

Abstract
Background
Autosomal recessive intellectual disability (ARID) is vastly heterogeneous. Truncating mutations of TRAPPC9 were reported in 8 ARID families. Autosomal recessive primary microcephaly (MCPH) represents another subgroup of ARID, itself very heterogeneous, where the size of the brain is very small since birth. MCPH1 plays a role at the centrosome via a BRCT1 domain, and in DNA Damage Repair (DDR) via BRCT2 and BRCT3, and it is not clear which of these two mechanisms causes MCPH in man.
Methods
We studied the phenotype and sequenced the exome in two siblings with MCPH and their unaffected sister.
Results
Homozygous mutations of TRAPPC9 (p.Leu178Pro) and of MCPH1 (p.Arg741X) were found in both affected siblings. Brain MRI showed anomalies previously associated with TRAPPC9 defects, supporting the implication of TRAPPC9 in the phenotype. Importantly, the asymptomatic sister with normal head size was homozygous for the MCPH1 truncating mutation and heterozygous for the TRAPPC9 mutation.
Conclusion
The affected siblings represent the first ARID cases with a TRAPPC9 missense mutation and with microcephaly of prenatal onset of. Furthermore, their unaffected sister represents strong evidence that the lack of MCPH1 BRCT3 domain does not cause MCPH in man, supporting a bifunctional model of MCPH1 where the centrosomal function is involved in brain volumic development and not the DDR function.
Keywords: centrosome, consanguinity, DNA damage repair, exome, microcephaly
1. INTRODUCTION
Autosomal recessive intellectual deficiencies (ARID) are a very heterogeneous subgroup of intellectual deficiencies (Najmabadi et al., 2011). TRAPPC9 mutations and MCPH1 mutations each cause a type of ARID.
TRAPPC9 deficiency was identified as a cause of ARID in eight families worldwide (Khattak & Mir, 2014). Additional features are microcephaly of postnatal onset, speech delay, and abnormalities of the corpus callosum, cerebellum, and white matter (Kakar et al., 2012). TRAPPC9 is part of the Trafficking Protein Particle (TRAPP) Complex II, and is involved in intra‐Golgi and endosomal trafficking via interactions with TRAPPC2 and TRAPPC10 (Zong et al., 2012). TRAPPC9 was also shown to play a role in nuclear factor kappaB (NF‐kB) activation (Philippe et al., 2009).
Contrary to microcephaly of postnatal onset (i.e., secondary microcephaly), primary microcephaly results from insufficient production of mature neurons during neurogenesis, and presents with a congenitally small brain (Passemard, Kaindl, & Verloes, 2013). MicroCephaly Primary Hereditary (MCPH) refer to a group of nonsyndromic, autosomal recessive primary microcephalies. MCPH is largely heterogeneous, with at least 16 different causal genes.
Microcephalin (MCPH1) is one of the first genes associated with MCPH. MCPH1 is expressed at the centrosome and plays roles in mitotic spindle alignment and mitotic cycle checkpoints. MCPH1 deficiency uncouples mitosis and the centrosomal cycle, causing premature mitotic entry, with premature neurogenic production and depletion of neural progenitors (Pulvers, Journiac, Arai, & Nardelli, 2015). Interestingly, MCPH1 also plays a role in DNA damage repair (DDR). MCPH1 contains three BRCT domains that were predicted to be crucial for MCPH1 function (Pulvers et al., 2015). N‐terminal BRCT1 is necessary for centrosomal localization in chicken cells, whereas C‐terminal BRCT2 and BRCT3 are required for ionizing radiation‐induced nuclear foci (IRIF) formation (Pulvers et al., 2015). BRCT2 and BRCT3 bind E2F1 to form a complex that transactivates BRCA1 and CHK1 (Yang, Lin, & Lin, 2008). BRCT2 and BRCT3 also interact with Cdc27, a subunit of the anaphase‐promoting complex (Singh, Wiltshire, Thompson, Mer, & Couch, 2012) and H2AX (Singh et al., 2012). Other DDR genes are also associated with primary microcephaly, for example, BLM or LIG4 (Maciejczyk et al. (2016), Berg et al. (2011)), so the exact mechanism by which MCPH1 mutations cause microcephaly, either centrosomal or via DDR, is not clear.
