

Abstract
The normal pattern of keratin expression in epidermis is altered in carcinomas as well as in nonmalignant diseases such as psoriasis and wound healing. Under these circumstances, the transcription of differentiation-specific keratins K1 and K10 is suppressed, whereas the activation- and hyperproliferation-associated keratins K6 and K16 are induced. Very little is known regarding transcriptional regulators involved in this switch. To investigate the nuclear factors that participate in regulation of expression of the K6 gene, we have characterized the binding sites for nuclear proteins on the promoter DNA of the K6 gene by gel retardation assays and site-specific deletion mutagenesis. We found four nuclear protein binding sites in the K6 gene promoter. Two are near the TATA box, but their ability to bind HeLa or keratinocyte nuclear extracts is independent of the TATA box-binding protein complex. The third binding site is a large palindrome. The sequences of these three sites do not correspond to any described target sequences for characterized transcriptional factors. The fourth is an AP-1 site, the target sequence for the proto-oncoproteins fos and jun. All four sites are independent of the previously characterized epidermal growth factorresponsive element, EGF-RE. These findings suggest that there may be two parallel pathways of induction of K6 transcription. One proceeds through the EGF-RE, which may be involved in nonmalignant hyperproliferation processes; the other, through the AP-1 site and the fos-jun protooncoproteins, may be related to induction in malignant processes.
Carcinomas are by far the most common of human neoplasms, representing fully one-third of all malignancies (Silverberg and Lubera, 1989). Epidermal basal cell and squamous cell carcinomas arise by a multistep process that can be reproduced in vitro in a murine model (Yuspa et al., 1992). For example, expression of fos and ras oncogenes is sufficient for the transformation of epidermal keratinocytes (Greenhalgh et al., 1990). A characteristic of epidermal carcinomas is the alteration of patterns of gene expression due, at least in part, to oncogenes activated during carcinogenesis.
Among the markers of epidermal carcinomas are keratins K6 and K16. These keratins are not found in normal epidermis. However, they are expressed in diseases associated with hyperproliferation—not only carcinomas, but also wound healing and psoriasis—where they generally replace keratins K1 and K10 in the suprabasal layers (Weiss et al., 1984). In healthy tissue, K6 and K16 are found in the outer root sheet of the hair follicle (Heid et al., 1988), fungi-form and filiform papillae of the tongue (Rentrop et al., 1986), and developing mammary epithelium (Smith et al., 1990). When cells of stratified epithelial origin are placed in culture, they rapidly induce synthesis of these keratins (Schweitzer et al., 1984; Roop et al., 1987; Schermer et al., 1989; Surya et al., 1990).
Expression of an oncogene in the epidermis of transgenic mice resulted in hyperplasia and induction of the K6 keratin (Wilson et al., 1990). Interestingly, the human genome contains two closely related genes for K6 keratin. They seem to be co-expressed, but the significance of the gene duplication is presently unknown (Tyner et al., 1985). An epithelium specific enhancer has been identified upstream from the bovine K6 gene (Blessing et al., 1990). The regulators shown to induce specifically the transcription of K6 are EGF and TGFα (Jiang et al., 1993). On the other hand, retinoic acid and thyroid hormones, through the action of their receptors, can suppress the expression of the K6 promoter (Tomic et al., 1990).
In this report we focus specifically on the molecular components that regulate expression of the K6 keratin gene. We pay special attention to the sequences potentially regulated by oncogenes because of the association of K6 expression with carcinomas. We have found four specific protein–DNA interactions in the K6 promoter. Two proteins bind to adjacent sites immediately upstream from the TATA box. The third binding site constitutes a large palindrome. The fourth is an AP1 site, which is recognized by fos and jun proto-oncoproteins (Franza et al, 1988; Rausher et al., 1988). We show below that AP1 protein is important for K6 expression in keratinocytes, thus demonstrating functionality of the AP1 protein in the epidermis and providing a link between carcinogenesis and altered gene expression in human carcinomas.
Materials and methods
DNA constructs
The plasmid pK6CAT was obtained by subcloning 381 bp of the K6 promoter into pGCAT C vector upstream from the chloramphenicol acetyl transferase (CAT) bacterial reporter gene (Jiang et al., 1991). DNA subfragments were PCR-amplified using DNA primers synthesized on a Pharmacia Gene Assembler Plus (Jiang et al., 1991). To make internal deletions, we used two rounds of PCR. This procedure replaces the site to be deleted by an Xba I site (Fig. 1). Sequences of the oligonucleotides used are presented in Table 1. Control plasmids pSV2CAT and pRSVZ have been described previously (Jiang et al., 1990).
Figure 1.
