

Abstract
The European Academy of Otology and Neurotology in collaboration with the Japanese Otological Society (EAONO/JOS) recently produced a joint consensus document outlining the definitions, classification and staging of middle ear cholesteatoma. The goals were to provide terminologies in the description of cholesteatoma, classify cholesteatoma into distinct categories to facilitate the comparison of surgical outcomes and to provide a staging system that reflects the severity, difficulty of complete removal and restoration of normal function. Cholesteatoma is considered a benign, expanding and destructive epithelial lesion of the temporal bone that is the result of a multifactorial process. If undetected and left treated, cholesteatoma may lead to significant complications including hearing loss, temporal bone destruction and cranial invasion. Recent advances in imaging modalities have allowed for high sensitivity and specificity in identifying the presence of cholesteatoma. Despite these advances, deficiencies exist around the world with access to health care facilities meaning cholesteatoma remains a serious and challenging entity to manage whether found within the pediatric or adult population. Proper diagnosis and management of each form of cholesteatoma is achieved by a thorough understanding of the etiology, classification, clinical presentation and histology, thereby facilitating prevention, early detection and appropriate treatment.
Keywords: Cholesteatoma, Middle ear cholesteatoma, Congenital cholesteatoma, Acquired cholesteatoma
Introduction and Background
Cholesteatomas are lesions that most often arise within pneumatized portions of the temporal bone to include the middle ear and mastoid, or both, and are only rarely found within the external auditory canal. They are non-neoplastic, often destructive, locally invasive masses that present mainly as unilateral lesions. In the most simple of descriptions, they are depicted as a cystic structure created by accumulation of desquamated keratin and squamous debris that is surrounded by a fibrous matrix and a usual finding of an inflammatory reaction [1].
The term cholesteatoma (“chole” representing cholesterol, “steat” representing fat and “oma” meaning “tumor”) as we now know, is a misnamed entity that was originally termed by Johannes Müller in 1838 under the flawed belief that the lesion was primarily a tumor of adipose tissue [2] but Müller may have been influenced by an original report of De Verney in 1683 that he described as a “steatoma” [3]. It should be noted early on that neither cholesterol nor fat is found within these lesions. They are occasionally referred to as “keratomas” to this day both in clinical as well as pathologic terms. This is also a misnomer as the term “keratoma” may be confused with keratosis obturans—an occlusive keratin plug of the external auditory canal.
It is estimated that over 20 million people worldwide are afflicted with otitis media. Of these, one-fourth (about 5 million) have a cholesteatoma [4], although the overall number of cases of acquired cholesteatoma seems to be in decline [5, 6]. The annual incidence of cholesteatoma is reported as 3 per 100,000 in children and 9.2 per 100,000 adults. Males slightly outnumber females in a ratio of 1.4:1 and cholesteatomas that present in the middle ear are more frequently found in persons younger than 50 years of age [7]. Caucasian persons show the highest prevalence but cholesteatoma is infrequently found in Inuit, Native American and Asian populations [8]. A number of reports reviewed by Jennings et al. [9] evaluated familial clustering and inheritability of cholesteatoma and found that incomplete penetrance exists and may depend on a combination of environmental and genetic factors for the formation of an acquired cholesteatoma. Jennings also states that evidence from syndromic cases suggests genes controlling ear morphology may be risk factors for congenital or acquired cholesteatoma formation [9]. Syndromes where a diagnosis of cholesteatoma has been a reported finding include Turner Syndrome [10–12], Treacher Collins Syndrome [13], Down Syndrome [14–17] and Focal Dermal Hypoplasia [18]. Numerous reports of patients presenting with cleft palate and cholesteatoma [19–25] have been presented with the rate of incidence approaching 6% in that population [23]. When compared to children who did not develop a cleft palate, those with a cleft palate face a 100–200 times greater likelihood of developing a cholesteatoma [23, 25]. Additionally, a link between allergic rhinitis and the development of cholesteatoma was recently discovered in that patients with allergic rhinitis presented with a significantly lower 10-year cholesteatoma disease free rate [26].
Classification, Etiology and Pathogenesis
Cholesteatomas are subdivided into three categories: the congenital form which is specific to children, the acquired type which affects both adults and children and the unclassifiable type which is a cholesteatoma whose origin cannot be accurately determined [1].
Congenital cholesteatoma is typically an expanding cystic mass of keratinizing squamous epithelium located medial to the intact tympanic membrane. It is assumed to be present at birth but, is usually diagnosed during infancy or in early childhood in patients with no prior history of otorrhea, perforation of the tympanic membrane, or previous ear surgery. In contradiction to this, the European Academy of Otology and Neurotology and the Japanese Otological Society (EAONO/JOS) working group recently stated that a history of previous bouts of otitis media or effusions does not exclude congenital cholesteatoma [1]. Additional criteria that are generally agreed upon for defining a congenital cholesteatoma is the finding that they do not show continuity with the external auditory canal and that the pars flaccida and pars tensa do not show retraction [27].
Currently, no single accepted modality for the formation of congenital cholesteatoma is universally accepted as the cause is not completely understood. Probably the most popular theory is the epithelial rest theory. Rests of epithelial cells, otherwise referred to as an epibranchial placode, are located behind an intact tympanic membrane and fail to involute [28]. In most instances, these rests are normally thought to resorb near the 33rd week of gestation but, a congenital cholesteatoma may form if involution does not happen due to persistent irritation [29]. According to Bennett et al. [5], this theory does not explain the existence of congenital cholesteatomas outside the anterior superior quadrant of the tympanic membrane where these rests are found. He additionally states reports have noted that they may also be found in the posterosuperior, posteroinferior, and anteroinferior regions of the lateral wall of the tympanic cavity which may explain the persistence of additional theories of formation [5]. The EAONO/JOS working group stated that congenital cholesteatoma is usually located at the anterosuperior quadrant of the middle ear, however; it may be located at the posterosuperior quadrant or other locations [1].
An additional theory of formation, the invagination theory, holds that squamous epithelium from the external canal migrates through the tympanic ring and into the middle ear, eventually forming into a congenital cholesteatoma [30]. In utero or during childhood, an inflammatory injury to the tympanic membrane near the neck of the malleus induces invagination leading to development of a congenital lesion. In this case, the tympanic membrane is adherent to the malleus or incus and leaves behind a remnant of keratinized epithelium which over time then forms a cholesteatoma [5, 31]. This theory does help to explain the existence of lesions that are not located in the anterosuperior quadrant of the tympanic membrane as the invagination process could occur anywhere along the tympanic membrane.
The acquired type cholesteatoma is presumed to arise due to Eustachian tube dysfunction following prior bouts of middle ear disease. In contrast to the congenital cholesteatoma, the acquired type is not presumed to be present at birth and, as a general statement, the common denominator is that keratinizing stratified squamous epithelium has grown beyond expected anatomic boundaries. In order to provide clarity, the EAONO/JOS states that acquired cholesteatoma is characterized by clinical signs and symptoms that are the result of growth with or without destruction of the adjacent structures, with or without tympanic membrane retraction and/or perforation, with or without otorrhea, with or without hearing deterioration and/or CT/MRI findings (soft tissue masses, focal areas of bony erosion of the middle ear, and mastoid) [1].
