

Abstract
Ultrafast-folding proteins are important for combining experiment and simulation to give complete descriptions of folding pathways. The WW domain family comprises small proteins with a three-stranded antiparallel β-sheet topology. Previous studies on the 57-residue YAP 65 WW domain indicate the presence of residual structure in the chemically denatured state. Here we analyze three minimal core WW domains of 38–44 residues. There was little spectroscopic or thermodynamic evidence for residual structure in either their chemically or thermally denatured states. Folding and unfolding kinetics, studied by using rapid temperature-jump and continuous-flow techniques, show that each domain folds and unfolds very rapidly in a two-state transition through a highly compact transition state. Folding half-times were as short as 17 μs at 25°C, within an order of magnitude of the predicted maximal rate of loop formation. The small size and topological simplicity of these domains, in conjunction with their very rapid two-state folding, may allow us to reduce the difference in time scale between experiment and theoretical simulation.
The combination of high-resolution experimental data with detailed computer simulation is required to solve the pathway of protein folding at atomic resolution. Unfortunately, there is an incompatibility between the time regime for folding and unfolding of most proteins [the millisecond to second range (1)] and that of current molecular dynamics (MD) simulations, which are typically conducted at high temperatures (>200°C) and last for less than 1 μs (2). Thus, the folding and unfolding pathways of the majority of real proteins are currently not testable by MD simulations under physiological conditions, although the αhelical engrailed homeodomain folds and unfolds so rapidly that simulations could be performed close to experimentally accessible conditions (3).
To continue this benchmarking of simulation with experiment, we are studying the all-β-sheet WW domains, a large family of small single-domain proteins (4) (typically 38–44 amino acids in length) that contain a distorted three-stranded antiparallel β-sheet (5). In contrast to all α-helical proteins, where the secondary structure is defined more by local interactions and the rules governing folding and stability are fairly well understood (6), analogous processes in β-sheet proteins are less clearly defined. As a number of WW domains have well defined monomeric native structures (7–9) that lack cofactors, prosthetic groups, disulfide linkages, and cis prolines that can complicate kinetic analyses, these domains are suitable model systems for understanding the different factors that contribute to β-sheet folding and stability.
Previous studies focused on a 57-residue human Yes kinase associated protein (YAP 65), where folding and unfolding are reversible and very rapid (10). Evidence was presented for a residual aromatic cluster in the chemically, but not the thermally, denatured state (11). To circumvent potential problems that may be associated with the extended termini of the 57-residue YAP 65 WW domain, we chose to study a series of shortened WW domain constructs 38–44 residues in length (Fig. 1). These correspond to the core domains of the human Yes-associated protein, the murine formin-binding protein 28 (FBP28), and a de novo designed WW domain (WW prototype) (7). We present here a detailed characterization of the equilibrium and kinetic behavior of these truncated WW domains and show that they fold more rapidly at room temperature than any previously reported β-sheet protein. This accelerated folding apparently does not depend on significant residual aromatic structure in the denatured state.
Figure 1.

Sequence alignment of the YAP 65, FBP28, and prototype WW domains. Black boxes indicate identical residues. Asterisks mark the highly conserved tryptophan residues. In conjunction with other highly conserved residues (black circles), the tryptophan residues form the hydrophobic core (5). The positions of Strands I–III, connected by β-hairpins, are indicated above the sequence alignment.
Materials and Methods
Reagents.
Recombinant WW prototype, wild-type FBP28, and FBP28 W30A domain were expressed as previously described (5). The YAP 65 WW domain was synthesized by using standard fluorenylmethoxycarbonyl chemistry and purified to >95% homogeneity (by G. Bloomberg, Department of Biochemistry, University of Bristol, Bristol, U.K.) by using reverse-phase HPLC. The mass and identity of peptides were confirmed by using matrix-assisted laser desorption ionization mass spectrometry. Calculated extinction coefficients (12) were used to determine protein concentration. The following buffers were used (free acid and sodium salt in 18 MΩ water), with the ionic strength kept constant at 150 mM by using NaCl [excluding solutions containing guanidinium chloride (GdmCl)]: 20 mM sodium formate buffer (pH 2.75, 3.0, and 3.5); 20 mM sodium acetate (pH 4.0, 4.5, and 5.0); 20 mM sodium phosphate pH 7.0; 20 mM sodium 3-[N-morpholino]propanesulphonic acid (Mops), pH 7.0. Concentrations of urea and guanidine hydrochloride solutions were determined from their refractive index (13).
Equilibrium Measurements.
