

Abstract
We report a customized gene panel assay based on multiplex long-PCR followed by third generation sequencing on nanopore technology (MinION), designed to analyze five frequently mutated genes in chronic lymphocytic leukemia (CLL): TP53, NOTCH1, BIRC3, SF3B1 and MYD88. For this purpose, 12 patients were selected according to specific cytogenetic and molecular features significantly associated with their mutational status. In addition, simultaneous analysis of the targets genes was performed by molecular assays or Sanger Sequencing. Data analysis included mapping to the GRCh37 human reference genome, variant calling and annotation, and average sequencing depth/error rate analysis. The sequencing depth resulted on average higher for smaller amplicons, and the final breadth of coverage of the panel was 94.1%. The error rate was about 6% and 2% for insertions/deletions and single nucleotide variants, respectively. Our gene panel allows analysis of the prognostically relevant genes in CLL, with two PCRs per patient. This strategy offers an easy and affordable workflow, although further advances are required to improve the accuracy of the technology and its use in the clinical field. Nevertheless, the rapid and constant development of nanopore technology, in terms of chemistry advances, more accurate basecallers and analysis software, offers promise for a wide use of MinION in the future.
Introduction
Chronic lymphocytic leukemia (CLL) is a disease with a highly heterogeneous clinical course, characterized by the accumulation and proliferation of clonal B cells in the blood, bone marrow and lymph nodes. Next–generation sequencing (NGS) technologies applied to genome-wide association studies (GWAS) have revealed several recurrent gene mutations associated with CLL that, in association with the high level of genetic heterogeneity among patients, is consistent with the large degree of clinical variability characteristic of this disease. In fact, a number of recurrent gene mutations have been reported in 2–10% of newly diagnosed CLL cases1, and clinical evidence revealed the prognostic role of specific mutated genes in defining different molecular subtypes of CLL. They may help to predict the potential evolution of the disease or the response to therapy. Identifying the most clinically and biologically relevant clones is crucial for evaluating CLL disease progression; in this context, NGS targeted gene sequencing allows driver mutations to be detected, at diagnosis and/or responsible for progression and relapse2,3, which could be used as predictive or prognostic biomarkers in CLL4.
However, the introduction of NGS technologies in clinical diagnostics requires a high initial investment when purchasing the sequencer, that is a limitation for local research facilities in developing countries and small research centers and hospitals. A valid alternative could be the use of MinION, the first commercially available sequencer based on nanopore technology. MinION has already been successfully used to detect mutations of the TP53 and ABL1 genes in CLL and in chronic myeloid leukemia (CML) patients, respectively5,6. Moreover, the very low costs, the ease of use, and the length of the reads make MinION an ideal tool for targeted sequencing of genes5–7.
In this report we describe the development of a customized gene panel, MinION-based, for targeted sequencing of the five genes TP53, BIRC3, NOTCH1, SF3B1 and MYD88, recurrently mutated in CLL, and analysis of its performance.
Results
Real-time data monitoring and error rate analysis
In our experiment, the library consisting of 12 barcoded samples was loaded for sequencing on MinION, employing a flowcell with 1,170 active pores. A total of 106,543 fast5 total files was produced, containing raw electric signals, for a total of about 65 Mb. Fast5 files were then uploaded in Metrichor for basecalling and demultiplexing during the run. Of the total reads produced, 48,599 passed 2D filters, and 46,451 had a recognizable barcode. Only “pass” reads were used in the study; the number of 2D passed reads for each sample is illustrated in Supplementary Table S1.
Thanks to the real-time analysis of the fast5 files produced, after 24 hours from the start of the sequencing we observed that the minimum sequencing depth of the targets for case #7 was lower, on average, than for the other CLL samples. Therefore, we decided to prepare and reload a new pool comprising all the libraries at different weight ratios according to the preliminary results obtained with real-time sequencing monitoring. Employing this expedient, the total pass 2D reads count was greater than 2,000 for each sample except for case #7 (Table 1). Plotting the distribution of read lengths, all passed 2D reads of each sample had a length corresponding to the expected amplicons size (Supplementary Fig. S2).
Table 1.
CLL patients clinical data and reads mapping analysis (del(11q), 11q deletion; del(13q). 13q deletion; del(17p), 17p deletion).
