , Seeringer, A. , Kerl, K. , Buechner, J. , Boos, J. , … Frühwald, M. C. (2016). Improved 6‐year overall survival in AT/RT ‐ results of the registry study Rhabdoid 2007. Cancer Medicine, 5, 1765–1775. 10.1002/cam4.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer, P. , Mautner, V. F. , Bäumer, T. , Schuhmann, M. U. , Tatagiba, M. , Heiland, S. , … Pham, M. (2013). Accumulation of non‐compressive fascicular lesions underlies NF2 polyneuropathy. Journal of Neurology, 260, 38–46. 10.1007/s00415-012-6581-8 [DOI] [PubMed] [Google Scholar]
- Biegel, J. A. , Zhou, J. Y. , Rorke, L. B. , Stenstrom, C. , Wainwright, L. M. , & Fogelgren, B. (1999). Germ‐line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Research, 59, 74–79. [PubMed] [Google Scholar]
- Bourdeaut, F. , Lequin, D. , Brugières, L. , Reynaud, S. , Dufour, C. , Doz, F. , … Delattre, O. (2011). Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clinical Cancer Research, 17, 31–38. 10.1158/1078-0432.CCR-10-1795 [DOI] [PubMed] [Google Scholar]
- Boyd, C. , Smith, M. J. , Kluwe, L. , Balogh, A. , MacCollin, M. , & Plotkin, S. R. (2008). Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clinical Genetics, 74, 358–366. 10.1111/j.1399-0004.2008.01060.x [DOI] [PubMed] [Google Scholar]
- Bruggers, C. S. , Bleyl, S. B. , Pysher, T. , Barnette, P. , Afify, Z. , Walker, M. , & Biegel, J. A. (2011). Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatric Blood and Cancer, 56, 1026–1031. 10.1002/pbc.22757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, J. M. , O'Hara, C. , Dundas, G. , Gilchrist, D. , Collins, M. S. , Eaton, K. , … Folpe, A. L. (2012). Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma‐like” schwannomatosis and a novel germline SMARCB1 mutation. American Journal of Surgical Pathology, 36, 154–160. 10.1097/PAS.0b013e3182380802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer, R. Z. , Donohue, D. E. , Mudunuri, U. S. , Temiz, N. A. , Loss, M. A. , Starner, N. J. , … Stephens, R. M. (2013). Non‐B DB v2.0: A database of predicted non‐B DNA‐forming motifs and its associated tools. Nucleic Acids Research, 41, D94–D100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dho, Y. S. , Kim, S. K. , Cheon, J. E. , Park, S. H. , Wang, K. C. , Lee, J. Y. , & Phi, J. H. (2015). Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nervous System, 31, 1305–1311. 10.1007/s00381-015-2739-x [DOI] [PubMed] [Google Scholar]
- Eaton, K. W. , Tooke, L. S. , Wainwright, L. M. , Judkins, A. R. , & Biegel, J. A. (2011). Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatric Blood and Cancer, 56, 7–15. 10.1002/pbc.22831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farschtschi, S. , Mautner, V. F. , Pham, M. , Nguyen, R. , Kehrer‐Sawatzki, H. , Hutter, S. , … Bäumer, P. (2016). Multifocal nerve lesions and LZTR1 germline mutations in segmental schwannomatosis. Annals of Neurology, 80, 625–628. 10.1002/ana.24753 [DOI] [PubMed] [Google Scholar]
- Foulkes, W. D. , Kamihara, J. , Evans, D. G. R. , Brugières, L. , Bourdeaut, F. , Molenaar, J. J. , … Diller, L. (2017). Cancer surveillance in Gorlin syndrome and Rhabdoid tumor predisposition syndrome. Clinical Cancer Research, 23, e62–e67. 10.1158/1078-0432.CCR-17-0595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante, L. , Paganini, I. , Frontali, M. , Ciabattoni, S. , Sangiuolo, F. C. , & Papi, L. (2016). Rhabdoid tumor predisposition syndrome caused by SMARCB1 constitutional deletion: Prenatal detection of new case of recurrence in siblings due to gonadal mosaicism. Familial Cancer, 15, 123–126. 10.1007/s10689-015-9836-6 [DOI] [PubMed] [Google Scholar]
- Hadfield, K. D. , Newman, W. G. , Bowers, N. L. , Wallace, A. , Bolger, C. , Colley, A. , … Evans, D. G. (2008). Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. Journal of Medical Genetics, 45, 332–339. 10.1136/jmg.2007.056499 [DOI] [PubMed] [Google Scholar]