Fluorescence spectra of samples were measured in a 4 × 10-mm cell thermostated to ±0.1°C in an Aminco-Bowman SLM Series 2 luminescence spectrometer (Aminco-SLM). Excitation of intrinsic fluorophores along the short pathlength was at 280 nm (4-nm bandwidth), with measurement of fluorescence emission from 300 to 450 nm (4-nm bandwidth, 1-nm/s scan rate). Chemical denaturation was measured by using separate filtered samples that contained 2–10 μM WW domain in 20 mM sodium phosphate buffer, pH 7.0 (or Mops, pH 7.0, where GdmCl is used), with varying final concentrations of urea or guanidinium hydrochloride. Samples were equilibrated at the appropriate temperature for 3–16 h before measurement.
A more accurate automated procedure was adopted for urea-induced denaturation by using the titration facility of an Aviv 202SF spectropolarimeter (Aviv Associates, Lakewood, NJ). Samples (1.4 ml) containing 10–20 μM WW domain in 20 mM sodium phosphate buffer, pH 7.0, were thermostated at 25 ± 0.1°C with constant stirring. The urea concentration was increased in 0.04–0.1 M increments by removal of solution from the cuvette and subsequent replacement with an equal volume of concentrated urea solution containing an equimolar concentration of WW domain. The time between injections, varied from 3 to 10 min, made no difference to the observed data and gave consistent results to denaturation experiments performed manually. Total fluorescence emission at >320 nm was collected on excitation at either 280 nm (fluorescence emission only studies) or 230 nm [simultaneous collection of far-UV circular dichroism (CD) and fluorescence emission data]. Equilibrium denaturation curves were fitted to a two-state model (14).
Steady-state far- and near-UV CD measurements were made by using either a Jasco-J720 (Easton, MD) or the Aviv 202SF spectropolarimeter, with thermostatting to within ±0.1°C of the desired temperature. CD spectra and thermal melts were measured in the far-UV region by using a 1-mm pathlength cell containing 65 μM protein in the appropriate buffer, whereas in the near-UV region, a 4-mm pathlength cell containing 75 μM protein was used. For spectral acquisition, 16 scans were typically recorded by using a bandwidth of 1 nm and scan rates of 10–50 nm/min. The high-protein, amino acid, and denaturant concentrations used in these experiments prevented accurate spectra measurement below 212 nm.
Degassed solutions were used in thermal denaturation experiments, and the ellipticity at 230 nm was measured between 4 and 98°C (by using scan rates of 50°C/h). Thermal denaturation curves were fitted to standard equations describing a two-state transition, assuming the excess heat capacity for unfolding (ΔC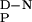 ) to be temperature independent (15). Thermal melts were highly reproducible, with the thermal midpoint (Tm) and the enthalpy for unfolding (ΔHD-N) typically varying by less than 1.2 K and 9 kJ/mol, respectively, in repeat measurements.
) to be temperature independent (15). Thermal melts were highly reproducible, with the thermal midpoint (Tm) and the enthalpy for unfolding (ΔHD-N) typically varying by less than 1.2 K and 9 kJ/mol, respectively, in repeat measurements.
Kinetic Studies.
Temperature-jump measurements in a 0.8-ml cell were made by using a DIA-RT capacitor-discharge T-jump apparatus (Dia-Log, Düsseldorf, Germany). Samples containing 5–50 μM WW domain in 20 mM sodium phosphate buffer, pH 7.0, were filtered and thoroughly degassed before the cell was thermostated. Discharges (10–30 kV) were used to effect 1–3°C jumps [calibrated as previously described (3)] to a final temperature of 25 ± 0.1°C, with complete heating within 0.5 μs under optimal conditions.
Typically, 8–30 traces were averaged and analyzed by using kaleidagraph (Synergy Software, Reading, PA), having removed or included the intrinsic change in the tryptophan fluorescence associated with the temperature jump. Relaxation reactions fitted well to single exponential functions, except when the intrinsic change in fluorescence after temperature jumps was included and the data were fitted to double exponential functions. The observed rate constant (kobs) for relaxation was independent of the starting temperature, protein concentration, temperature-jump size, capacitor used, or whether the observed data were fitted to single or double exponential functions.
Continuous-flow measurements were made on an instrument similar to that described by Shastry et al. (16), except that the titanium mixing ball was replaced by a 150- to 200-μm-diameter glass bead, and the flow rate was calculated for each acquisition by using a calibrated electronic timing device. The quartz flow cuvette was illuminated at 280 nm by using a Hg/Xe light source and monochromator, with the fluorescence intensity collected at >320 nm being recorded by using a Kodak KAF1401E charge-coupled device in a MicroMax Interline digital camera (Princeton Instruments, Trenton, NJ). Data were corrected and normalized as previously described (16). The dead time of the instrument, measured by using a standard quenching reaction (17), was 75 μs at the fastest flow rates examined (≈1 ml/s). Lower flow rates (≈0.65 ml/s) were used to acquire data where concentrated urea solutions (up to 9 M) were being mixed or slower rates measured, and under these conditions, the dead time of the instrument was increased to ≈175 μs. Folding and unfolding of the YAP 65 WW domain were measured at a final concentration of 10 μM. Rate constants were determined by fitting data to single exponential functions by using kaleidagraph software. Replicate measurements were highly superimposable.