Like the congenital type, multiple pathophysiologic theories have been proffered to explain the formation of acquired cholesteatomas, although as yet, no single process has been completely accepted as the definitive mechanism of formation for all cases [27]. Many believe that the pathogenesis involved incorporates a complex mix of mechanisms as opposed to a singular method due to the fact that all factors of presentation and growth cannot be fully and simply explained with an as yet proposed singular method. These theories differ from the congenital type as it is based on the presence or absence of a perforation and subsequent migration of epithelium into the middle ear through that perforation. Acquired cholesteatoma is further subclassified as a retraction pocket variant of cholesteatoma and a non-retraction pocket variant [1].
The retraction pocket theory of formation, occasionally referred to as a primary type cholesteatoma, forms due to an underlying Eustachian tube problem that results in poor aeration of the epitympanic space. The pars flaccida, pars tensa or both are then drawn medially by retraction to the top of the malleus neck, thereby forming a retraction pocket as a result of negative pressure in the middle ear [32, 33]. The development of this pocket restricts the normal migratory pattern of the tympanic membrane thereby losing its ability of self-cleaning and further enhancing the potential for keratin debris accumulation, allowing the formed sac to slowly enlarge. The resultant masses are classified as a pars flaccida or attic cholesteatoma, pars tensa cholesteatoma or a combination of pars flaccida and pars tensa type cholesteatomas [1].
Non-retraction pocket cholesteatomas, often referred to as secondary acquired cholesteatomas, are commonly found in patients with acute otitis media. These lesions are thought to develop under three possible lines of thought: the epithelial migration theory, squamous metaplasia theory, and basal cell hyperplasia theory [32]. The epithelial migration theory assumes that tympanic membrane perforations act as a precursor and that squamous epithelium of the tympanic membrane then migrates into the middle ear. This site of traumatic injury can come about as a result of surgery, blast injury, and foreign body or iatrogenic causes, all leading to the formation of a cholesteatoma [1, 34]. It is thought that the edges of the perforation migrate as well due to the fact that tympanic membrane epithelium and cholesteatoma epithelium have similar properties.
The squamous metaplasia theory proposes that desquamated middle ear epithelium transforms into keratinizing stratified squamous epithelium secondary to chronic or recurrent otitis media [32], thereby leading to the formation of a cholesteatoma. Advocates of the basal cell hyperplasia theory propose that papillations, pseudopods or microcysts filled with keratin formed in the basal cell layer of the pars flaccida epithelium, invade the subepithelium of Prussack’s space [32, 34]. An inflammatory reaction, possibly due to poor ventilation may cause a break in the basal membrane allowing a cord of epithelium to start inward proliferation.
A third cholesteatoma category is given the designation as unclassifiable for those lesions whose origin cannot be accurately determined [1]. In certain large or open cases, it may not be possible to categorize the lesions under the heading of congenital or acquired cholesteatoma therefore, the unclassifiable designation.
Cholesteatomas that present post-surgically may be considered as a fourth category [1]. This category may arise as residual or recurrent lesions, although these are not mutually exclusive [1]. Residual cholesteatoma results from the incomplete surgical removal of the cholesteatoma matrix and a recurrent cholesteatoma results from the reformation of the retraction pocket after a complete previous surgical removal [1].
Clinical Features
Risk factors for development of a cholesteatoma include middle ear disease, prior surgery, traumatic injury or congenital anomalies. Cholesteatomas have been noted to persist in a static state for many years prior to manifestation of significantly destructive events as they are unresponsive to antimicrobial therapy although it is estimated that the growth rate of congenital lesions enlarges the mass by approximately 1 mm in diameter per year [24].
As opposed to acquired cholesteatoma in adults, congenital cholesteatoma progresses in a relatively muted fashion as unilateral hearing loss passes unnoticed in children thereby allowing larger lesions to remain undetected until incidental identification during a routine physical examination [25]. Hearing loss may be evident when the cholesteatoma is large enough to fill the middle ear or when the ossicles have eroded [1]. The ossicular chain is the first structure to be damaged and the most commonly damaged ossicle is the anvil, approaching 100% of the time in cases where damage occurred [35].
The first and most frequent symptom of an acquired cholesteatoma is otorrhea (66.5%) [4] that can be unrelenting or periodically recurrent in nature. Patients note scant possible purulent discharge that may be foul smelling. On clinical examination, the presence of abundant granulation tissue may accompany the drainage. Pain (which may be an indicator of advanced disease) and an earache are common symptoms prior to episodes of purulent discharge. The combination of otorrhea, tinnitus and hearing impairment (hypoacusis) seen in 23.3% of patients, and solely hypoacusis seen at a rate of 7.6%, also denote frequently experienced symptoms [4]. Hearing loss experienced by cholesteatoma patients can be progressive conductive or sensorineural with conductive hearing loss resulting from impaired movement of the ossicles [28]. Tinnitus is a common clinical complaint that may be the result of sigmoid sinus compression by a cholesteatoma [35] or can be a resultant effect of damage to the cochlea leading to irreparable sensorineural hearing loss [28].
Examination of the tympanic membrane to evaluate for acquired cholesteatoma should include inspection of the pars flaccida [24]. During the examination, the anterosuperior quadrant of the mesotympanum must be inspected for the presence of a round, white, compressible lesion under the tympanic membrane as this finding is considered pathognomic [24]. The differential diagnosis may include a finding such as tympanosclerosis, defined as hyalinization of fibrous connective tissues of the tympanic membrane and middle ear, followed by calcification that may result in degraded hearing. In comparison to cholesteatoma, tympanosclerosis presents as hard white plaques with an irregularly sharp edge as opposed to the smooth, rounded and curved edge of cholesteatoma [36].
Imaging
Given its composition, the extent of a cholesteatoma is still difficult to detect with modern imaging modalities especially in the case of ears that have previously undergone surgery. Computed tomography (CT) has been the standard modality as it does provide for the ability of identifying bony changes. Owing to excellent ability of spatial resolution, CT has a high sensitivity but low specificity in the case of a mass lesion because it may be misinterpreted as granulation tissue, cholesterol granuloma or other soft tissue neoplasms [8, 37]. Typical findings on CT include a sharply marginated and expansile soft tissue lesion, retraction of the tympanic membrane, scutum blunting and erosion of the tympanic tegmen and ossicles [8]. Within pneumatized areas of the temporal bone, normal aeration is lost and CT often shows evidence of bony erosion due to scalloped margins being observed [38]. Specific examples of typical findings seen on CT are provided in Figs. 1 and 2. CT has limited value in the post-operative period due to the high negative predictive value when it shows a well-aerated, disease free middle ear and mastoid with no evidence of soft tissue present [38]. However, in patients who have undergone previous tympanomastoidectomy, interpreting bony erosion is made nearly impossible when attempting to differentiate between surgical changes and pathologic bony destruction due to cholesteatoma [38]. In this case, the sensitivity and specificity of detecting residual or recurrent cholesteatoma drops dramatically.