| Sample | Sex/Age | FISH | IgVH status | Sample type | 2D mapped reads count/ total 2D reads count |
|---|---|---|---|---|---|
| CLL#1 | F/63 | 88% del(13q), 96,5% del(11q) | Unmutated | Peripheral blood | 3013/3066 (98.27%) |
| CLL#2 | M/73 | 83% del(13q), 8% del(17p), 66% del(11q) | Unmutated | Peripheral blood | 4752/4859 (97.80%) |
| CLL#3 | F/62 | 42% del(11q) | Unmutated | Peripheral blood | 2254/2272 (99.21%) |
| CLL#4 | M/72 | 50% del(11q), 80% +12q13 | Unmutated | Peripheral blood | 3207/3234 (99.17%) |
| CLL#5 | M/50 | 71% del(13q), 97,1% del(11q) | Unmutated | Bone marrow | 3845/3919 (98.11%) |
| CLL#6 | F/53 | 51% +12q13 | Unmutated | Peripheral blood | 4432/4593 (96.49%) |
| CLL#7 | M/74 | 65% +12q13 | Mutated | Peripheral blood | 1450/1460 (99.32%) |
| CLL#8 | F/71 | 89% del(13q), 91% del(17p) | Unmutated | Peripheral blood | 5392/5483 (98.34%) |
| CLL#9 | M/79 | 87% del(13q) (biallelic), 84% del(17p) | Mutated | Peripheral blood | 4959/5106 (97.12%) |
| CLL#10 | M/58 | 74% del(13q) | Mutated | Peripheral blood | 3428/3653 (93.84%) |
| CLL#11 | M/59 | 62% +12q13 | Unmutated | Peripheral blood | 3232/3345 (96.62%) |
| CLL#12 | M/62 | negative for del(13q), del(17p), del(11q), +12q13 | Unmutated | Peripheral blood | 2480/2535 (97.83%) |
NanoOK analysis was performed to calculate the error rate for each amplicon. In Table 2, the mean values of identical, inserted and deleted bases, and substitutions per 100 aligned bases (including indels) are shown for each amplicon. As observed, separate error rate analysis for insertions, deletions and substitutions revealed higher error values for deletions, in line with historically known nanopore error rate data8–10.
Table 2.
Error rate analysis of MinION sequencing data. As observed, for all the targets of the CLL panel the mean error rate calculated was higher for deletions than for insertions and substitutions (Indels: insertions/deletions).
| Targets | Mean identity per 100 aligned bases (including indels) | Mean insertion error rate | Mean deletion error rate | Mean substitution error rate |
|---|---|---|---|---|
| BIRC3 exons 7–9 | 92.86% | 0.91% | 4.47% | 1.76% |
| TP53 exons 10–11 | 90.83% | 0.90% | 6.26% | 2.02% |
| NOTCH1 exon 34–3′ UTR | 90.50% | 0.86% | 6.58% | 2.09% |
| SF3B1 exons 14–16 | 92.75% | 0.89% | 4.56% | 1.80% |
| TP53 exons 2–9 | 90.09% | 0.80% | 7.19% | 1.93% |
| MYD88 exons 3–5 | 91.09% | 0.89% | 5.98% | 2.04% |
| BIRC3 exon 6 | 93.45% | 0.74% | 4.56% | 1.25% |
| Mean | 91.65% | 0.86% | 5.66% | 1.84% |
Sequencing depth analysis
Analysis of NanoOK data showed that the range of sequencing depth was more uniform for longer amplicons, and, notably, inversely related to the amplicon size, the smaller amplicons having a greater depth (up to about 2100x) (Fig. 1). These data suggest that, in the case of amplicons of variable size and smaller than 3 kb, further adjustments should be made in the adapter ligation step of MinION library preparation, in order to ensure a more uniform depth rate. In any case, except for case #7, the minimum sequencing depth value was never below 50x for all the amplicons analyzed; therefore, we decided to exclude this sample from the subsequent analyses.
Figure 1.
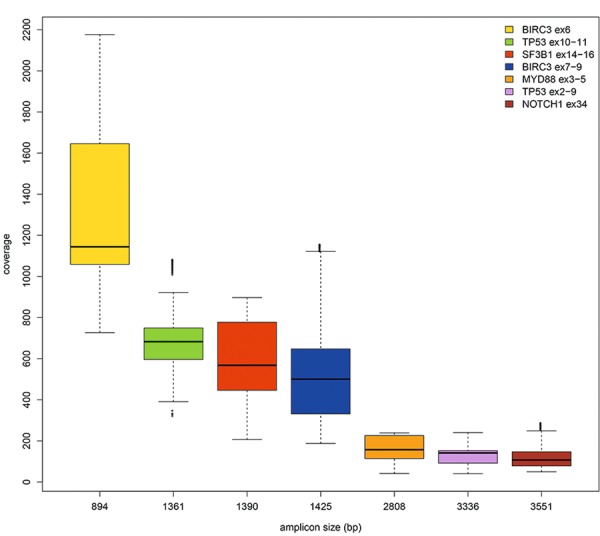
Boxplot of sequencing depth data and amplicons size (bp). The range of read depth was more uniform for longer amplicons and inversely related to the amplicon size, the smaller amplicons showing a higher sequencing depth.
Performance evaluation of MinION sequencing for CLL hotspot mutations
We also focused on the known hotspot mutations in CLL, NOTCH1 p.P2514fs*411,12, SF3B1 p.K700E, p.G742D, p.K666 and p.H66212,13, and MYD88 p.L265P14, and calculated the average depth of coverage in these specific genomic positions. For all hotspot mutations, the minimum sequencing depth value was above 50×, and reflected the range of read depth related to amplicons sizes. Examining the mean and substitutions/deletions/insertions error rates in these positions, we observed a comparable trend to the general error rate analysis, with higher error rates for deletions than for substitutions or insertions. Supplementary Table S2 reports the error rate/depth of coverage data of the known hotspots.
Variant calling, filtering and annotation
We performed two parallel analyses of MinION sequencing data, uploading the fastq files extracted from fast5 files to Galaxy platform or analyzing them with the Nanopolish variant calling pipeline15, by applying similar settings (see Material and Methods). In detail, we created and used a complete workflow (https://usegalaxy.org/u/ematlab/w/mutation-detection-llc-panel), including read mapping, variant calling and annotation. Similarly, the Nanopolish pipeline was applied starting from fast5 files to variant calling, and then the ANNOVAR annotation tool16 from Galaxy was used to annotate them. For both the workflows, SNV and indels analysis was performed for each CLL case.