Curve Fitting.
The mD-N value in equilibrium denaturation experiments, which defines the dependence of the free energy for unfolding (ΔGD-N) on urea concentration (δΔGD-N/δ [denaturant]), was constrained to the average value determined by free-fitting curves for 15 independent experiments (1.6 ± 0.04 kJ/mol). The dependence of log kobs for the YAP 65 and FBP28 W30A WW domains on urea concentration was fitted to an equation describing a two-state transition, where kobs displays a strictly linear dependence on urea concentration (14). A second-order polynomial term was included to allow fitting of the curvature in the observed kinetic data for the WW prototype (18).
Results
Far-UV CD Spectroscopy.
Contributions from aromatic amino acids, rather than the intrinsic secondary structure, dominate the far-UV CD spectrum of the native conformation of each WW domain (11), with a strong peak of positive ellipticity centered at ≈230 nm (Fig. 2A). These aromatic contributions result in residual ellipticity in the far-UV CD spectrum of both the GdmCl- and urea-denatured states (not shown) of each WW domain. The spectra of the chemically denatured states were significantly different from those of the corresponding thermally denatured states, which resemble that expected for a random coil conformation. This residual ellipticity could misleadingly be interpreted as evidence for the chemically and thermally denatured states being distinct species. However, the residual ellipticity of an appropriate mixture containing free aromatic amino acids in denaturant had greater intrinsic positive ellipticity than the chemically denatured state of each WW domain (Fig. 2A). Subtracting these reference spectra from that of the chemically denatured state of each WW domain yielded a difference spectrum qualitatively similar to that of the thermally denatured state and approximated to that of a random coil at 298 K. Control experiments showed that the far-UV CD spectrum of the free aromatic amino acid mixture changes only marginally as a function of temperature, and subtracting this spectrum from that of the thermally denatured state increased its qualitative similarity to that of chemically denatured state (data not shown). The different amplitudes of the far-UV CD spectra for the chemically and thermally denatured states are attributed to the linear temperature dependence of the peptide bond far-UV CD signal in the random coil conformation. Therefore, the far-UV CD data provide no evidence for significant structural differences between the chemically and thermally denatured states of each WW domain.
Figure 2.

CD spectra of the YAP 65 WW domain under native and denaturing conditions. (A) Far-UV CD spectra of the YAP 65 WW domain at 298 K in 20 mM Mops, pH 7.0 (upper solid line); 371 K in 20 mM Mops, pH 7.0 (lower solid line); 298 K in 5.6 M GdmCl/20 mM Mops, pH 7.0 (- - - - -). Spectrum of solutions at 298 K containing free aromatic amino acids (N-acetyl tryptophanamide, N-acetyl tyrosinamide, and N-acetyl phenylalanine) at equimolar concentrations to the aromatic content of the YAP 65 WW domain, in the presence (— – — –) or absence (· · · ·) of 5.6 M GdmCl/20 mM Mops, pH 7.0. The difference spectrum (– · · · –) generated by subtracting the contribution of the free aromatic amino acids in 5.6 M GdmCl and at 298 K from that of the YAP 65 WW denatured state under identical conditions. Similar results were obtained for the wild-type FBP28 and prototype WW domains. (B) Near-UV spectra of the wild-type FBP28 WW domain under native and denaturing conditions at 298 K. The near-UV CD spectrum acquired in 20 mM Mops, pH 7.0 (· · · ·) and in 5.6 M GdmCl/20 mM Mops, pH 7.0 (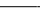 ). The near-UV CD spectrum of free aromatic amino acids at equimolar concentrations to the aromatic content of wild-type protein, in the presence (- - - - -) and absence of 5.6 M GdmCl (– · · · –), is similar to that of the chemically denatured domain. Similar results were obtained for the prototype WW domain.
). The near-UV CD spectrum of free aromatic amino acids at equimolar concentrations to the aromatic content of wild-type protein, in the presence (- - - - -) and absence of 5.6 M GdmCl (– · · · –), is similar to that of the chemically denatured domain. Similar results were obtained for the prototype WW domain.
Near-UV CD Spectroscopy.
The near-UV CD spectrum of the native conformation of each WW domain had significant positive ellipticity (Fig. 2B), consistent with a chiral environment around one or more aromatic amino acids. Most, but not all, of this ellipticity was lost when the WW domains were dissolved in concentrated denaturant solutions. The residual ellipticity is suggested to be indicative of a residual aromatic cluster in the chemically denatured state of the 57-residue YAP 65 WW domain (11). For the prototype and FBP28 WW domains, the ellipticity of the GdmCl-denatured state was very similar to that of a mixture of free aromatic amino acids under identical solvent conditions (Fig. 2B) and very close to that of the thermally denatured state (data not shown). Thus, again there appears to be little reliable spectroscopic evidence for structured or distinct chemically and thermally denatured states.