Fig. 1.

Coronal images from temporal bone CTs in four different patients with right cholesteatoma. a Small cholesteatoma in Prussak’s space (red arrow) without bony erosion. This is a common site for pars flaccida retraction and acquired cholesteatoma formation. b Cholesteatoma in the left mesotympanum to hypotympanum (red arrow), which is a less common site. c Cholesteatoma in the right epitympanum (red arrow) with blunting or erosion of the right scutum (green arrow). This lesion probably started in Prussak’s space adjacent to the bony scutum. d Large cholesteatoma in the right epitympanum, mesotympanum, and hypotympanum (red arrows), with bony erosion of the scutum and malleus/ossicles (green arrow)
Fig. 2.

Temporal bone CT and brain MRI in 41-year-old male after transcanal endoscopic resection of a right epitympanic cholesteatoma. a Axial CT shows the resection cavity in the right Prussak’s space (red arrow), with residual cholesteatoma in the right mastoid antrum (yellow arrow). b Coronal CT shows the resection cavity at the right lateral epitympanum (red arrow) plus the surgical approach for a transcanal atticotomy (yellow arrow). An alternative approach would be via the mastoid antrum (mastoidectomy). c Axial T2-weighted MRI shows the small residual cholesteatoma (red arrow) to be of similar intensity with fluid, e.g. prepontine cistern and fourth ventricle (yellow arrows). d Axial diffusion-weighted MRI shows “restricted diffusion” in the cholesteatoma (red arrow), much brighter than free fluid (yellow arrows)
Due to the small size of congenital cholesteatoma, CT is not mandatory as there may be little benefit to justify the involved radiation and anesthetic procedure that may be necessary in a pediatric case. Additionally, due to ionizing radiation that is produced when performing CT, cone beam CT (CBCT) could be considered as an alternative as significantly less ionizing radiation is introduced. For some, the information received in a CBCT rivals that of CT.
Diffusion weighted imaging MRI (DWI-MRI) can be a valuable tool in the detection of cholesteatoma as advantages exist over CT such as a shorter examination time than delayed contrast material-enhanced imaging and no need for a contrast media injection prior to the examination [39]. The examination may be lacking the detailed information of bony structures though. MRI sequences show a cholesteatoma to be dark on T1 weighted images and bright on T2 weighted images when compared to brain tissue. As cholesteatomas do not take in contrast media, to overcome this finding, the use of delayed contrast techniques have been in use as this technique takes advantage of the fact that other tissues will possibly take up more contrast media given a proper amount of time [37, 38]. T1 images are obtained 30–45 min after contrast administration. Inflamed mucosa, granulation tissue and fibrosis will show enhancement whereas, a lesion in the proper location that does not enhance may suggest a cholesteatoma is present [40].
Advancements in DWI-MRI allows for identifying the presence of small collections of keratin debris that would otherwise be interpreted as fluid or edematous mucosa on CT and is proficient in detecting recurrent cholesteatoma due to its high sensitivity and specificity [38, 39]. Cholesteatomas appear to have a high signal intensity, mainly attributable to restricted water diffusion due to the oily consistency of the retained fluid [8]. According to Vercruysse et al., DWI revealed sensitivity of 81% and a specificity of 100% with a positive predictive value of 40% in patients who had not undergone surgery as yet [40]. DWI is a valuable assessment tool for the presence of erosion of the semicircular canal, invasion of the membranous labyrinth or middle cranial fossa and to assess abscess formations [39]. Non-echo planar imaging DWI techniques provide for less image distortion and less artefacts are seen than with other DWI techniques. EPI DWI has a deficiency whereby artefacts can be seen at a junction with numerous anatomic tissues leading to distortion. Non-EPI DWI provides for higher resolution and thinner slices (2 mm) can be obtained with less false positive findings noted [41]. As a result, DWI can prevent unnecessary second look surgeries for suspected residual or recurrent cholesteatoma in cases that are found to be inaccessible to clinical otomicroscopy, which includes those lesions in the mastoid cavity, ones deep to reconstructive materials and those cases growing around structures where cholesteatoma may have been missed in the primary procedure [38].
Gross Pathologic Findings
At the gross level, cholesteatomas present as primarily white, compressible, ovoid lesions which when intact, are surrounded by a thin wall (Fig. 3). Often, the surface of the “capsule” is disrupted to show a shaggy, irregular and friable surface with edematous edges. The contents of the sac then spill externally to display flaky, greasy or cheese-like keratinaceous debris (Fig. 4). Often the specimen is submitted as numerous layered, white flakes that not only represent keratin, but may include diminutive fragments of bone. All contents encountered in the surgical procedure should be submitted in their entirety for histopathologic examination.
Fig. 3.
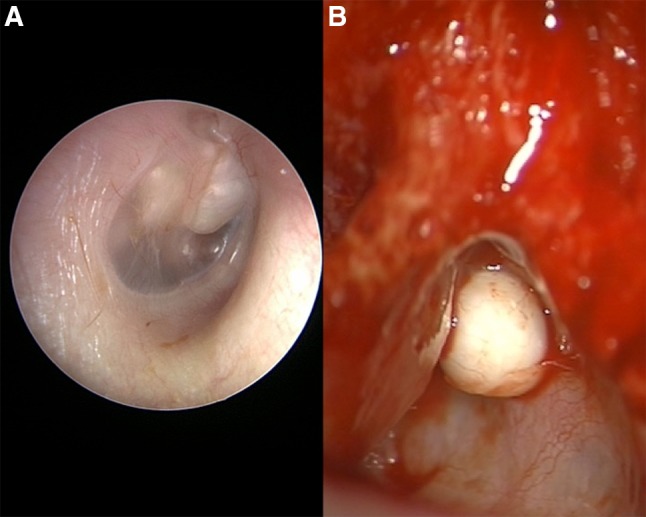
a Right ear with advanced congenital cholesteatoma showing involvement of the mesotympanum anterior and posterior to the malleus that appears to separate the lesion into halves. b Discrete nodule of left ear cholesteatoma at anterosuperior portion of mastoid found in the epitympanic region-aditus ad antrum, wedged between the posterior bony external auditory canal wall (at left) and the tegmen mastoideum (at right)
Fig. 4.