- Hilden, J. M. , Meerbaum, S. , Burger, P. , Finlay, J. , Janss, A. , Scheithauer, B. W. , … Biegel, J. A. (2004). Central nervous system atypical teratoid/rhabdoid tumor: Results of therapy in children enrolled in a registry. Journal of Clinical Oncology, 22, 2877–2884. 10.1200/JCO.2004.07.073 [DOI] [PubMed] [Google Scholar]
- Hsiao, M. C. , Piotrowski, A. , Callens, T. , Fu, C. , Wimmer, K. , Claes, K. B. , & Messiaen, L. (2015). Decoding NF1 intragenic copy‐number variations. American Journal of Human Genetics, 97, 238–249. 10.1016/j.ajhg.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebos, T. J. , Plomp, A. S. , Wolterman, R. A. , Robanus‐Maandag, E. C. , Baas, F. , & Wesseling, P. (2007). Germline mutation of INI1/SMARCB1 in familial schwannomatosis. American Journal of Human Genetics, 80, 805–810. 10.1086/513207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter, S. , Piro, R. M. , Reuss, D. E. , Hovestadt, V. , Sahm, F. , Farschtschi, S. , … Mautner, V. F. (2014). Whole exome sequencing reveals that the majority of schwannomatosis cases remain unexplained after excluding SMARCB1 and LZTR1 germline variants. Acta Neuropathologica, 128, 449–452. 10.1007/s00401-014-1311-1 [DOI] [PubMed] [Google Scholar]
- Janson, K. , Nedzi, L. A. , David, O. , Schorin, M. , Walsh, J. W. , Bhattacharjee, M. , … Biegel, J. A. (2006). Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatric Blood and Cancer, 47, 279–284. 10.1002/(ISSN)1545-5017 [DOI] [PubMed] [Google Scholar]
- Kehrer‐Sawatzki, H. , Farschtschi, S. , Mautner, V. F. , & Cooper, D. N. (2017). The molecular pathogenesis of schwannomatosis, a paradigm for the co‐involvement of multiple tumour suppressor genes in tumorigenesis. Human Genetics, 136, 129–148. 10.1007/s00439-016-1753-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz, N. A. , Wiens, A. L. , Agarwal, A. , Hingtgen, C. M. , Emerson, R. E. , & Mosier, K. M. (2013). Schwannomatosis: The overlooked neurofibromatosis? American Journal of Roentgenology, 200, W646–W653. 10.2214/AJR.12.8577 [DOI] [PubMed] [Google Scholar]
- Kordes, U. , Bartelheim, K. , Modena, P. , Massimino, M. , Biassoni, V. , Reinhard, H. , … Frühwald, M. C. (2014). Favorable outcome of patients affected by rhabdoid tumors due to rhabdoid tumor predisposition syndrome (RTPS). Pediatric Blood and Cancer, 61, 919–921. 10.1002/pbc.24793 [DOI] [PubMed] [Google Scholar]
- Kordes, U. , Gesk, S. , Frühwald, M. C. , Graf, N. , Leuschner, I. , Hasselblatt, M. , … Schneppenheim, R. (2010). Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer, 49, 176–181. 10.1002/gcc.20729 [DOI] [PubMed] [Google Scholar]
- Louvrier, C. , Pasmant, E. , Briand‐Suleau, A. , Cohen, J. , Nitschké, P. , Nectoux, J. , … Parfait, B. (2018). Targeted next‐generation sequencing for differential diagnosis of neurofibromatosis type 2, schwannomatosis, and meningiomatosis. Neuro‐Oncology. 2018 Feb 2. 10.1093/neuonc/noy009. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S. , Wang, G. , Bacolla, A. , Zhao, J. , Spitser, S. , & Vasquez, K. M. (2015). Short inverted repeats are hotspots for genetic instability: Relevance to cancer genomes. Cell Reports, 10, 1674–1680. 10.1016/j.celrep.2015.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker, V. L. , Esparza, S. , Smith, M. J. , Stemmer‐Rachamimov, A. , & Plotkin, S. R. (2015). Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist, 17, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganini, I. , Chang, V. Y. , Capone, G. L. , Vitte, J. , Benelli, M. , Barbetti, L. , … Papi, L. (2015). Expanding the mutational spectrum of LZTR1 in schwannomatosis. European Journal of Human Genetics, 23, 963–968. 10.1038/ejhg.2014.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski, A. , Xie, J. , Liu, Y. F. , Poplawski, A. B. , Gomes, A. R. , Madanecki, P. , … Messiaen, L. M. (2012). Germline loss‐of‐function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nature Genetics, 46, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorke, L. B. , Packer, R. J. , & Biegel, J. A. (1996). Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: Definition of an entity. Journal of Neurosurgery, 85, 56–65. 10.3171/jns.1996.85.1.0056 [DOI] [PubMed] [Google Scholar]