It is less clear-cut whether there is residual aromatic structure in the chemically denatured state of the shortened YAP 65 WW domain. There were small differences between the near-UV CD spectrum of the chemically denatured state and a mixture of aromatic amino acids in concentrated GdmCl solutions (not shown). However, the signal-to-noise ratio of these experiments was very low, with the small spectral differences being observed only at higher protein concentrations (180 μM) than required to characterize the spectra of the other WW domains (75 μM). Thus, it is presently unclear whether the residual ellipticity observed in the near UV CD region reflects genuine asymmetry in the environment of core aromatic residues or whether this may have resulted from intermolecular association or aggregation.
Thermal Denaturation Measurements.
Thermal denaturation of the WW domains, measured by using far-UV CD spectroscopy, was highly reversible (Fig. 3). Differential scanning calorimetry (DSC) was hampered by the low unfolding enthalpies of these domains. There was aggregation of wild-type FBP28 and prototype WW domains at the higher protein concentrations needed to increase the sensitivity of DSC experiments. Fortunately, this was not encountered with the YAP 65 WW domain, where the calorimetric Tm was found to be consistent with the values determined by using far-UV and near-UV CD spectroscopy (not shown).
Figure 3.

Thermal denaturation of the YAP 65 WW domain. Thick solid lines indicate the best fit of repeated thermal denaturation curves to a two-state transition assuming the experimentally determined ΔC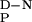 value (see Inset). The parameters from these fits were used to define the extrapolated baseline for the thermally denatured state (thin solid line). The denatured baseline coincides with the intrinsic ellipticity at 230 nm of the chemically denatured state (filled circle) after the intrinsic ellipticity of the aromatic acids has been subtracted (data from Fig. 2A used). Similar results were observed for the other WW domains. (Inset) Dependence of ΔHD-N and Tm values (hollow circles) on pH for the wild-type FBP28 WW domain. Data were determined from thermal denaturation experiments monitored by using far-UV CD spectroscopy in the pH range 2.75–7.0. The gradient of the best line fit to the data (dashed line), weighted by the errors, defines the ΔC
value (see Inset). The parameters from these fits were used to define the extrapolated baseline for the thermally denatured state (thin solid line). The denatured baseline coincides with the intrinsic ellipticity at 230 nm of the chemically denatured state (filled circle) after the intrinsic ellipticity of the aromatic acids has been subtracted (data from Fig. 2A used). Similar results were observed for the other WW domains. (Inset) Dependence of ΔHD-N and Tm values (hollow circles) on pH for the wild-type FBP28 WW domain. Data were determined from thermal denaturation experiments monitored by using far-UV CD spectroscopy in the pH range 2.75–7.0. The gradient of the best line fit to the data (dashed line), weighted by the errors, defines the ΔC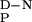 .
.
The change in heat capacity on unfolding, a key thermodynamic parameter relating the amount of hydrophobic area that becomes solvent exposed on unfolding (19), was calculated from the variation of ΔHD-N and Tm of the wild-type FBP28 WW domain with pH (Fig. 3). The experimentally determined ΔC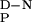 of 1,745 ± 67 J/mol⋅K (r = 0.9898) was consistent with the value expected for proteins of this size and in good agreement with values determined by using empirical relationships (19). As the ΔC
of 1,745 ± 67 J/mol⋅K (r = 0.9898) was consistent with the value expected for proteins of this size and in good agreement with values determined by using empirical relationships (19). As the ΔC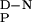 , the Tm values were well defined, and the slope of the denatured state baselines coincides with the random coil values at 298 K (Fig. 3), it was possible to calculate the thermodynamic parameters for thermal denaturation accurately (Table 1).
, the Tm values were well defined, and the slope of the denatured state baselines coincides with the random coil values at 298 K (Fig. 3), it was possible to calculate the thermodynamic parameters for thermal denaturation accurately (Table 1).
Table 1.