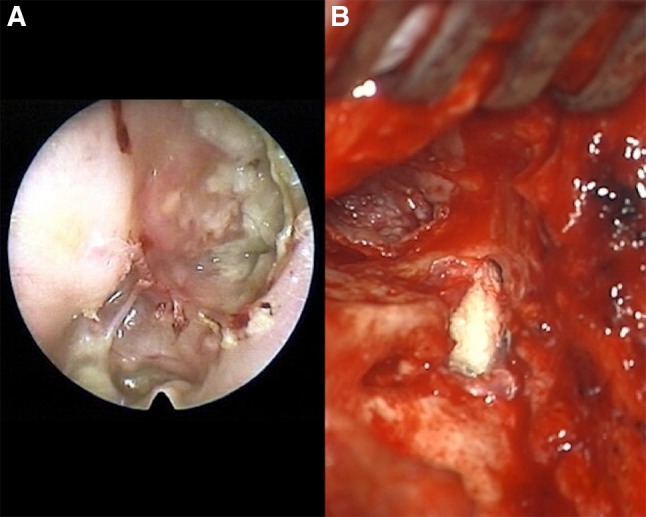
a Extensive right ear attic cholesteatoma with rupture and spilled keratinaceous contents. b Surgical entry via mastoidectomy approach where a tympanic membrane retraction pocket formed a sac into the mastoid cavity and eroded lateral canal wall. The top left of photo looks down the ear canal to the eardrum
Histopathologic Findings
The requirements for a pathologic diagnosis of cholesteatoma include a combination of squamous epithelium, granulation tissue and keratinaceous debris. At face value, a finding of squamous epithelium in the middle ear differs from the normal glandular epithelial lining, and is therefore deemed abnormal [42]. The components of cholesteatomas have been characterized as the perimatrix, matrix and cystic contents. The perimatrix, or most peripheral portion of a cholesteatoma, primarily consists of an abundance of granulation tissue with the immediate subepithelial area occasionally consisting of occasionally cellular and thickened, dense fibrous connective tissue (Fig. 5). This layer differs depending on the age of the patient. Congenital cholesteatomas show less of a connective tissue layer and more granulation tissue, whereas adult patients are more prone to possess dense connective tissue in this area [43]. It is this layer that is in close contact with middle ear epithelium and bone whether it is the ossicles or wall of the ear canal, and some consider it responsible for their erosion and destruction. Small fragments of degenerate bone and acellular basophilic osteoid are also occasionally identified within the perimatrix or cyst lumen and may represent portions of eroded ossicles or canal wall (Fig. 6).
Fig. 5.
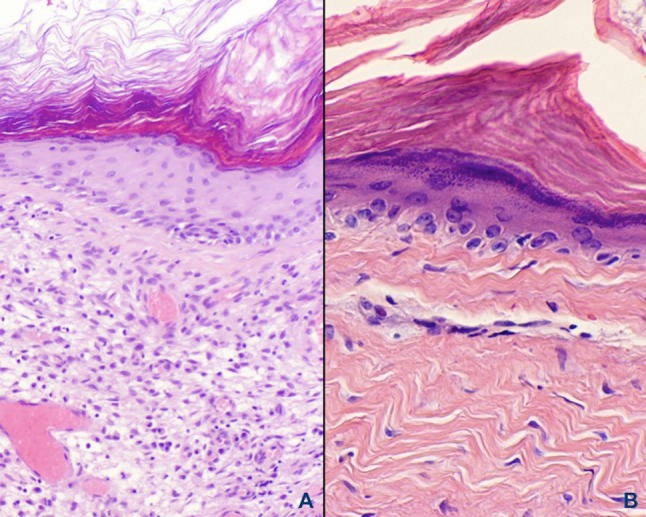
a Abundant granulation tissue peripheral to the squamous epithelial lining comprises the perimatrix. Note that rete peg formation is absent but a prominent granular cell layer is present. b Dense fibrous connective tissue adjacent to the epithelium is present with a chronic inflammatory cell infiltrate found in place of granulation tissue
Fig. 6.
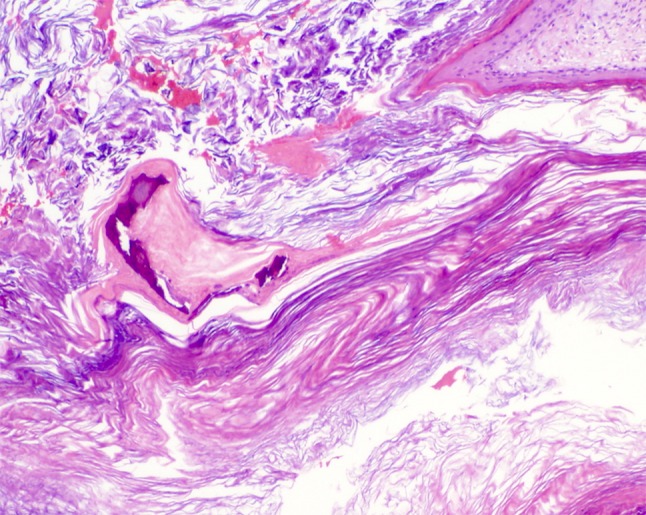
Irregular acellular basophilic calcifications that may represent eroded ossicles or bone of the middle ear canal are seen in conjunction with desquamated keratin of the cystic lumen
The essential diagnostic feature of matrix is that it consists of keratinizing, cytologically bland stratified squamous epithelium arranged in convoluted cyst-like formations (Fig. 7). This epithelium ranges from thin to atrophic, lacks rete ridges and shows a prominent granular cell layer with abundant keratohyaline granules. Given this finding, the histology of cholesteatomas is not unlike that seen in epidermal inclusion cysts of the skin, although keratohyaline granules are not as prevalent in epidermal inclusion cysts of the skin. These cyst-like structures may collapse or decompress upon surgical removal to histologically compose an undulating, ribbon-like appearance of the epithelium whereby it folds upon itself. The basal cell layer of cholesteatoma may demonstrate architectural atypia in the basal epithelium as well as mild cytologic atypia without dysplasia. Although squamous cell carcinoma has been reported as arising from cholesteatoma, it is an extremely rare event [44].
Fig. 7.
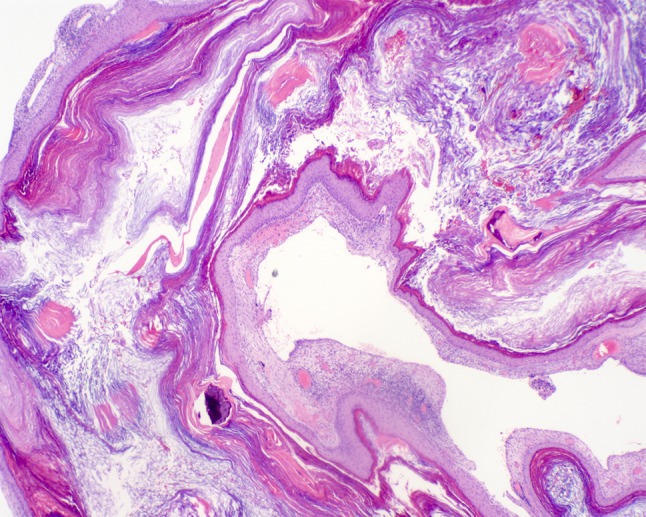
Cystic formation of bland squamous epithelium surrounding abundant layers of laminated, desquamated and anucleate keratin filling the cystic luminal area
The cystic contents are contained within a luminal area that is comprised of abundant layers of laminated, free-flowing or aggregated, desquamated, anucleate keratin. When rupture of the cystic contents occurs, a foreign body giant cell reaction ensues in response to keratinous debris. Awareness by the pathologist is needed as additional histologic findings may include small nerve fibers within the specimen. It may be surmised that the facial nerve may be incorporated in the surgical procedure, but most often these small nerve twigs represent chorda tympani, whether intended or not.