We here report on two siblings with severe ID, microcephaly and hypoplasia of the corpus callosum, a homozygous TRAPPC9 mutation, and a homozygous MCPH1 truncation of BRCT3, and their normal sister with the homozygous MCPH1 mutation only.
2. MATERIAL AND METHODS
The two affected siblings, a boy and a girl, and their nonaffected sister, were born to Moroccan parents originating from the same small village, and likely consanguineous.
The affected son was referred at 16 years of age for psychomotor delay and severe ID, microcephaly and hyperkinesia. The HC was 52 cm (−2.8SD), height was 161 cm and weight 61.6 kg. He had sphincter control and could express himself with a few words. Brain MRI showed hypoplasia of the corpus callosum and mild colpocephaly. Head circumference (HC) at birth was not known.
A younger sister was referred at 10 years of age for psychomotor delay and severe ID, microcephaly, hyperkinesia, and epilepsy. HC was <P3, height at P10, and weight at P25. She was not toilet‐trained, not able to speak, avoided ocular contact, and walked with an enlarged basis of sustentation. Her head was reportedly small at birth. At 13 years, her HC of 48.5 cm (−4SD). MRI showed atrophy of the corpus callosum and cerebellum, and some abnormal signals in the supratentorial white matter. Fundi were normal with intermittent strabismus. A metabolic work‐up, standard karyotype, and FISH analysis of all subtelomeric regions were normal.
HC and intellect of the parents and nonaffected sister were strictly normal: 59 cm in father, 57 cm in mother, and 57 cm in nonaffected sister whose height was 160 cm.
SNP genotyping revealed a large homozygous region of 14 Mb at 8q24 encompassing TRAPPC9, and a small homozygous region of 0.4 Mb in 8p23 encompassing MCPH1.
For whole‐exome sequencing, the two probands’ genomic DNAs were sheared and exonic sequences enriched using Roche SeqCap EZ Human Exome v3.0 (64 Mb) DNA capture. Sequencing was performed in the affected boy by AROS applied biotechnology (http://arosab.com/), and in the affected girl on a HiSeq1500 Illumina sequencer at the BRIGHTcore BRussels Interuniversity Genomics High Throughput core (http://www.ngs.brightcore.brussels/). Raw sequences were aligned to the reference genome GRCh37 using BWA algorithm version 0.7.10 (Li and Durbin (2009)), duplicated reads were then marked using Picard version 1.97 (http://broadinstitute.github.io/picard/), alignment quality was improved using the GATK (DePristo et al. (2011)) realigner and base recalibrator version 2.7, and finally, variants were called using GATK Haplotype Caller version 2.7. The resulting variant set was annotated and filtered using the Highlander software (http://sites.uclouvain.be/highlander/). Variants were filtered for quality criteria, allelic frequency <0.5%, functional impact, homozygous genotype, and cosegregation in the two affected sibs. Variants were then sorted by decreasing Combined Annotation Dependent Depletion (CADD) score (Kircher et al., 2014). The variants of interest were confirmed by Sanger sequencing. DNA was amplified using a standard Polymerase Chain Reaction (TRAPPC9 exon 2, forward primer: CTCCCAGGGTAGGCTCTCAG, reverse primer: AAGAGCCGGGAGTCATACAG; MCPH1 exon 13, forward primer: TTCGCCTACGCTATGGAGACT, reverse primer: ATCTGGACCACACCACAGCG). The PCR product was purified with Exosap‐IT (Affymetrix), and sequenced by the company Beckman Coulter Genomics.
All procedures complied with the ethical guidelines of Hôpital Erasme—Université Libre de Bruxelles, whose Ethics Committee approved our study protocol under reference P2005/076 (Ethics Committee Erasme Hospital, OMO21). Written informed consent to participate in our study was obtained from the patient's representative.