Equilibrium denaturation parameters for the WW domains at 298 K and pH 7.0
| Protein | Buffer (pH 7.0) | mD-N*, kJ/mol⋅M | [Denaturant]50%, M | ΔG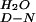 †, kJ/mol †, kJ/mol |
Tm, K | ΔH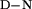 ‡, kJ/mol ‡, kJ/mol |
ΔG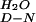 §, kJ/mol §, kJ/mol |
|---|---|---|---|---|---|---|---|
| YAP | Phosphate | 1.6 ± 0.04 | 2.96 ± 0.06 | 4.74 ± 0.10 | 316.6 ± 0.6 | 100 ± 4.6 | 4.32 ± 0.85 |
| YAP | Mops | ND | ND | ND | 313.3 ± 0.6 | 93.3 ± 4.3 | 3.85 ± 0.96 |
| Prototype | Phosphate | 1.6 ± 0.04 | 3.15 ± 0.11 | 5.04 ± 0.16 | 320 ± 0.3 | 104.6 ± 2.5 | 5.46 ± 0.56 |
| Prototype | Mops | ND | ND | ND | 320 ± 0.2 | 88.7 ± 1.7 | 4.81 ± 0.38 |
| FBP28 | Phosphate | 1.6 ± 0.04 | ND | ND | 334.1 ± 0.4 | 110.5 ± 1.3 | 8.32 ± 0.29 |
| FBP28 | Mops | 3.55 ± 0.11¶ | 1.96 ± 0.06¶ | 6.96 ± 0.19 | 330 ± 0.4 | 103.8 ± 2.9 | 7.19 ± 0.66 |
| FBP28 W30A | Phosphate | 1.6 ± 0.04 | 4.68 ± 0.26 | 7.49 ± 0.34‖ | 329 ± 0.2 | 101.7 ± 2.1 | 6.88 ± 0.47 |
| FBP28 W30A | Mops | 3.73 ± 0.15¶ | 1.40 ± 0.12¶ | 5.22 ± 0.29 | 327.4 ± 0.3 | 92.9 ± 2.5 | 5.90 ± 0.56 |
ND, not determined. The denaturant midpoint is too low or too high for the data to be fitted accurately.
The average mD-N value was used as a constrained parameter in fitting urea denaturation curves.
The free energy for unfolding in water (ΔG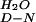 ) was calculated by using the mD-N and midpoint denaturant concentration ([Denaturant]50%) values, as previously described (14).
) was calculated by using the mD-N and midpoint denaturant concentration ([Denaturant]50%) values, as previously described (14).
ΔH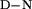 refers to the enthalpy of unfolding at the Tm.
refers to the enthalpy of unfolding at the Tm.
Calculated by using standard thermodynamic relationships. The ΔC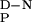 measured for the wild-type FBP28 WW domain (1,745 ± 67 J/mol⋅K) was assumed to fit the thermal denaturation curves of the other WW domains.
measured for the wild-type FBP28 WW domain (1,745 ± 67 J/mol⋅K) was assumed to fit the thermal denaturation curves of the other WW domains.
GdmCl was used as the denaturant in these experiments.
This value may be an overestimate, as the average mD-N value was used to calculate the ΔG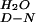 . The W30A mutation may reduce the mD-N value and, by corollary, the ΔG
. The W30A mutation may reduce the mD-N value and, by corollary, the ΔG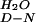 value.
value.
Chemical Denaturation Measurements.
The transition regions of the urea equilibrium denaturation curves of the YAP 65, FBP28, and prototype WW domains were very broad, as expected for small proteins. The unfolding transition for each protein was almost coincident when measured by either far-UV CD or fluorescence emission spectroscopy (Fig. 4A). Fluorescence was the preferred probe, as the slope of denatured baselines was generally defined better and less prone to fitting errors than in far-UV CD experiments. The free energy of unfolding for the YAP 65 and prototype WW domains in water were very similar (Table 1). Unfortunately, it was not possible to effect complete denaturation of the wild-type FBP28 WW domain by using urea (not shown). Consequently, a destabilized mutant (W30A) (7) was used as a pseudo-wild-type domain in equilibrium urea denaturation studies (data not shown; Table 1). Equilibrium denaturation of both FBP28 WW domains could also be measured under similar conditions by using GdmCl, a stronger denaturant (Fig. 4B). Reassuringly, the calculated ΔGD-N values in water for each WW domain were very similar and essentially independent of the buffer or denaturant used (heat, urea, or GdmCl). Thus, in addition to the earlier spectroscopic evidence that suggests that the chemically and thermally denatured states are structurally similar, it appears they are also thermodynamically equivalent.
Figure 4.

Urea-induced equilibrium denaturation of the prototype WW domain at 298 K. (A) Urea-induced denaturation of 10 μM protein in 20 mM sodium phosphate buffer, pH 7.0, was monitored by total fluorescence emission at >320 nm (filled circles) and far-UV CD spectroscopy at 230 nm (hollow circles). (B) GdmCl-induced equilibrium denaturation of wild-type (filled circles) and W30A mutant FBP28 WW domains (hollow circles) at 298 K. These data are normalized to allow plotting on a common scale. Solid lines indicate the best fit of each data set to a two-state transition. The ΔGD-N and mD-N values were within error irrespective of whether these data were fitted assuming nonsloping or sloping baselines (35) (Table 1).
Kinetic Measurements.
Submillisecond relaxation kinetics were measured after microsecond temperature jumps. Rates (Fig. 5A) and amplitudes (Fig. 5B) for relaxation to the new equilibrium position were accurately measured over a very wide range of urea concentrations. Rate constants from temperature-jump experiments were verified by using continuous-flow apparatus (Fig. 5C).
Figure 5.