SNV and indels were filtered for hotspot mutations or genomic location (exonic and splicing variants), functional effect (nonsynonymous variants), and frequency based on minor allele frequency (maf) from the 1000 Genomes Project (<1%). Variant allele frequency cut-off was set at 10% and 15% for SNV and indels, respectively, in agreement with the recent results of serial dilution for MinION somatic mutation analysis17. The filtered variants were also analysed in silico by the pathogenicity prediction program SIFT incorporated in ANNOVAR.
It is well-known that nanopore sequencing reads still have a high error rate, especially in homopolymer contexts9. To reduce the risk of producing false positive results due to sequencing errors, we checked if the same variants were called in different patients. To this aim, we calculated the recurrence of all SNV and indels and found some recurrent variants in several samples, and even in all 12 CLL cases. Except for the known CLL mutation hotspots, and the polymorphisms identified, considering the low likelihood of finding the same variant simultaneously in a small cohort, we decided to exclude the variants occurring in multiple samples from further validation analyses. In total 256 genomic recurrently mutated positions were therefore determined; most of them (91%) were sites of indels, in line with the results of the error rate analysis (see Material and Methods), whereas 9 were sites of both SNV/indels and 14 only of SNV (Fig. 2, Supplementary Table S3).
Figure 2.
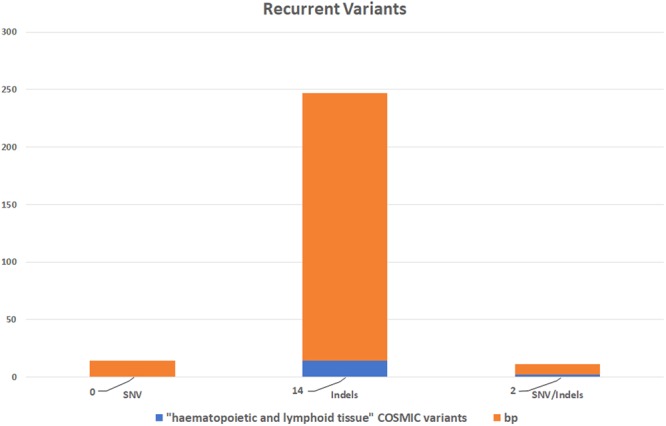
Plot of the recurrent variants identified in the CLL patients analyzed and filtered out from the analysis. Most of them are sites of indels (nucleotides count orange). A small fraction of these critical positions (about 7%) are annotated in COSMIC v81 as identified in haematopoietic and lymphoid tissue (blue).
Excluding these critical positions from the analysis, the actual breadth of coverage of the custom panel dropped from 100% of the long amplicons covering all the target regions to 94.7%. Indeed, 5.3% of the genomic positions covered with the amplicons design were sites of recurrent errors, determining a high fraction of false positive results in these positions. These data are not related to panel design, but rather to the chemistry and base calling algorithms used (See Material and Methods).
We also verified whether these critical positions are known to be sites of experimentally observed mutations in haematopoetic and lymphoid tissue and found that about 7% of them were COSMIC (v81) variants annotated in haematological malignancies (Fig. 2). These filtered-out COSMIC variants did not include the known hotspots listed above.
We also retrieved the nucleotide sequences flanking of all the SNV/indels called and observed that most of these mutations suggested a context-specific error frequently caused by homopolymer sequences (Supplementary Table S3).
For these reasons, we decided to focus on the non-recurrent variants and known hotspots: 18 SNV and 6 indels (Supplementary Table S4). We compared these mutations revealed by MinION with the results of SS and molecular assays: 8 pathogenic mutations were finally confirmed by SS or other molecular assays in 6 patients, with 2 patients concurrently harboring 2 mutations. In detail, 6 mutations were SNV, whereas the main hotspot deletion of NOTCH1 p.P2514fs*4 was detected in 2 patients (Table 3). These mutations were simultaneously identified with SS except for 2 variants having a mutation allelic ratio below the detection limit of SS: for case #4, one mutation in the coding region of BIRC3, previously not easily detected by SS electropherogram analysis, was detected by visual inspection on the indications generated by MinION, whereas in case #2 a mutation in SF3B1 was validated by ASO-PCR (Fig. 3, Supplementary Fig. S3).
Table 3.
Genomic description and annotation of mutations detected by MinION sequencing or other molecular assays (ASO-PCR: Allele Specific Oligonucleotide PCR; SS: Sanger Sequencing).
| Patient | Gene | Variant | Hotspot mutation | Mutation type | Protein description | Impact | Allelic Ratio (%) | Validation method | Varscan detection | Nanopolish detection | Depth of coverage |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CLL#2 | SF3B1 | chr2:198267360T>C | yes | SNV | p.K666R | MODERATE | 11.75 | ASO-PCR | yes | no | 722 |
| CLL#2 | NOTCH1 | chr9:139390721G>C | yes | SNV | p.Y2490X | HIGH | 27.14 | SS | yes | no | 73 |
| CLL#4 | BIRC3 | chr11:102206703T>G | no | SNV | p.L444X | HIGH | 10.04 | SS | yes | no | 323 |
| CLL#5 | TP53 | chr17:7577022G>A | no | SNV | p.R306* | HIGH | 84.14 | SS | yes | yes | 156 |
| CLL#8 | TP53 | chr17:7577121G>T | yes | SNV | p.R273G | MODERATE | 47.54 | SS | yes | yes | 70 |
| CLL#8 | NOTCH1 | chr9:139390649AG/— | no | INDEL | p.P2514fs*4 | HIGH | 20.69 | SS | yes | no | 62 |
| CLL#9 | TP53 | chr17:7578478G>C | no | SNV | p.P19R | MODERATE | 57.50 | SS | yes | yes | 91 |
| CLL#12 | NOTCH1 | chr9:139390649AG/— | yes | INDEL | p.P2514fs*4 | HIGH | 20.69 | SS | yes | no | 86 |
Figure 3.

