

Abstract
Differentiation of atrial fibroblasts into myofibroblasts plays a critical role in atrial fibrosis. Sodium tanshinone IIA sulfonate (DS-201), a water-soluble derivative of tanshinone IIA, has been shown to have potent antifibrotic properties. However, the protective effects of DS-201 on angiotensin II- (Ang II-) induced differentiation of atrial fibroblasts into myofibroblasts remain to be elucidated. In this study, human atrial fibroblasts were stimulated with Ang II in the presence or absence of DS-201. Then, α-smooth muscle actin (α-SMA), collagen I, and collagen III expression and reactive oxygen species (ROS) generation were measured. The expression of transforming growth factor-β1 (TGF-β1) and the downstream signaling of TGF-β1, such as phosphorylation of Smad2/3, were also determined. The results demonstrated that DS-201 significantly prevented Ang II-induced human atrial fibroblast migration and decreased Ang II-induced α-SMA, collagen I, and collagen III expression. Furthermore, increased production of ROS and expression of TGF-β1 stimulated by Ang II were also significantly inhibited by DS-201. Consistent with these results, DS-201 significantly inhibited Ang II-evoked Smad2/3 phosphorylation and periostin expression. These results and the experiments involving N-acetyl cysteine (antioxidant) and an anti-TGF-β1 antibody suggest that DS-201 prevent Ang II-induced differentiation of atrial fibroblasts to myofibroblasts, at least in part, through suppressing oxidative stress and inhibiting the activation of TGF-β1 signaling pathway. All of these data indicate the potential utility of DS-201 for the treatment of cardiac fibrosis.
1. Introduction
Atrial fibrillation (AF), one of the most common arrhythmias, has become a serious epidemic across the world. Although atrial pathophysiology has been extensively studied, there are limited available therapies for patients with AF [1]. It is well known that atrial fibrosis which leads to atrial structural remodeling plays a pivotal role in the development and maintenance of AF [2, 3]. Inhibition of atrial fibrosis might be a plausible approach for AF prevention and therapy.
It has been demonstrated that atrial fibrosis is generally originated from nonmyocyte growth and extracellular matrix (ECM) protein deposition [4–6]. As the main nonmyocytes, fibroblasts undergo transdifferentiation into myofibroblasts, characterized by expression of contractile proteins, such as α-smooth muscle actin (α-SMA), and production of large amount of ECM components, such as collagen I and III, which are critically involved in atrial fibrosis [7, 8]. In addition, the development of atrial fibrosis and the myofibroblast differentiation have been shown to be highly regulated by angiotensin II (Ang II) and downstream activation of signaling pathway via soluble cytokines such as transforming growth factor-β1 (TGF-β1) [6, 9–11]. Therefore, the inhibition of Ang II-induced myofibroblast differentiation may be an important means for curing atrial fibrosis.
Danshen, also known as Salvia miltiorrhiza, is a traditional Chinese herbal medicine that has been widely used for many years to treat various diseases including coronary artery disease, myocardial infarction, atherosclerosis, and cerebrovascular disorders [12, 13]. Sodium tanshinone IIA sulfonate (DS-201, molecular structure is shown in Figure 1) is a water-soluble derivative of tanshinone IIA which is one of the most pharmacologically active monomers extracted from danshen. It has been reported that DS-201 or tanshinone IIA can induce vasodilation [14–16], inhibit inflammatory response [17–19], and prevent atherosclerosis [20–23], cardiac injury [24, 25], and hypertrophy [26–28]. In addition, DS-201 or tanshinone IIA has been demonstrated to inhibit renal fibrosis [29, 30], bladder fibrosis [31], and pulmonary fibrosis [32, 33] and to prevent TGF-β-, radiation-, and hypertension-induced cardiac fibrosis [34–36]. Furthermore, tanshinone IIA has been clinically proven effective in treating the patients with liver fibrosis and severe pneumonia [37]. However, there are no studies that have examined the effects of DS-201 on Ang II-induced differentiation of atrial fibroblasts into myofibroblasts.
Figure 1.
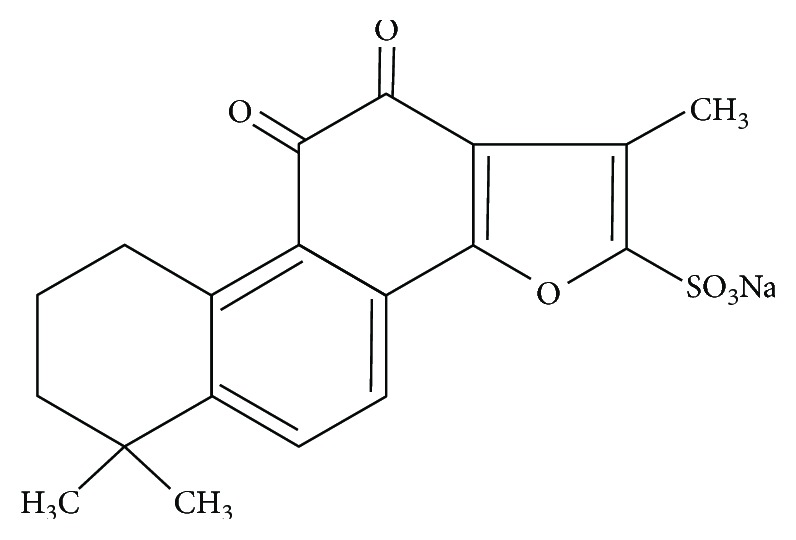
The molecular structure of DS-201.