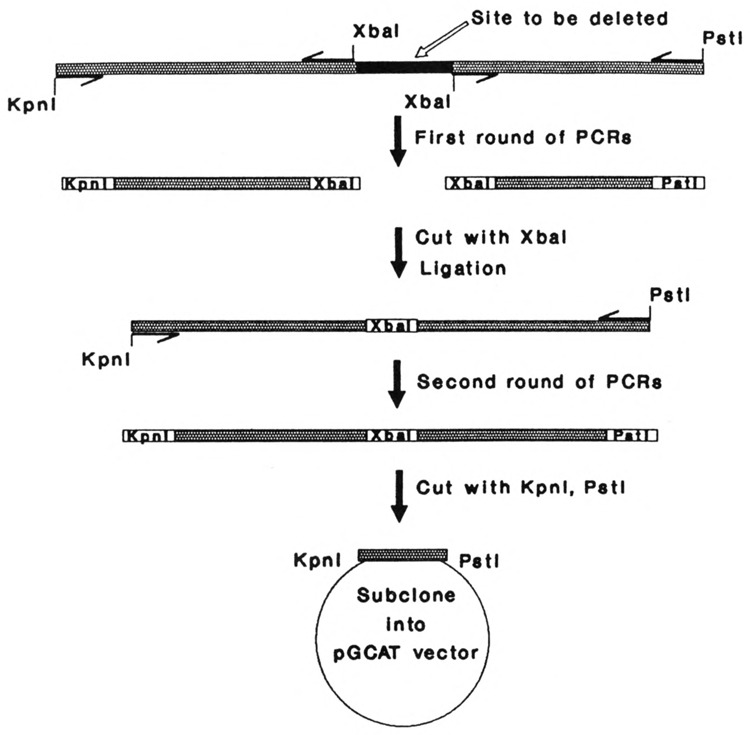
The strategy to create internal deletions. Half-headed arrows represent the oligonucleotides used as primers in PCR. The restriction sites Pst I, Kpn I, and Xba I contained in the oligonucleotides are indicated. The products of the first round of PCR, after appropriate restriction and ligation, serve as a template for the second round. For details see Materials and Methods.
Table 1.
List of oligonucleotides.
| All oligonucleotides are indicated in the 5′ to 3′ orientation. F: forward; R: reverse. Restriction sites: X: Xba I; P: Pst I; K: Kpn I. | ||||
| Oligonucleotides used in the PCR reactions | ||||
| −131XF | TTTTCTACATTCCAGGACTAGGGC | −201 PR | TTTCTGCAGGTTATGAAATACCGA | |
| −8PR | TTTCTGCAGGAGATGAGAGGGCTT | −191 PR | TTTCTGCAGTATTACAAAAGTTAT | |
| −251XF | TTTTCTAGAATTTTGCCCTGACTA | −181 PR | TTTCTGCAGACACCTGCATTATTA | |
| −131PR | TTTCTGCAGAGGTGAGCTTGCAGG | −171 PR | TTTCTGCAGAGTGAGATTCACACC | |
| −381XF | TTTTCTAGACACTCAGGGCATTGT | −271 PR | TTTCTGCAGACTGGAGATCTCAGC | |
| −251PR | TTTCTGCAGTGTTCCTGCCTCCAG | −291 PR | TTTCTGCAGAAGAGTATTCTCAAT | |
| −44PR | TTTCTGCAGAGGCGAGAGGGAGGA | −311 PR | TTTCTGCAGAAGTTGGAATTAGTC | |
| −87XF | TTTTCTAGATACTGGAGTCCGATT | −331 PR | TTTCTGCAGCTTCAGCAAAGGTTC | |
| −100PR | TTTCTGCAGTGGAGAGCATGGGCT | −331XF | TTTTCTAGAGACAGTGACTAATTC | |
| −151XF | TTTTCTAGATCCCAACCTGCAAGC | −311XF | TTTTCTAGATCATGAATTGAGAAT | |
| −161XF | TTTTCTAGAGCCCAGCCCTTCCAA | −381KF | TTTGGTACCCACTCAGGGCATTGT | |
| −171XF | TTTTCTAGATATTTGTAAAGCCCA | −211XR | TTTTCTAGAACCGAGATTGCATTT | |
| −181XF | TTTTCTAGATGAATCTCACTATTT | −191XF | TTTTCTAGAAATGCAGGTGTGAAT | |
| −191XF | TTTTCTAGAAATGCAGGTCTGAAT | −331XR | TTTTCTAGACTTCAGCAAAGGTTC | |
| −201XF | TTTTCTAGACTTTTGTAATAATGC | −134XR | TTTTCTAGATGAGCTTGCAGGTTG | |
| −211XF | TTTTCTAGATATTTCATAACTTTT | −99XF | TTTTCTAGATATATAAGCTGCTAC | |
| −281XF | TTTTCTAGAAGATCTCCAGTCAAA | −115XR | TTTTCTAGAGGGCCCTAGTCCTGG | |
| −221PR | TTTCTGCAGCATTTTTCGCTTCCT | −115XF | TTTTCTAGACAGCCCATGCTCTCC | |
| −211PR | TTTCTGCAGACCGAGATTGCATTT | −108XR | TTTTCTAGAATGGGCTGGGCCCTA | |
| Oligonucleotides used as probes or competitors | ||||
| −331 to −311 | 5′GACAGTGACTAATTCC | |||
| ACTGATTAAGGTTGAA5′ | ||||
| −211 to −191 | 5′GGTATTTCATAACTTTTGTA | |||
| AAGTATTGAAAACATTATGG5′ | ||||
| −131 to −89 | 5′TTCCAGGACTAGGGCCCAGCCCCATGCTCTCCATATATA | |||
| CTGATCCCGGGTCGGGTACGAGAGGTATATATTCGAC5′ | ||||
| −126 to −89 | 5′GGACTAGGGCCCAGCCCATGCTCTCCATATATA | |||
| TCCCGGGTCGGGTACGAGAGGTATATATTCGAC5′ | ||||
| −120 to −89 | 5′GGGCCCAGCCCATGCTCTCCATATATA | |||
| GTCGGGTACGAGAGGTATATATTCGAC5′ | ||||
| −115 to −89 | 5′CAGCCCATGCTCTCCATATATA | |||
| GTACGAGAGGTATATATTCGAC5′ | ||||
| −131 to −99 | 5′TTCCAGGACTAGGGCCCAGCCCATGCTC | |||
| CTGATCCCGGGTCGGGTACGAGAGGTA5′ | ||||
| AP-1 | 5′CTAGTGATGAGTCAGCCGGATC | |||
| GATCACTACTCAGTCGGCCTAG5′ | ||||
Probes and competitors for gel retardation assays