Relaxation kinetics for unfolding of the WW prototype domain in 2.0 M urea/20 mM sodium phosphate, pH 7.0. (A) Jumps (3°C) from 22 to 25°C were initiated by 30 kV discharges from a 20 nF capacitor. The average of eight traces is shown, omitting the initial fast change in signal that arises from the intrinsic temperature dependence of tryptophan fluorescence. The subsequent relaxation to the new equilibrium fits to a single exponential function (residuals, Inset) with kobs = 5,210 ± 7 s−1. (B) The urea dependence of the kinetic unfolding amplitudes for the prototype WW domain. The kinetic amplitudes expected were simulated (broken line) by using only the equilibrium ΔC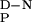 , ΔHD-N, Tm, and mD-N values and were highly consistent with the measured kinetic amplitudes (triangles). (C) Unfolding of the YAP 65 WW domain at 298 K measured by continuous-flow fluorescence spectroscopy. The fit of an individual unfolding trace (10 μM YAP 65 WW domain in 8.45 M urea, 20 mM phosphate buffer, pH 7.0) to a single exponential function is shown with kobs = 2,265 ± 8 s−1 (residuals, Inset).
, ΔHD-N, Tm, and mD-N values and were highly consistent with the measured kinetic amplitudes (triangles). (C) Unfolding of the YAP 65 WW domain at 298 K measured by continuous-flow fluorescence spectroscopy. The fit of an individual unfolding trace (10 μM YAP 65 WW domain in 8.45 M urea, 20 mM phosphate buffer, pH 7.0) to a single exponential function is shown with kobs = 2,265 ± 8 s−1 (residuals, Inset).
The urea dependence of kobs for the YAP 65 and prototype WW domains (Fig. 6) fitted well to a two-state transition (see Materials and Methods), although there was an unfolding rollover in the chevron plot for the WW prototype. This rollover is consistent with a change in either the native or transition state ensemble of the prototype domain as a function of urea concentration (20–23). Nonlinear urea activity may also contribute partially to this curvature (18). The limited kinetic data on wild-type FBP28 WW show that it folds much faster than the other WW domains (extrapolated kobs of ≈32,000 s−1 in water). The more complete kinetic data set for the FBP28 W30A mutant showed faster folding than for the wild-type protein, with a half-time in water of ≈17 μs. The folding rate enhancement, in conjunction with the destabilization of the protein, suggests that W30 may make unfavorable interactions in the transition state (24) of the wild-type protein (φF ≈ −0.4), resulting in a relative retardation of the folding and unfolding rate compared with the alanine mutant.
Figure 6.

Dependence of log kobs on urea concentration for different WW domains at 298 K and pH 7.0. The data sets for the YAP 65 (triangles), prototype (squares), and W30A FBP28 (filled circles) WW domains were fitted to two-state transitions (as described in Materials and Methods). The high denaturation midpoint of the wild-type FBP28 WW domain (hollow circles) prevents the observed data from being accurately fitted. Filled triangles refer to YAP 65 WW kinetic data obtained by using continuous-flow measurements, hollow triangles to temperature-jump measurements.
The calculated free energy of unfolding and mD-N values determined in equilibrium and kinetic measurements agree fairly well (Tables 1 and 2). Thus, a two-state model is apparently sufficient to describe the folding and unfolding of all of the characterized WW domains. Interestingly, the βTS values for the folding transition states of the WW domains, a direct index of their solvent exposure and thus compactness, are very similar and highly collapsed at ≈0.7 (Table 2), in common with many other β-sheet proteins (1).
Table 2.
Kinetic parameters for different WW domains at 298 K and pH 7.0
| Protein |
k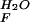 , s−1 , s−1
|
mF*, kJ/mol⋅M |
k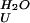 , s−1 , s−1
|
mU*, kJ/mol⋅M | ΔG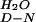 ,† kJ/mol ,† kJ/mol |
βTS‡ |
|---|---|---|---|---|---|---|
| YAP | 4,300 ± 600 | 1.13 ± 0.03 | 780 ± 110 | 0.31 ± 0.01 | 4.24 ± 0.49 | 0.78 |
| Prototype | 7,000 ± 300 | 0.89 ± 0.01 | 1,200 ± 60 | 0.50 ± 0.05 | 4.38 ± 0.16 | 0.64 |
| FBP28 W30A | 41,000 ± 3800 | 0.80 ± 0.01 | 5,240 ± 290 | 0.28 ± 0.01 | 5.05 ± 0.27 | 0.74 |
X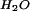 refers to the extrapolated value of parameter in water.
refers to the extrapolated value of parameter in water.
mF and mU refer to the urea dependence of the free energy of activation for folding and unfolding, respectively.
Calculated from the extrapolated rate constants for folding and unfolding in water (ΔG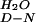 = −RT⋅ln(k
= −RT⋅ln(k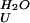 /k
/k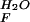 ), as previously described (14).
), as previously described (14).
βTS = mF/(mU + mF). This parameter is an index of the denaturant-binding capacity of the transition state relative to that of the native conformation.