Thus, in the present study, we investigated the effects of DS-201 on Ang II-induced myofibroblast transdifferentiation, and our data indicate that DS-201 prevents Ang II-induced myofibroblast transdifferentiation via suppressing oxidative stress and TGF-β1 signaling pathway in human atrial fibroblasts.
2. Materials and Methods
2.1. Chemicals and Reagents
DS-201 (purity ≥ 98%) was obtained from the National Institutes for Food and Drug Control (Beijing, China). Ang II was purchased from Sigma (St Louis, MO, USA). Human adult atrial fibroblasts and fibroblast medium-2 were purchased from ScienCell (Carlsbad, CA, USA). Antibodies to α-SMA, phosphorylated Smad2/3, and total Smad2/3 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to TGF-β1 and GAPDH were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies to collagen I, collagen III, and periostin were obtained from Abcam (Cambridge, MA, USA). Cell Counting Kit 8 (CCK8) solution, 2′,7′-dichlorofluorescein diacetate (DCFH-DA), total superoxide dismutase (SOD) assay kits, catalase (CAT) assay kits, and n-acetyl cysteine (NAC) were purchased from Beyotime (Shanghai, China).
2.2. Cell Culture
Human adult atrial fibroblasts were cultured in fibroblast medium-2, and cells used were passaged 3–7 times.
2.3. Cell Viability Assays
Human atrial fibroblasts (1 × 104/well) were seeded on 96-well plates and grown to confluence. Then, cells were starved of serum for 12 h, followed by stimulation with DS-201 (0, 5, 25, 50, 100, and 200 μM) for 24 h. Cell viability was measured with CCK8 solution at 450 nm using a Spectra Max M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
2.4. Cell Proliferation Assays
Human atrial fibroblasts (1 × 104/well) were seeded on 96-well plates and grown to 50% confluence. Then, cells were starved of serum for 12 h, followed by stimulation with Ang II in the absence or in the presence of DS-201 (5, 25, 50, and 100 μM) for 24 h. The cell proliferation was measured by adding 10 μL CCK8 solutions into the wells and following 4 h incubation at 37°C. Then, the OD values of each well were measured by a Spectra Max M5 microplate reader at the absorbance of 450 nm.
2.5. Immunoblotting
Human atrial fibroblasts (3 × 105/well) were plated on 3.5 cm wells, grown to confluence, and starved of serum for 12 h. Then, cells were exposed to Ang II (0.5 μM) without or with DS-201 (5, 25, 50, and 100 μM) for 24 h. In some experiments, fibroblasts were pretreated with NAC (10 mM) or an anti-TGF-β1 antibody (2 μg/mL), followed by stimulation with Ang II (0.5 μM) for 24 h. Then, cells were harvested and lysed in ice-cold RIPA lysis buffer (Beyotime) supplemented with protease and phosphatase inhibitors. The protein samples were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% nonfat dry milk solution for 1 h at room temperature. The blocked membranes were probed with antibodies against α-SMA (1 : 200), collagen I (1 : 1000), collagen III (1 : 1000), TGF-β1 (1 : 1000), phosphorylated Smad2/3 (1 : 200), total Smad2/3 (1 : 200), periostin (1 : 1000), and GAPDH (1 : 1000) overnight at 4°C. After washing with phosphate-buffered saline containing 0.05% Tween 20, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) specific to the primary antibody. Then, the membranes were treated with enhanced chemiluminescence reagents (Merck Millipore, Watford, UK), and protein signal was imaged using ChemiDoc XRS (Bio-Rad Laboratories). ImageJ was used to measure the density of bands.
2.6. Cell Migration Assays
Cell migration was measured using transwell migration chambers with an 8.0 μm-sized porous membrane (Corning Costar, Corning, NY, USA). Human atrial fibroblasts (3 × 104) were added to the upper chambers and exposed to Ang II (0.5 μM) without or with DS-201 (5, 25, 50, and 100 μM) for 24 h. Then, the cells remaining in the upper chamber were removed using a cotton swab, and the membranes were fixed and then stained with 0.5% crystal violet. The cells that had migrated to the lower chamber were counted.
2.7. Measurement of Intracellular Reactive Oxygen Species (ROS)
Human atrial fibroblasts were exposed to Ang II (0.5 μM) without or with DS-201 (5, 25, 50, and 100 μM) for 1 h. Then, cells were washed with fibroblast medium-2 and subsequently incubated with DCFH-DA (10 μM) for 30 min at 37°C. After incubation, the fluorescence intensity of the cells was determined using a Spectra Max M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA) (488/525 nm), and the fluorescence images were also captured with an EVOS inverted microscope (AMG, Mill Creek, WA, USA).
2.8. Measurement of Intracellular SOD and CAT Level
Human atrial fibroblasts were exposed to Ang II (0.5 μM) without or with DS-201 (5, 25, 50, and 100 μM) for 1 h. Cell lysates were prepared, and the protein concentrations were determined using a BCA protein assay. Then, the level of SOD and CAT was measured by the respective kits according to the manufacturer's instructions.
2.9. Data Analysis
All data were expressed as mean ± standard deviation (SD) of the mean. Results were analyzed by one-way analysis of variance (ANOVA) followed by post hoc comparison. P < 0.05 was considered to be significantly different.
3. Results
3.1. Effects of DS-201 on Cell Viability
To identify whether DS-201 treatment induced cell death, the effects of DS-201 on fibroblast viability were detected. Human atrial fibroblasts were stimulated with DS-201 (0, 5, 25, 50, 100, and 200 μM) for 24 h, and cell viability assay was performed with a CCK8 solution. The results showed no significant differences among the groups (Figure 2), indicating that at the maximum concentration of 200 μM, DS-201 did not affect fibroblast viability. Therefore, the highest concentration of DS-201 we used was 100 μM in the follow-up experiments.
Figure 2.
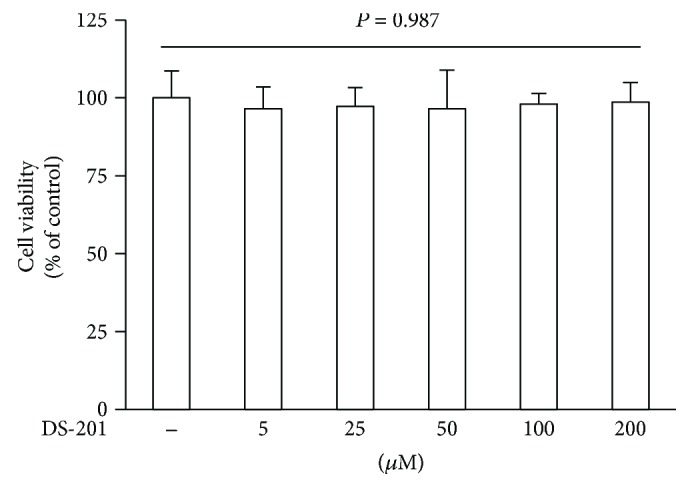
Effects of DS-201 on cell viability. Human atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50,100, and 200 μM) for 24 h. Cell viability was measured using the CCK8 assay. Data shown are mean ± SD of 4 independent experiments, presented as % of the control value (first bar).
3.2. DS-201 Prevents Ang II-Induced Fibrotic Response in Human Atrial Fibroblasts
Fibrotic response in atrial fibroblasts, involving fibroblast differentiation to myofibroblast, fibroblast-derived ECM protein deposition, fibroblast migration, and proliferation, plays an import role in atrial fibrosis. To explore the effects of DS-201 on Ang II-induced fibrotic response, myofibroblast differentiation and ECM protein deposition were firstly investigated. Atrial fibroblasts were exposed to Ang II (0.5 μM) in the absence or presence of DS-201 (0, 5, 25, 50, and 100 μM) for 24 h. Then, cell lysates were prepared, and the expression of α-SMA, collagen I, and collagen III was determined with western blotting. The data showed that α-SMA, collagen I, and collagen III expression significantly increased with Ang II stimulation but decreased by DS-201 treatment (Figure 3). We also examined the effects of DS-201 alone on α-SMA, collagen I, and collagen III expression, and the results showed no significant differences among the groups (Supplementary Figure 1 and Supplementary Figure 2). The results indicate that DS-201 prevents Ang II-induced myofibroblast differentiation and ECM protein expression.
Figure 3.

DS-201 prevents Ang II-induced fibrotic response in atrial fibroblasts. Atrial fibroblasts were exposed to Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM) for 24 h. (a) Expression of α-SMA was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of α-SMA normalized to GAPDH was calculated. (b) Expression of collagen I was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of collagen I normalized to GAPDH was calculated. (c) Expression of collagen III was also analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of collagen III normalized to GAPDH was calculated. All data shown are mean values ± SD and are expressed as fold changes. ∗∗ P < 0.01 versus control (first bar); # P < 0.05 versus Ang II; ## P < 0.01 versus Ang II.
The migration of fibroblast was also detected using a transwell chamber migration assay. Human atrial fibroblasts were added to the upper chamber containing porous filters and treated with Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM). After 24 h, the cells that had migrated through the membrane were counted. The results showed that Ang II significantly increased the migrated cells compared with that in the vehicle control group, and DS-201 treatment significantly inhibited Ang II-induced fibroblast migration (Figures 4(a) and 4(b)). The effects of DS-201 on Ang II-induced fibroblast proliferation were also determined. The data showed that the cell proliferation significantly increased by Ang II stimulation but decreased with DS-201 treatment (Figure 4(c)). As a whole, all of these results suggest that DS-201 prevent Ang II-induced fibrotic response in human atrial fibroblasts.
Figure 4.

DS-201 prevents Ang II-induced cell migration and cell proliferation. (a) Atrial fibroblasts were added to upper chambers containing porous filters and then treated with Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM). After 24 h, the cells were fixed and were stained with crystal violet. The cells that had migrated into the lower chambers were counted. Representative images of cell migration are shown (scale bar, 200 μm). (b) Quantitative assessment of 3 independent cell migration experiments was performed. (c) Atrial fibroblasts were stimulated with Ang II in the absence or in the presence of DS-201 (5, 25, 50, and 100 μM) for 24 h. Then, cell proliferation was measured by CCK8 assay. All data shown are mean values ± SD and are expressed as % of the control value (first bar). ∗∗ P < 0.01 versus control (first bar); ## P < 0.01 versus Ang II.
3.3. DS-201 Prevents Ang II-Induced Oxidative Stress
Since overproduction of ROS has been demonstrated to contribute to atrial fibrosis [38, 39], the effects of DS-201 on Ang II-induced ROS generation were detected. Atrial fibroblasts were stimulated with Ang II (0.5 μM) in the absence or presence of DS-201 (0, 5, 25, 50, and 100 μM) for 1 h. Then, cells were stained with DCFH-DA, and the fluorescence intensity of DCF was measured at 488/525 nm using a microplate reader. The results showed that intracellular ROS significantly increased in Ang II-treated fibroblast but decreased by DS-201 treatment (Figure 5(a)). Similar results were also shown in fluorescence images (Figure 5(b)). The activity of the antioxidant enzymes, such as SOD and CAT, was also measured. The data showed that the SOD and CAT levels significantly decreased by Ang II stimulation but restored with DS-201 treatment (Figures 5(c) and 5(d)). We also examined the effects of DS-201 alone on ROS generation, and the results showed no significant differences among the groups (Supplementary Figure 3). These data indicate that DS-201 prevents Ang II-induced oxidative stress.
Figure 5.

DS-201 inhibits Ang II-induced oxidative stress. Atrial fibroblasts were exposed to Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM) for 1 h. (a) Cells were stained with DCFH-DA and the fluorescence intensity of DCF was measured at 488/525 nm using a microplate reader. (b) The DCF fluorescence was also monitored by fluorescence microscopy, and representative images of 3 independent experiments are shown (scale bar, 100 μm). (c, d) The level of SOD and CAT was measured by the respective kits according to the manufacturer's instructions. All data shown are mean ± SD for 3 independent experiments and presented as % of the control value (first bar). ∗∗ P < 0.01 versus Con; # P < 0.05 versus Ang II; ## P < 0.01 versus Ang II.
To further determine the effects of antioxidant on Ang II-induced fibrotic response in atrial fibroblasts, the cells were pretreated with NAC for 1 h, followed by stimulation with Ang II for 24 h. The results demonstrated that NAC significantly inhibited Ang II-induced α-SMA expression and ECM deposition (Figure 6). As a whole, these results indicate that DS-201 prevents Ang II-induced differentiation of atrial fibroblasts to myofibroblasts through inhibiting oxidative stress.
Figure 6.

NAC inhibits Ang II-induced fibrotic response in atrial fibroblasts. Atrial fibroblasts were pretreated with NAC (10 mM) for 1 h, followed by stimulation with Ang II (0.5 μM) for 24 h. Expression of α-SMA, collagen I, and collagen III was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of α-SMA, collagen I, and collagen III normalized to GAPDH was calculated. All data shown are mean values ± SD and are expressed as fold changes. ∗∗ P < 0.01 versus control (first bar); ## P < 0.01 versus Ang II.
3.4. DS-201 Prevents Ang II-Induced TGF-β1 Activation
Previous studies have demonstrated that TGF-β1 plays a key role in atrial fibrosis [11, 39]. Specially, it was shown that in the absence of TGF-β1, Ang II was not able to induce cardiac hypertrophy and fibrosis in vivo [40]. Therefore, the effects of DS-201 on Ang II-induced TGF-β1 activation were investigated. Atrial fibroblasts were exposed to Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM) for 24 h. Cell lysates were prepared, and the protein was analyzed using western blotting. The results demonstrated that the expression of TGF-β1 significantly increased with Ang II stimulation but decreased in response to DS-201 treatment (Figure 7(a)). We also found that DS-201 alone had no effects on TGF-β1 expression (Supplementary Figure 4).
Figure 7.

DS-201 prevents Ang II-induced TGF-β1 activation in human atrial fibroblasts. Atrial fibroblasts were exposed to Ang II (0.5 μM) with or without DS-201 (0, 5, 25, 50, and 100 μM) for 24 h. (a) Expression of TGF-β1 was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of TGF-β1 normalized to GAPDH was calculated. (b) Phosphorylation (p) of Smad2/3 was analyzed by western blotting. Representative images of 3 independent experiments and densitometric analysis of phosphorylated Smad2/3 normalized to total Smad2/3 are shown. (c) Expression of periostin was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ration of periostin normalized to GAPDH was calculated. ∗∗ P < 0.01 versus control (first bar); ## P < 0.01 versus Ang II.
The phosphorylation of Smad2/3, the downstream signaling of TGF-β1, was also determined. The data showed that DS-201 significantly inhibited Ang II-evoked Smad2/3 phosphorylation (Figure 7(b)). Furthermore, periostin is well known to be a TGF-β1-inducible matrix protein. We then investigated whether DS-201 affects periostin expression. The results showed that the expression of periostin significantly increased in response to Ang II, whereas DS-201 decreased its expression (Figure 7(c)). Taken together, these results suggest that DS-201 prevents Ang II-induced TGF-β1 activation.
To further identify the effects of blockade of TGF-β1 on Ang II-induced fibrotic response in atrial fibroblasts, the cells were pretreated with an anti-TGF-β1 antibody for 1 h, followed by stimulation with Ang II for 24 h. The results demonstrated that the anti-TGF-β1 antibody significantly inhibited Ang II-induced α-SMA expression and ECM deposition (Figure 8). As a whole, these results indicate that DS-201 prevents Ang II-induced differentiation of atrial fibroblasts to myofibroblasts through inhibiting the activation of TGF-β1 signaling pathway.
Figure 8.

The blockade of TGF-β1 inhibits Ang II-induced fibrotic response in atrial fibroblasts. Atrial fibroblasts were pretreated with an anti-TGF-β1 antibody (2 μg/mL) for 1 h, followed by stimulation with Ang II (0.5 μM) for 24 h. Expression of α-SMA, collagen I, and collagen III was analyzed by western blotting, and representative images of 3 independent experiments are shown. The ratio of α-SMA, collagen I, and collagen III normalized to GAPDH was calculated. All data shown are mean values ± SD and are expressed as fold changes. ∗∗ P < 0.01 versus control (first bar); ## P < 0.01 versus Ang II.
4. Discussion
It is well known that Ang II, which has been reported to be activated in various cardiovascular diseases, such as myocardial infarction and hypertension [41, 42], plays a major role in atrial fibrosis by promoting differentiation of atrial fibroblasts into myofibroblasts [43]. Therefore, the identification of new preventive and therapeutic approaches targeting Ang II-induced myofibroblast differentiation has great clinical implications. In the present study, we evaluated the effects of DS-201 on Ang II-induced atrial fibrosis, and the results demonstrated that DS-201 prevented Ang II-induced myofibroblast differentiation via inhibiting oxidative stress and downregulating TGF-β1 signaling pathway in human atrial fibroblasts.
The differentiation of fibroblasts into myofibroblasts is characterized by α-SMA expression and ECM protein deposition. Previous reports [44–46] and our data in this study demonstrated that Ang II significantly increased fibroblast migration, α-SMA expression, and collagen production. However, the treatment with DS201 prevented Ang II-induced fibrotic response in atrial fibroblasts in a dose-dependent manner. These results were consistent with previous studies which showed that DS-201 inhibited TGF-β-, radiation-, and hypertension-induced cardiac fibrosis [34–36].
Growing evidence has highlighted oxidative stress as an important mechanism in pathologic cardiac remodeling [38, 39]. Previous reports [44, 47] and our results demonstrated that Ang II significantly increased intracellular ROS generation in fibroblasts. The present results clearly showed that DS-201 significantly inhibited Ang II-induced ROS generation and increased the activation of enzymes which can scavenge ROS such as SOD and CAT in atrial fibroblasts, which was in line with these reported by other investigators [48, 49]. Although the mechanisms by which ROS mediates the differentiation of atrial fibroblasts into myofibroblasts remain unclear, our experiments involving NAC suggest that the inhibition of oxidative stress significantly prevents Ang II-induced fibrotic response in atrial fibroblasts. These results were consistent with previous studies which have shown that ROS is necessary for Ang II- or TGF-β1-induced α-SMA expression and myofibroblast differentiation [44, 50]. Thus, the present findings strongly suggest that DS-201 prevents Ang II-induced differentiation of atrial fibroblasts to myofibroblasts by blocking oxidative stress.
TGF-β1 plays an important role in the pathogenesis of cardiac remodeling and fibrosis. In genetic modification studies, TGF-β1 overexpression in the mouse heart was associated with fibrosis [51, 52]. In addition, extensive evidence has suggested a direct link between the renin-angiotensin system and TGF-β1, indicating that TGF-β1 acts downstream of Ang II [53]. Consistent with previous studies, our data also showed that Ang II significantly increased TGF-β1 expression and Smad2/3 phosphorylation, whereas treatment with DS-201 dose-dependently inhibited increased the expression of TGF-β1 and activation of Smad2/3. Periostin, a TGF-β-inducible matrix protein, has been demonstrated to contribute to fibrosis by regulating ECM molecules such as collagen and fibronectin [54]. Periostin knockout resulted in reduced fibrosis and hypertrophy after pressure overload, whereas periostin-overexpressing transgenic mice develop spontaneous hypertrophy with aging [55, 56]. Consistent with these reports, our results showed that periostin expression was massively increased by Ang II stimulation, but its expression was decreased with a concomitant reduction in fibrosis after DS-201 treatment.
Although few previous studies focused the effects of DS-201 on Ang II-induced TGF-β1 signaling pathway activation, previous reports have indicated that tanshinone IIA attenuated TGF-β1 expression in pulmonary fibrosis [32] and in chronic kidney disease [57], which were partly in agreement with our results. Furthermore, our experiments involving an anti-TGF-β1 antibody indicate that the blockade of TGF-β1 signaling pathway significantly inhibits Ang II-induced fibrotic response. Taken together, the present results suggest that DS-201 prevents Ang II-induced differentiation of atrial fibroblasts to myofibroblasts through downregulating TGF-β1 signaling pathway.
Based on our results in the present study and those of published reports, it is conceivable that DS-201 has implications for atrial fibrosis prevention and therapy. However, the underlying mechanisms, especially, the target of DS-201, have not been fully elucidated. Therefore, further investigations are needed to better explore the therapeutic potential of DS-201.
5. Conclusions
In summary, we have described a protective role of DS-201 in Ang II-induced differentiation of atrial fibroblasts to myofibroblasts and the molecular mechanism involved in the present study. Our data demonstrated that DS-201 prevented Ang II-induced atrial fibrosis through inhibiting oxidative stress and suppressing TGF-β1 signaling pathway. These novel findings indicate the potential application of DS-201 for the prevention and treatment of fibrosis disease in the clinical practice.
Acknowledgments
This work was supported by the Foundation of Office of Science & Technology of Luzhou City (2015-S-42). The authors gratefully acknowledge Dr. Yan Yang (Key Laboratory of Ministry of Education for Medical Electrophysiology and the Institute of Cardiovascular Research, Southwest Medical University) for providing the sodium tanshinone IIA sulfonate.
Contributor Information
Jun Cheng, Email: lzcj1221@swmu.edu.cn.
Liqun Wang, Email: yy000213@163.com.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there is no conflict of interests.
Authors' Contributions
Tangting Chen, Miaoling Li, and Xuehui Fan carried out the experiments. Jun Cheng performed the data analysis. Liqun Wang and Jun Cheng designed the experiments and wrote the manuscript.
Supplementary Materials
This supplementary material file contains 4 figures. These results show the effects of DS-201 on α-SMA, collagen I, collagen III, and TGF-β1 expression and ROS generation. Supplementary Figure 1: the effects of DS-201 on α-SMA expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of α-SMA was analyzed by western blotting. Supplementary Figure 2: the effects of DS-201 on collagen I and collagen III expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of collagen I and collagen III was analyzed by western blotting. Supplementary Figure 3: the effects of DS-201 on ROS generation. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 1 h, and then the production of ROS was measured by DCFH-DA staining. Supplementary Figure 4: the effects of DS-201 on TGF-β1 expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of TGF-β1 was analyzed by western blotting.
References
- 1.Musa H., Kaur K., O’Connell R., et al. Inhibition of platelet-derived growth factor-AB signaling prevents electromechanical remodeling of adult atrial myocytes that contact myofibroblasts. Heart Rhythm. 2013;10(7):1044–1051. doi: 10.1016/j.hrthm.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu Y. F., Chen Y. J., Lin Y. J., Chen S. A. Inflammation and the pathogenesis of atrial fibrillation. Nature Reviews Cardiology. 2015;12(4):230–243. doi: 10.1038/nrcardio.2015.2. [DOI] [PubMed] [Google Scholar]
- 3.Lau D. H., Schotten U., Mahajan R., et al. Novel mechanisms in the pathogenesis of atrial fibrillation: practical applications. European Heart Journal. 2016;37(20):1573–1581. doi: 10.1093/eurheartj/ehv375. [DOI] [PubMed] [Google Scholar]
- 4.Pellman J., Lyon R. C., Sheikh F. Extracellular matrix remodeling in atrial fibrosis: mechanisms and implications in atrial fibrillation. Journal of Molecular and Cellular Cardiology. 2010;48(3):461–467. doi: 10.1016/j.yjmcc.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan A. Y., Zimetbaum P. Atrial fibrillation and atrial fibrosis. Journal of Cardiovascular Pharmacology. 2011;57(6):625–629. doi: 10.1097/FJC.0b013e3182073c78. [DOI] [PubMed] [Google Scholar]
- 6.Thanigaimani S., Lau D. H., Agbaedeng T., Elliott A. D., Mahajan R., Sanders P. Molecular mechanisms of atrial fibrosis: implications for the clinic. Expert Review of Cardiovascular Therapy. 2017;15(4):247–256. doi: 10.1080/14779072.2017.1299005. [DOI] [PubMed] [Google Scholar]
- 7.Petrov V. V., Fagard R. H., Lijnen P. J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39(2):258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 8.Weber K. T. Fibrosis in hypertensive heart disease: focus on cardiac fibroblasts. Journal of Hypertension. 2004;22(1):47–50. doi: 10.1097/00004872-200401000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Novo G., Guttilla D., Fazio G., Cooper D., Novo S. The role of the renin–angiotensin system in atrial fibrillation and the therapeutic effects of ACE-Is and ARBS. British Journal of Clinical Pharmacology. 2008;66(3):345–351. doi: 10.1111/j.1365-2125.2008.03234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine M. J., Schweitzer P. Angiotensin converting enzyme inhibitors and angiotensin II receptor blockers in atrial fibrillation. Vnitrni Lekarstvi. 2010;56(11):1138–1141. [PubMed] [Google Scholar]
- 11.Dobaczewski M., Chen W., Frangogiannis N. G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. Journal of Molecular and Cellular Cardiology. 2011;51(4):600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J. Y., Fan J. Y., Horie Y., et al. Ameliorating effects of compounds derived from Salvia miltiorrhiza root extract on microcirculatory disturbance and target organ injury by ischemia and reperfusion. Pharmacology & Therapeutics. 2008;117(2):280–295. doi: 10.1016/j.pharmthera.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L., Zuo Z., Chow M. S. S. Danshen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. The Journal of Clinical Pharmacology. 2005;45(12):1345–1359. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]
- 14.Fan G., Zhu Y., Guo H., Wang X., Wang H., Gao X. Direct vasorelaxation by a novel phytoestrogen tanshinone IIA is mediated by nongenomic action of estrogen receptor through endothelial nitric oxide synthase activation and calcium mobilization. Journal of Cardiovascular Pharmacology. 2011;57(3):340–347. doi: 10.1097/FJC.0b013e31820a0da1. [DOI] [PubMed] [Google Scholar]
- 15.Morton J. S., Andersson I. J., Cheung P. Y., Baker P., Davidge S. T. The vascular effects of sodium tanshinone IIA sulphonate in rodent and human pregnancy. PLoS ONE. 2015;10(3, article e0121897) doi: 10.1371/journal.pone.0121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu G. B., Zhou E. X., Qing D. X. Tanshinone II(A) elicited vasodilation in rat coronary arteriole: roles of nitric oxide and potassium channels. European Journal of Pharmacology. 2009;617(1–3):102–107. doi: 10.1016/j.ejphar.2009.06.046. [DOI] [PubMed] [Google Scholar]
- 17.Li W., Zhang Y., Xing C., Zhang M. Tanshinone IIA represses inflammatory response and reduces radiculopathic pain by inhibiting IRAK-1 and NF-κB/p38/JNK signaling. International Immunopharmacology. 2015;28(1):382–389. doi: 10.1016/j.intimp.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 18.Lu B. L., Li J., Zhou J., Li W. W., Wu H. F. Tanshinone IIA decreases the levels of inflammation induced by Aβ1-42 in brain tissues of Alzheimer’s disease model rats. Neuroreport. 2016;27(12):883–893. doi: 10.1097/WNR.0000000000000618. [DOI] [PubMed] [Google Scholar]
- 19.Yang J.-X., Pan Y. Y., Ge J. H., et al. Tanshinone II A attenuates TNF-α-induced expression of VCAM-1 and ICAM-1 in endothelial progenitor cells by blocking activation of NF-κB. Cellular Physiology and Biochemistry. 2016;40(1-2):195–206. doi: 10.1159/000452537. [DOI] [PubMed] [Google Scholar]
- 20.Chang C. C., Chu C. F., Wang C. N., et al. The anti-atherosclerotic effect of tanshinone IIA is associated with the inhibition of TNF-α-induced VCAM-1, ICAM-1 and CX3CL1 expression. Phytomedicine. 2014;21(3):207–216. doi: 10.1016/j.phymed.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Chen W., Tang F., Xie B., Chen S., Huang H., Liu P. Amelioration of atherosclerosis by tanshinone IIA in hyperlipidemic rabbits through attenuation of oxidative stress. European Journal of Pharmacology. 2012;674(2-3):359–364. doi: 10.1016/j.ejphar.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 22.Chen Z., Xu H. Anti-inflammatory and immunomodulatory mechanism of tanshinone IIA for atherosclerosis. Evidence-Based Complementary and Alternative Medicine. 2014;2014:6. doi: 10.1155/2014/267976.267976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang Z. Y., Lin R., Yuan B. X., Yang G. D., Liu Y., Zhang H. Tanshinone IIA downregulates the CD40 expression and decreases MMP-2 activity on atherosclerosis induced by high fatty diet in rabbit. Journal of Ethnopharmacology. 2008;115(2):217–222. doi: 10.1016/j.jep.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 24.Gao S., Liu Z., Li H., Little P. J., Liu P., Xu S. Cardiovascular actions and therapeutic potential of tanshinone IIA. Atherosclerosis. 2012;220(1):3–10. doi: 10.1016/j.atherosclerosis.2011.06.041. [DOI] [PubMed] [Google Scholar]
- 25.Jiang B., Zhang L., Wang Y., et al. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food and Chemical Toxicology. 2009;47(7):1538–1544. doi: 10.1016/j.fct.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 26.Feng J., Zheng Z. Effect of sodium tanshinone II A sulfonate on cardiac myocyte hypertrophy and its underlying mechanism. Chinese Journal of Integrative Medicine. 2008;14(3):197–201. doi: 10.1007/s11655-008-0197-y. [DOI] [PubMed] [Google Scholar]
- 27.Weng Y. S., Wang H. F., Pai P. Y., et al. Tanshinone IIA prevents Leu27IGF-II-induced cardiomyocyte hypertrophy mediated by estrogen receptor and subsequent Akt activation. American Journal of Chinese Medicine. 2015;43(8):1567–1591. doi: 10.1142/S0192415X15500895. [DOI] [PubMed] [Google Scholar]
- 28.Yang L., Zou X., Liang Q., et al. Sodium tanshinone IIA sulfonate depresses angiotensin II-induced cardiomyocyte hypertrophy through MEK/ERK pathway. Experimental and Molecular Medicine. 2007;39(1):65–73. doi: 10.1038/emm.2007.8. [DOI] [PubMed] [Google Scholar]
- 29.Jiang C., Shao Q., Jin B., Gong R., Zhang M., Xu B. Tanshinone IIA attenuates renal fibrosis after acute kidney injury in a mouse model through inhibition of fibrocytes recruitment. Biomed Research International. 2015;2015:10. doi: 10.1155/2015/867140.867140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D. T., Huang R. H., Cheng X., Zhang Z. H., Yang Y. J., Lin X. Tanshinone IIA attenuates renal fibrosis and inflammation via altering expression of TGF-β/Smad and NF-κB signaling pathway in 5/6 nephrectomized rats. International Immunopharmacology. 2015;26(1):4–12. doi: 10.1016/j.intimp.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 31.Jiang X., Chen Y., Zhu H., et al. Sodium tanshinone IIA sulfonate ameliorates bladder fibrosis in a rat model of partial bladder outlet obstruction by inhibiting the TGF-β/Smad pathway activation. PLoS ONE. 2015;10(6, article e0129655) doi: 10.1371/journal.pone.0129655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H., Li Y., Wang Y., et al. Tanshinone IIA attenuates bleomycin-induced pulmonary fibrosis via modulating angiotensin-converting enzyme 2/ angiotensin-(1-7) axis in rats. International Journal of Medical Sciences. 2014;11(6):578–586. doi: 10.7150/ijms.8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang H., He H., Ji H., et al. Tanshinone IIA ameliorates bleomycin-induced pulmonary fibrosis and inhibits transforming growth factor-beta-β-dependent epithelial to mesenchymal transition. Journal of Surgical Research. 2015;197(1):167–175. doi: 10.1016/j.jss.2015.02.062. [DOI] [PubMed] [Google Scholar]
- 34.Zhan C. Y., Tang J. H., Zhou D. X., Li Z. H. Effects of tanshinone IIA on the transforming growth factor β1/Smad signaling pathway in rat cardiac fibroblasts. Indian Journal of Pharmacology. 2014;46(6):633–638. doi: 10.4103/0253-7613.144933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu J., Li H. L., Wu H. Y., et al. Sodium tanshinone IIA sulfonate attenuates radiation-induced fibrosis damage in cardiac fibroblasts. Journal of Asian Natural Products Research. 2014;16(9):941–952. doi: 10.1080/10286020.2014.935769. [DOI] [PubMed] [Google Scholar]
- 36.Wang P., Wu X., Bao Y., et al. Tanshinone IIA prevents cardiac remodeling through attenuating NAD(P)H oxidase-derived reactive oxygen species production in hypertensive rats. Pharmazie. 2011;66(7):517–524. [PubMed] [Google Scholar]
- 37.Li L. C., Kan L. D. Traditional Chinese medicine for pulmonary fibrosis therapy: progress and future prospects. Journal of Ethnopharmacology. 2017;198:45–63. doi: 10.1016/j.jep.2016.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takimoto E., Kass D. A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 39.Purnomo Y., Piccart Y., Coenen T., Prihadi J., Lijnen P. Oxidative stress and transforming growth factor-β 1-induced cardiac fibrosis. Cardiovascular & Hematological Disorders-Drug Targets. 2013;13(2):165–172. doi: 10.2174/1871529X11313020010. [DOI] [PubMed] [Google Scholar]
- 40.Schultz J. E. J., Witt S. A., Glascock B. J., et al. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. Journal of Clinical Investigation. 2002;109(6):787–796. doi: 10.1172/JCI0214190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holtz J. Pathophysiology of heart failure and the renin-angiotensin-system. In: Grobecker H., Heusch G., Strauer B. E., editors. Angiotensin and the Heart. Steinkopff; 1993. pp. 183–201. [DOI] [PubMed] [Google Scholar]
- 42.Ma T. K., Kam K. K., Yan B. P., Lam Y. Y. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. British Journal of Pharmacology. 2010;160(6):1273–1292. doi: 10.1111/j.1476-5381.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circulation Research. 2015;116(7):1269–1276. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 44.Bai J., Zhang N., Hua Y., et al. Metformin inhibits angiotensin II-induced differentiation of cardiac fibroblasts into myofibroblasts. PLoS One. 2013;8(9, article e72120) doi: 10.1371/journal.pone.0072120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siddesha J. M., Valente A. J., Sakamuri S. S. V. P., et al. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. Journal of Molecular and Cellular Cardiology. 2013;65:9–18. doi: 10.1016/j.yjmcc.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J. J., Huang N., Lu Y., et al. Improving vagal activity ameliorates cardiac fibrosis induced by angiotensin II: in vivo and in vitro . Scientific Reports. 2015;5(1, article 17108) doi: 10.1038/srep17108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia Y., Xu J., Yu Y., et al. Nifedipine inhibits angiotensin II-induced cardiac fibrosis via downregulating Nox4-derived ROS generation and suppressing ERK1/2, JNK signaling pathways. Pharmazie. 2013;68(6):435–441. [PubMed] [Google Scholar]
- 48.Hong H. J., Liu J. C., Cheng T. H., Chan P. Tanshinone IIA attenuates angiotensin II-induced apoptosis via Akt pathway in neonatal rat cardiomyocytes. Acta Pharmacologica Sinica. 2010;31(12):1569–1575. doi: 10.1038/aps.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan P., Liu J. C., Lin L. J., et al. Tanshinone IIA inhibits angiotensin II-induced cell proliferation in rat cardiac fibroblasts. American Journal of Chinese Medicine. 2011;39(2):381–394. doi: 10.1142/S0192415X11008890. [DOI] [PubMed] [Google Scholar]
- 50.Cucoranu I., Clempus R., Dikalova A., et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation Research. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 51.Nakajima H., Nakajima H. O., Salcher O., et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circulation Research. 2000;86(5):571–579. doi: 10.1161/01.RES.86.5.571. [DOI] [PubMed] [Google Scholar]
- 52.Verheule S., Sato T., Everett T, et al. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-β1. Circulation Research. 2004;94(11):1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovascular Research. 2004;63(3):423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 54.Conway S. J., Izuhara K., Kudo Y., et al. The role of periostin in tissue remodeling across health and disease. Cellular and Molecular Life Sciences. 2014;71(7):1279–1288. doi: 10.1007/s00018-013-1494-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oka T., Xu J., Kaiser R. A., et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circulation Research. 2007;101(3):313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lorts A., Schwanekamp J. A., Elrod J. W., Sargent M. A., Molkentin J. D. Genetic manipulation of periostin expression in the heart does not affect myocyte content, cell cycle activity, or cardiac repair. Circulation Research. 2009;104(1):e1–e7. doi: 10.1161/CIRCRESAHA.108.188649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahn Y. M., Kim S. K., Lee S. H., et al. Renoprotective effect of tanshinone IIA, an active component of Salvia miltiorrhiza, on rats with chronic kidney disease. Phytotherapy Research. 2010;24(12):1886–1892. doi: 10.1002/ptr.3347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This supplementary material file contains 4 figures. These results show the effects of DS-201 on α-SMA, collagen I, collagen III, and TGF-β1 expression and ROS generation. Supplementary Figure 1: the effects of DS-201 on α-SMA expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of α-SMA was analyzed by western blotting. Supplementary Figure 2: the effects of DS-201 on collagen I and collagen III expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of collagen I and collagen III was analyzed by western blotting. Supplementary Figure 3: the effects of DS-201 on ROS generation. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 1 h, and then the production of ROS was measured by DCFH-DA staining. Supplementary Figure 4: the effects of DS-201 on TGF-β1 expression. Atrial fibroblasts were exposed to DS-201 (0, 5, 25, 50, and 100 μM) for 24 h, and then the expression of TGF-β1 was analyzed by western blotting.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.