PCR products were purified by electroelution after separation on 1.5% agarose gel. DNA sequences to be used as probes were digested with Xba I restriction enzyme to provide a 3′ recessed end and then end-labeled with Klenow fragment and [α32P]dCTP (40 μCi per reaction). Radiolabeled probes were purified by gel filtration using Sephadex G50 columns (Excellulose GF5; Pierce). Small DNA fragments (<50 bp) were synthesized in the oligonucleotide synthesizer (Table 1). Those to be used as probes were designed to contain 5′ overhangs for [α32P]dCTP end-labeling. Double-stranded fragments were generated by annealing the two complementary strands in 10 mM Tris, 1 mM EDTA, 150 mM NaCl. Synthetic AP-1 consensus DNA was purchased from Stratagene (Stratagene Hot Foot Kit).
Cells and transfections
All DNA used in transfections was purified through two CsC1-ethidium bromide equilibrium gradient centrifugations.
HeLa cells were maintained in Dulbecco-modified Eagle’s medium (DMEM) supplemented with 10% calf serum. The transfection procedure of Chen and Okayama (1987) was slightly modified (Jiang et al., 1990 and 1991).
Normal human epidermal keratinocytes (HEK) were purchased from Clonetics and grown in defined serum-free keratinocyte medium (Gibco) supplemented with epidermal growth factor and bovine pituitary extract provided by the manufacturer Cells were expanded through two 1:4 passages before transfection. Subconfluent cultures were transfected at 80% confluency using polybrene and DMSO shock as previously described (Jiang et al., 1991). Forty-eight hours after transfection, cells were washed twice with phosphate-buffered saline and harvested by scraping. The sonic disruption combined with freeze thaw cycles as well as the β-galactosidase and CAT assays procedures have been described (Jiang et al., 1991). Relative CAT activity values represent the promoter activities corrected for the efficiency of transfection.
Nuclear extracts
Nuclear extracts from HeLa cells were prepared according to the method of Dignam et al. (1983). For the preparation of the nuclear extracts of keratinocytes, the same method was modified as follows. Pelleted cells (5 minutes, 3,000 rpm, 4°C) were resuspended in 2 volumes of buffer containing 1.5 mM MgCl2, 10 mM KC1, 0.5 mM DTT, 10 mM Hepes, pH 7.9, and then broken by 20 to 30 strokes of a B pestle in a glass Dounce homogenizer. The resulting nuclei were pelleted by 17,000 rpm centrifugation in SW41 rotor at 4°C, resuspended in 3 ml of buffer containing 420 mM NaCl, 225 mM EDTA, 1.5 mM MgCl2, 25% glycerol, 20 mM Hepes, 0.5 mM DTT, 1 μg/ml pepstatine A, 0.5 μg/ml leupeptin, 1 mM benzamidine, and 0.5 mM PMSF. Nuclei were broken by 15 to 20 strokes of the B pestle in a Dounce homogenizer and gently stirred at 4°C for 30 minutes Cell debris was removed by 16,500 rpm centrifugation at 4°C (SW41). Clear supernatant was dialyzed against 1 liter of buffer containing 200 mM EDTA 100 mM KC1, 20% glycerol, 0.5 mM DTT, 20 mM HEPES, and the same concentrations of protease inhibitors as above, for 5 hours at 4°C.