Discussion
WW Domains as Model Folding Systems.
The WW domains are excellent model systems for characterizing the folding and stability of β-sheet proteins (11). Reversible chemical and thermal denaturation, in conjunction with very broad unfolding transitions, make them highly suitable for kinetic relaxation studies. Our data are consistent with the truncated WW domains folding by two-state transitions under both equilibrium and presteady-state conditions. The ground state for folding appears to be a random coil ensemble for the prototype and FBP28 WW domains, although the situation is ambiguous for the YAP 65 domain. The rapid folding and unfolding of these domains make them ideal candidates for MD simulations.
The higher stability of the FBP28 WW domain makes it particularly amenable to protein engineering studies (25), which should allow the folding transition state to be characterized in detail. The WW prototype is also particularly interesting as being a designed (7), rather than a naturally evolved, protein: it has little evolutionary or functional constraints placed on its sequence. This may allow us to determine whether proteins with virtually identical topologies, but low sequence identity, fold in similar ways as recently suggested (26–30).
We find little evidence for residual structure in the chemically denatured state of the prototype and both FBP28 WW domains. The observed differences in chirality in both the far- and near-UV CD spectra of the chemically and thermally denatured states of these domains are not structural, but instead reflect dominating contributions from aromatic amino acids, which have significant intrinsic chirality even in the absence of higher-order structure. The origin of the differences between the near-UV CD spectra of the chemically and thermally denatured states of the YAP 65 WW domain are unclear. These species have very similar CD spectra (when proper controls are implemented) and are apparently thermodynamically equivalent. Thus, we believe that the apparent chirality in the near-UV CD spectrum of chemically denatured state is artifactual and may have resulted from intermolecular association or aggregation, although residual structure has not yet been fully excluded.
It is interesting, however, that the domains that apparently lack residual structure in the denatured ensemble clearly fold faster than the YAP 65 WW domain. This suggests that there may be no requirement per se for residual structure in denatured β-sheet proteins to achieve ultra-fast folding, with the possibility that such structure may actually slow folding reactions. Interestingly, the destabilized W30A mutant of the FBP28 WW domain folds faster than the wild-type protein and has a negative ΦF value (≈−0.4). This is consistent with results from MD simulations of the unfolding of this protein (31), where the residues that comprise the third β-strand (27–30) were predicted to form nonnative interactions before the transition state for folding. Thus, the W30A mutation may perturb these nonnative or unfavorable interactions during folding of the FBP28 WW domain, resulting in the accelerated folding observed. Comparative analyses of the WW domains may allow us to resolve which residues and sequence positions are important in defining the folding pathway and stability of these proteins.
Fast Folding.
The folding rate constant in water for the FBP28 W30A domain is the fastest yet reported for an all-β-sheet protein at room temperature (kF = 41,000 ± 3,800 s−1). This rate constant is only marginally lower than that for formation of an isolated β-hairpin (115,000 s−1) (32) and within an order of magnitude of the predicted maximum rate of loop formation (33). Perhaps folding of the WW domains is close to the speed limit for β-sheet folding and is restricted only by the rate of β-hairpin formation, with subsequent steps of the reaction occurring very rapidly. Preliminary Φ-value analyses of the YAP 65 (31) and Pin (34) WW domains are consistent with this hypothesis, although it has yet to be determined whether this is a generic feature of WW domain folding. If this is the case, then the observed differences in the folding rates of these proteins may simply reflect differences in the propensity to form secondary structure elements such as the β-hairpins, with possible effects on the stability of each domain.
The small size of the WW domains, combined with their very rapid folding and unfolding reactions, may mean that the time scale of β-sheet formation and breakdown could soon overlap with that of computer simulations, allowing a direct and iterative test of theory and experiment. In the accompanying paper (31), we show that the folding and unfolding transition states of the WW domains have very similar features, irrespective of whether they are characterized by using experimental methods or ab initio theoretical simulations. Thus, accurate, atomic level descriptions of folding pathways may be a realistic prospect for the not-too-distant future, making it possible to test and validate current models of protein folding.
Acknowledgments
We thank Heiner Roder and Rama Shastry for help and advice during the construction of the continuous-flow instrument, Dimitry Veprintsev (Centrë for Protein Engineering) for writing software to control both the continuous-flow and temperature-jump apparatus, and Ugo Mayor for helpful discussions. This work was funded by the Medical Research Council, Cambridge, U.K.