3. RESULTS
The exome sequencing data from the two affected children were filtered for rare (allele frequency <0.5%), nonsynonymous or splice‐junction variants that were homozygous in both affected siblings. Three such variants were identified in the MCPH1 gene (c.2221C>T p.Arg741X), the TRAPPC9 gene (c.533T>C p.Leu178Pro) and the COL22A1 gene (c.1793G>A p.Arg598Gln) (Figure 1). The latter variant was predicted to be tolerated by several effect prediction programs and was thus not further considered.
Figure 1.
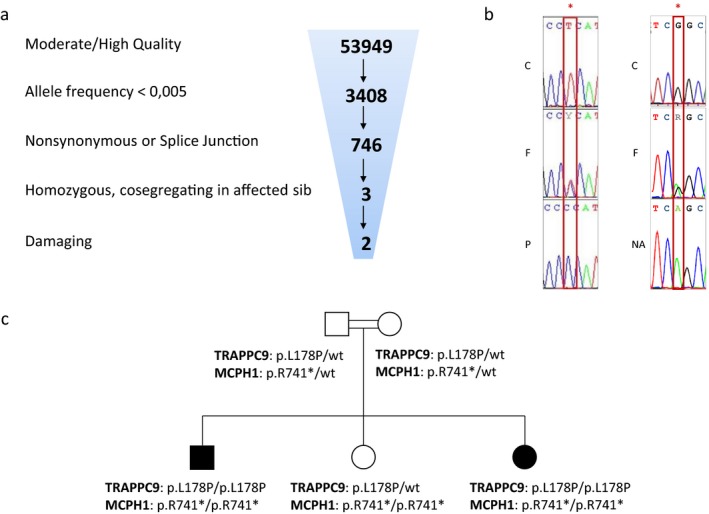
TRAPPC9 and MCPH1 mutations. (a) Exome sequencing data from one of the probands, showing the filtering parameters used to sort through the variant dataset. (b) Sanger sequencing of part of exon 2 of the TRAPPC9 gene (left panel), and of part of exon 13 of the MCPH1 gene (right panel). The TRAPPC9 T to C mutation (*) at position 533 of the coding DNA sequence was found homozygous in the proband (P), and heterozygous in his father (F); a normal sequence is shown in an unrelated control subject (C). The MCPH1 C to T mutation (*) at position 2221 of the coding DNA sequence was found homozygous in the nonaffected sib (NA), and heterozygous in their father (F); a normal sequence is shown in an unrelated control subject (C). (c) Family tree showing the consanguineous parents, the two probands and the nonaffected child, and their genotypes for TRAPPC9 and MCPH1 mutations
The c.2221C>T homozygous truncating mutation occurred in exon 13 of the 14‐exons MCPH1 gene (transcript_ensembl ENST00000344683) changing the Arginine at position 741 of the polypeptide into a stop codon, p.Arg741X (Figure 1). This change, at chr8:6478981, was encompassed in a short 0.4 Mb homozygous stretch (chr8:6538837‐6906207). The variant frequency was 2.5 × 10−5 in the Exome Aggregation Consortium (Lek et al., 2016) with only 2 alleles reported, both heterozygous. The variant was absent from 1000G, GoNL, and our in‐house database. The mutation was located just upstream of the BRCT3 domain of the protein which spans amino acid residues 751 through 833, resulting in a truncation of this domain (Figure 2). Sanger sequencing confirmed homozygosity of the mutation in the two probands and heterozygosity in both parents. Importantly, Sanger sequencing also showed homozygosity of the MCPH1 mutation in the nonaffected sister, who had a normal phenotype with a normal head size (Figure 1).
Figure 2.