Studies evaluating markers as indicators of the aggressive nature and bone destruction capacity have recently shown that some cholesteatomas overexpress Ki-67, a cell proliferation marker of tumors and non-neoplastic proliferative disorders that includes cholesteatoma. CK17, a marker of keratinocyte differentiation, has been shown to be a predictor of aggressiveness and invasive capability when present [45]. Hamed et al. observed that overexpression of both markers were seen in the epithelial portion of cholesteatoma in cases of invasive cholesteatoma where severe ossicular destruction was present whereas, the non-invasive group expressed each marker in an inactive form [45].
Other Histopathologic Findings
Although most cases sent for histopathologic diagnosis following suspicion for a cholesteatoma are diagnosed as such, the pathologist should be vigilant for potential concurrent diagnoses and differential diagnostic possibilities that may arise in this area even if the histology may be dramatically different such as with a paraganglioma. A spectrum of concurrent diseases reported in association with cholesteatoma of the middle ear includes cholesterol granuloma, otic polyp, tympanosclerosis, acquired encephalocele, middle ear paraganglioma, schwannoma and neuroendocrine adenoma of the middle ear.
Microscopic Differential Diagnosis
The pathologist should be able to differentiate those microscopic entities that could provide microscopic dilemmas with cholesteatoma such as unaffected tympanic membrane, cholesterol granuloma, granulation tissue of otitis media and squamous cell carcinoma.
Normal tympanic membrane is a consideration as it presents on the external surface with a thin epidermal layer composed of stratified squamous epithelium adjacent to an underlying band of connective tissue. Type I and type II collagen as well as elastic fibers comprises this area. The inner component is composed of simple cuboidal epithelium that is continuous with the lining of the tympanic cavity.
As additional histologic considerations, the histologic presentations of cholesterol granuloma and otitis media are very similar. Cholesterol granuloma is thought to present following trauma and resultant hemorrhage that leads to the formation of cholesterol crystals following rupture of red blood cells. The break down of the erythrocyte membrane of releases lipids resulting in numerous clefts scattered within abundant granulation and foreign body giant cells. Otitis media includes a variably sized collection of chronic inflammatory cells including plasma cells, lymphocytes, histiocytes, foamy histiocytes and multinucleated giant cells. Cholesterol clefts are also noted scattered within background granulation tissue. In addition, glands demonstrating cuboidal epithelium with or without cilia small foci of calcifications and reactive bone are also occasionally included.
Squamous cell carcinoma is always a component of the histologic differential diagnosis when epithelium is involved. In middle ear lesions, a lack of epithelial maturation will be present along with hallmarks of significant pleomorphism, numerous mitoses which are often atypical, individual cell keratinization and the formation of keratin pearls.
Co-factors in Cholesteatoma Formation
Several factors stimulate the bone resorption that is an inherent aspect of cholesteatoma formation. These include inflammation, local pressure, and specific cytokeratins [46]. Granulation tissue next to ossicles may produce several enzymes and mediators that may accelerate ossicle resorption including lysosomal enzymes, collagenases, and prostaglandins [47]. Recently, analyses have revealed an excessive host response to inflammation in the form of paracrine and autocrine secretions that then leads to the progression of cholesteatomas [27, 46–48]. Additionally, angiogenic growth factors such as cyclooxygenase-2, interleukin-8 platelet-derived growth factor epidermal growth factor and vascular endothelial growth factor released by inflammatory cell populations in the matrix and perimatrix, allow for angiogenesis thereby paving the way for migration of keratinocytes into the middle ear via a vibrant vascular network [49, 50]. These factors, combined with the inflammatory cells in the granulation tissue are a potent stimulus for epithelial migration and provide somewhat of a substrate. Upregulated cytokines such as interleukin-1, interleukin-6, interleukin-17 and interferon beta have been shown to promote inflammatory bone resorption [36, 46, 51, 52]. Bone resorption itself may explain the increase in osteolysis associated with acquired cholesteatoma [27]. Recent studies have identified that matrix metalloproteinase 9 (MMP-9), which is able to degrade type IV collagen, plays a key role in inflammatory cell migration adding to the destructive behavior of cholesteatomas [53]. Receptor activator overexpression of nuclear factor-κB ligand (RANKL) and low expression of osteoprotegerin (OPG) are typical features found in middle ear cholesteatoma patients, specifically in the cholesteatomas perimatrix [50]. The altered RANKL/OPG protein ratio suggests that the RANKL–OPG pathway may play a major role in the inflammation associated with middle ear cholesteatoma [54]. Overall, a host inflammatory response could be beneficial in instances of insult but, an excessive inflammatory response may promote the growth and destructive effects of cholesteatomas.
Inherent to the formation and progression of cholesteatoma is the colonization by bacteria within the middle ear and the formation of biofilm that aids in the persistence of inflammation. Numerous pathogens to include Gram positive, Gram negative and various fungal elements have been identified in the middle ear in association with cholesteatoma tissues. Significant difficulty exists when attempting to treat these elements as delivery of systemic antibiotics is hampered by the lack of blood flow to the lesion and the fact that topical antibiotics may not penetrate as deep as necessary to eradicate pathogens that may be the root cause of the general inflammatory stimulus that leads to progression of cholesteatomas. Advanced infection can progress to significant complications such as cavernous sinus thrombosis, meningitis, brain abscess and mastoiditis [27].
Staging
The EAONO/JOS working group developed a staging system that applies to four categories of middle ear cholesteatoma: pars flaccida cholesteatoma, pars tensa cholesteatoma, congenital cholesteatoma, and cholesteatoma secondary to a tensa perforation. The utility of this system is that it may be used for evaluating initial pathology in a standardized fashion and for standardization in reporting of surgical outcomes throughout the otologic community.
It additionally may be practical for adjusting for the severity of the condition during outcome evaluations as well as provide information that is useful for the counseling of patients. Their findings are outlined below [1]:
-
Stage I: Localized cholesteatoma
The site of cholesteatoma origin, i.e., the attic for pars flaccida cholesteatoma, the tympanic cavity for pars tensa cholesteatoma, congenital cholesteatoma, and cholesteatoma secondary to a tensa perforation.
Stage II: Cholesteatoma involving two or more sites.
-
Stage III: Cholesteatoma with extracranial complications or pathologic conditions
Includes: facial palsy, labyrinthine fistula: with conditions at risk of membranous labyrinth, labyrinthitis, postauricular abscess or fistula, zygomatic abscess, neck abscess, canal wall destruction more than half the length of the bony ear canal, destruction of the tegmen: with a defect that requires surgical repair, and adhesive otitis: total adhesion of the pars tensa.