Abbreviations
- MD
molecular dynamics
- GdmCl
guanidinium chloride
- CD
circular dichroism
- YAP 65
human yes kinase-associated protein
- FBP
murine formin-binding protein
- Mops
3-[N-morpholino]propanesulphonic acid
References
- 1.Jackson S E. Folding Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 2.Daggett V. Curr Opin Struct Biol. 2000;10:160–164. doi: 10.1016/s0959-440x(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 3.Mayor U, Johnson C M, Daggett V, Fersht A R. Proc Natl Acad Sci USA. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. . (First Published November 21, 2000; 10.1073/pnas.250473497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sudol M. Prog Biophys Mol Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- 5.Macias M J, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Nature (London) 1996;382:646–649. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- 6.Villegas V, Viguera A R, Aviles F X, Serrano L. Folding Des. 1996;1:29–34. [PubMed] [Google Scholar]
- 7.Macias M J, Gervais V, Civera C, Oschkinat H. Nat Struct Biol. 2000;7:375–379. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 8.Ranganathan R, Lu K P, Hunter T, Noel J P. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 9.Kanelis V, Rotin D, Forman-Kay J D. Nat Struct Biol. 2001;8:407–412. doi: 10.1038/87562. [DOI] [PubMed] [Google Scholar]
- 10.Crane J C, Koepf E K, Kelly J W, Gruebele M. J Mol Biol. 2000;298:283–292. doi: 10.1006/jmbi.2000.3665. [DOI] [PubMed] [Google Scholar]
- 11.Koepf E K, Petrassi H M, Sudol M, Kelly J W. Protein Sci. 1999;8:841–853. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gill S C, von Hippell P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 13.Pace C N. Methods Enzymol. 1986;131:3286–3299. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 14.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 15.Nicholson E M, Scholtz J M. Biochemistry. 1996;35:11369–11378. doi: 10.1021/bi960863y. [DOI] [PubMed] [Google Scholar]
- 16.Shastry M C R, Luck S D, Roder H. Biophys J. 1998;74:2714–2721. doi: 10.1016/S0006-3495(98)77977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterman B F. Anal Biochem. 1979;93:442–444. doi: 10.1016/s0003-2697(79)80176-1. [DOI] [PubMed] [Google Scholar]
- 18.Matouschek A, Matthews J M, Johnson C M, Fersht A R. Protein Eng. 1994;7:1089–1095. doi: 10.1093/protein/7.9.1089. [DOI] [PubMed] [Google Scholar]
- 19.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matouschek A, Fersht A R. Proc Natl Acad Sci USA. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matouschek A, Otzen D E, Itzhaki L S, Jackson S E, Fersht A R. Biochemistry. 1995;34:13656–13662. doi: 10.1021/bi00041a047. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson N, Li W, Capaldi A P, Kleanthous C, Radford S E. J Mol Biol. 2001;307:393–405. doi: 10.1006/jmbi.2000.4492. [DOI] [PubMed] [Google Scholar]
- 23.Silow M, Tan Y J, Fersht A R, Oliveberg M. Biochemistry. 1999;38:13006–13012. doi: 10.1021/bi9909997. [DOI] [PubMed] [Google Scholar]
- 24.Serrano L, Matouschek A, Fersht A R. J Mol Biol. 1992;224:805–818. doi: 10.1016/0022-2836(92)90563-y. [DOI] [PubMed] [Google Scholar]
- 25.Matouschek A, Fersht A R. Methods Enzymol. 1991;202:82–112. doi: 10.1016/0076-6879(91)02008-w. [DOI] [PubMed] [Google Scholar]
- 26.Chiti F, Taddei N, White P M, Bucciantini M, Magherini F, Stefani M, Dobson C M. Nat Struct Biol. 1999;6:1005–1009. doi: 10.1038/14890. [DOI] [PubMed] [Google Scholar]
- 27.Martinez J C, Serrano L. Nat Struct Biol. 1999;6:1010–1016. doi: 10.1038/14896. [DOI] [PubMed] [Google Scholar]
- 28.Villegas V, Martinez J C, Aviles F X, Serrano L. J Mol Biol. 1998;283:1027–1036. doi: 10.1006/jmbi.1998.2158. [DOI] [PubMed] [Google Scholar]
- 29.Riddle D S, Grantcharova V P, Santiago J V, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 30.Clarke J, Cota E, Fowler S B, Hamill S J. Structure (London) 1999;7:1145–1153. doi: 10.1016/s0969-2126(99)80181-6. [DOI] [PubMed] [Google Scholar]
- 31.Ferguson N, Pires J R, Toepert F, Johnson C M, Pan Y P, Daggett V, Oschkinat H, Fersht A R. Proc Natl Acad Sci USA. 2001;98:13008–13013. doi: 10.1073/pnas.221467398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munoz V, Thompson P A, Hofrichter J, Eaton W A. Nature (London) 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 33.Eaton W A, Munoz V, Thompson P A, Chan C K, Hofrichter J. Curr Opin Struct Biol. 1997;7:10–14. doi: 10.1016/s0959-440x(97)80003-6. [DOI] [PubMed] [Google Scholar]
- 34.Jager M, Nguyen H, Crane J C, Kelly J W, Greubele M. J Mol Biol. 2001;311:373–393. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 35.Santoro M M, Bolen D W. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]