Gel retardation
Approximately 7 μg HEK or HeLa nuclear extract were first incubated 15 minutes on ice with or without a 200-fold molar excess of double-stranded DNA used as an unlabeled competitor in binding buffer containing 2 μg poly(dI-dC), 10% glycerol, 5 mM MgCl2, 1 mM DTT, 100 mM NaCl, 20 mM Tris-HCl, pH 8, 2% polyvinyl alcohol, 0.1 mM EDTA, and 4 mM spermidine. Then 15,000 cpm of 32P-labeled PCR amplified DNA probe or 80,000 cpm of oligonucleotide probe were added and incubated for an additional 15 minutes on ice. Double-stranded salmon sperm DNA fragmented by sonication was used as a nonspecific competitor (Maniatis et al., 1982). Probe bound to nuclear protein was resolved from free probe through a 4% poly-acrylamide gel (acrylamide: bis-acrylamide = 29:1). After drying, gels were autoradiographed overnight at −70°C on XAR 5 film (Kodak) with intensifier screens.
Supershift assays were run as described above with different amounts of affinity-purified antiserum added at the beginning of the first incubation. Antibody directed against fos family members (Vosatka et al., 1989) was generously supplied by Dr. E. Ziff (New York University Medical Center).
Results
Sequence of the K6 gene promoter region
The sequence of the K6 promoter region available for study contains 381 bp upstream from the translation start site (Fig. 2). Although relatively short, this segment contains several important regulatory elements, including cell type-specific and hormone-responsive factors (Jiang et al., 1991; Tomic et al., 1990). Perusal of this sequence allowed identification of a TATA box and an AP-2 site (Leask et al., 1991). Our earlier study (Jiang et al., 1993) demonstrated the presence of an epidermal growth factor-responsive element (EGF-RE) that did not correspond to any sequence of eukaryotic transcription regulatory sites previously described (Faisst and Meyer, 1992).
Figure 2.

Nucleotide sequence of the promoter of the human K6 gene. Nucleotides are numbered from the translation initiation codon (underlined). The TATA box is in boldface, and the protein–DNA binding sites described in Results are doubly underlined. The two other binding sites described previously, AP-2 (Leask et al., 1991) and EGF-RE (Jiang et al., 1993) sites, are overlined. The three fragments of the K6 promoter region used as probes in gel retardation experiments (I, II, and III) are schematically represented at the bottom of the figure. The translation start site is indicated by an arrow. The protein binding sites described in this study are indicated by ovals.
To determine the sites that actually bind nuclear proteins, we divided the promoter region into three segments, radiolabeled each one, and used them in gel retardation assays. As sources of binding proteins, we used nuclear extracts of HeLa cells and normal HEK. We chose these two epithelial cell types because HEK express K6 in culture, whereas HeLa cells do not (Moll et al., 1982). Correspondingly, the K6 gene promoter was active when transfected into HEK, but not HeLa cells (Jiang et al., 1991).
All three DNA segments of the K6 promoter bound specific nuclear proteins. The protein binding sites were localized using competitors of various lengths. We also compared the patterns of gel retardation obtained with nuclear extracts of HeLa cells to those of keratinocytes. Finally, we identified the sites precisely by using short, synthetic, double-stranded oligonucleotides as probes in the gel retardation assay.
The function of each protein binding site was determined by creating a specific, internal deletion from each site in the K6 promoter. The mutant DNAs were cloned upstream of the CAT reporter gene and transfected into HEK and HeLa cell cultures.
The two TATA box-proximal binding sites
Using fragment I (bp −131 to −8) as a probe, we observed a difference between the nuclear extracts from HeLa cells and those from keratinocytes. Four retarded bands were observed with HeLa, while only three were apparent with HEK extracts (Fig. 3A). We do not know whether the difference is due to a cell type-specific protein or to artifacts such as proteolysis.
Figure 3.