-
Stage IV: Cholesteatoma with intracranial complications
Includes: purulent meningitis, epidural abscess, subdural abscess, brain abscess, sinus thrombosis, and brain herniation into the mastoid cavity.
The staging system does not apply to petrous bone cholesteatoma but applies to cholesteatoma in attic, cholesteatoma in pars tensa or secondary to pars tensa perforation and to congenital cholesteatoma.
Treatment
Any initiation of treatment should begin with a thorough patient history to include a detailed otologic history, as pertinent information as to the course of events and symptoms experienced could play a key role in evaluation and accurate diagnosis. A complete and detailed head and neck examination to most importantly include otomicroscopy, is paramount in the evaluation process of cholesteatoma and subsequent disease extent. It should also be understood that no purely medical or non-surgical options exist for the treatment of cholesteatoma, therefore surgical excision is the mainstay of treatment. Surgical treatment has the ideal goal of the patient remaining with complete eradication of the cholesteatoma, a dry ear free of recurrence, preserved or improved hearing and a cosmetic outcome. In the case of children, special attention should be given due to the fact that the temporal bone of children possess well-pneumatized air cells which provide for adequate space for spread of disease and thereby intensifying the aggressive nature of cholesteatomas [27].
Although additional advanced techniques such as the Tos-modified CWU mastoidectomy for cholesteatoma surgery exist, the main treatment options consist of standardized microscopic surgical procedures: canal wall down (CWD) and canal wall up (CWU) mastoidectomy. Controversy still exists over the use of these procedures with the point of contention being the preservation of the posterior canal wall, complete eradication of tumor and recidivism rates with each procedure. The CWD approach (which usually infers a modified radical mastoidectomy) consists of the removal of the bony posterior wall such that the canal and the mastoid are considered a common cavity [55]. The radical CWD mastoidectomy employs removing mastoid air cells, total exteriorization of the middle ear, attic and mastoid cavity in addition to obliteration of the Eustachian tube [55]. CWD for the pediatric population is contraindicated because the temporal bone is still a developing entity [57]. Recurrence rates of pediatric cholesteatomas approach 27% [56]. Indications for use of a CWD procedure include use in an only hearing ear, intracranial involvement of a labyrinthine fistula or a sclerotic mastoid [57]. Wound healing and cavity problems to include accumulation of keratin debris and wax necessitating life long ear cleaning are inherent problems with a CWD procedure. Avoidance of exposure to water along with decreased resonance due to decreased middle ear volume resulting in poor hearing are common complaints noted with the CWD procedure. Advocates of the CWD procedure claim that CWD exposes the entire lesion allowing for complete removal. CWU does not adequately expose portions of the middle ear cleft which ultimately results in far higher residual disease rates [57].
The closed, or CWU procedure is much more commonly used in practice today as it overcomes the problems and necessary maintenance following a CWD procedure. Like the CWD procedure, all mastoid air cells are removed but the hallmark is that the posterior bony external auditory canal is preserved along with the integrity of the ear canal contours through preservation of the posterior wall or reconstruction of a defect following disease removal [55]. Benefits are that less structural damage ensues during the surgical procedure which can lessen would healing problems. In addition to facilitating the fitting of hearing aids post-operatively, cavity problems such as accumulation of debris and the need to avoid exposure to water are avoided with a CWU procedure [57]. Detractors of this procedure argue that the procedure insufficiently exposes the middle ear cleft while at the same time sacrifices a number of healthy mastoid air cells [27]. This inadequate exposure is a cause of higher incidents of residual disease in patients undergoing CWU procedures that ranges from 20% of patients who underwent a CWU procedure to an overall relapse rate as high as 70% [58]. This eventuality necessitates a planned second-look procedure 6–12 months following initial CWU surgery. In total, CWU patients show a nearly 3 times greater likelihood of recurrence than with CWD surgery [59].
Recent advances in the use of the endoscope and in optical technology have led to new treatment modalities for middle ear surgery as the endoscope allows viewing of hidden areas that are difficult when utilizing microscopic vision [58]. Utilization of high definition visualization, such as a 4K magnification endoscope and narrow band imaging filter has been shown to improve visualization of tissue based on varying degrees of vascularity allowing for better differentiation between pathology and normal anatomy [58]. This may then aid or promote the choice of a CWU procedure and lead to a decrease in the number of recurrences and residual disease associated with this procedure [58].
Conclusion
As with any disease process, careful and early evaluation is the key to early diagnosis through the use of modern as well as traditional evaluation methods in order to facilitate treatment and management. Inherent in that process is the need for thorough knowledge and complete awareness of important anatomic structures, how they relate to function and management of the middle ear, the histology involved and the specific surgical process that may require customization to each patient depending on the disease extent involved, making for less complications of cholesteatomas and preserved hearing.
Acknowledgements
Thanks and gratitude to Dr. Robert Shih for his tremendous assistance in radiographic image selection and interpretation and to Drs. Caroline Schlocker and George Conley for their intraoperative images and interpretation.
Compliance with Ethical Standards
Conflict of interest
James T. Castle declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Disclaimer: The opinions and assertions expressed herein are those of the authors and are not to be construed as official or representing the views of the Department of the Navy or the Department of Defense. I certify that all individuals who qualify as authors have been listed; each has participated in the conception and design of this work, the writing of the document, and the approval of the submission of this version; that the document represents valid work; that if we used information derived from another source, we obtained all necessary approvals to use it and made appropriate acknowledgements in the document; and that each takes public responsibility for it.
References
- 1.Yung M, Tono T, Olszewska E, et al. EAONO/JOS Joint consensus statements on the definitions, classification and staging of middle ear cholesteatoma. J Int Adv Otol. 2017;13:1–8. doi: 10.5152/iao.2017.3363. [DOI] [PubMed] [Google Scholar]
- 2.Müller J. Ueber den feineren bau und die formen der krankhaften geschwulste. Berlin, G Reimer. 1838. Folio.