The SF3B1 mutation identified in case#2 thanks to MinION and validated by ASO-PCR. Aligned reads are visualized by the Integrative Genomics Viewer (IGV) browser (on the left). The genomic position, and the depth of sequencing for each base are reported as a gray bar. Variants with an allelic ratio >10% are reported as colored depth of sequencing bars, where each color represents the specific base fraction. As shown, the mutation was barely visible in SS (on the right) and was validated by ASO-PCR (at the bottom). The original image of the gel is provided as Supplementary Fig. S4.
Applying the Nanopolish pipeline, we confirmed only 3 of the 8 variants previously called with Varscan, suggesting that Nanopolish is more stringent for variants detection, less sensitive than Varscan, and can give false negatives (Table 3). The lower sensitivity of Nanopolish for detecting variants was due to the computation of an improved consensus sequence before variant calling rather than to the more stringent parameters used. Indeed, this step is part of the Nanopolish pipeline, and probably makes the identification of low allele-frequency variants more difficult. In accordance with this, all the variants called in our samples with Nanopolish showed high allele frequencies (Table 3).
Discussion
MinION is a low-cost, handheld sequencer that produces long reads (recently up to megabases) in real-time18,19. Nowadays, MinION can generate up to around 5–10Gbp of DNA sequence data, and has been used in particular to sequence and assemble microbial, yeast and fungi genomes20,21. Recently, MinION has also been used to sequence Ebola, chikungunya, and hepatitis C viruses using an unbiased metagenomic approach with randomly amplified cDNA22. Analysis of genomic mutations in cancer5,6,17,23,24 and in the infectious diseases field25 has been reported using MinION. In these studies nanopore technology was employed to detect mutations in a single gene target, or in several targets after PCR products pooling, applying slightly different methods17,25.
To the best of our knowledge, this is the first report of targeted gene sequencing based on a custom panel of pre-pooled primers for multiplex long-PCR on MinION.
Multiplex gene panels are one of the strategies most commonly used to enrich the genomic regions to be sequenced, and are widely employed in NGS technologies. This approach, applied to MinION, allowed us to enrich all the gene regions of interest, setting up only 2 PCR for each sample and thus simplifying the experimental procedures.
Thanks to MinION real-time sequencing, reads can be analysed as they are generated, significantly speeding up the analysis of results and modifying the experimental conditions if necessary; this advantage allowed us to intervene in real-time on the quality of case #7 sequencing. Other advantages of MinION include its portability and easy setting for library preparation and sequencing as compared to second-generation sequencers, as well as its cost-effectiveness.
Thanks to analyses of the utilization and impact of NGS in CLL, in the last years several custom/academic or commercial CLL target panels have been developed, most of them based on the amplicon sequencing approach. For example, in 2013 Kolhman et al. described their 13-gene panel for investigating CLL NGS, proposing it in routine diagnostics; the authors declared that using their panel the detection of the main NOTCH1 hotspot p.P2514fs*4 was successful, but that it was not able to identify larger indels26. Currently, only one commercial amplicon-based CLL gene panel is available which allows the detection of both SNV and copy number variant analysis for the nine most prognostically relevant genes in CLL2. Nonetheless, attempts to design a comprehensive gene panel useful for CLL prognostication and therapy-effect prediction, are still continuing2.
All these approaches have surely proven more accurate than nanopore sequencing, allowing even low allele frequency mutations (subclonal) to be detected, because of their lower error rate. Furthermore, higher throughput allows the analysis of more samples and/or more genes per sample. However, some considerations about short-read sequencing platforms should be made. Firstly, although their accuracy is very high (>99.5%), they are still affected by specific sequencing errors: Illumina displays a tendency towards substitution errors, whereas Ion Torrent technology fails when measuring homopolymers27. Secondly, some weaknesses related to the general short-read sequencing approach are well known: the technical limit of detecting larger indels and other structural variations (SV), and the impossibility of conducting the phasing analysis of mutations. In fact, SV larger than the read length cannot be detected and, using genomic DNA, the mutations may be too distant to be detected on the same read.
By exploiting the long reads produced by MinION sequencing, it is possible to evaluate the phasing of mutations, clarifying the allelic context of mutations affecting the same gene, but too geographically distant to be detected with short reads sequencing; this analysis may be particularly useful for CLL patients in the context of disease progression and therapy resistance. For example, in the case of no co-occurrence between mutated TP53 and del(17p)12, phasing analysis of multiple mutations would allow the physician to establish whether both TP53 alleles are mutated or not28.