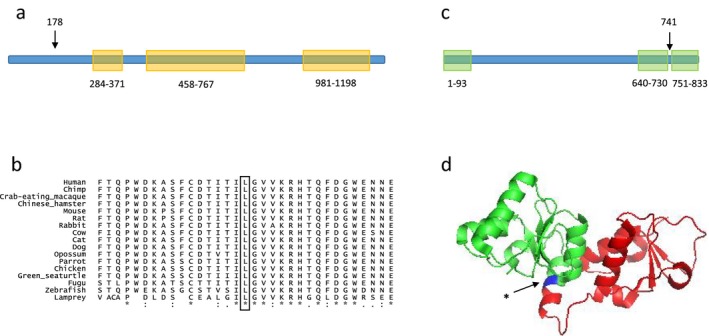
TRAPPC9 and MCPH1 conservation and domains. (a) Linear presentation of the TRAPPC9 protein, showing the three Trs120 domains (residues 284–371, 458–767, 981–1198). Arrow, position of the Leu178Pro mutation. (b) Alignment of TRAPPC9 amino acids sequence (residues 162–194) in multiple species (UCSC). “*” residue identical in all species. “:” conserved substitutions. “.” semiconserved substitutions. (c) Linear presentation of the MCPH1 protein, showing the three BRCT domains (residues 1–93, 640–730, 751–833). Arrow, position of the Arg741X mutation. (d) Crystal structure (PDB 3t1n Singh et al. (2012)) of MCPH1 tandem BRCT domains (residues 640–835). The portion of the protein truncated by the mutation appears on the right side of the arrow.
A nonsynonymous homozygous mutation was found in exon 2 of the 23‐exons TRAPPC9 gene, c.533T>C (transcript_ensembl ENST00000389328) changing the Leucine at position 178 of the polypeptide into a Proline, p.Leu178Pro (Figure 1). This change, at chr8:141461234 (GRCh37), was encompassed by a 14 Mb homozygous stretch (chr8:128066556‐142227859), consistent with homozygosity by descent (autozygosity) for this chromosomal segment. The variant was not present in the public databases ExAC, 1000G, GoNL, nor in our in‐house database. The variant was predicted to be deleterious by 5 different programs (damaging in SIFT, probably_damaging in pph2, deleterious in LRT, disease_causing in mutation_taster, medium in mutation_assessor), and had a CADD score of 22. Leu178 is very conserved in vertebrates; UCSC Multiz Alignments of 100 Vertebrates (https://genome.ucsc.edu/) showed the presence of a Leucine in all species at this position (Figure 2). The mutation was located in the TRAPP II complex Trs120 domain of the TRAPPC9 protein which spans amino acid residues 99 to 1206. TRAPPC9 is predicted to be intolerant to variation by ExAC (Lek et al., 2016; z score = 1.64) and RVIS programs (percentile 2.77%, meaning that only 2.77% of all the genes are more intolerant to variation than TRAPPC9) (not shown). In particular, exon 2 as well as the Trs120 domain of TRAPPC9 gene were predicted to be extremely intolerant to variation by the subRVIS program (percentiles being, respectively, 1.556% and 0.066%) (exon 2 Supplementary File S1; Trs120 domain not shown) (Gussow, Petrovski, Wang, Allen, & Goldstein, 2016). Sanger sequencing confirmed homozygosity of the mutation in the two probands and heterozygosity in both parents as well as in the nonaffected sister (Figure 1).
4. DISCUSSION
We report on two siblings with microcephaly and severe ID in whom exome sequencing revealed homozygous mutations in two genes, TRAPPC9 and MCPH1.
The patients’ phenotype was consistent with a TRAPPC9 defect, which is characterized by severe intellectual deficiency (ID), postnatal microcephaly, abnormalities of the corpus callosum, cerebellum, and white matter, as found in our patients. Together with the intolerance to variation in TRAPPC9 exon 2; the absolute conservation of Leu178 in vertebrates; and the predicted pathogenicity of the p.Leu178Pro protein change, consistency of the phenotype provides strong evidence for causality of this genetic variant. To date, all reported TRAPPC9 mutations were nonsense and splice variants (Supplementary File S2), and this is to our knowledge the first missense mutation of TRAPPC9. It is also the first case with congenital microcephaly (see below).
TRAPPC9 is expressed in neurons, and TRAPPC9 mutations could impair neuronal development via defective vesicular trafficking. Alternatively, or perhaps in addition, the mechanism might involve nuclear factor kappaB (NF‐kB) activation. A defective activation of the NF‐kB pathway has been reported in skin fibroblasts from a patient carrying a biallelic TRAPPC9 mutation (Philippe et al., 2009). According to ELM (Dinkel et al. (2016)), Leu178 is part of a kinase interaction motif, so this region may have a role in interaction with IKBKB or NIK and activation of the NF‐kB pathway.