- 3.De Verney JG. Traité de l’organie de l’ouïe. Paris: E. Michallet; 1683. [Google Scholar]
- 4.Aquino JE, Cruz Filho NA, de Aquino JN. Epidemiology of middle ear and mastoid cholesteatomas: study of 1146 cases. Braz J Otorhinolaryngol. 2011;77:341–347. doi: 10.1590/S1808-86942011000300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett M, Warren F, Jackson GC. Congenital cholesteatoma: theories, facts and 53 patients. Otolaryngol Clin N Am. 2006;39:1081–1094. doi: 10.1016/j.otc.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Shohet JA, De Jong AI. The management of pediatric cholesteatoms. Otolaryngol Clin N Am. 2002;35:841–851. doi: 10.1016/S0030-6665(02)00052-X. [DOI] [PubMed] [Google Scholar]
- 7.Olszewska E, Wagner M, Bernal-Sprekelsen M, et al. Etiopathogenesis of cholesteatoma. Eur Arch Otorhinolaryngol. 2004;261:6–24. doi: 10.1007/s00405-003-0623-x. [DOI] [PubMed] [Google Scholar]
- 8.Barath K, Huber AM, Stampfil P, et al. Neuroradiology of cholesteatomas. AJNR Am J Neuroradiol. 2011;32:221–229. doi: 10.3174/ajnr.A2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jennings BA, Prinsley P, Philpott C, et al. The genetics of cholesteatoma. A systematic review using narrative synthesis. Clin Otolaryngol. 2017;43:55–67. doi: 10.1111/coa.12900. [DOI] [PubMed] [Google Scholar]
- 10.Bois E, Nassar M, Zenaty D, et al. Otologic disorders in Turner syndrome. Eur Ann Otorhinolaryngol Head Neck Dis. 2017;135:21–24. doi: 10.1016/j.anorl.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Lim DBN, Gault EJ, Kubba H, et al. Cholesteatoma has a high prevalence in Turner syndrome, highlighting the need for earlier diagnosis and the potential benefits of otoscopy training for paediatricians. Acta Paediatr. 2014;103:e282–e287. doi: 10.1111/apa.12450. [DOI] [PubMed] [Google Scholar]
- 12.Hall JE, Richter GT, Choo DI. Surgical management of otologic disease in pediatric patients with Turner syndrome. Int J Pediatr Otorhinolaryngol. 2009;73:57–65. doi: 10.1016/j.ijporl.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 13.Mann W, Al-Nawas B, Wriedt S. Cholesteatoma of the hypotympanum in a patient with Treacher Collins syndrome. Auris Nasus Larynx. 2014;41:101–104. doi: 10.1016/j.anl.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Nash R, Possamai V, Maskell S, et al. Canal wall reconstruction and preservation in the surgical management of cholesteatoma in children with Down’s syndrome. Int J Pediatr Otorhinolaryngol. 2014;78:1747–1751. doi: 10.1016/j.ijporl.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 15.Paulson LM, Weaver TS, Macarthur CJ. Outcomes of tympanostomy tube placement in children with Down syndrome—a retrospective review. Int J Pediatr Otorhinolaryngol. 2014;78:223–226. doi: 10.1016/j.ijporl.2013.10.062. [DOI] [PubMed] [Google Scholar]
- 16.Bacciu A, Pasanisi E, Vincenti V, et al. Surgical treatment of middle ear cholesteatoma in children with Down syndrome. Otol Neurotol. 2005;26:1007–1010. doi: 10.1097/01.mao.0000185042.46523.9b. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki C, Ohtani I. Bone destruction resulting from rupture of a cholesteatoma sac: temporal bone pathology. Otol Neurotol. 2004;25:674–677. doi: 10.1097/00129492-200409000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Büchner SA, Itin P. Focal dermal hypoplasia syndrome in a male patient. Report of a case and histologic and immunohistochemical studies. Arch Dermatol. 1992;128:1078–1082. doi: 10.1001/archderm.1992.01680180072008. [DOI] [PubMed] [Google Scholar]
- 19.Imbery TE, Sobin LB, Commesso E, et al. Long-term otologic and audiometric outcomes in patients with cleft palate. Otolaryngol Head Neck Surg. 2017;157:676–682. doi: 10.1177/0194599817707514. [DOI] [PubMed] [Google Scholar]
- 20.Kopcsányi G, Vincze O, Bagdán V, et al. Retrospective analysis of tympanoplasty in children with cleft palate: a 24-year experience. II. Cholesteatomatous cases. Int J Pediatr Otorhinolaryngol. 2015;79:698–706. doi: 10.1016/j.ijporl.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Djurhuus BD, Skytthe A, Faber CE, Christensen K. Cholesteatoma risk in 8,593 orofacial cleft cases and 6,989 siblings: a nationwide study. Laryngoscope. 2015;125:1225–1229. doi: 10.1002/lary.25022. [DOI] [PubMed] [Google Scholar]
- 22.Lau CC, Loh KK, Kunaratnam N. Middle ear diseases in cleft palate patients in Singapore. Ann Acad Med Singap. 1988;17:372–374. [PubMed] [Google Scholar]
- 23.Kuo CL, Lien CF, Chu CH, et al. Otitis media with effusion in children with cleft lip and palate: a narrative review. Int J Pediatr Otorhinolaryngol. 2013;77:1403–1409. doi: 10.1016/j.ijporl.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 24.James AL, Papsin BC. Some considerations in congenital cholesteatoma. Curr Opin Otolaryngol Head Neck Surg. 2013;21:431–439. doi: 10.1097/MOO.0b013e328364b457. [DOI] [PubMed] [Google Scholar]
- 25.Harris L, Cushing SL, Hubbard B, et al. Impact of cleft palate type on the incidence of acquired cholesteatoma. Int J Pediatr Otorhinolaryngol. 2013;77:695–698. doi: 10.1016/j.ijporl.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Kuo CL, Shiao AS, Wen HC, et al. Increased risk of cholesteatoma among patients with allergic rhinitis: a nationwide investigation. Laryngoscope. 2017;128:547–553. doi: 10.1002/lary.26220. [DOI] [PubMed] [Google Scholar]
- 27.Kuo CL, Shiao AS, Yung M, et al. Updates and knowledge gaps in cholesteatoma research. Biomed Res Int. 2015;2015:1–17. doi: 10.1155/2015/854024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michaels L. Origin of congenital cholesteatoma from a normally occurring epidermoid rest in the developing middle ear. Int J Pediatr Otorhinolaryngol. 1988;15:51–65. doi: 10.1016/0165-5876(88)90050-X. [DOI] [PubMed] [Google Scholar]
- 29.Levenson MJ, Michaels L, Parisier SC, et al. Congenital cholesteatomas in children: an embryologic correlation. Laryngoscope. 1988;98:949–955. doi: 10.1288/00005537-198809000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Aimi K. Role of the tympanic ring in the pathogenesis of congenital cholesteatoma. Laryngoscope. 1983;93:1140–1146. doi: 10.1288/00005537-198309000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Tos M. A new pathogenesis of mesotympanic (congenital) cholesteatoma. Laryngoscope. 2000;110:1890–1897. doi: 10.1097/00005537-200011000-00023. [DOI] [PubMed] [Google Scholar]