In perspective, we could also consider implementing our panel to identify even the structural alterations frequently identified in CLL patients, obtaining the complete CLL mutational spectrum in a single MinION experiment.
All genes included in our panel have a prognostic significance in CLL. Mutations of TP53 are found in 4–37% of CLL patients, generally occur within the DNA-binding motifs (exons 4–8), and have been associated with very poor prognosis in several studies29,30. In a recent report our group demonstrated that nanopore technology shows a correlation with SS results but is more sensitive, and has therefore proven to be a useful tool for TP53 gene mutation detection5. NOTCH1 gene mutations in CLL mainly cluster within the p.P2514fs*4 hotspot, and have independently conferred adverse overall survival (OS) in multiple studies, whereas their impact on progression-free survival has been inconsistent31–33. Moreover, NOTCH1 mutations have also been identified as a predictive marker for patients who are less likely to respond to the addition of rituximab to therapeutic regimens34. Mutations in the SF3B1 gene cluster in the selected HEAT repeats of the SF3B1 protein, and have been identified in 4–12% of CLL patients, with a higher frequency at the time of disease progression13; SF3B1 mutations are correlated with a shorter duration of remission after treatment, fludarabine-refractory disease, and poorer OS35,36. BIRC3 and MYD88 gene mutations have been detected in up to 4% of CLL cases14,37; while gene mutations in the former are associated with other adverse prognostic factors that predict chemo-refractoriness and poorer prognosis37, the MYD88 gene mutations, mostly L265P, identify CLL patients with a more favorable outcome14.
Our proof-of-concept demonstration of targeted gene sequencing in CLL using the MinION sequencer reveals the feasibility of this approach but identifies specific challenges to be dealt with in future projects. Indeed, in the last 2 years five different MinION chemistry versions (R6.0, R7.0, R7.3, R9 and R9.4), and several bioinformatics tools have been presented, improving MinION’s performance, but, despite these efforts and progress, the main weak point of MinION, namely its high error rate especially in homopolymeric sequences, still persists. In this context, basecalling is a crucial step, because it can hugely affect MinION sequencing data. To date, several basecallers have been developed to improve read accuracy38, and new bioinformatic pipelines are being tested, also for variant calling analysis23,39.
Regarding the MinION error rate, we approached this issue by excluding from the analysis those genomic positions recurrently detected as mutated in the samples analyzed, admitting the inability to evaluate the mutational status of these genomic positions with the data analysis tools currently available. By retrieving the adjacent bases of these genomic positions, most of the false positive were within or next to homopolymer sequences. This strategy clearly reduced the total actual breadth of coverage, but produced more reliable results, and did not interfere with the detection of the CLL mutation hotspots. The main NOTCH1 hotspot p.P2514fs*4 is a particular case because it involves an “AG” dinucleotide deletion followed by a “G” homopolymer which does not seem to affect its correct identification.
Today, other single-molecule platforms are also being developed in order to overcome the limit of the nanopore error rate and detect very low-frequencies point mutations40,41. In the future, the implementation of these innovative approaches in MinION sequencing may contribute to better discriminate between mutated and wild-type DNA molecules and will be extended to achieve the concurrent detection of multiple biomarker mutations.
In conclusion, although the sample number in our study is relatively small, and an extension of the case series is warranted to correctly estimate the sensitivity and specificity of our assay, our data showed that the CLL gene panel provides a satisfactory performance. We propose a workflow that can potentially enable laboratories equipped with only basic molecular biology techniques to perform detailed targeted gene sequencing analysis in CLL patients. In the next future, we plan to evaluate how we can scale up the target panel, including more amplicons in a larger patients sample size using MinION. The constant improvement of nanopore technology will likely result inits widespread application in clinical practice.
Material and Methods
Patients
The study included 12 newly diagnosed CLL patients. Patients were selected according to the presence of the genetic aberrations 11q, 13q and 17p deletions (del(11q), del(13q), del(17p)), and trisomy 12, frequently associated with CLL and identified by Fluorescent In Situ Hybridization (FISH), as previously reported42–44, and the immunoglobulin heavy-chain variable (IgHV) mutational status. These clinical parameters are significantly associated with the mutational status of the genes included in our panel11,35. Only one case, CLL#12, showed none of the frequent genetic aberrations analyzed in FISH analysis. All samples included in the study were characterized for the NOTCH1 hotspot p.P2514fs*4 and MYD88 hotspot p.L265P by molecular assays. For the exons of BIRC3, TP53 and SF3B1 included in the panel, mutational status was verified by Sanger Sequencing (SS). These evaluations were performed in concomitance with the MinION sequencing analysis. All 12 samples were analyzed by MinION and SS or molecular assays in a blinded manner.
Genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen) and the DNA concentration and purity were checked using the Qubit 2.0 fluorometer (Life Technologies) and Nanodrop UV-Vis spectrometer (Thermo Fisher Scientific).
Patients clinical and biological characteristics are summarized in Table 1, which also illustrates the ratio of the 2D mapped reads count to the total 2D reads count for each sample.
The study complies with the current ethical guidelines, and has been approved by the Azienda Ospedaliero-Universitaria Consorziale Policlinico of Bari (Bari, Italy). The written informed consent was obtained from the patients included in this study, and patient records/information was anonymized and de-identified prior to analysis.