MCPH1 is composed of 14 exons, and all human mutations reported to date were found in exons 1 to 11, consisting of large deletions, small insertions or deletions, nonsense and missense mutations (Pulvers et al., 2015). No disease‐causing variant has been reported in the last three exons.
Experiments on chicken cells using various truncated forms of chicken Mcph1 showed that BRCT1 is necessary for centrosomal localization, whereas BRCT2 and BRCT3 are required for IRIF formation and response to DNA damage (Pulvers et al., 2015). The study of the two major transcripts of MCPH1 (full‐length MCPH1, and a transcript lacking exons 9 to 14) also showed that the C‐terminus of the protein was necessary for DNA damage response. Indeed, the truncated isoform was able to complement the defective chromosome condensation in human MCPH1‐deficient cells, but was not able to form MCPH1 foci after irradiation (Gavvovidis et al., 2012). Similarly, in a mouse model lacking only the BRCT3 domain, body size and brain weight were normal, and the PCC phenotype was not present cytogenetically (Trimborn et al., 2010).
Taken together, these data suggest that deletion of BRCT3 does not cause human microcephaly. Homozygous truncation of this domain, or any biallelic truncating mutations, have never been reported in normal subjects, however. Interestingly, ExAC mentions one homozygous genotype for a MCPH1 gene mutation, p.Glu521Ter, which is predicted to truncate BRCT2 and BRCT3. The phenotype, whether strictly normal or microcephalic, is not reported, however.
Here we show a biallelic truncation of the BRCT3 domain in the normal, unaffected sister. Our observation is consistent with the full‐length MCPH1 isoform not being required for brain volumic development, and hence that the mechanism causing MCPH consists of a centrosomal localization defect, and not a DNA damage response defect. It supports a bifunctional model of MCPH1 where the centrosomal function only is involved in brain volumic development.
Congenital, primary microcephaly in our patients suggests that TRAPPC9, like MCPH‐associated genes, plays a role in the production of the pool of neural progenitors that will allow for volumic development of the brain. We cannot rule out, however, that congenital microcephaly might have resulted from a combined effect of the TRAPPC9 and the MCPH1 mutations, either via a digenic interaction, or independently with the resulting phenotype being an overlap of features of both defects. The normal phenotype in the MCPH1‐mutated sister nevertheless strongly argues against the latter hypothesis.
5. CONCLUSION
Our observation shows that truncation of the third BRCT domain of MCPH1 is consistent with a normal phenotype, arguing strongly for the hypothesis that MCPH1 mutations cause MCPH by a centrosomal defect rather than by a DNA damage response defect. We also describe the first ID‐associated missense mutation in TRAPPC9.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
ACKNOWLEDGMENTS
SD is a Research Fellow of the FRS‐FNRS and is supported by the Fonds Erasme. MA is supported by an E‐Rare ERA‐Net, and the orphan disease grant Jean Van Damme.
We thank Marianne Rooman for critical discussion of protein interactions.