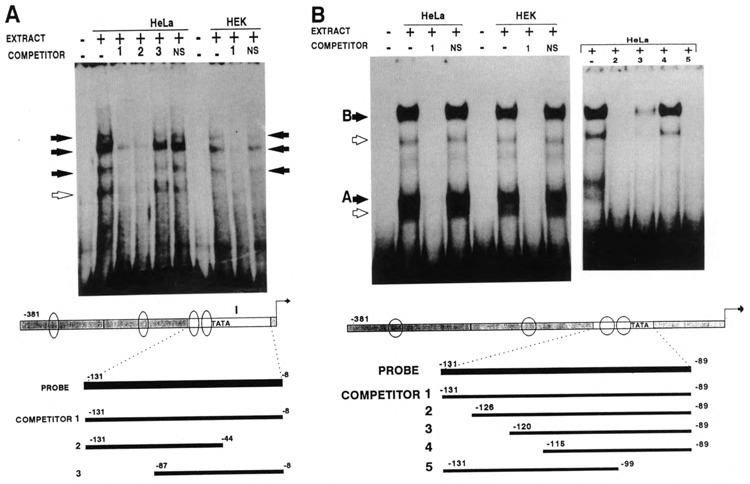
The binding sites in fragment I. A. Gel retardation of fragment I using either HeLa or HEK nuclear extracts. The PCR-engineered DNA probe and the unlabeled competitors are schematically represented at the bottom of the figure. Black arrows show the bands which comigrate with extracts from both cell types. The open arrow shows the additional band observed with HeLa extract only. B. Gel retardation of a synthetic 40 bp oligonucleotide (−131 to −89). Comparison of the binding of HEK and HeLa nuclear extracts shows that the pattern observed with both extracts is similar. The right panel shows mapping results using the unlabeled oligonucleotide competitors identified at the bottom. Note that site A is competed by all oligonucleotides, including competitor 5, which lacks the TATA box. Site B is completely competed by oligonucleotide 2 and 5, partially by 3, and not competed at all by 4. The proteins binding onto sites A and B are therefore independent of each other and also of the TATA box-binding proteins. Open arrows mark satellite bands, which are not observed consistently. NS: nonspecific DNA.
We used three competitors to map the binding sites. One corresponds to fragment I itself, and the two others divide the fragment into thirds. As competitors 1 (−131 to −8) and 2 (−131 to −87) abolish the binding, whereas competitor 3 (−87 to −8) does not, we deduced that the binding sites are within the distal third, nucleotides −131 to −87 (Fig. 3A). The gel shift assay using the corresponding oligonucleotide probe (−131 to −89) showed that these 42 bp are able to bind nuclear proteins from HeLa cells or HEK (Fig. 3B). Only two major protein–DNA complexes were observed with both cell types extracts, which means that the longer fragment can bind additional proteins that the 42 bp probe cannot. Each of the two bands is occasionally associated with a higher-mobility satellite band (open arrows in Fig. 3B). The satellites are not present in all preparations. Their intensities vary, and we suspect that they contain proteolytic derivatives of the two major bands.
The two complexes observed could be due either to two different protein binding sites, or to two different complexes binding to the same site. To resolve this question and characterize the binding sites within the 40 bp oligonucleotide probe, we designed several shorter, synthetic oligonucleotide DNA fragments corresponding to various portions of this probe (Fig. 3B) Because the TATA-box sequence is included within the 3′ portion of the 40 bp probe (positions −98 to −92), we wanted to determine first whether either of the two bands was due to binding of the TFIID protein complex to the TATA box (Nakajima et al., 1988). Therefore, we designed an oligonucleotide missing the TATA box (−131 to −99) and used it as a competitor in a gel retardation assay. This oligonucleotide was able to abolish both protein-DNA complexes, which means that both are due to proteins binding to sites other than the TATA box.
The mapping experiments on the 40 bp oligonucleotide probe that follow led us to identify, in fact, two different protein binding sites, both independent of each other and independent of the TATA box. Competition of the two protein–DNA complexes reveals that competitor −126 to −89 is able to abolish both complexes. The competitor −120 to −89 allows partial formation of the upper complex but abolishes the lower one. Competitor −115 to −89 does not compete at all with the upper complex yet abolishes the lower one. Therefore, one of the two protein binding sites, the one we call site A, is located within the sequence CAGCCCATGCTCTCCAT between nucleotides −115 to −99. The other one, site B, is located within the sequence GGACTAGGGCCC between nucleotides −126 to −115. Oligonucleotides comprising these two sequences retain the ability to bind nuclear proteins from HeLa or HEK extracts (data not shown).
The large palindromic site PAL
Using the fragment II of the K6 promoter (− 251 to −131) as a probe and HeLa nuclear extracts, we found three specific retarded bands (Fig. 4B). Two competitors containing the proximal part of fragment II, −201 to −100 and −211 to −100, abolished all the retarded bands. Using a series of competitors encompassing the distal part of fragment II, we found that competitors −281 to −221 and −281 to −211 were not able to compete with the binding, but competitors −281 to −201, −281 to −191, and −281 to −181 abolished the retarded bands (data not shown). Interestingly, both −281 to −201 and −201 to −100 competed for the binding, although the two competitors have only one base pair in common Therefore the binding site present in fragment II is contained between nucleotides −211 and −191. The same 20 bp binding sequence can be recognized by nuclear proteins from HEK extracts (data not shown).
Figure 4.
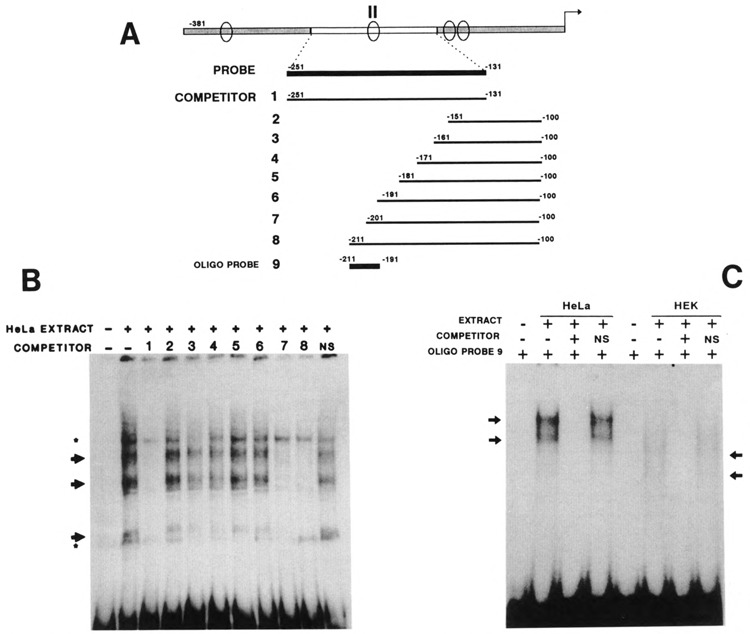
Determination of the protein binding site in fragment II. A. Schematic representation of the PCR-engineered DNA used as probes or unlabeled competitors to map the protein binding site in fragment II. Oligonucleotide probe 9 (−211 to −191) is used in C. B. Gel retardation of fragment II using HeLa nuclear extract. Arrows indicate the specific, stars the nonspecific bands. The numbers correspond to the competitors described in A. NS: nonspecific competitor. C. Oligonucleotide probe 9 binds proteins from HeLa and HEK nuclear extracts. The binding was competed by the same unlabeled oligonucleotide. Note the differences in pattern between nuclear extracts of the two cell types.
A synthetic oligonucleotide probe corresponding to these 20 bp was used with both HeLa and HEK nuclear extracts, confirming the ability of this sequence to bind nuclear proteins of both cell types (Fig. 4C). The differences in the banding pattern may be related to a difference in the binding proteins or to proteolysis in one or both of the extracts.
The sequence of this binding site, TATTTCATAACTTTTGTAATA, is palindromic. It does not correspond to any known binding sequence (Faisst and Meyer, 1992). If the palindromic nature of this sequence is important for protein binding, we would expect the contacts between the protein and DNA to be symmetrical. Indeed, as mentioned above, either half of the palindrome can compete for the binding protein.
AP-1 protein binding site in fragment III
With fragment III (−381 to −251) used as a probe and nuclear extracts from HEK, we found one major retarded band (Fig. 5B). The mapping experiment using unlabeled DNA subfragments allowed us to localize the binding site within nucleotides −331 to −311 (Fig. 5A and B). Comparing the binding ability of HeLa and HEK nuclear extracts to fragment III, we found that the retarded band migrated to the same position with extracts of both cell types (not shown). The ability of this 20 bp sequence, separated from its DNA context, to bind HeLa or HEK nuclear extracts (Fig. 5C) has been demonstrated using the corresponding oligonucleotide probe (−331 to −311).
Figure 5.
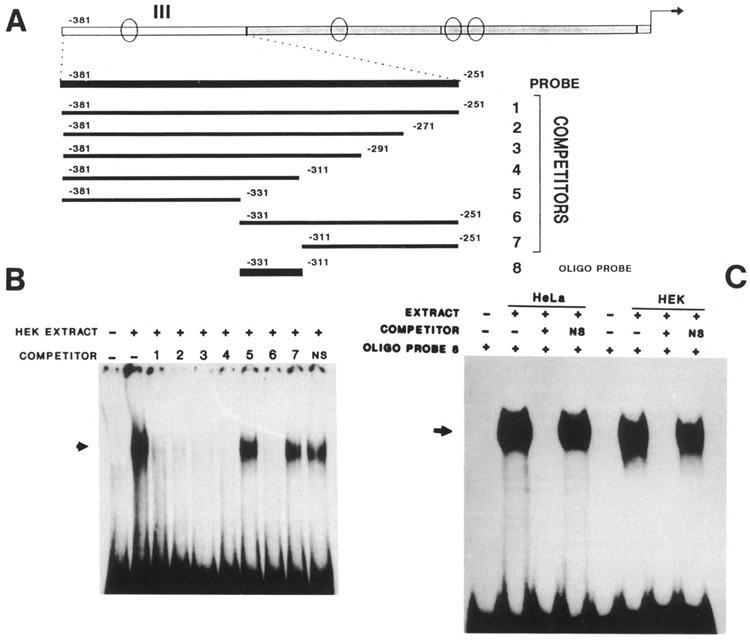
Determination of the binding site present in fragment III. A. Schematic representation of the PCR-engineered DNA used as probes or unlabeled competitors in the mapping of binding sites in fragment III of the K6 promoter. Oligonucleotide 8 (−331 to −311) is used as a probe in C. B. Mapping experiment of fragment III using HEK nuclear extract. The competition occurs with competitors 1, 2, 3, 4, and 6, but not 5 and 7. The binding site is therefore located between nucleotides −331 and −311. NS: nonspecific competitor. C. Comparison of HeLa and HEK nuclear extracts binding to oligonucleotide probe 8. The competitor is the unlabeled oligonucleotide −331 to −311.
Examination of this DNA sequence reveals in its core close similarity to the consensus recognition element for the AP-1 transcription factor (Faisst and Meyer, 1992) and identity with the AP-1 sequence found in the endothelin-1 gene (Lee et al., 1991). The AP-1 site sequence in the K6 promoter, TGACTAA, is located between nucleotides −326 and −320. To test whether the binding is due to the AP-1 protein, we used increasing amounts of the AP-1 consensus sequence as a competitor. The dose-response competition curve showed that an amount of competitor as low as 0.25 ng, 2.5-fold molar excess, was able to reduce the intensity of the retarded band significantly (Fig. 6), while 5 to 10 ng of the AP-1 consensus competitor completely abolished the protein–DNA complex. These results strongly support the hypothesis that an AP-1 site is contained in the 20 bp DNA sequence.
Figure 6.
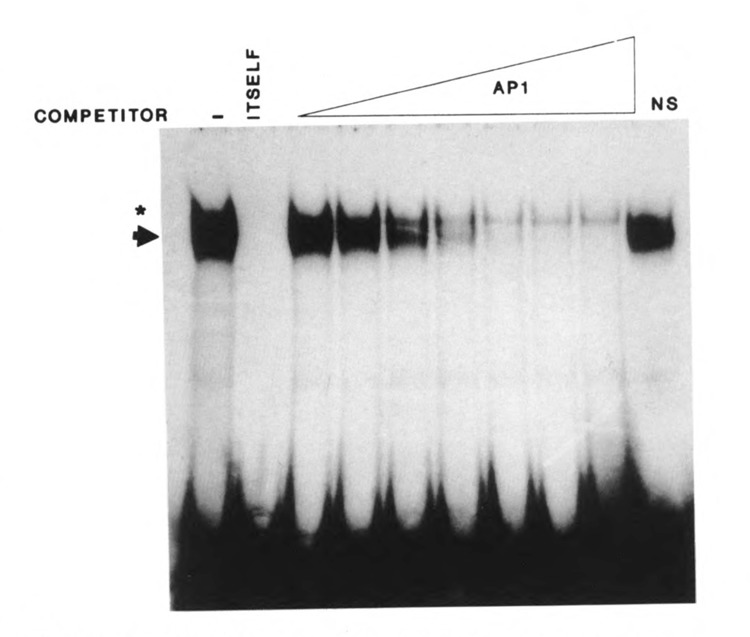
Identification of the AP-1 site in fragment III of the K6 promoter. Oligonucleotide probe 8 (see Fig. 5) used with HeLa nuclear extract reveals only one major retarded band (dark arrow), which is completely competed by itself. The star indicates a nonspecific band. NS: nonspecific competitor. Increasing amounts of an oligonucleotide containing the consensus AP-1 sequence progressively abolished the binding to the probe. Amounts of the AP-1 consensus competitor oligonucleotide ranged from 0.1 to 25 ng.
To confirm that the binding protein is indeed AP-1, we performed a “supershift” assay. The HeLa nuclear extracts were incubated with increasing amounts of fos-specific antiserum, prior to addition of the radiolabeled oligonucleotide probe (−331 to −311). Because fos cannot bind DNA alone, but only as a dimer with jun (Kouzarides and Ziff, 1988), a supershift with fos-specific antibody is prima facie evidence of jun binding as well. The intensity of the original retarded band observed without the anti-serum decreased progressively when increasing amounts of the antiserum were added (Fig. 7). Concomitantly with the disappearance of the retarded band, a new band at an even slower mobility appeared, and its intensity increased as a function of the amount of the antiserum added. The ability of this anti-fos antiserum to further retard the migration of the DNA–protein complex indicates that the binding protein contains a member of the fos family and is therefore AP-1.
Figure 7.