CLL panel preparation and testing
Our customized CLL panel included the known mutation hotspots of SF3B112,13, NOTCH112,32 and MYD8814, and the genomic regions of the five genes reported in Table 4. The enrichment strategy we adopted to primarily select our targets was multiplex long-PCR. Amplicons were designed to cover one or multiple exons of the five genes TP53, BIRC3, NOTCH1, SF3B1 and MYD88; primer design was performed using Primer3 software (http://primer3.ut.ee/) with the following non-default parameters (Tmin 59 °C, Topt 60 °C, Tmax 61 °C), to yield product sizes greater than 800 bp. The FastPCR (http://primerdigital.com/fastpcr.html) and Multiple Primer Analyzer (Thermo Fisher Scientific) tools were used to analyze and compare multiple primer sequences simultaneously. Overall, seven pairs of primers were selected for the selected target genes and analyzed with the Multiplex 2.1 tool (http://bioinfo.ut.ee/multiplx/) using the default parameters, to evaluate primers compatibility and find the best primers pooling solution. Two primers pools were thus identified: pool 1 included primers for BIRC3 exons 7–9, TP53 exons 10–11, SF3B1 exons 14–16 and NOTCH1 exon 34 including 3′UTR, while pool 2 was assembled with primers for TP53 exons 2–9, BIRC3 exon 6 and MYD88 exons 3–5. The total panel size was about 15 kb. In Table 4 the composition of the two primer pools and the respective amplicon sizes are shown; primers sequences are shown in Supplementary Table S5.
Table 4.
Composition of the 2 pools of the custom CLL gene panel, with the relative size of the corresponding amplicons.
| Gene target | Amplicon size (bp) | |
|---|---|---|
| Pool1 | BIRC3 exons 7–9 | 1425 |
| TP53 exons 10–11 | 1361 | |
| NOTCH1 exon 34–3′ UTR | 3551 | |
| SF3B1 exons 14–16 | 1390 | |
| Pool2 | TP53 exons 2–9 | 3336 |
| MYD88 exons 3–5 | 2808 | |
| BIRC3 exon 6 | 894 |
Multiplex long-PCR target enrichment
For each CLL sample two multiplex long-PCRs were performed with the two primer pools using PrimeSTAR GXL DNA Polymerase (Takara Bio Inc.), 70 ng of genomic DNA, in a final volume of 25 uL. Thermal cycling conditions were 98 °C for 10 minutes, 60 °C for 15 seconds, 68 °C for 4 minutes (35 cycles) and 12 °C hold for both the two long-PCR.
Since 3 amplicons of pool 1 had a very similar size of about 1.3 kb and were not easily distinguishable by 1% agarose gel electrophoresis, a restriction enzyme of these critical amplicons was made, and the BglII restriction enzyme (10,000 units/mL, New England BioLabs Inc.) was finally selected to verify their successful amplification and discriminate them. In detail, 5 uL of the primer pool 1 PCR products were incubated with 0.5 units of BglII and 1uL of NEBuffer 3.1 in 1 hour at 37 °C. Digestion products were visualized by SYBR Safe on agarose gel 1%.
PCR products from both the long PCR were purified with the QIAquick PCR Purification Kit (Qiagen) in an elution volume of 30 uL, and the DNA concentration and purity was measured with a Qubit 2.0 fluorometer (Life Technologies) and Nanodrop UV-Vis spectrometer (Thermo Fisher Scientific).
Two uL of pool 1 and pool 2 purified amplicons were visualized by SYBR Safe on agarose gel 1.0% (Supplementary Fig. S1).
Library preparation and sequencing
For each sample, 300 ng of PCR purified products from each long-PCR were pooled, in a final volume of 25 uL in nuclease-free water and used for library preparation. According to the 2D Native barcoding genomic DNA (SQK-LSK 208) protocol, the amplicons were end-prepared using the NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs Inc.) and barcoded with the ligation of nanopore-specific Native Barcodes (NB01-NB12) using Blunt/TA Ligase Master Mix (New England Biolabs Inc.). Equimolar amounts of each barcoded amplicon were then pulled and 540 ng of the final pool were diluted to 58 μl in nuclease-free water and prepared for sequencing with the ligation of Native Barcoding adapters and the tether using NEBNext Quick Ligation Module (New England Biolabs Inc.). All purifications were performed with Agencourt AMPure XP beads (Beckman Coulter Inc.). Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) were used to elute the library in the pre-sequencing mix.
After the Platform QC run and the priming of the flowcell, the sequencing mix (37.5 μl of Running Buffer with Fuel Mix, 25.5 μl of Library Loading Bead Kit, 12 μl of the Pre-sequencing Mix) was loaded and the MAP_48Hr_Sequencing_Run.py protocol was started (MinION flowcell: FLO-MAP106).
Real-time analysis of the sequencing files generated by MinION was performed using a custom script including fast5 extraction, mapping with the BWA-MEM aligner and counting of the mapped reads; for each amplicon, the minimum number of reads per amplicon considered for the real-time analysis was 100x.
The sequence data from this study have been submitted to the NCBI Short Read Archive (https://www.ncbi.nlm.nih.gov/sra) under accession n° SRP133465.
Data Analysis
During MinION sequencing, DNA bases were called using a cloud-based software (Metrichor), generating FAST5 files, from which FASTQ files were extracted with the Poretools toolkit 0.6.0.
The NanoOK tool (v.1.14) was employed to analyze the read depth and error rate, using the FASTA sequences of the target amplicons as reference. Depth of coverage and error rates of the known hotspots were calculated with bam-readcount program and by visual inspection of bam files with the Integrative Genomics Viewer (IGV) browser45,46.
Data analysis from reads mapping to variant calling was performed in parallel using Galaxy, a web-based platform for processing NGS data (https://usegalaxy.org/), and Nanopolish, a package specific for signal-level analysis of nanopore sequencing data (https://github.com/jts/nanopolish).
In Galaxy, reads were aligned on GRCh37 human reference genome with the BWA-MEM method using specific Nanopore platform parameters (https://github.com/lh3/bwa/blob/master/NEWS.md/#release-079-19-may-2014); the leftalign utility from FreeBayes package was applied to homogenize the positional distribution of insertions and deletions. The BAM files obtained were visualized. Single nucleotide variants (SNV) and insertions/deletions (indels) detection was separately performed with the Varscan software47 (minimum read depths supporting variants: 5, minimum read depth:20, p-value threshold for calling variants:0.99), and the VCF files obtained were annotated with ANNOVAR16. The pipeline implemented in Galaxy is reported in Supplementary File S1.
Similarly, in the Nanopolish pipeline, reads mapping was executed with BWA-MEM, and the “nanopolish variants” subprogram was used to simultaneously call SNV and indels, which were annotated as described above (-min-candidate-frequency 0.1, -min-candidate-depth = 20).
The ChIPpeakAnno48 and BSgenome.Hsapiens.UCSC.hg19 R packages were used to retrieve the sequences flanking the variants detected, in order to evaluate the presence of homopolymer contexts and their potential association with MinION sequencing errors.
Validation of sequencing variants
The filtered variants were then compared with the results obtained using SS; electropherograms were then analyzed by visual inspection, glass free software for SS analysis data (http://shiny.bat.infspire.org/igcllglass/) and GeneScreen49.
For one SNV present at a level below the sensitivity threshold of SS (about 11%), an Allele Specific Oligonucleotide PCR (ASO-PCR) assay was accomplished. The Web-based Allele Specific Primer (WASP) designing tool (http://bioinfo.biotec.or.th/WASP) was used to design the allele-specific primers for detecting the mutated and wild-type site, using the default parameters.
ASO-PCR was performed with PlatinumTM Taq DNA Polymerase (Invitrogen), using 100 ng of genomic DNA, in a final volume of 50 uL. Thermal cycling conditions were 95 °C for 30 seconds, 95 °C for 30 seconds, 65 °C for 30 seconds, 72 °C for 30 seconds (35 cycles) and 4 °C hold.
Data availability
The sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession n° SRP133465.
Electronic supplementary material
Acknowledgements
This work was supported by “Associazione Italiana contro le Leucemie (AIL)-BARI”. The authors would like to thank Ms. MVC Pragnell, B.A. for language revision of the manuscript.
Author Contributions
P.O. and F.A. conceived and designed the study, and wrote the manuscript. P.O. and C.C. performed the Long-PCRs, Barcoding and nanopore experiments. P.O. and C.F.M. performed all bioinformatics analysis. P.C. performed conventional cytogenetic analysis. L.A., A.Z., N.C. and G.T. conducted FISH experiments and interpreted data. A.M., L.I., E.P. and C.B. performed diagnostic molecular analysis. A.G. provided clinical data. G.S. and F.A. supervised the manuscript preparation. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-30330-y.
References
- 1.Wang L, et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodríguez-Vicente AE, et al. Next-generation sequencing in chronic lymphocytic leukemia: recent findings and new horizons. Oncotarget. 2017;8:71234–71248. doi: 10.18632/oncotarget.19525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutton L-A, Rosenquist R. Deciphering the molecular landscape in chronic lymphocytic leukemia: time frame of disease evolution. Haematologica. 2015;100:7–16. doi: 10.3324/haematol.2014.115923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. Am. Soc. Hematol. Educ. Progr. 2017;2017:329–337. doi: 10.1182/asheducation-2017.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minervini CF, et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016;11:96. doi: 10.1186/s13000-016-0550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minervini CF, et al. Mutational analysis in BCR-ABL1 positive leukemia by deep sequencing based on nanopore MinION technology. Exp. Mol. Pathol. 2017;103:33–37. doi: 10.1016/j.yexmp.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Cumbo C, et al. Genomic BCR-ABL1 breakpoint characterization by a multi-strategy approach for ‘personalized monitoring’ of residual disease in chronic myeloid leukemia patients. Oncotarget. 2018;9:10978–10986. doi: 10.18632/oncotarget.23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magi, A., Semeraro, R., Mingrino, A., Giusti, B. & D’Aurizio, R. Nanopore sequencing data analysis: state of the art, applications and challenges. Brief. Bioinform. 10.1093/bib/bbx062 (2017). [DOI] [PubMed]
- 9.O’Donnell CR, Wang H, Dunbar WB. Error analysis of idealized nanopore sequencing. Electrophoresis. 2013;34:2137–44. doi: 10.1002/elps.201300174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain M, et al. Improved data analysis for the MinION nanopore sequencer. Nat. Methods. 2015;12:351–356. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baliakas P, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29:329–336. doi: 10.1038/leu.2014.196. [DOI] [PubMed] [Google Scholar]
- 12.Rossi D, Gaidano G. Molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev. Hematol. 2012;5:593–602. doi: 10.1586/ehm.12.58. [DOI] [PubMed] [Google Scholar]
- 13.Wan Y, Wu CJ. SF3B1 mutations in chronic lymphocytic leukemia. Blood. 2013;121:4627–34. doi: 10.1182/blood-2013-02-427641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez-Trillos A, et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood. 2014;123:3790–6. doi: 10.1182/blood-2013-12-543306. [DOI] [PubMed] [Google Scholar]
- 15.Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods. 2015;12:733–5. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki A, et al. Sequencing and phasing cancer mutations in lung cancers using a long-read portable sequencer. DNA Res. 2017;24:585–596. doi: 10.1093/dnares/dsx027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansen HJ, et al. Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 2017;7:7213. doi: 10.1038/s41598-017-07650-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 10.1038/nbt.4060 (2018). [DOI] [PMC free article] [PubMed]
- 20.Istace B, et al. De novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience. 2017;6:1–13. doi: 10.1093/gigascience/giw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. genomics. 2017;3:e000132. doi: 10.1099/mgen.0.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greninger AL, et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015;7:99. doi: 10.1186/s13073-015-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Euskirchen P, et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017;134:691–703. doi: 10.1007/s00401-017-1743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norris AL, Workman RE, Fan Y, Eshleman JR, Timp W. Nanopore sequencing detects structural variants in cancer. Cancer Biol. Ther. 2016;17:246–53. doi: 10.1080/15384047.2016.1139236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkmann A, et al. Development and preliminary evaluation of a multiplexed amplification and next generation sequencing method for viral hemorrhagic fever diagnostics. PLoS Negl. Trop. Dis. 2017;11:e0006075. doi: 10.1371/journal.pntd.0006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roller, A. et al. A 13-Gene Panel Targeted To Investigate CLL By Next-Generation Amplicon Deep-Sequencing Can Be Successfully Implemented In Routine Diagnostics. Blood122 (2013).
- 27.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malcikova J, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114:5307–5314. doi: 10.1182/blood-2009-07-234708. [DOI] [PubMed] [Google Scholar]
- 29.Zenz T, et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia. 2010;24:2072–2079. doi: 10.1038/leu.2010.208. [DOI] [PubMed] [Google Scholar]
- 30.International CLL-IPI working group An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet. Oncol. 2016;17:779–790. doi: 10.1016/S1470-2045(16)30029-8. [DOI] [PubMed] [Google Scholar]
- 31.Minervini A, et al. Droplet digital PCR analysis of NOTCH1 gene mutations in chronic lymphocytic leukemia. Oncotarget. 2016;7:86469–86479. doi: 10.18632/oncotarget.13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossi D, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–529. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansouri L, et al. NOTCH1 and SF3B1 mutations can be added to the hierarchical prognostic classification in chronic lymphocytic leukemia. Leukemia. 2013;27:512–4. doi: 10.1038/leu.2012.307. [DOI] [PubMed] [Google Scholar]
- 34.Pozzo F, et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia. 2016;30:182–9. doi: 10.1038/leu.2015.182. [DOI] [PubMed] [Google Scholar]
- 35.Montserrat E, Bauman T, Delgado J. Present and future of personalized medicine in CLL. Best Pract. Res. Clin. Haematol. 2016;29:100–110. doi: 10.1016/j.beha.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Rossi D, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118:6904–8. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rossi D, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119:2854–62. doi: 10.1182/blood-2011-12-395673. [DOI] [PubMed] [Google Scholar]
- 38.Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C. & Mutlu, O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform. 10.1093/bib/bby017 (2018). [DOI] [PMC free article] [PubMed]
- 39.Szalay T, Golovchenko JA. De novo sequencing and variant calling with nanopores using PoreSeq. Nat. Biotechnol. 2015;33:1087–1091. doi: 10.1038/nbt.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu L-Q, Gates KS, Wang MX, Li G. What is the potential of nanolock- and nanocross-nanopore technology in cancer diagnosis? Expert Rev. Mol. Diagn. 2018;18:113–117. doi: 10.1080/14737159.2018.1410060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, et al. Nanolock-Nanopore Facilitated Digital Diagnostics of Cancer Driver Mutation in Tumor Tissue. ACS sensors. 2017;2:975–981. doi: 10.1021/acssensors.7b00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albano F, et al. Decreased TET2 gene expression during chronic myeloid leukemia progression. Leuk. Res. 2011;35:e220–2. doi: 10.1016/j.leukres.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Specchia G, et al. Insertions generating the 5′RUNX1/3′CBFA2T1 gene in acute myeloid leukemia cases show variable breakpoints. Genes. Chromosomes Cancer. 2004;41:86–91. doi: 10.1002/gcc.20061. [DOI] [PubMed] [Google Scholar]
- 44.Zagaria A, et al. A fluorescence in situ hybridization study of complex t(9;22) in two chronic myelocytic leukemia cases with a masked Philadelphia chromosome. Cancer Genet. Cytogenet. 2004;150:81–5. doi: 10.1016/j.cancergencyto.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu LJ, et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carr IM, et al. GeneScreen: a program for high-throughput mutation detection in DNA sequence electropherograms. J. Med. Genet. 2011;48:123–30. doi: 10.1136/jmg.2010.082081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession n° SRP133465.