- 32.Kuo CL. Etiopathogenesis of acquired cholesteatoma: prominent theories and recent advances in biomolecular research. Laryngoscope. 2015;125:234–240. doi: 10.1002/lary.24890. [DOI] [PubMed] [Google Scholar]
- 33.Sudhoff H, Tos M. Pathogenesis of attic cholesteatoma: clinical and immunohistochemical support for combination of retraction theory and proliferation theory. Am J Otol. 2000;21:786–792. [PubMed] [Google Scholar]
- 34.Karmody CS, Northrop C. The pathogenesis of acquired cholesteatoma of the human middle ear: support for the migration hypothesis. Otol Neurotol. 2012;33:42–47. doi: 10.1097/MAO.0b013e31823c919c. [DOI] [PubMed] [Google Scholar]
- 35.Falcioni M, Taibah A, Rohit Pulsatile tinnitus as a rare presenting symptom of residual cholesteatoma. J Laryngol Otol. 2004;118:165–166. doi: 10.1258/002221504772784694. [DOI] [PubMed] [Google Scholar]
- 36.Ahn JM, Huang CC, Abramson M. Localization of interleukin-1 in human cholesteatoma. Am J Otolaryngol. 1990;11:71–77. doi: 10.1016/0196-0709(90)90001-C. [DOI] [PubMed] [Google Scholar]
- 37.Venail F, Bonafe A, Poirrier V, et al. Comparison of echo-planar diffusion-weighted imaging and delayed postcontrast T1-weighted MR imaging for the detection of residual cholesteatoma. AJNR Am J Neuroradiol. 2008;29:1363–1368. doi: 10.3174/ajnr.A1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corrales CE, Blevins NH. Imaging for evaluation of cholesteatoma: current concepts and future directions. Curr Opin Otolaryngol Head Neck Surg. 2013;21:461–467. doi: 10.1097/MOO.0b013e328364b473. [DOI] [PubMed] [Google Scholar]
- 39.Henninger B, Kremser C. Diffusion weighted imaging for the detection and evaluation of cholesteatoma. World J Radiol. 2017;28:217–222. doi: 10.4329/wjr.v9.i5.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vercruysse JP, De Foer B, Pouillon M, et al. The value of diffusion-weighted MR imaging in the diagnosis of primary acquired and residual cholesteatoma: a surgical verified study of 100 patients. Eur Radiol. 2006;16:1461–1467. doi: 10.1007/s00330-006-0160-2. [DOI] [PubMed] [Google Scholar]
- 41.Más-Estellés F, Mateos-Fernández M, Carrascosa-Bisquert B, et al. Contemporary non-echo-planar diffusion-weighted imaging of middle ear cholesteatomas. Radiographics. 2012;32:1197–1213. doi: 10.1148/rg.324115109. [DOI] [PubMed] [Google Scholar]
- 42.Caponetti G, Thompson LD, Pantanowitz L. Cholesteatoma. Ear Nose Throat J. 2009;88:1196–1198. [PubMed] [Google Scholar]
- 43.Bassiouny M, Badour N, Omran A, et al. Histopathological and immunohistochemical characteristics of acquired cholesteatoma in children and adults. EJENTAS. 2012;13:7–12. [Google Scholar]
- 44.Rothschild S, Ciernik IF, Hartmann M, et al. Cholesteatoma triggering squamous cell carcinoma: case report and literature review of a rare tumor. Am J Otolaryngol. 2009;30:256–260. doi: 10.1016/j.amjoto.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Hamed MA, Nakata S, Shiogama K, et al. Cytokeratin 13, Cytokeratin 17, and Ki-67 expression in human acquired cholesteatoma and their correlation with its destructive capacity. Clin Exp Otorhinolaryngol. 2017;10:213–220. doi: 10.21053/ceo.2016.01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chole RA, Tinling SP. Basal lamina breaks in the histogenesis of cholesteatoma. Laryngoscope. 1985;95:270–275. doi: 10.1288/00005537-198503000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Ferlito O, Devaney KO, Rinaldo A, et al. Clinicopathological consultation ear cholesteatoma versus cholesterol granuloma. Ann Otol Rhinol Laryngol. 1997;106:79–85. doi: 10.1177/000348949710600114. [DOI] [PubMed] [Google Scholar]
- 48.Preciado DA. Biology of cholesteatoma: special considerations in pediatric patients. Int J Pediatr Otorhinolaryngol. 2012;76:319–321. doi: 10.1016/j.ijporl.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 49.Maniu A, Harabagiu O, Perde Schrepler M, et al. Molecular biology of cholesteatoma. Rom J Morphol Embryol. 2014;55:7–13. [PubMed] [Google Scholar]
- 50.Sudhoff H, Dazert S, Gonzales AM, et al. Angiogenesis and angiogenic growth factors in middle ear cholesteatoma. Am J Otol. 2000;21:793–798. [PubMed] [Google Scholar]
- 51.Fukudome S, Wang C, Hamajima Y, et al. Regulation of the angiogenesis of acquired middle ear cholesteatomas by inhibitor of DNA binding transcription factor. JAMA Otolaryngol Head Neck Surg. 2013;139:273–278. doi: 10.1001/jamaoto.2013.1750. [DOI] [PubMed] [Google Scholar]
- 52.Haruyama T, Furukawa M, Kusunoki T, et al. Expression of IL-17 and its role in bone destruction in human middle ear cholesteatoma. J Otorhinolaryngol Relat Spec. 2010;72:325–331. doi: 10.1159/000319897. [DOI] [PubMed] [Google Scholar]
- 53.Olszewska E, Matulka M, Mroczko B, et al. Diagnostic value of matrix metalloproteinase 9 and tissue inhibitor of matrix metalloproteinases 1 in cholesteatoma. Histol Histopathol. 2016;31:307–315. doi: 10.14670/HH-11-677. [DOI] [PubMed] [Google Scholar]
- 54.Chen AP, Wang B, Zhong F, et al. Expression levels of receptor activator of nuclear factor-κB ligand and osteoprotegerin are associated with middle ear cholesteatoma risk. Acta Otolaryngol. 2015;135:655–666. doi: 10.3109/00016489.2015.1011789. [DOI] [PubMed] [Google Scholar]
- 55.Kuo CL, Liao WH, Shiao AS. A review of current progress in acquired cholesteatoma management. Eur Arch Otorhinolaryngol. 2015;272:3601–3609. doi: 10.1007/s00405-014-3291-0. [DOI] [PubMed] [Google Scholar]
- 56.Morita Y, Takahashi K, Izumi S, et al. Risk factors of recurrence in pediatric congenital cholesteatoma. Otol Neurotol. 2017;38:1463–1469. doi: 10.1097/MAO.0000000000001587. [DOI] [PubMed] [Google Scholar]
- 57.Vital V. Pediatric cholesteatoma: personal experience and review of the literature. Otorhinolaryngol Head Neck Surg. 2011;45:5–14. [Google Scholar]
- 58.Zhang H, Wong PY, Magos T, et al. Use of narrow band imaging and 4K technology in otology and neuro-otology: preliminary experience and feasibility study. Eur Arch Otorhinolaryngol. 2017;275:301–305. doi: 10.1007/s00405-017-4783-5. [DOI] [PubMed] [Google Scholar]
- 59.Tomlin J, Chang D, McCutcheon B, et al. Surgical technique and recurrence in cholesteatoma: a meta-analysis. Audiol Neurootol. 2013;18:135–142. doi: 10.1159/000346140. [DOI] [PubMed] [Google Scholar]