Duerinckx S, Meuwissen M, Perazzolo C, Desmyter L, Pirson I, Abramowicz M. Phenotypes in siblings with homozygous mutations of TRAPPC9 and/or MCPH1 support a bifunctional model of MCPH1. Mol Genet Genomic Med. 2018;6:660–665. 10.1002/mgg3.400
REFERENCES
- Berg, E. , Christensen, M. O. , Dalla Rosa, I. , Wannagat, E. , Jänicke, R. U. , Rösner, L. M. , … Mielke, C. (2011). XRCC4 controls nuclear import and distribution of Ligase IV and exchanges faster at damaged DNA in complex with Ligase IV. DNA Repair, 10, 1232–1242. 10.1016/j.dnarep.2011.09.012 [DOI] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , … McKenna, A. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43, 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel, H. , Van Roey, K. , Michael, S. , Kumar, M. , Uyar, B. , Altenberg, B. , … Dahl, S. L. (2016). ELM 2016 – Data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Research, 44(D1), D294–D300. 10.1093/nar/gkv1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavvovidis, I. , Rost, I. , Trimborn, M. , Kaiser, F. J. , Purps, J. , Wiek, C. , … Schindler, D. (2012). A novel MCPH1 isoform complements the defective chromosome condensation of human MCPH1‐deficient cells. PLoS ONE, 7, e40387 10.1371/journal.pone.0040387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow, A. B. , Petrovski, S. , Wang, Q. , Allen, A. S. , & Goldstein, D. B. (2016). The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biology, 17, 9 10.1186/s13059-016-0869-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar, N. , Goebel, I. , Daud, S. , Nürnberg, G. , Agha, N. , Ahmad, A. , … Borck, G. (2012). A homozygous splice site mutation in TRAPPC9 causes intellectual disability and microcephaly. European Journal of Medical Genetics, 55, 727–731. 10.1016/j.ejmg.2012.08.010 [DOI] [PubMed] [Google Scholar]
- Khattak, N. A. , & Mir, A. (2014). Computational analysis of TRAPPC9: Candidate gene for autosomal recessive non‐syndromic mental retardation. CNS & Neurological Disorders Drug Targets, 13, 699–711. [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Tukiainen, T. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformaticas (Oxford, England), 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejczyk, M. , Mikoluc, B. , Pietrucha, B. , Heropolitanska‐Pliszka, E. , Pac, M. , Motkowski, R. , & Car, H. (2016). Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia‐telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biology, 11, 375–383. 10.1016/j.redox.2016.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi, H. , Hu, H. , Garshasbi, M. , Zemojtel, T. , Abedini, S. S. , Chen, W. , … Zecha, A. . (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478, 57–63. 10.1038/nature10423 [DOI] [PubMed] [Google Scholar]
- Passemard, S. , Kaindl, A. M. , & Verloes, A. (2013). Microcephaly. Handbook of Clinical Neurology, 111, 129–141. 10.1016/B978-0-444-52891-9.00013-0 [DOI] [PubMed] [Google Scholar]
- Philippe, O. , Rio, M. , Carioux, A. , Plaza, J. M. , Guigue, P. , Molinari, F. , … Munnich, A. (2009). Combination of linkage mapping and microarray‐expression analysis identifies NF‐kappaB signaling defect as a cause of autosomal‐recessive mental retardation. American Journal of Human Genetics, 85, 903–908. 10.1016/j.ajhg.2009.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulvers, J. N. , Journiac, N. , Arai, Y. , & Nardelli, J. (2015). MCPH1: A window into brain development and evolution. Frontiers in Cellular Neuroscience, 9, 92 10.3389/fncel.2015.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, N. , Basnet, H. , Wiltshire, T. D. , Mohammad, D. H. , Thompson, J. R. , Héroux, A. , … Mer, G. (2012). Dual recognition of phosphoserine and phosphotyrosine in histone variant H2A.X by DNA damage response protein MCPH1. Proceedings of the National Academy of Sciences of the United States of America, 109, 14381–14386. 10.1073/pnas.1212366109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, N. , Wiltshire, T. D. , Thompson, J. R. , Mer, G. , & Couch, F. J. (2012). Molecular basis for the association of microcephalin (MCPH1) protein with the cell division cycle protein 27 (Cdc27) subunit of the anaphase‐promoting complex. Journal of Biological Chemistry, 287, 2854–2862. 10.1074/jbc.M111.307868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimborn, M. , Ghani, M. , Walther, D. J. , Dopatka, M. , Dutrannoy, V. , Busche, A. , … Klose, J. (2010). Establishment of a mouse model with misregulated chromosome condensation due to defective Mcph1 function. PLoS ONE, 5, e9242 10.1371/journal.pone.0009242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S.‐Z. , Lin, F.‐T. , & Lin, W.‐C. (2008). MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint. DNA repair and apoptosis. EMBO Reports, 9, 907–915. 10.1038/embor.2008.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, M. , Satoh, A. , Yu, M. K. , Siu, K. Y. , Ng, W. Y. , Chan, H. C. , … Yu, S. (2012). TRAPPC9 mediates the interaction between p150 and COPII vesicles at the target membrane. PLoS ONE, 7, e29995 10.1371/journal.pone.0029995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials