

Abstract
Binding sites for the transcription factor Rap1 are widespread in the yeast genome. With respect to many, but not all, genes, Rap1p has an apparent activation function. Whether Rap1 is itself a transcriptional activator, or whether it is in some way required for activation by additional factors, is not clear. We have identified a previously unrecognized Rap1p binding site in the internal regulatory region of Ty1 elements. We demonstrate that this site is capable of binding Rap1 in vitro and that, in vivo, Rap1p plays an important regulatory role in Ty1 and Ty1-mediated adjacent gene expression. Our data suggest that in Ty1 elements, maximal levels of RAP1-mediated activation depend on the formation of a complex with Mcm1, an independent DNA-binding protein that functions in transcription as well as in DNA replication, and with a third factor, IBF, previously identified as a binding activity with a site situated between the Rap1p and Mcm1p binding sites in this region of Ty1 elements.
Rap1 is a multifunctional DNA-binding protein with binding sites in the promoters of a large number of unrelated genes (Shore and Nasmyth, 1987; Shore et al., 1987; Buchman et al., 1988; Buchman et al., 1988; Hurd and Roberts, 1989; Henry et al., 1990). Studies of hybrid Rap1 derivatives in which RAP1 sequences are fused to heterologous DNA binding motifs have shown that the Rap1 protein contains regions capable of transcriptional activation (Hardy et al., 1992). It is presumed that Rap1p plays the role of a transcriptional activator in regulating the expression of most genes with promoters containing Rap1 binding sites. Rap1p has an apparently distinct silencing function; specific mutations in the RAP1 gene result in derepression of silent mating type loci (Hardy et al., 1992). Mutations in the RAP1 gene also affect telomere length, due to the presence of telomeric Rap1 binding sites (Lustig et al., 1990).
Studies of a Rap1 binding site at the HIS4 locus suggest another possible function for Rap1 protein. In the HIS4 promoter, the Rap1 binding site overlaps one of the major binding sites for the transcriptional activator, Gcn4. Under standard in vitro conditions, Rap1 but not Gcn4 binding can be detected. At high Gcn4 concentrations (in vitro), Rap1 protein is competed from its binding site. In vivo, a functional Rap1 binding site is required for GCN4-mediated activity as well as for BAS1 and BAS2-mediated basal expression of the HIS4 gene in vivo (Arndt and Fink, 1986; Devlin et al., 1991). Comparisons of micrococcal nuclease sensitivity of HIS4 promoter derivatives containing or lacking a Rap1 binding site led to the suggestion of a role for Rap1 in establishment or maintenance of a chromatin configuration permissive for the binding of the factors that allow regulated HIS4 expression (Devlin et al., 1991). It is possible that all RAP1-mediated transcriptional activation is the indirect result of a requirement for Rap1p in chromatin clearance. Alternatively, Rap1 may have distinct roles in transcriptional activation that vary with the locus.
Molecular analysis of sequences adjacent to the block II Ty1 enhancer region (Fig. 1; Errede et al., 1985) revealed sequence homology to a consensus Rap1 binding site. To investigate the use of this Rap1 binding site and its potential significance in regulation of Ty and adjacent gene expression, we have examined this region of a Ty1 element in three different contexts: in isolation, in combination with neighboring sequence elements, and as part of an intact Ty1 element. Our studies reveal that the Ty1 Rap1 binding site is capable of binding Rap1p, that Rap1 binding has a role in activating Ty1 and Ty1-mediated adjacent gene transcription, and that RAP1 mediated activation of Ty1-mediated transcription is tied to the presence and function of the positive transcription factor Mcm1, as well as to an uncharacterized DNA-binding protein whose binding site is situated adjacent to the Mcm1 binding site and overlaps that of Rap1.
Figure 1.
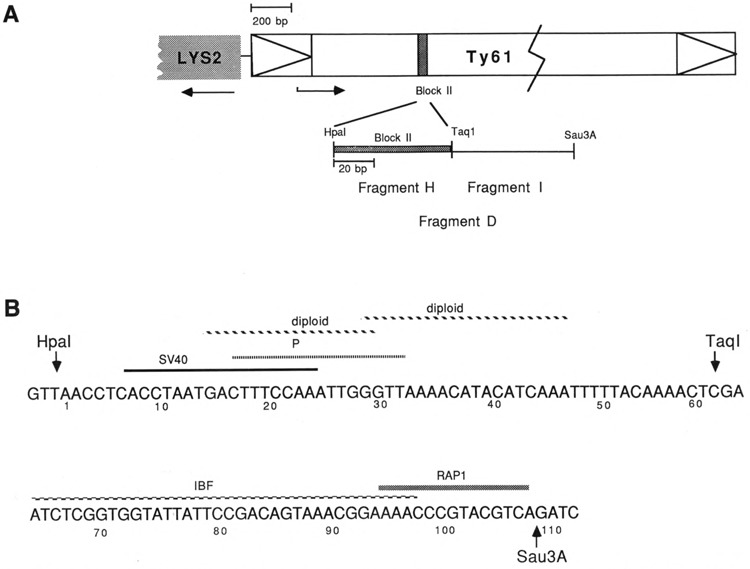
Details of the block II and adjacent region of the Ty61 element. A. Location of the block II sequences within the Ty element. The arrows drawn in the terminal repeats (δ) indicate the direction of transcription of the element. The promoter for Ty transcription is located in the leftmost repeat. The arrows drawn beneath the Ty element and the adjacent LYS2 gene indicate the direction of Ty and LYS2 transcription. The first 1,000 bases of the Ty element are drawn to scale; the remainder of the 5.9 kb element is condensed. The Hpa I–Sau3A fragment (below) illustrates the relationships between restriction fragments D, H, and I. B. Sequence of the Hpa I–Sau3A fragment D. Sequence similarities are indicated. Abbreviations: SV40, similarity to the enhancer core region from within the SV40 72-bp repeats; P, similarity to the consensus Mcm1 binding site; diploid, similarity to the a1/α2 control sequence or its complement; IBF, region protected by an unknown factor, here designated I binding factor; RAP1, similarity to the consensus Rap1p binding site.
Materials and methods
Strains
Yeast strains used in these studies are listed in Table 1. All strains are from our collection or were constructed for this work, unless otherwise indicated. The mcm1-1 mutation was introduced into our strain background by crosses with strain SY1433 (RM9-2C from L. Bruhn and G. Sprague; Passmore et al.. 1988). The rap1-1, rap1-2, and rap1-5 mutations were constructed in vitro (Kurtz and Shore, 1991) and introduced into our strain background by crosses with strains YDS409 and YDS410 (from S. Kurtz and D. Shore). The rap1-100 mutation was isolated by M. Breitenbach and introduced into our strain background by crosses with strain DG175 (from D. Geisman and K. Tatchell).
Table 1.
Yeast strains.
| Strain | Genotype (Source)a |
|---|---|
| JF819 | MATa ura3-52 lys2-128δ his4-917 leu2-1 |
| JF820 | MATα ura3-52 lys2-128δ his4-917 leu2-1 |
| JF1019 | MATa spt13-1 ura3-52 lys2-128δ his4-917 leu2-3,112 |
| JF1096 | MATα mcm1-1 b ura3-52 leu2-3,112 his4-917 |
| JF1165 | MATα rap1-1 b ura3-1 his4-917 leu2-1 trp1-1 ade2-1 |
| JF1166 | MATa rap1-2 b lys2-128δ his4-917 leu2-1 ade2-1 |
| JF1167 | MATα rap1-2 b 2 ura3-1 his4-917 leu2-3,112 trp1-1 ade2-1 |
| JF1170 | MATa rap1-5 b ura3-1 lys2-128δ his4-917 leu2-1 ade2-1 |
| JF1171 | MATα rap1-5 b ura3-1 leu2-3,112 his4-917 trp1-1 ade2-1 |
| JF1175 | MATα spt13-1 rap1-2 ura3-1 leu2-3,112 trp1-1 his4-917 |
| JF1179 | MATα spt13-1 rap1-5 his4-917 ura3-1 leu2-3,112 trp1-1 |
| JF1206 | MATα spt13-203::TRP1 his4-917 lys2-128δ leu2 ura3-52 trp1Δ1 |
| GA127 | MATα his3-11,15 trp1-1 leu2-3,112 ura3-1 ade2-1 mcm1Δ::LEU2 pombeADH1-MCM1-98::URA3 (G. Ammerer) |
| GA129 | MATα his3-11, 15 trp1-1 leu2-3,112 ura3-1 ade2-1 |
Strains are from our laboratory collection unless otherwise indicated.
Media and genetic procedures
All media were prepared as described by Sherman et al. (1978) and included synthetic complete media (SC) lacking one or more specific amino acids and rich media (YPD). Plasmids were introduced into appropriate strains by LiAc-based transformation (Ito et al., 1983).
Ty1 fragments
For amplification, Ty1 fragment D (originally isolated from the Ty61 element in pGS5; Simchen et al., 1984; Fassler and Winston, 1989; Yu and Fassler, 1993) was subcloned into the Sma I site of a pUC19 plasmid derivative in which the EcoR I site had been previously converted to an Xho I site. Fragment D was released for further subcloning by digestion with Xho I plus Sal I. The Taq I site in the pUC19 fragment D subclone was subsequently converted to an Xho I site to facilitate the subcloning of fragment I. Subcloning of fragment H has been previously described (Yu and Fassler, 1993).
Plasmids
One or more copies of a restriction fragment containing the Ty1 Rap1 binding site and adjacent sequences were cloned into the unique Xho I site of the UAS-less reporter plasmid pLG670Z (Guarente and Ptashne, 1981), to create CYC1-lacZ fusion derivatives with Ty1-derived UAS sequences. Ty1 fragments were also substituted for the CYC1 UAS in vector pLGΔ312 (Guarente and Ptashne, 1981). The construct pLGΔ312 is a derivative of the CYC1-lacZ vector pLG669Z (Guarente and Ptashne, 1981), from which 1.5 kb of CYC1 sequence upstream of the CYC1 UAS have been deleted. Orientation and number of inserts in each clone were assessed by DNA sequence analysis. A CYC1 primer (sequence: 5′-CACCAGTGAGACGGGCAAC-3′) was used for this purpose.
Construction of plasmid YCplys2-61Z has been described previously (Fassler and Winston, 1989). In the present study, this plasmid was introduced into wild-type, rap1, and mcm1 mutant strains to evaluate the role of the Rap1 and Mcm1 proteins in Ty and adjacent gene expression.
Probes for Northern (RNA) hybridization analysis were lacZ, a Pst I fragment isolated from plasmid pMC1871 (Casadaban et al., 1983); LYS2, an EcoR V to Eag I fragment from plasmid FB119 (F. Winston); Ty1, a Hind III-Eag I fragment from plasmid B161 (R. Surosky, B.-K. Tye, and G. R. Fink, unpublished data); and DED1, a Pst I–BamH I fragment isolated from plasmid JH(L)122 (J. Hirschman and F. Winston).
Plasmid pBG51, containing a Rap1 protein of novel length, was constructed to facilitate the positive identification of Rap1p in particular DNA–protein complexes. pBG51 was constructed by the in-frame deletion of an internal 438 bp BstB I–PpuM I fragment from plasmid D943 (D. Shore), followed by subcloning of the RAP1 gene derivative into the 2 μ plasmid YEp24. The resulting Rap1 protein is shortened by 146 aa; nevertheless, plasmid pBG51 is capable of complementing the temperature-sensitive phenotype of rap1 mutants. The mobility of DNA complexes containing this truncated Rap1 protein is distinct from the mobility of complexes containing the natural Rap1 protein.
Site-directed mutagenesis
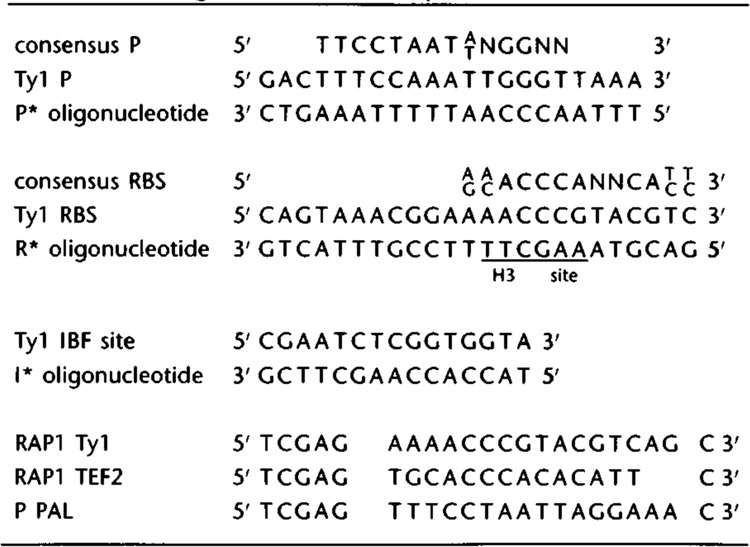
Appropriate Xho I–Sal I fragments were sub-cloned into a modified pGEM5Zf vector (Promega) in which the EcoR V site was converted into an Xho I site using Xho I linkers. Single-stranded uracil-containing template was prepared essentially by standard procedures (Kunkel et al., 1987). A single mutagenic oligonucleotide containing a convenient restriction endonuclease recognition site was annealed to the template and extended in the presence of deoxyribonucleotides, T4 DNA polymerase, and T4 DNA ligase. Transformants were screened by restriction analysis for the presence of the new sequence. Where indicated, the sequence was confirmed by using additional oligonucleotides as primers in standard dideoxy sequencing reactions. The mutagenized fragments, I*R, D*R, D*P, and D*I, containing mutations in either the Rap1p (R), Mcm1p (P), or IBF (I) binding sites, were subcloned into the pLG670Z or pLGΔ312 reporter plasmids for functional analysis. The sequences of the oligonucleotides used to generate binding site mutations are given in Table 2.
Table 2.
Oligonucleotide sequences.
Quantitative RNA and DNA hybridization analysis
Methods for preparation and analysis of RNA were as described previously (Fassler and Winston, 1988). Yeast DNA was prepared as described by Hoffman and Winston (1987) with minor modifications. Southern hybridization conditions (50% formamide, 42°C) were as prescribed in the Nytran manual. Where quantitation was necessary, several exposures were prepared with preflashed film and a single intensification screen. After densitometry, peaks were weighed and normalized to expression of the DED1 gene.
Electrophoretic mobility shift assays
Procedures for making yeast extracts, protein–DNA binding reactions, and electrophoretic fractionation of complexes were described previously (Company et al., 1988) with modifications (Yu and Fassler, 1993). Extracts were prepared from log phase cultures grown at 30°C with the exception of rap1-1 cultures, which were grown at room temperature and shifted to 37°C for 12 hours where indicated. These incubation conditions were previously shown to eliminate the binding activity of the Rap1-1 protein (Kurtz and Shore, 1991). Binding buffer was 5 mM Trisacetate (pH 7.9), 50 mM potassium-acetate, 1 mM magnesium-acetate, 1 mM dithiothreitol, 5% glycerol. 2–4 μg of yeast extract and 2 μg poly(dI-dC) were routinely added to 20 μl binding reactions. Reactions were incubated at 30°C for 15 minutes to allow protein–DNA complex formation. Specific competitor DNA was added to 50-fold molar excess.
DNA probes were isolated as restriction fragments (see plasmids above) or were synthetic oligonucleotides containing Xho I cohesive ends (Table 2). Radiolabeled DNA fragments were generated for use as probes by treatment with E. coli DNA polymerase large subunit and deoxyribonucleotides, including [α32P]dCTP. 0.2 ng of labeled probe were added to each reaction.
Gels for resolving protein–DNA complexes contained 4% polyacrylamide (29:1) in TBE buffer (89 mM Tris, 89 mM borate, 2.4 mM EDTA; pH 8.0). Gels were prerun for 20 minutes at 150 V. Complexes were resolved after electrophoresis for 1 hour at 175 V.
β-galactosidase assays
Yeast protein extracts were prepared by glass bead lysis from cultures grown at 30°C and harvested at a density of 1 × 107 cells/ml as described previously (Company et al., 1988). Protein concentration was determined using the Bradford assay (Bio-Rad). A single preparation of bovine serum albumin (Promega) was used to construct standard protein curves. β-galactosidase activities were calculated in Miller units (Miller, 1972). Due to fluctuations in absolute activities in measurements of transformants on different days, activities were normalized to the activity of an ACT1-lacZ fusion gene (Yu and Fassler, 1993) expressed in wild-type cells and measured in each experiment. Activities for each construct are the average of three to fifteen different transformants. Standard deviations did not exceed 30% of the average normalized value.
Synthetic oligonucleotides
Synthetic oligonucleotides were prepared by C. Olson with an Applied Biosystems DNA synthesizer (Department of Biological Sciences, University of Iowa). Both strands of the sequence were synthesized for use in cloning experiments or for use as probe in electrophoretic mobility shift experiments. Annealing and cloning were accomplished as previously described (Yu and Fassler, 1993). Sequences of synthetic oligonucleotides used in these studies are shown in Table 2.
Results
The Ty1 enhancer region contains multiple binding sites, including a putative RAP1 binding site
Insertion of Ty transposable elements of yeast into the 5′ noncoding regions of genes can result in either inactivation or cell type-dependent activation of adjacent gene expression (Errede et al., 1980; Roeder et al., 1980; Williamson et al., 1981; Eibel and Philippsen, 1984; Simchen et al., 1984). Mutational and molecular analyses of Ty1 and Ty2 elements indicate that multiple internal cis-acting regions contribute to these regulatory effects (Errede et al., 1985; Roeder et al., 1985; Liao et al., 1987). In Ty1 elements, one such region is the block II sequence originally identified by its homology to the enhancer of the DNA tumor virus SV40 (Errede et al., 1985). The block II region of Ty1 elements is contained within a 112 bp Hpa I–Sau3A fragment designated fragment D (Company and Errede, 1987), which includes the binding sites for a minimum of four factors (Fig. 1). A central Taq I site subdivides D into two halves, which have been designated H and I (Fig. 1).
Analysis of the H fragment revealed the presence of a functional Mcm1 binding, or P, site (Company and Errede, 1988; Errede, 1993). It has been shown previously that the P site alone is capable of activating transcription (Company and Errede, 1988; Yu and Fassler, 1993). Multimerization of this site increased the activation of a reporter gene seven- to eightfold (Company and Errede, 1988), and mutations in the SPT13 (GAL11) gene result in further increases in P site-mediated activation (Yu and Fassler, 1993).
We show here that fragment I (Fig. 1) also has transcriptional activity (Fig. 2). This activity was high when fragment I was present in single copy in either orientation, and was increased 4.6-fold when three copies of the fragment were present in the pLG670Z vector. Oddly, in the pLG670Z vector, the activity of fragment I exceeded the activity of the intact fragment D. (This point is further considered in the Discussion.) Previous DNase I footprinting demonstrated the presence of an unidentified factor bound to sequences in fragment I (Company and Errede, 1988; Fig. 1). For convenience, we have tentatively designated this previously unnamed footprinting activity IBF (I binding factor). In addition to the IBF site, fragment I contains 85% (11/13) identity to a consensus Rap1p binding site.
Figure 2.
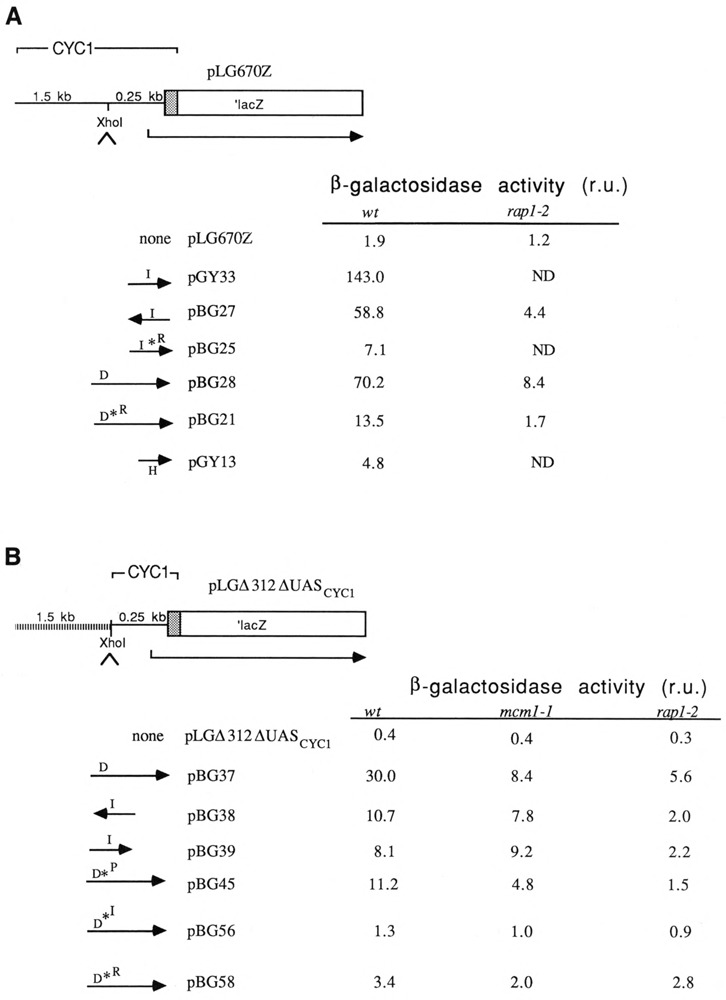
β-galactosidase activities of Ty1 fragment I and D and H clones in the pLG670Z and the pLGΔ312 vectors. Vector pLG670Z contains a unique Xho I cloning site 250 bp upstream from the CYC1 ATG. 1.5 kb of CYC1 promoter sequence are present upstream of the cloning site. Transcription starts at the normal CYC1 initiation sites. pLGΔ312 differs from pLG670Z only in the absence of the 1.5 kb of CYC1 sequence upstream of the Xho I cloning site. Orientation of I and D inserts are illustrated. The I*R and D*R fragments contain a mutation in the RAP1 binding site; the D*P and D*I fragments contain mutations in the Mcm1 and IBF binding sites, respectively (Table 2). β-galactosidase activity for each construct was measured at 30°C and is presented as relative units (r.u.) in which the activity of each construct was normalized to expression (see Materials and Methods for additional details) from plasmid pGY63 carrying an ACT1-lacZ fusion gene measured in the wild-type strain according to the following formula: 1000 × [β-gal activity (Miller units) construct X in strain Y]/[β-gal activity (Miller units) construct pGY63 in JF820]. The ACT1-lacZ control plasmid, pGY63, averaged 5751 units in JF820. Between three and six transformants of each construct (with the exception of pGY13, for which n = 4) were measured. Southern hybridization analysis indicated that the rap1-2 mutation did not significantly affect plasmid copy number. A twofold correction was made to all values obtained in mcm1-1 strains to compensate for the twofold reduction in copy number due to the mcm1-1 mutation. Standard deviations of the normalized values were less than 30% of the average value. The wild-type strain was JF820; the rap1-2 mutant was JF1167. ND: not determined.
Role of Rap1p in Ty and adjacent gene expression
The presence of a putative Rap1p binding site in Ty1 elements suggested that RAP1 might contribute to the regulation of Ty elements and/or the expression of genes adjacent to Ty elements. Two genetic observations support this contention. Strains carrying the his4-917 Ty2 insertion mutation in the promoter of the HIS4 gene (Roeder et al., 1980) exhibit a tight His− phenotype (Fig. 3, top panel, A) due to reduced transcription of the HIS4 gene. In his4-917 mutants, the 6 kb Ty element is located between the HIS4 transcription initiation site and TATA, displacing and rendering nonfunctional all upstream regulatory information, including the Gcn4p, Rap1p, and Bas1/2p recognition elements. Suppressor of Ty insertion mutations (spt), such as spt13, restore transcription to the HIS4 gene and confer a His+ phenotype (Fig. 3, top panel, C) by affecting transcriptional signals within the Ty element. When rap1 mutations were introduced into his4-917 backgrounds, a weak His+ phenotype was observed (Fig. 3, top panel, B). Hence, rap1 mutants showed a weak Spt phenotype. Adjacent gene expression was previously found to be stimulated in some situations in which Ty transcription is diminished (Winston et al., 1984). The effect of rap1 mutations on adjacent gene expression suggested the possibility that RAP1 may normally contribute positively to Ty transcription.
Figure 3.
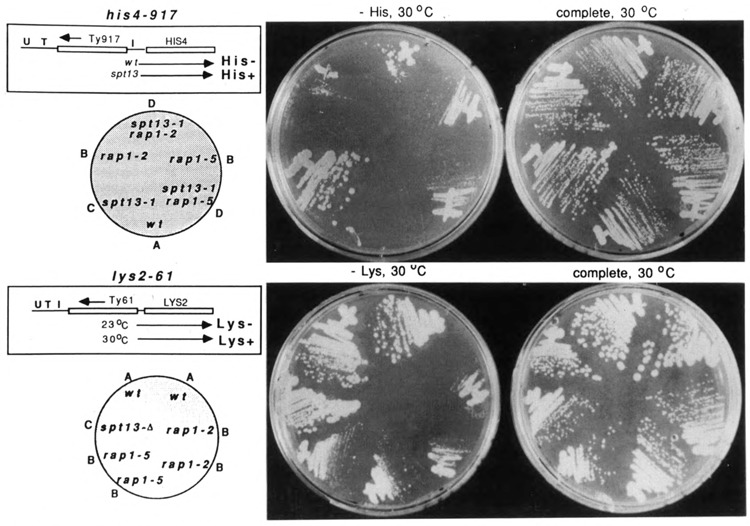
Effect of rap1 mutations on the his4-917 Ty insertion mutation and on Ty-mediated gene expression. Top: phenotype of the his4-917 Ty insertion mutation in wild-type cells (A); suppression by the rap1-2 and rap1-5 mutants (B); and inhibition of spt13-mediated suppression of his4-917 by the same mutations (C, D). Strains were streaked on SC-His and SC complete plates and incubated at 30°C for 8–10 days. spt13-1, JF1019; wild-type, JF820; rap1-2, JF1167; rap1-5, JF1170; spt13-1 rap1-2, JF1175; spt13-1 rap1-5, JF1179. Bottom: phenotype of the lys2-61 insertion mutation at 30°C in wild-type (A) and rap1 mutants (B) carrying the YCplys2-61Z plasmid. Strains were streaked on SC-Lys and SC complete plates and incubated at 30°C for approximately 7 days. Wild-type, JF820; spt13-Δ, JF1206; rap1-2, JF1167; rap1-5, JF1170. All strains carry the lys2-128δ insertion mutation, which renders them phenotypically Lys− in the absence of the plys2-61Z plasmid. U, T, and I in the diagrams of his4-917 and lys2-61 Ty insertion mutations signify the UAS, TATA, and initiation sites of the adjacent gene, respectively.
We further found that rap1 mutations interfered with normal suppression by spt13 mutations. In Figure 3 (top panel, D), the negative impact of rap1-2 and rap1-5 mutations on spt13-mediated suppression of the his4-917 Ty insertion can be seen. This suggests that in addition to activating Ty transcription, RAP1 may have a second role in activating adjacent gene expression, and that some of the adjacent gene transcription seen in spt13 mutants is RAP1-dependent.
To examine directly the effect of rap1 mutations on Ty transcript levels, we took advantage of a marked Ty1 construct. The plasmid plys2-61Z, containing the lys2-61 Ty insertion mutation with lacZ sequences inserted into the body of the Ty61 element located adjacent to LYS2 (Fassler and Winston, 1989), was introduced into wild-type and rap1 mutant strains. (These hosts are Lys− due to the presence of a δ insertion mutation, lys2-128δ, at LYS2.) Using a lacZ probe, we were able to examine expression of that single Ty element. A fourfold decrease in transcription of the Ty61 element was observed in rap1-2 mutants at 30°C (Fig. 4). Total Ty1 transcript levels were similarly (threefold) affected.
Figure 4.
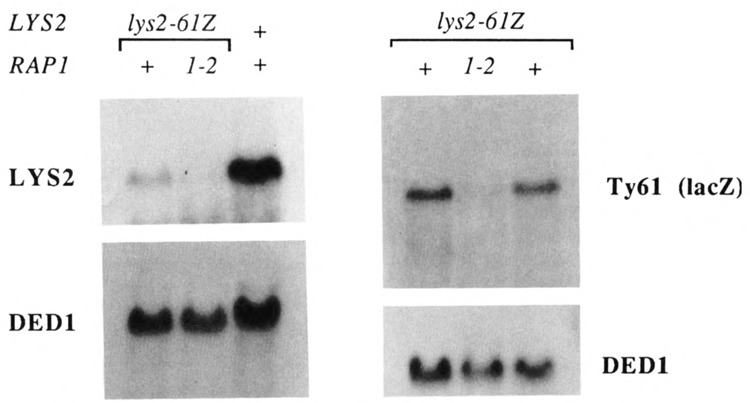
Northern (RNA) hybridization analysis of Ty61 and adjacent gene (LYS2) transcription in wild-type and rap1 mutants. RNA was prepared from 30°C cultures of YCplys2-61Z transformants of wild-type (JF820) and rap1-2 (JF1166 and JF1167) strains. The rightmost lane in the Ty61 Northern (right panel) contained RNA from the mcm1-1 mutant, JF1096. Ty61 expression was detected with a lacZ probe that hybridized to a small fragment of the E. coli lacZ gene inserted within the Ty element. LYS2 expression (left panel) was detected with an internal fragment of the LYS2 gene (see Materials and Methods). Hybridization to the DED1 gene was used for comparing amounts of RNA loaded in each lane. Strains transformed with the plys2-61Z plasmid contain the lys2-128δ insertion mutation, which confers a Lys− phenotype. In lys2-128δ strains, the internal LYS2 probe used in these experiments detects a weakly hybridizing transcript of a novel size (Clark-Adams et al., 1988) that does not interfere with visualization of the lys2-61 transcript. The lys2-128δ transcript is not seen in this figure.
The lys2-61 allele is a cold-sensitive Ty insertion mutation (Lys+ at 30°C and Lys− at 23°C). At 30 °C, low levels of LYS2 transcription are apparent by Northern analysis (Fig. 4, left). Hence, in this case we were able to examine the dependence of adjacent gene transcription on RAP1 in the absence of spt mutations. The rap1-2 mutation diminished transcription of the LYS2 gene at 30°C by approximately 50% (Fig. 4, left). Normal growth of a wild-type strain carrying the lys2-61 insertion mutation on media lacking lysine at 30°C was likewise compromised in rap1 mutants (Fig. 3, bottom panel; compare B with A). This observation is consistent with our finding (above) that his4-917 suppression was compromised in the spt13 rap1 double mutants (Fig. 3, top panel, D) and supports the idea that RAP1 must be at least partially responsible for adjacent gene expression.
Rap1 protein binds to Ty1 elements
To examine the putative Ty1 Rap1p binding site in greater detail, fragment I or an oligonucleotide containing the Ty1 Rap1p binding site (Table 2) was radiolabeled and tested in electrophoretic mobility shift assays with protein extracts from wild-type yeast strains. A single protein complex, I-1 (Fig. 5, lanes 1 and 9), was observed. Formation of this complex could be specifically inhibited with a 50-fold molar excess of unlabeled fragment I (Fig. 5, lane 2) or with an oligonucleotide duplex containing the high-affinity RAP1 binding site from the TEF2 promoter (TEF2; Fig. 5, lane 3). In contrast, a 50-fold molar excess of pGEM5Zf (Promega) polylinker DNA had no inhibitory effect (Fig. 5, lane 4). Experiments in which a mutant I fragment containing an altered Rap1p binding site (I*R) was employed as the DNA probe (Fig. 5, lane 5) confirmed that complex I-1 depends on an intact Rap1p binding site (RBS).
Figure 5.
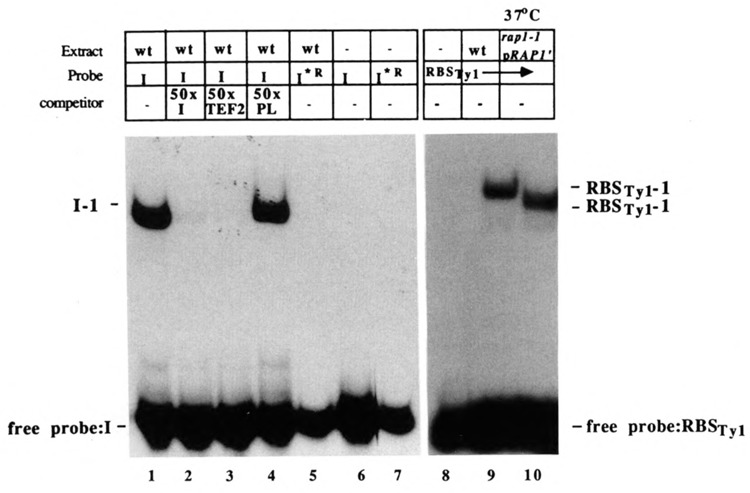
Factor binding to a Ty1 fragment I (approximately 50 bp) containing either a wild-type (I; lanes 1–4) or mutant (I*R; lane 5) RAP1 binding site and to a 22 bp oligonucleotide containing the Ty1 RAP1 binding site (lanes 8–11). Binding reaction mixtures contained 2–4 μg of protein from wild-type (JF820; lanes 1–5, 9) or rap1-1 (JF1165; lane 10) extracts. The rap1-1 strain was transformed with the RAP1 deletion plasmid pBG51 (see Materials and Methods), and extracts were prepared from cultures grown at room temperature and shifted to 37°C (lane 10). Reactions were performed in the absence of specific competitor (none) or with a 50-fold molar excess of fragment I (lane 2), oligonucleotide duplex containing the RAP1-binding site from the TEF2 gene (TEF2; lane 3), or a 465 bp Pvu II restriction fragment containing polylinker (PL) sequences from plasmid pGEM5Zf (Promega; lane 4). Positions of free probe and complex 1 (I-1 or RBSTy1-1) are indicated to the left of the autoradiogram. In lanes 6, 7, and 8, the extract was omitted. Lanes 1–7 and lanes 8–10 are taken from separate gels. Note also that a different radiolabeled probe was used in the two experiments.
To determine whether Rap1 protein is contained in complex I-1 or whether this complex is merely dependent on RAP1 for its formation or maintenance, electrophoretic mobility shift assays were performed using extracts from temperature-sensitive rap1-1 mutant strains carrying a plasmid-borne allele of RAP1 encoding a truncated Rap1 protein. When the rap1-1 strain is incubated for a prolonged period at 37°C, the binding activity of the Rap1-1 protein is eliminated (Kurtz and Shore, 1991; W. M. Gray, unpublished observations), and all remaining binding activity is due to the presence of the truncated Rap1 protein. Hence, the shift in mobility of complex RBSTy1-1 in lane 10 relative to lane 9 (Fig. 5) confirmed the presence of the Rap1 protein in this complex.
The Ty1 RBS contributes to activation of a reporter gene
Possible functions for Rap1 binding to the Ty1 site were investigated by subcloning the Ty1 I fragment in the promoter of a CYC1-lacZ reporter gene lacking a natural UAS. As previously mentioned and shown in Figure 2, fragment I directed a significant level of β-galactosidase activity. This activity was reduced 13-fold in a rap1 mutant background (rap1-2), and 20-fold when the cloned fragment I contained a mutation in the Rap1p binding site (I*R; compare pGY33 and pBG25 in Figure 2A). Hence, I-mediated activation is largely attributable to Rap1. Fragment D-mediated activation showed similar RAP1 dependence (Fig. 2). The Rap1p binding site mutation does not completely eliminate Rap1 binding (Gray and Fassler, unpublished data); hence the combined effect of the binding mutation and the trans-acting mutation in the RAP1 gene results in lower activity than either mutation alone.
Mcm1p is required for activation by fragment D
Located adjacent to fragment I (in fragment H) is a series of regulatory sequences that include an Mcm1p binding (P) site, a1/α2 diploid control sites, and homology to the SV40 enhancer core (Fig. 1; Errede et al., 1985; Errede, 1993). To begin to investigate the relationship between Rap1 and other proteins bound to the region, the activities of CYC1-lacZ reporter plasmids containing fragment D were compared in wild-type and in mcm1-1 mutant backgrounds (Fig. 2, pBG37). For these assays, Ty1 fragments were cloned into the pLGΔ312 vector. pLGΔ312 is related to pLG670Z but lacks CYC1 sequences upstream of the Xho I cloning site. Although pLG670Z exhibits very low levels of β-galactosidase activity in the absence of cloned sequences (Fig. 2, vector alone), and is therefore thought to be “dead,” we found that vector sequences (presumably upstream of the Xho I site) amplified the activity of cloned Rap1 binding sites in an Mcm1-dependent manner (data not shown). In contrast, the activity of a cloned I fragment showed no Mcm1 dependence in the pLGΔ312 vector (Fig. 2).
The activity of the pLGΔ312 fragment D clone was reduced 3.6-fold in the mcm1-1 background. Since the activity of pLG670Z fragment H clones (containing the P site) constitutes a small fraction (<10%; Fig. 2, pGY13) of the activity of pLG670Z fragment D clones, the magnitude of the dependence of D-mediated activation on MCM1 was unanticipated.
To distinguish between the requirement for Mcm1 protein bound to the P site, and one possible alternative, that Mcm1p activity might be required for the modification of Rap1p to a more active conformation, we examined the activity of a D fragment that had been altered to contain a mutant P site. P site mutations were introduced into fragment D by site-directed mutagenesis, and the mutant D*P fragment was introduced into the pLGΔ312 vector. The P site mutation reduced D-mediated activity approximately threefold (Fig. 2, pBG45). This result argues that the physical presence of Mcm1p and not mere MCM1 function is required for maximal levels of RAP1-mediated activation. A further twofold decrease in activity in the mcm1-1 mutant background probably reflects residual binding of Mcm1 at the mutant P site.
IBF activity is also required for D-mediated activation
The presence of a large DNase I-protected region (IBF site) between the Rap1p and Mcm1p binding sites suggested an involvement of IBF in D-mediated activation. To investigate this possibility, a mutagenic oligonucleotide was designed in which two of the strongly protected bases in the IBF footprint were altered (see Table 2). After in vitro mutagenesis of the D fragment using the I* oligo, the D*I fragment was cloned into pLGΔ312 and tested for activity. The I* mutation resulted in a 23-fold decrease in D-mediated activation (Fig. 2). We conclude that IBF, along with the Mcm1 and Rap1 proteins, is a necessary component of the D fragment activation complex.
Rap1 and Mcm1 are not mutually dependent for binding to the Ty1 element
The physical relationship between Mcm1, Rap1, and IBF was investigated using electrophoretic mobility shift assays to examine the ability of the three proteins to bind to fragment D in the presence and absence of one another. Three complexes were detected when fragment D was used as a probe (Fig. 6, lane 1). To determine which if any of these complexes contained Rap1 protein, a derivative of the D fragment containing a mutant Rap1 binding site (D*R) was generated. Complexes D-1 and D-2 were absent when D*R was used as a probe (Fig. 6, lane 5). The D-3 complex was unaffected by the RBS mutation. To confirm the presence of Rap1p in complexes D-1 and D-2, extracts were prepared from rap1-1 strains carrying a plasmid (pBG51) encoding a truncated Rap1 protein. In these trials, rap1-1 binding was eliminated due to incubation of the extract cultures at the nonpermissive temperature (37°C, see Materials and Methods), and complexes D-1 and D-2 were shifted in mobility due to binding of the truncated form of Rap1p (Fig. 7, lane 7). As expected, complex D-3 was unaffected by the presence of the Rap1 truncation protein.
Figure 6.
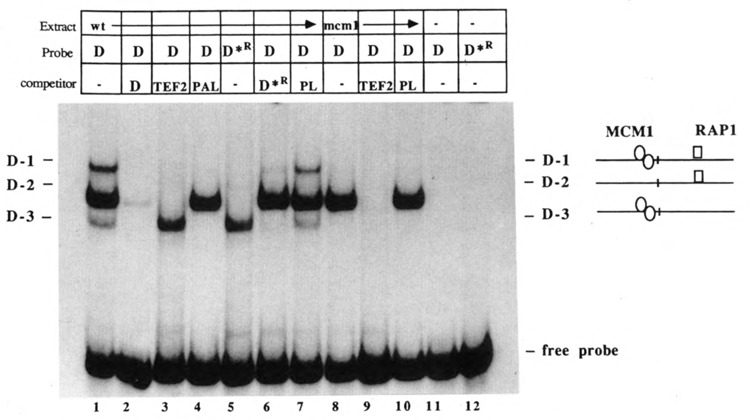
Factor binding to Ty1 fragment D containing either a wild-type (D) or a mutant (D*R) RAP1 binding site. Binding reaction mixtures contained 2–4 μg of protein from wild-type (JF820) or mcm1-1 (JF1096) strains. Reactions were performed in the absence of competitor (–; lanes 1, 5, and 8); or in the presence of a 50-fold molar excess of fragment D (lane 2); oligonucleotide duplex containing the RAP1 binding site from the TEF2 gene (TEF2; lanes 3, 9); oligonucleotide duplex containing the palindromic Mcm1 binding site (PAL; lane 4); a 465 bp Pvu II restriction fragment containing polylinker (PL) sequences from plasmid pGEM5Zf (Promega; lanes 7, 10); or fragment D*R containing a mutation in the RAP1 binding site (lane 6). In lanes 11 and 12, extract was omitted. Positions of the free probe, complex D-1, D-2, and D-3 are indicated on each side of the autoradiogram. To the right is a schematic of the proteins known to be present in each complex.
Figure 7.
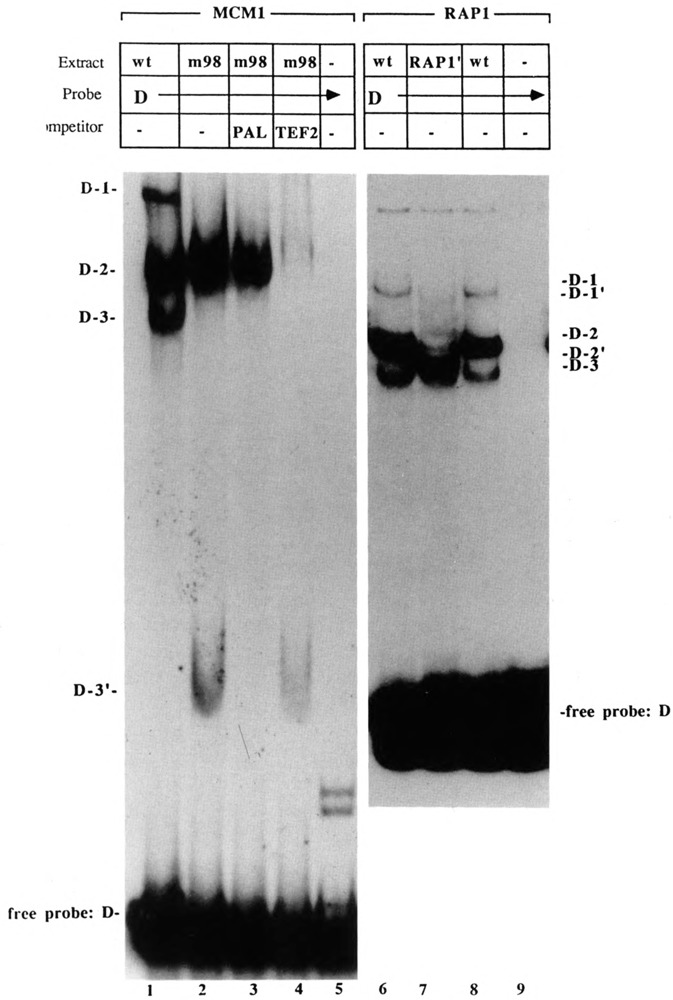
Factor binding assay to the Ty1 fragment D. Binding reaction mixtures contained 2–4 μg of extract from wild-type (GA129, lane 1; andJF820, lane 6 and 8), mcm1-98 (GA127), or rap1-1 (JF1165) strains. In lane 7, extract was from a rap1-1 mutant (JF1165) carrying the pBG51 RAP1 deletion plasmid. Reactions were performed in the absence (–) or presence of competitor DNA as indicated. The positions of the normal D complexes (D-#) and shifted complexes (D-#′) due to the Rap1 truncation are indicated to the right of the autoradiogram. The positions of shifted D complexes due to the truncated Mcm1 protein are indicated to the left. Abbreviations: PAL, oligonucleotide containing consensus MCM1 binding site; m98, extract prepared from a strain carrying the mcm1-98 truncation allele; pRAP1′, plasmid pBG51 carrying the RAP1 truncation allele.
The absence of D-1 and D-3 in mcm1-1 mutant extracts (Fig. 6, lane 8) suggested the presence of Mcm1 protein in these complexes. This was confirmed in separate experiments in which extract was prepared from a strain (GA127) in which the gene encoding the wild-type Mcm1 protein was replaced with the mcm1-98 allele encoding a truncated Mcm1 derivative. Figure 7 (lane 2) shows the absence of D-1 and D-3 from their normal positions and the presence of a complex of new, faster mobility. Since D-1 normally contains both Mcm1 and Rap1 proteins, formation of D-1′ should be inhibited by the presence of both PAL and TEF2 competitor DNA in the binding reaction. Formation of D-3′ (Mcm1p), on the other hand, is expected to be inhibited by the inclusion of PAL competitor DNA but not by TEF2. On the basis of this type of competition analysis (Fig. 7, lanes 3 and 4), we believe that the new complex is D-3′. D-1′ may be comigrating with D-2.
The Mcm1 complex (D-3) showed no dependence on the Rap1 binding site, and the Rap1 complex (D-2) showed no dependence on MCM1. The lack of dependence of Rap1p binding on MCM1 and of Mcm1p binding on RAP1 was not surprising, since our previous results indicated that MCM1 was capable of binding to a fragment (H) lacking the RBS, and Rap1 was capable of binding to a fragment (I) lacking the P (Mcm1 binding) site.
In previous footprinting experiments (Company and Errede, 1988), the IBF protein was shown to be present in complexes formed on fragment D The binding conditions in our analysis differed from those in the studies in which IBF was originally reported (see Materials and Methods). When the mutant D*I oligonucleotide was used as a probe in electrophoretic mobility shift assays, there was no change in the mobility of any of the three DNA–protein complexes. To test directly for the presence of IBF in complexes D-1, D-2, and D-3, each complex was isolated and subjected to hydroxyl radical and DNase I footprinting. The footprinting experiments confirmed the presence of the Mcm1 and Rap1 proteins in the three complexes as originally assigned, but provided no evidence for the presence of IBF (data not shown) in any of these complexes. The absence of IBF from our in vitro complexes suggests that IBF is not essential for binding or recruitment of Mcm1 or Rap1 proteins to their respective sites, nor is it required for the formation of the Mcm1–Rap1 complex in vitro. Nevertheless, since β-galactosidase assays indicate that IBF is necessary for D-mediated activation, we further conclude that conditions exist in vivo in which the IBF protein will be present.
Discussion
Rap1p binds to Ty1 elements
Our analysis of one region of Ty1 elements involved in transcriptional regulation has led to the identification of a previously unrecognized Rap1 binding site. Previous studies of this region of Ty1 elements identified two protein complexes (Company and Errede, 1988). Previous DNase I protection studies (Company and Errede, 1988) showed that the two complexes were related, one containing a single factor protecting a binding site for an unidentified protein (IBF), and the second containing IBF plus Mcm1p. Our electrophoretic mobility shift assays revealed three protein complexes. We find that two of the three complexes contain Rap1p. Hence fragment D contains at least four factor binding sites: the three discussed in this work (the P site, Rap1 binding site, and IBF binding site), and a fourth located adjacent to Mcm1p (Yu and Fassler, 1993). The abundance of consensus sequences shown in Figure 1 suggests that additional proteins may be involved.
Ty1 fragment I activates transcription
The isolated fragment I, containing the Ty1 Rap1 and IBF binding sites, was capable of directing high levels of RAP1-dependent transcriptional activity when cloned in front of a heterologous reporter gene. The extent of this activation was vector-dependent; levels of fragment I-mediated β-galactosidase activity were substantially reduced in the pLGΔ312 vector relative to the related pLG670Z, which contains upstream CYC1 sequences (Fig. 2). These studies suggest the presence of sequences in the CYC1 promoter that specifically enhance RAP1 activation. We conclude that, at least in some cases, Rap1 associates with additional proteins to confer maximal levels of activation. In the case of pLG670Z-I clones, the associated protein(s) are unknown, and in the context of the Ty1 element, one associated protein is Mcm1.
Maximal activation requires the concerted function of the Rap1, Mcm1, and IBF proteins
Using a combination of cis- and trans-mutations, we have shown that the activity of Ty1 fragment D depends on binding of the Mcm1, Rap1, and IBF proteins. The activity of each protein alone can be deduced from Figure 2B. β-galactosidase activity directed by the D*I sequences in the mcm1-1 background (1.0 u) represents activation due to Rap1 binding. Likewise, β-galactosidase activity directed by D*I in the rap1-2 background (0.9 u) represents activation due to binding of Mcm1p. β-galactosidase activity directed by D*R in the mcm1-1 background (2.0 u) is presumed to reflect IBF activation. Since the activity attributable to the individual proteins is much less than the activity of fragment D (30 u), we conclude that maximal D-mediated activation is due to the concerted function of these three proteins. We cannot presently eliminate the possibility that additional proteins may also be involved.
The role of IBF in D-mediated activation
Due to the absence of IBF from our extracts, we have not yet been able to test the effect of each protein on binding of the others. But several possible relationships among the proteins can be imagined. All three proteins may bind simultaneously to the Ty1 element enhancer to mediate activation. Alternatively, as has been suggested for the TPI promoter, where Rap1p appears to recruit the Gcr1 transcriptional activator (Scott and Baker, 1993), Rap1p may be required for recruitment of IBF. Finally, analogous to the relationship between the Rap1 and Gcn4 proteins proposed for the HIS4 promoter (Devlin et al., 1991), IBF may ultimately displace Rap1p from its overlapping binding site. In this case, IBF and not RAP1 is expected to be the functional activator. The future identification and characterization of the IBF gene product will be instructive in this regard.
Vector effects
Several aspects of this analysis were initially puzzling. The activity of fragment pLG670Z D clones was less than that of the cloned I fragment. In addition, although a fragment with a mutant P site (D*P) was expected to mediate levels of activity comparable to that of I clones, this was not true in the pLG670Z vector (data not shown). Furthermore, I-mediated activation in pLG670Z appeared to require MCM1 activity (data not shown). On the basis of extensive analysis of the activities of these clones in the pLG670Z and pLGΔ312 vectors, we are led to conclude that upstream CYC1 sequences present in pLG670Z but not in pLGΔ312 are capable of enhancing activation mediated by Ty1 fragment I sequences. This is clear from the difference in relative activities in the two vectors (143 vs. 8.1 and 58.8 vs. 10.7; Fig. 2, compare pBG27 with pBG38 and pGY33 with pBG39). From these data, it is also clear that the enhancement is somewhat dependent on the orientation of the insert. We speculate that additional proteins bound to fragment D may prevent the hypothetical interaction between Rap1 and factors bound to upstream CYC1 sequences, thus largely insulating fragment D from these influences.
Role of Rap1 in Ty1 and adjacent gene transcription
rap1 mutants have a weak Spt phenotype, in that the His− phenotype due to insertion of the Ty917 element in the promoter of the HIS4 gene is partially suppressed in rap1 mutant backgrounds. Some SPT genes (SPT3 and SPT14, for example) are involved in promoting transcription of the Ty element itself (Winston et al., 1984; Fassler et al., 1991). In these spt mutants, restoration of adjacent gene expression appears to be an indirect result of reductions in the level of expression of the divergently transcribed Ty element. Our observation that the rap1-2 mutations decreased Ty61 transcript levels (Fig. 4) suggests that rap1-mediated suppression may likewise be indirect. Our failure to detect suppression of the lys2-61 Ty insertion mutation by the rap1-2 mutation was presumably due to the fact that this temperature-sensitive rap1 allele is not defective at the temperature (room temperature) at which theTy61 insertion causes a Lys− phenotype.
The observation that rap1 mutations interfere with spt13-mediated suppression of his4-917 and with the temperature-dependent read-through expression of the LYS2 gene in the lys2-61 insertion indicates that RAP1 plays a second role in activation of adjacent gene expression. The related observation that the mcm1-1 mutation also interferes with spt13-mediated suppression of the his4-917 Ty insertion mutation (Yu and Fassler, 1993) is consistent with the view that the Rap1 and Mcm1 proteins function jointly in adjacent gene activation, as we demonstrated using a heterologous reporter gene. In contrast, we suggest that Mcm1p is probably not required in RAP1-mediated activation of Ty expression. This view is based on the observation that, in contrast to the decrease in Ty61 transcription observed in rap1-2 mutants, the mcm1-1 mutation does not result in decreased transcription of this element (Fig. 4).
By our analysis, the Rap1 binding site is one element of a very complex Ty1 enhancer. Although our experiments do not eliminate the possibility that additional Rap1 binding sites within Ty1 elements also play a role in modulating Ty1 and adjacent gene expression, such sites are presumably located (like the site in fragment D) within the transcriptional unit. There is no evidence for a standard UAS in Ty1 elements (Fulton et al., 1988), and Ty1 sequences upstream of the major Ty1 initiation site contain no close matches to a consensus Rap1 binding site. Hence, while Rap1 binding sites implicated in gene activation are generally located in the 5′ nontranscribed region of genes, Rap1 regulation of Ty1 transcript levels probably occurs from a position downstream of the initiation site. In one view, RAP1-mediated activation of Ty transcription may be distinguished from RAP1-mediated activation of other genes by virtue of the as yet uncharacterized IBF protein. Alternatively, additional roles for the Rap1 protein in downstream regulation may yet be discovered.
Acknowledgments
We thank R. Deschenes, G. Gussin, F. Winston, and D. Weeks sor valuable comments on the manuscriptt S. Plattner for photographic assistance, G. Gingerich for technical support, members of the laboratory for helpful discussions, and Guoying Yu for the donation of materials and plasmids. We are grateful to D. Shore, K. Tatchell, F. Winston, and G. Sprague for strains and plasmids.
This work was supported in part by grants from the National Institutes of Health (GM40306 and HG00003) and by an American Cancer Society seed grant from the University of Iowa.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Arndt K. T. and Fink G. R. (1986), Proc Natl Acad Sci USA 83, 8516–8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A. R., Kimmerly W. J., Rine J., and Kornberg R. D. (1988), Mol Cell Biol 8, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A. R., Lue N. F., and Kornberg R. D. (1988), Mol Cell Biol 8, 5086–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban M. J., Martinez-Arias A., Sharira S. K., and Chou J. (1983), Methods Enzymol 100, 293–308. [DOI] [PubMed] [Google Scholar]
- Clark Adams C. D., Norris D., Osley M. A., Fassler J. S., and Winston F. (1988), Genes Dev 2, 150–159. [DOI] [PubMed] [Google Scholar]
- Company M., Adler C., and Errede B. (1988), Mol Cell Biol 8, 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Company M. and Errede B. (1987), Mol Cell Biol 7, 3205–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Company M. and Errede B. (1988), Mol Cell Biol 8, 5299–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin C., Tice-Baldwin K., Shore D., and Arndt K. T. (1991), Mol Cell Biol 11, 3642–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibel H. and Philippsen P. (1984), Nature 307, 386–388. [DOI] [PubMed] [Google Scholar]
- Errede B. (1993), Mol Cell Biol 13, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede B., Cardillo T. S., Sherman F., Dubois E., Deschamps J., and Wiame J. M. (1980), Cell 22, 427–436. [DOI] [PubMed] [Google Scholar]
- Errede B., Company M., Ferschak J. D., Hutchison C. A., and Yarnell W. S. (1985), Proc Natl Acad Sci USA 82, 5423–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler J. S., Gray W., Lee J. P., Yu G., and Gingerich G. (1991), Mol Gen Genet 230, 310–320. [DOI] [PubMed] [Google Scholar]
- Fassler J. S. and Winston F. (1988), Genetics 118, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler J. S. and Winston F. (1989), Mol Cell Biol 9, 5602–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton A. M., Rathjen P. D., Kingsman S. M., and Kingsman A. J. (1988), Nucleic Acids Res 16, 5439–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. and Ptashne M. (1981), Proc Natl Acad Sci USA 78, 2199–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy C. F. J., Balderes D., and Shore D. (1992), Mol Cell Biol 12, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y. A., Chamber A., Tsang J. S. H., Kingsman A. J., and Kingsman S. M. (1990), Nucleic Acids Res 18, 2617–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman C. S. and Winston F. (1987), Gene 57, 267–272. [DOI] [PubMed] [Google Scholar]
- Hurd H. K. and Roberts J. W. (1989), Mol Cell Biol 9, 5339–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., Fukuda Y., Murata K., and Kimura A. (1983), J Bacteriol 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T. A., Roberts J. D., and Zakour R. A. (1987), Methods Enzymol 154, 367–382. [DOI] [PubMed] [Google Scholar]
- Kurtz S. and Shore D. (1991), Genes Dev 5, 616–628. [DOI] [PubMed] [Google Scholar]
- Liao X.-B., Clare J. J., and Farabaugh P. J. (1987), Proc Natl Acad Sci USA 84, 8520–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig A. J., Kurtz S., and Shore D. (1990), Science 250, 549–553. [DOI] [PubMed] [Google Scholar]
- Miller J. H. (1972), Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Passmore S., Maine G. T., Elble R., Christ C., and Tye B.-K. (1988), J Mol Biol 204, 593–606. [DOI] [PubMed] [Google Scholar]
- Roeder G. S., Farabaugh P. J., Chaleff D. T., and Fink G. R. (1980), Science 209, 1375–1380. [DOI] [PubMed] [Google Scholar]
- Roeder G. S., Rose A. B., and Pearlman R. E. (1985), Proc Natl Acad Sci USA 82, 5428–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott E. W. and Baker H. V. (1993), Mol Cell Biol 13, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F., Fink G. R., and Lawrence C. W. (1978), Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Shore D. and Nasmyth K. (1987), Cell 51, 721–732. [DOI] [PubMed] [Google Scholar]
- Shore D., Stillman D. J., Brand A. H., and Nasmyth K. A. (1987), EMBO J 6, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchen G., Winston F., Sty1es C. A., and Fink G. R. (1984), Proc Natl Acad Sci USA 81, 2431–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson V. M., Young E. T., and Ciriacy M. (1981), Cell 23, 605–614. [DOI] [PubMed] [Google Scholar]
- Winston F., Durbin K. J., and Fink G. R. (1984), Cell 39, 675–682. [DOI] [PubMed] [Google Scholar]
- Yu G. and Fassler J. S. (1993), Mol Cell Biol 13, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]