- Rousseau, G. , Noguchi, T. , Bourdon, V. , Sobol, H. , & Olschwang, S. (2011). SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurology, 11, 9 10.1186/1471-2377-11-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestini, R. , Bacci, C. , Provenzano, A. , Genuardi, M. , & Papi, L. (2008). Evidence of a four‐hit mechanism involving SMARCB1 and NF2 in schwannomatosis associated schwannomas. Human Mutation, 29, 227–231. 10.1002/humu.20679 [DOI] [PubMed] [Google Scholar]
- Sévenet, N. , Sheridan, E. , Amram, D. , Schneider, P. , Handgretinger, R. , & Delattre, O. (1999). Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. American Journal of Human Genetics, 65, 1342–1348. 10.1086/302639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. J. , Isidor, B. , Beetz, C. , Williams, S. G. , Bhaskar, S. S. , Richer, W. , … Evans, D. G. (2015). Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology, 84, 141–147. 10.1212/WNL.0000000000001129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. J. , Wallace, A. J. , Bowers, N. L. , Eaton, H. , & Evans, D. G. (2014). SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genetics, 207, 373–378. 10.1016/j.cancergen.2014.04.001 [DOI] [PubMed] [Google Scholar]
- Smith, M. J. , Wallace, A. J. , Bowers, N. L. , Rustad, C. F. , Woods, C. G. , Leschziner, G. D. , … Evans, D. G. (2012). Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics, 13, 141–145. 10.1007/s10048-012-0319-8 [DOI] [PubMed] [Google Scholar]
- Squire, S. E. , Chan, M. D. , & Marcus, K. J. (2007). Atypical teratoid/rhabdoid tumor: The controversy behind radiation therapy. Journal of Neuro‐oncology, 81, 97–111. [DOI] [PubMed] [Google Scholar]
- Sredni, S. T. , & Tomita, T. (2015). Rhabdoid tumor predisposition syndrome. Pediatric and Developmental Pathology, 18, 49–58. 10.2350/14-07-1531-MISC.1 [DOI] [PubMed] [Google Scholar]
- Swensen, J. J. , Keyser, J. , Coffin, C. M. , Biegel, J. A. , Viskochil, D. H. , & Williams, M. S. (2009). Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1 . Journal of Medical Genetics, 46, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekautz, T. M. , Fuller, C. E. , Blaney, S. , Fouladi, M. , Broniscer, A. , Merchant, T. E. , … Gajjar, A. (2005). Atypical teratoid/rhabdoid tumors (ATRT): Improved survival in children 3 years of age and older with radiation therapy and high‐dose alkylator‐based chemotherapy. Journal of Clinical Oncology, 23, 1491–1499. 10.1200/JCO.2005.05.187 [DOI] [PubMed] [Google Scholar]
- Versteege, I. , Sévenet, N. , Lange, J. , Rousseau‐Merck, M. F. , Ambros, P. , Handgretinger, R. , … Delattre, O. (1998). Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature, 394, 203–206. 10.1038/28212 [DOI] [PubMed] [Google Scholar]
- Vitte, J. , Gao, F. , Coppola, G. , Judkins, A. R. , & Giovannini, M. (2017). Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nature Communications, 8, 300 10.1038/s41467-017-00346-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Khajavi, M. , Connolly, A. M. , Towne, C. F. , Batish, S. D. , & Lupski, J. R. (2009). The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nature Genetics, 41, 849–853. 10.1038/ng.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Seeman, P. , Liu, P. , Weterman, M. A. , Gonzaga‐Jauregui, C. , Towne, C. F. , … Lupski, J. R. (2010). Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: Rare CNVs as a cause for missing heritability. American Journal of Human Genetics, 86, 892–903. 10.1016/j.ajhg.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials