

Abstract
Chemical synthesis allows the incorporation of nonnatural amino acids into proteins that may provide previously untried probes of their folding pathway and thermodynamic stability. We have used a flexible thioether linker as a loop mimetic in the human yes kinase-associated protein (YAP 65) WW domain, a three-stranded, 44-residue, β-sheet protein. This linkage avoids problems of incorporating sequences that constrain loops to the extent that they significantly change the nature of the denatured state with concomitant effects on the folding kinetics. An NMR solution structure shows that the thioether linker had little effect on the global fold of the domain, although the loop is apparently more dynamic. The thioether variants are destabilized by up to 1.4 kcal/mol (1 cal = 4.18 J). Preliminary Φ-value analysis showed that the first loop is highly structured in the folding transition state, and the second loop is essentially unstructured. These data are consistent with results from simulated unfolding and detailed protein-engineering studies of structurally homologous WW domains. Previously, Φ-value analysis was limited to studying side-chain interactions. The linkers used here extend the protein engineering method directly to secondary-structure interactions.
Understanding how proteins spontaneously fold to a stable three-dimensional structure is exceptionally difficult. This difficulty applies especially to β-sheet proteins, whose secondary structure is stabilized more by long-range sequence-distant interactions than is that in α-helical proteins. Small β-sheet proteins are useful paradigms for understanding the search problem. There is increasing evidence of the importance of β-hairpins and loops in driving β-sheet folding (1–6). The stability of these loops may also influence the overall stability of β-sheet proteins in two ways. One way is by contributing to the overall thermodynamic stability of the system (7–9). The other way is in the effect on the folding pathway (2, 3), such that folding may or may not be facilitated by loop or hairpin formation, with concomitant effects on the folding rate and, thus, on stability.
The WW domains are a family of small, triple-stranded, antiparallel β-sheet proteins (typically, of 34–44 amino acids) with a small hydrophobic core involving residues from both the N and C termini (10). The loop structures and overall bend of the β-sheet are well conserved in the different members of this family (11–13). A number of these domains are well suited for biophysical studies of β-sheet folding and loop formation as they reversibly denature, are amenable to mutagenesis, and are easily synthesized by using both recombinant and organic chemistry techniques (12, 14, 15). Thus, these domains can be synthesized with both natural and nonnatural amino acids in their sequences, allowing for sophisticated experiments that cannot easily be performed with recombinant proteins. The effects of substitutions on the stabilities of WW domains have been studied in the context of a designed WW-domain prototype (12) by incorporating β-turn mimetics out of context in loop 1 (14) and also by detailed protein engineering (5).
Here, we study three YAP 65 WW domain analogues that contain a (2-amino-ethylsulfanyl)-acetic acid building block (Fig. 1) in either the first or the second loop. This linker is equivalent to two glycine residues joined by a thioether (–S–CH2–) instead of a peptide bond (–N–CO–), in this way exchanging a hydrogen-bond donor and acceptor by silent groups with higher internal flexibility. In YAP ΔS24G25, this thioether linkage replaces the S24 and G25 of the β-hairpin between strands 1 and 2, resulting in the deletion of one important hydrogen bond in the secondary structure. In YAP ΔS24 and ΔI33, the thioether-linked fragment replaces S24 in loop 1 or I33 in loop 2, respectively, resulting in an insertion of one amino acid equivalent with reduced conformational restrictions, as compared with glycine. The structure of one of these variants, in conjunction with ligand-binding data and a preliminary Φ-value analysis on the influence of loops on folding kinetics and stability, are presented. Molecular dynamics (MD) simulations also were performed to investigate the unfolding of a structurally homologous WW domain, and the results are in excellent agreement with these and other experimental data.
Figure 1.

Amino acid sequence of the YAP 65 WW domain and its thioether analogues ΔS24, ΔS24G25 and ΔI33. In the sequences, Z refers to the nonnatural amino acid (2-amino-ethylsulfanyl)-acetic acid.
Materials and Methods
Synthesis of (N-Fmoc-2-amino-ethylsulfanyl)-Acetic Acid.
Bromoacetic acid tert-butylester (5.85 g, 30 mmol) were added to a solution of cysteamine (2.31 g, 30 mmol) in dimethylformamide at 60°C. The mixture was stirred for 1 h and then cooled to room temperature. Fmoc-OSu (9.45 g, 28 mmol) and diisopropylethylamine were added, and the mixture was stirred for 12 h at room temperature and thereafter concentrated in vacuo. The solid was dissolved in CH2Cl2, and the solution was extracted three times with 5% citric acid, 0.1 M sodium hydrogen carbonate, and brine. The organic layer was dried with Na2SO4, concentrated in vacuo, and treated with a solution of 50% trifluoroacetic acid in CH2Cl2 for 2 h. After evaporation, the resulting crude solid was extracted with ethyl acetate. The product was isolated after evaporation to yield 8.5 g (79%) as an amorphous white solid. The purity of the product was characterized by analytical HPLC.
Peptide Synthesis.
The WW–thioether derivatives were synthesized on solid phase (50 μmol scale) on a Tentagel SRam resin (Rapp Polymere, Tübingen, Germany) by using PyBOP activation and a standard Fmoc-chemistry-based protocol of an AMS 422 Peptide Synthesizer (Abimed, Langenfeld, Germany). In the protocol, the (N-Fmoc-2-amino-ethylsulfanyl)-acetic acid building block was used like a normal Fmoc amino acid. Side-chain protections of amino acids are as follows: Glu, Asp (Ot Bu); Ser, Thr, Tyr, Trp (t Bu); His, Lys (Boc); Asn, Gln (Trt); Arg (Pbf). Trifluoroacetic acid (TFA)/phenol/triisopropylsilane/H2O (9.4:0.1:0.3:0.2) was used for resin cleavage and side-chain deblocking. The crude peptides were purified to homogeneity by RP-HPLC using the linear solvent gradient 5–60% B in A for 30 min, with A = 0.05% TFA in water, and B = 0.05% TFA in acetonitrile. The HPLC had the UV detector at 214 nm, a Vydac C18 column of 20 × 250 mm, and a flow rate of 10 ml/min. The MS were performed on a matrix-assisted laser desorption ionization-time of flight mass spectrometer (Laser BenchTopII, Applied Biosystems). The purity of the product was characterized by analytical HPLC.
Far-UV CD Spectroscopy.
Far-UV CD spectra and thermal denaturation experiments were acquired as described (16) by using a 1-mm pathlength cell containing 65 μM protein in 20 mM sodium phosphate buffer (pH 7.0) at an ionic strength of 150 mM. Repeat scans were highly reversible with 95% of the signal recovered after thermal denaturation. Thermal melts were fitted to a two-state transition, with the change in heat capacity upon unfolding (ΔCpD−N) assumed to be temperature-independent (17) and constrained to the value determined for the homologous FBP28 WW domain (ΔCpD−N = 417 ± 17 cal/mol K; ref. 16).
Temperature-Jump Experiments.
Temperature-jump measurements were made as described (16), except that 30-kV discharges from a 20-nF capacitor were used to initiate 3°C jumps to 25°C (±0.1); a final protein concentration of 25 μM was used. Traces (15–20) were acquired and the averaged data set fit to a single exponential function with kaleidagraph (Synergy Software, Reading, PA). The standard errors reported for kobs are the fitted errors determined from nonlinear regression analysis.
NMR Spectroscopy.
Two-dimensional nuclear Overhauser effect spectroscopy (NOESY; ref. 18) with a 130-ms mixing time (1.2 mM protein) and two-dimensional correlation spectroscopy (TOCSY; ref. 19) experiments with 20-and 70-ms spinlock were recorded for the WW domain-ligand complex (2.4 mM protein plus 2-fold molar excess of ligand) on a 600-MHz DRX spectrometer (Bruker, Karlsruhe, Germany). Experiments were carried out in 10 mM potassium phosphate buffer, pH 6/100 mM NaCl/0.1 mM DTT/0.1 mM EDTA in H2O and in D2O at 15°C. Spectra were processed with xwinnmr (Bruker) and analyzed with ansig (20).
Structure Calculations.
A total of 224 interresidue NOE restraints derived from the NOESY experiment were assigned to four classes of distances (2.5 Å − 0.70/+0.3; 3.0 Å − 1.2/+ 0.4; 4.0 Å − 2.2/+ 0.75; 5.0 Å − 3.2/+ 0.75). Structures were calculated by simulated annealing from random coordinates (21) at 2,000 K by using x-plor 3.1 (22) and floating stereospecific assignment. Force constants for NOE were 50 kcal mol−1 Å−2; bond lengths, bond angles, and improper angles were 1,000 kcal mol−1 Å−2, 500 kcal mol−1 rad−2, and 500 kcal mol−1 rad−2, respectively.
MD Simulations.
The simulations were performed with the program encad (23) and a previously described force field (24, 25). Model 1 of the NMR structures of FBP28WW (GATAVSEWTEYKTADGKTYYYNNRTLESTWEKPQELK) determined by Macias et al. (PDB accession no. 1E0L) (12) was used as the starting structure for the simulations. The Asp and Glu residues were protonated to mimic a low-pH environment. This starting structure was first minimized for 500 steps in vacuo. Then, the protein was solvated with a water box that extended at least 12 Å from any protein atom, yielding 3,415 water molecules. The density of the solvent was set to the experimental value for the temperature of interest: 0.983, 0.975, and 0.958 gm/ml at 333, 348, and 373 K, respectively (26). All atoms in the system were explicitly present. The system was equilibrated before production simulations as follows: 100 steps of minimization of the water molecules; 2,000 steps of dynamics of the water; 1,000 cycles of minimization of the water; 1,000 steps of minimization of the protein; and, finally, 500 steps of minimization of the full system. In the production simulations, the nonbonded cut-off was 8 Å. The nonbonded pair list was updated every two steps and a time step of 2 fs was used. Periodic boundary conditions were used to minimize edge effects and the microcanonical ensemble (NVE) was used. The simulations were carried out for 34, 20, and 16 ns at 333, 348, and 373 K, respectively, resulting in a total of 70 ns and 350,000 structures for analysis.
Results
Far-UV CD Spectroscopy.
The far-UV CD spectrum of the wild-type YAP 65 WW domain under native conditions exhibits a strong peak of positive ellipticity at 230 nm (15). This ellipticity is due to dominating contributions from the aromatic amino acids (Fig. 2a; ref. 16). The far-UV CD spectra of all three thioether variants under native conditions are qualitatively very similar to that of the wild-type protein, although the observed ellipticity is reduced in amplitude. The far-UV CD spectra of all four proteins are very similar at 371 K and consistent with that expected for a random-coil conformation.
Figure 2.

Far-UV CD properties of the wild-type YAP WW domain and thioether variants. (a) The far-UV CD spectrum of the wild type (triangles), YAP ΔS24 (circles), ΔS24G25 (diamonds), and ΔI33 (squares) WW domains at 277 K (white symbols) and 371 K (black symbols). (b) Thermal denaturation of the wild-type and thioether variant YAP WW domains measured at 230 nm with far-UV CD spectroscopy. Solid lines indicate the best fits to a two-state transition (17). The posttransition baselines of these domains coincide above 345 K, consistent with their unfolding to a similar denatured conformation as shown in a.
The large change in the far-UV CD signal that accompanies denaturation makes this technique a highly suitable probe for monitoring the reversible thermal denaturation of WW domains (Fig. 2b). The thermal unfolding transitions of these proteins fitted well to an equation describing a two-state transition. As the thioether variants were destabilized relative to the wild-type YAP WW domain, it was more difficult to obtain accurate thermodynamic parameters because of uncertainties in determining the slope of the pretransition baselines. The calculated midpoints for thermal unfolding (Tm) for the ΔS24, ΔS24G25, and ΔI33 variants were 310 ± 3.6, 298 ± 2, and 300 ± 1.8 K, respectively, which are significantly lower than for the wild-type protein (Tm = 316.6 ± 0.6 K). Thus, there is an approximately equal mixture of folded and unfolded conformations of the ΔI33 and ΔS24G25 YAP WW domains at room temperature under these conditions.
Solution NMR and Ligand Binding.
The proton NMR spectrum of the wild-type YAP 65 WW domain is well resolved with clearly defined upfield and downfield resonances for the native conformation (Fig. 3a), with NɛH proton resonances at 10.25 and 9.95 ppm for the N-terminal (W17) and C-terminal (W39) tryptophans, respectively. The respective signals of both tryptophans occur at 10.04 ppm in the unfolded state. The proton NMR spectrum of the YAP ΔI33 variant at 285 K (Fig. 3b, lower trace) is in close accord with the thermal denaturation data, with the resonances indicating an approximately equimolar mixture of native and unfolded species (45 vs. 55%). A 32% population of the folded state was observed for the YAP ΔS24G25 variant (Fig. 3c, lower trace), resulting in line broadening caused by the particular exchange situation with the unfolded state (27), and 34% folding for the variant ΔS24 (Fig. 3d, lower trace). A detailed analysis of the NMR line-broadening to determine the apparent rate constants for folding and unfolding (27) of these thioether YAP WW domain variants was not performed here, although this approach may have merit in the future. It is most probable that the slight differences in the calculated populations of native and unfolded states reported with NMR and thermal denaturation experiments are a consequence of the differing experimental conditions, although this result could be caused also by uncertainties in curve fitting and peak integration.
Figure 3.

Downfield region of the 1H NMR spectra of wild-type and thioether variant YAP WW domains. (a) The wild-type YAP 65 WW domain. (b) Construct ΔI33 without (lower trace) and with (upper trace) the ligand GTPPPPYTVG. (c) Construct ΔS24 without (lower trace) and with (upper trace) ligand. (d) Construct ΔS24G25 without (lower trace) and with (upper trace) ligand.
Addition of a proline-rich ligand (10) (Ac-GTPPPPYTVG-NH2) to the YAP ΔS24 and ΔS24G25 variants resulted in significant amide chemical-shift changes (Fig. 3 c and d, respectively, upper traces) and in considerable sharpening of the NMR signals, with chemical shifts higher than 8.5 ppm. Of the two tryptophans, only the functionally important W39 displayed a chemical-shift change upon ligand binding, consistent with previous studies on the larger 57-residue YAP 65 WW domain (28). The chemical-shift changes and peak sharpening upon ligand binding suggest that there is a shift in the equilibrium toward forming more native structure. Specifically, the integrated peak areas are in accord with native populations of 66 and 53% for YAP ΔS24 and YAP ΔS24G25 WW domains, respectively, indicating that the binding sites of these variant domains are intact. In contrast, no resonance shifts and line-sharpening effects were observed in the one-dimensional NMR spectrum of the YAP ΔI33 variant in the presence of an excess of the same peptide ligand (Fig. 3b, upper trace). The NMR results were corroborated by using surface plasmon resonance, where equilibrium dissociation constants (Kd) of ≈240 and 266 μM were determined for the YAP ΔS24 and YAP ΔS24G25 domains, respectively (data not shown). These values are somewhat lower than previously reported for the intact full-length YAP WW domain (Kd = ≈52 μM; ref. 10).
The structural properties of YAP ΔS24 WW domain in complex with the peptide ligand were investigated in detail to check on the intactness of the sheet and the properties of the modified loop. In this variant, a strong modification of the loop is made by the replacement of S24 Cα with the fragment CH2-CH2-S-CH2. The overall structure (Fig. 4) is essentially the same as the wild-type domain (10). The β-sheet extends to T22 and Q26, whereby an NOE between the amide protons of T22 and Q26 is missing, but contacts between the two respective side chains are observed. The NOE between T22 and the side chain of W39 are present in the same manner as in the wild type, indicating a similar overall bend of the sheet as in the native protein. There are few NOEs between residues in the loop, which is widened as compared with the structure of the wild-type domain. The poorer definition of the first loop suggests an increase in flexibility caused by the thioether moiety, consistent with its flexible nature and the disappearance of the NOE between the amide protons of T22 and Q26, although this apparent flexibility may be caused by a lack of appropriate NMR constraints.
Figure 4.

Solution structure of the YAP65 ΔS24 WW domain thioether variant. Stereo view showing the backbone superposition of the ensemble of the 10 structures of lowest energy calculated by restrained simulated annealing (Left). Cartoon representation of the minimal energy structure (Right).
Folding and Unfolding Kinetics.
The effects of the introduced thioether linkages on the kinetics for folding and unfolding of the YAP WW domain were assessed with a rapid temperature-jump apparatus (Fig. 5). The observed relaxation rate for the wild-type YAP WW domain after temperature jump-induced unfolding was very rapid (kobs = 4,870 ± 17 s−1), which is in accord with previous studies (kobs = 4,300 ± 600 s−1). Interestingly, the observed relaxation rate for the YAP ΔI33 thioether variant was significantly faster (kobs = 13,510 ± 33 s−1) than for the wild-type domain, in contrast to the relaxation rates for the YAP ΔS24 and YAP ΔS24G25 variants, which were significantly slower (kobs = 2,090 ± 3 and 2,727 ± 5 s−1, respectively).
Figure 5.

Relaxation kinetics for the temperature jump-induced unfolding of the wild-type and thioether variant WW domains. The initial temperature-dependent change in tryptophan fluorescence that accompanies temperature jumps is not shown. Fitting each trace to a single exponential function gave random residuals (not shown) and allowed the observed rate constant (kobs) for the unfolding of each domain to be determined.
Despite the difficulties in accurately determining the equilibrium constants for unfolding of the unstable thioether YAP variants, it was still possible to calculate the contribution of the rate constants for folding and unfolding in water (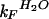 and
and  , respectively) to the observed rate at 298 K (Table 1). From the calculated rate constants, it was possible to determine ΦF values (29–31), which define the relative energetic contribution that residues make to the stability of the folding transition state and the native conformation. A ΦF value of 1 refers to an interaction that is as structured in the transition state as in the native state, whereas a ΦF value of 0 defines an interaction that is as unstructured in the transition state as in the denatured state. A ΦF value of greater than 1 refers to an interaction that stabilizes the transition state more than the native state. The rate constants for folding of YAP ΔS24 and YAP ΔS24G25 were retarded approximately three-fold relative to the wild-type domain (ΦF = 1.6 ± 0.2 and 0.5 ± 0.1, respectively), in contrast to the YAP ΔI33 domain, where there was a 1.5-fold rate enhancement (ΦF = −0.2 ± 0.1). These results were consistent with the first β-hairpin being highly structured in the folding transition state. In contrast, the second hairpin seemed to be unstructured or making unfavorable, possibly nonnative, interactions in the transition state.
, respectively) to the observed rate at 298 K (Table 1). From the calculated rate constants, it was possible to determine ΦF values (29–31), which define the relative energetic contribution that residues make to the stability of the folding transition state and the native conformation. A ΦF value of 1 refers to an interaction that is as structured in the transition state as in the native state, whereas a ΦF value of 0 defines an interaction that is as unstructured in the transition state as in the denatured state. A ΦF value of greater than 1 refers to an interaction that stabilizes the transition state more than the native state. The rate constants for folding of YAP ΔS24 and YAP ΔS24G25 were retarded approximately three-fold relative to the wild-type domain (ΦF = 1.6 ± 0.2 and 0.5 ± 0.1, respectively), in contrast to the YAP ΔI33 domain, where there was a 1.5-fold rate enhancement (ΦF = −0.2 ± 0.1). These results were consistent with the first β-hairpin being highly structured in the folding transition state. In contrast, the second hairpin seemed to be unstructured or making unfavorable, possibly nonnative, interactions in the transition state.
Table 1.
Equilibrium and kinetic parameters of wild-type YAP WW domain and thioether variants at 298 K and pH 7.0
| Protein | Tm, K* |
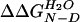 , kcal/mol† , kcal/mol†
|
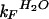 , s−1‡ , s−1‡
|
 , s−1‡ , s−1‡
|
ΦF§ |
|---|---|---|---|---|---|
| YAP wt | 316 ± 0.6 | – | 4300 ± 190 | 565 ± 190 | – |
| YAP ΔS24 | 310 ± 3.6 | −0.45 ± 0.01 | 1305 ± 230 | 785 ± 230 | 1.6 ± 0.2 |
| YAP ΔS24G25 | 298 ± 2.0 | −1.36 ± 0.02 | 1360 ± 445 | 1360 ± 445 | 0.5 ± 0.1 |
| YAP44 ΔI33 | 300 ± 1.8 | −1.21 ± 0.02 | 7225 ± 2245 | 6185 ± 2245 | −0.3 ± 0.1 |
The low stability of the thioether variants makes accurate curve fitting difficult and results in some uncertainty in the calculated Tm of these WW domains.
† The change in the free energy of folding upon mutation, relative to the wild-type YAP domain (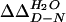 ), was calculated using
), was calculated using 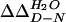 = 〈ΔTm〉·ΔSm as described (39), where ΔSm is the change in entropy upon folding of the wild-type YAP WW domain at the thermal midpoint (ΔSm = −75.6 ± 3.5 cal/mol·K).
= 〈ΔTm〉·ΔSm as described (39), where ΔSm is the change in entropy upon folding of the wild-type YAP WW domain at the thermal midpoint (ΔSm = −75.6 ± 3.5 cal/mol·K).  values calculated directly from the free energy of folding (
values calculated directly from the free energy of folding ( ) at 298 K for each protein were consistent with the values calculated using the above method but subject to higher errors (40).
) at 298 K for each protein were consistent with the values calculated using the above method but subject to higher errors (40).
‡ 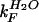 and
and 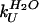 were determined from the equilibrium constant for unfolding (keq) and kobs using kF = kobs/(1 + Keq) and kU = kobs/(1 + 1/Keq).
were determined from the equilibrium constant for unfolding (keq) and kobs using kF = kobs/(1 + Keq) and kU = kobs/(1 + 1/Keq).
MD Simulations.
The thermal unfolding of the structurally homologous FBP28 WW domain at 333, 348, and 373 K was simulated independently of the experimental data (Fig. 6). At all temperatures, unfolding began with a loss of structure in the third β-strand. At 373 K, the remaining secondary structure then was lost to yield essentially a random coil. The only evidence for residual secondary structure was in the residues that define the third β-strand, where a highly unstable, nonnative turn formed between residues 27 and 30 (ESTW). It is possible that the unfolding simulation needs to run for a longer duration to fully equilibrate to the thermally denatured state, which previously has been shown to be unstructured by using both far- and near-UV CD spectroscopy (16). The pattern of unfolding was very similar at 333 and 348 K, except that the structure was lost more slowly than at the higher temperature, with the first hairpin persisting over a significant proportion of the unfolding trajectories. The persistence of the first hairpin was almost entirely caused by clustering of the tyrosine 11, 19, and 21 side chains; a dynamic, distorted β-sheet hydrogen-bonding network was present but the hairpin region lacked β-structure as defined by (φ,ϕ) angles (at least 3 consecutive residues with φ = −170 to −50° and ϕ = 80 to −170°). The excellent concordance of the simulated unfolding trajectories at different temperatures suggests that the unfolding mechanism is essentially independent of the temperature in the range studied. Given the good agreement between the simulations, we expect that the residual structure at 333 and 348 K will melt with extended simulation run times.
Figure 6.

Thermal unfolding simulations of the FBP28 WW domain. The β-strands are colored as follows: β1 (residues 8–12), red; β2 (res. 18–22), green; and β3 (res. 27–30), blue. This figure was made by using midasplus (38). The transition states for unfolding are near 2 ns.
Discussion
Structural Effects of Loop Mimetics.
The thioether moiety could be accommodated within both loops of the YAP WW domain, although it reduced the free energy of unfolding. The presence of this synthetic linker in the first hairpin (as in YAP ΔS24) did not affect the overall structure of the protein, but loop flexibility was increased. It was not possible to determine the structural effects of the thioether linkage on the second loop (as in YAP ΔI33), as ligand binding was abolished, and it is difficult to determine the solution structures of unstable WW domains except when in a stable complex with peptide ligands (10). The similarity of the far-UV CD and NMR spectra of the ΔI33 variant domain to those of the wild type and other thioether variants under native conditions suggested that their three-dimensional structures are similar.
Mass-Action Effects of Ligand Binding.
Interestingly, the combined NMR and ligand binding results for the YAP ΔS24 and YAP ΔS24G25 variants are qualitatively similar to those reported for the W17F mutant of the 57-residue YAP 65 WW domain (28). As the amide resonances of the W17F mutant were poorly defined, it was reported to be a “natively unfolded” protein, such that the unfolded conformation retains a functional binding site and ligand-binding initiates folding. However, we believe that the apparent lack of residual structure in the denatured state of a number of homologous WW domains (16), in conjunction with the ligand binding results for the thioether YAP variants and W17F mutant (28), are more consistent with changes in the position of equilibrium (in response to stabilization of the native state by ligand binding) rather than a discrete change in state induced by binding to the unfolded ensemble.
β-Hairpin Formation Early in Folding.
Residue 24 of the first hairpin was important to the folding and stability of the YAP WW domain (Table 1). Replacement of S24 and G25 with the thioether linkage (equivalent to G24G25), retards the folding rate by approximately three-fold as does replacement of S24 with the thioether moiety (as in YAP ΔS24). This retardation of the folding rate, in conjunction with the reduction in the free energy for folding, is consistent with S24 being highly structured in the folding transition state. By contrast, I33 of the second hairpin seems to be making few or unfavorable interactions in the folding transition state, as this variant is destabilized and folding is accelerated when residue 33 is substituted with the thioether linkage. There were similar results when other residues in the first and second loops of the YAP WW domain were replaced with thioether linkers (data not shown).
The results for these YAP thioether variants are consistent with recent work on the Pin1 WW domain (5), where the first loop was found to be highly structured in the folding transition state (ΦF = 1.4–1.8) and the second loop was significantly less structured (ΦF = −0.09–0.3). The sequence distribution of residues important to stabilizing the transition and native states of the Pin1 WW domain is polarized. Residues in the first loop apparently stabilize the folding transition state of the Pin1 WW domain more than the native state. By contrast, the native state is stabilized by a hydrophobic cluster that is composed of residues that have less importance in stabilizing the folding transition state near physiological temperatures.
The apparent division of residues between those that are kinetically important and those that are thermodynamically important may be a general feature of the WW domains and has implications for experimental design. An important feature of the loop mimetics used here is that they are flexible and do not introduce artificial constraints into the conformation of the denatured ensemble, although the polypeptide chain is increased in length by one amino acid equivalent in the YAP ΔS24 and ΔI33 variants. Nonnatural amino acids and synthetic-turn mimetics have been used to constrain and stabilize the conformation of isolated peptides into β-hairpins and β-sheet structures (6, 14, 32, 33). Interpreting the effects of constrained-turn mimetics upon the folding kinetics and stability is significantly more difficult than with the thioether linkers described here, as the former moieties have the side effect of overconstraining the conformation of the denatured ensemble. This constraint results in the turn being present in the denatured, transition, and native-state ensembles, such that the energetics are minimally changed upon folding, as observed in a recent study of the Pin WW domain (14). This problem necessitates the use of alternative linkers, such as the thioether moiety used here, to understand the sequence dependence of the kinetics and stability of β-sheet formation.
Agreement of Experiment and Simulation.
The qualitative agreement between these preliminary experimental data for the YAP WW domain, the detailed Φ analysis of the Pin WW domain (5) and the theoretical simulation results presented here is excellent. This agreement suggests that simulation may now be at the point where it can predict reliably experimental results. Residues R14, Y23, and F25 of the Pin WW domain all have fractional ΦF values (0.49–0.68) and are in the positions equivalent to the corresponding hydrophobic cluster (Y11, Y19, Y21) that is predicted to be transiently populated en route to the transition-state ensemble of the FBP28 WW domain. Furthermore, the accuracy of these theoretical simulations seems to extend to conformational states formed very late in the unfolding trajectory, which in the case of a two-state transition must be formed early in the folding reaction. Overall, the unstructured nature of structures over the last 10 ns of the 373 K simulation is consistent with both far- and near-UV CD spectroscopy (16) indicating that the denatured state is essentially a random coil; however, the unfolding simulations need to run longer to fully equilibrate to the thermally denatured state at the lower temperatures. Interestingly, residues 27–30 (ESTW), which define strand 3 of the FBP28 WW domain, were predicted to be involved in nonnative structure between the fully denatured ensemble and the unfolding transition state (Fig. 6). This prediction is consistent with previous kinetic studies that indicate W30 of this protein makes unfavorable, possibly nonnative interactions in the folding transition state (ΦF = −0.4; ref. 16). The general agreement between simulation and experimental results is very encouraging, particularly given the sequence differences. This agreement is consistent with recent suggestions that proteins with similar structures but low-sequence identity can fold in similar ways (34–36).
The results from the Φ-value analyses of the YAP and Pin WW domains also are consistent with results from other theoretical simulations of the folding of three-stranded β-sheet domains (2, 3), where β-hairpin formation was found to be rate-limiting to folding. MD was used to simulate the unfolding of the YAP WW domain (37) and also was used to indicate that the second and third strands were the first to separate during unfolding, which is consistent with both the early structuring in folding of the first hairpin and an energetically polarized transition state.
Future Directions.
The loop mimetics used in this study are an important complement to the use of natural mutations in Φ-value analyses. The systematic use of both natural and peptidomimetic substitutions will provide further benchmarks for combining with and testing simulation results. Importantly, we now have the opportunity to conduct experiments on the contributions of individual hydrogen bonds within secondary structure to the folding kinetics and stability of the protein under investigation (as in YAP ΔS24G25), experiments that simply cannot be performed on recombinant proteins. It is hoped that by using the strategy above, we will be able to determine what contributions hairpins and cross-strand interactions make to β-sheet folding and stability, the roles that sequence, hydrophobicity, and hydrogen bonding play in these processes, and whether structurally or energetically polarized transition states (1, 34) are general features of β-sheet folding.
Acknowledgments
This work was funded by the Medical Research Council, United Kingdom and the Deutsche Forschungsgemeinschaft, Grant SCHN 317/6–7. R.P. acknowledges a fellowship from the Deutsche Akademische Austauschdienst. Financial support for the computational studies was provided by National Institutes of Health Grant GM 50789 (to V.D.).
Abbreviations
- YAP 65
human yes kinase associated protein
- NOESY
nuclear Overhauser effect spectroscopy
- MD
molecular dynamics
- YAP ΔX
YAP 65 WW domain with residue(s) X replaced with a (2-amino-ethylsulfanyl)-acetic moiety
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1K5R).
References
- 1.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 2.Bursulaya B D, Brooks C L. J Am Chem Soc. 1999;121:9947–9951. [Google Scholar]
- 3.Ferrara P, Caflisch A. Proc Natl Acad Sci USA. 2000;97:10780–10785. doi: 10.1073/pnas.190324897. . (First Published September 12, 2000; 10.1073/pnas.190324897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCallister E L, Alm E, Baker D. Nat Struct Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 5.Jager M, Nguyen H, Crane J C, Kelly J W, Greubele M. J Mol Biol. 2001;311:373–393. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 6.Espinosa J F, Munoz V, Gellman S H. J Mol Biol. 2001;306:397–402. doi: 10.1006/jmbi.2000.4349. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez-Alvarado M, Blanco F J, Niemann H, Serrano L. J Mol Biol. 1997;273:898–912. doi: 10.1006/jmbi.1997.1347. [DOI] [PubMed] [Google Scholar]
- 8.Ramirez-Alvarado M, Kortemme T, Blanco F J, Serrano L. Bioorg Med Chem. 1999;7:93–103. doi: 10.1016/s0968-0896(98)00215-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H X X, Hoess R H, DeGrado W F. Nat Struct Biol. 1996;3:446–451. doi: 10.1038/nsb0596-446. [DOI] [PubMed] [Google Scholar]
- 10.Macias M J, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Nature (London) 1996;382:646–649. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- 11.Kanelis V, Rotin D, Forman-Kay J D. Nat Struct Biol. 2001;8:407–412. doi: 10.1038/87562. [DOI] [PubMed] [Google Scholar]
- 12.Macias M J, Gervais V, Civera C, Oschkinat H. Nat Struct Biol. 2000;7:375–379. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 13.Ranganathan R, Lu K P, Hunter T, Noel J P. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 14.Kaul R, Angeles A R, Jager M, Powers E T, Kelly J W. J Am Chem Soc. 2001;123:5206–5212. doi: 10.1021/ja0102890. [DOI] [PubMed] [Google Scholar]
- 15.Koepf E K, Petrassi H M, Sudol M, Kelly J W. Protein Sci. 1999;76:841–853. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson, N., Johnson, C. M., Macias, M., Oschkinat, H. & Fersht, A. R. (2001) Proc. Natl. Acad. Sci. USA98, (in press). [DOI] [PMC free article] [PubMed]
- 17.Nicholson E M, Scholtz J M. Biochemistry. 1996;35:11369–11378. doi: 10.1021/bi960863y. [DOI] [PubMed] [Google Scholar]
- 18.Jeener J, Meier B H, Bachmann P, Ernst R R. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 19.Braunschweiler L, Ernst R R. J Magn Reson. 1983;53:521–529. [Google Scholar]
- 20.Kraulis P J. J Magn Reson. 1989;84:627–633. [Google Scholar]
- 21.Kuszewski J, Nilges M, Brunger A T. J Biomol NMR. 1992;2:33–56. doi: 10.1007/BF02192799. [DOI] [PubMed] [Google Scholar]
- 22.Brünger T A. x-plor: A System for X-ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1993. [Google Scholar]
- 23.Levitt M. encadComputer Program, Energy Calculations and Dynamics. Palo Alto, CA: Molecular Applications Group; 1990. [Google Scholar]
- 24.Levitt M, Hirshberg M, Sharon R, Daggett V. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 25.Levitt M, Hirshberg M, Sharon R, Laidig K E, Daggett V. J Phys Chem B. 1997;101:5051–5061. [Google Scholar]
- 26.Kell G S. J Chem Eng. 1967;12:66–68. [Google Scholar]
- 27.Huang G S, Oas T G. Proc Natl Acad Sci USA. 1995;92:6878–6882. doi: 10.1073/pnas.92.15.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koepf E K, Petrassi H M, Ratnaswamy G, Huff M E, Sudol M, Kelly J W. Biochemistry. 1999;38:14338–14351. doi: 10.1021/bi991105l. [DOI] [PubMed] [Google Scholar]
- 29.Fersht A R, Leatherbarrow R J, Wells T N C. Biochemistry. 1987;26:6030–6038. doi: 10.1021/bi00393a013. [DOI] [PubMed] [Google Scholar]
- 30.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 31.Matouschek A, Kellis J T, Serrano L, Fersht A R. Nature (London) 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 32.Haque T S, Gellman S H. J Am Chem Soc. 1997;119:2303–2304. [Google Scholar]
- 33.Stanger H E, Gellman S H. J Am Chem Soc. 1998;120:4236–4237. [Google Scholar]
- 34.Riddle D S, Grantcharova V P, Santiago J V, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 35.Plaxco K W, Larson S, Ruczinski I, Riddle D S, Thayer E C, Buchwitz B, Davidson A R, Baker D. J Mol Biol. 2000;298:303–312. doi: 10.1006/jmbi.1999.3663. [DOI] [PubMed] [Google Scholar]
- 36.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 37.Ibragimova G T, Wade R C. Biophys J. 1999;77:2191–2198. doi: 10.1016/S0006-3495(99)77059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrin T E, Huang L E, Jarvis L E, Langridge R. J Mol Graph. 1988;6:13–27. [Google Scholar]
- 39.Becktel W J, Schellman J A. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 40.Jackson S E, Moracci M, Elmasry N, Johnson C M, Fersht A R. Biochemistry. 1993;32:11259–11269. doi: 10.1021/bi00093a001. [DOI] [PubMed] [Google Scholar]