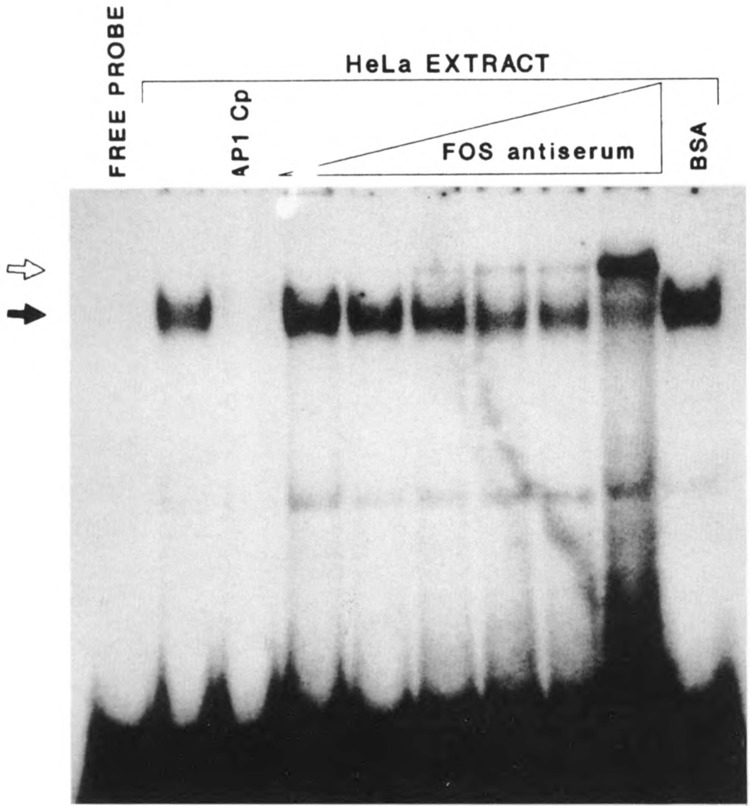
Fos binds to the AP-1 site of the K6 promoter. The gel retardation assay was performed using oligonucleotide probe 8 (−331 to −311) and HeLa nuclear extract. As is also shown in Figure 6, the competition of the specific retarded band (dark arrow) is complete with 5 ng of the AP-1 consensus oligonucleotide (AP-1 Cp). Preincubation of the nuclear extract with increasing amounts of affinity-purified anti-fos antiserum (0.5 to 15μl) resulted in a supershifted band (shaded arrow) consistent with the formation of a DNA/AP-1 protein/antiserum complex. Note that increasing amounts of antiserum led to a progressive decrease in intensity of the original band. Preincubation with 10 μg BSA had no effect.
Transfections and CAT assays
The functional involvement of each protein binding site identified in the K6 promoter was assessed according to the strategy described in Figure 1, by creating a series of internal deletion mutants. The mutant promoters with specifically deleted binding sites were cloned into the pGCAT reporter vector. The relative CAT activity of each construct was compared to that of the wild-type promoter after transfection into HEK or HeLa cells (Fig. 8).
Figure 8.
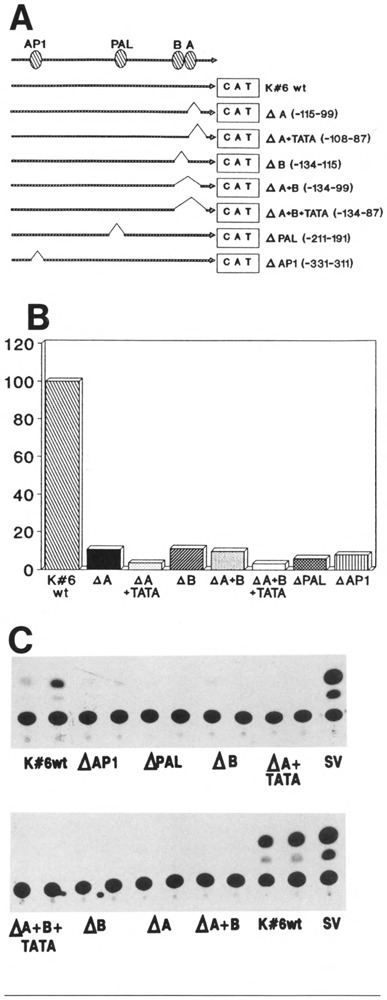
Functional assessment of the protein binding sites in the K6 promoter activity. A. Schematic representation of the K6 wild-type (wt) promoter and the mutant promoter constructs, linked to the CAT reporter gene (box). The numbers indicate the nucleotides bordering the deleted sites. The various protein binding sites are marked on the top line. B. Relative CAT activity of each of the constructs in A after transfection into HEK. The CAT levels were normalized to equivalent β-gal levels of the co-transfected pRSVZ vector. The relative CAT activity of the K6 wild-type promoter was designated 100%. Δ: deletion. C. Results of the CAT assays. Each DNA construct was transfected in duplicate into HEK. Equivalent amounts of the cell extract were used to determine CAT levels. The rightmost track on each thin layer chromatogram results from the positive control cells that were transfected with the pSV2CAT vector.
As previously described (Jiang et al., 1991), the K6 promoter is active only in HEK, not in HeLa cells. Accordingly, none of the mutant constructs exhibited any CAT activity in HeLa cells (data not shown).
Transfection of constructs with deletions of the TATA box led to undetectable CAT activity. Therefore, the presence of the TATA-box sequence is essential for basal K6 promoter transcription. In constructs missing either site A or site B or both, but containing the TATA box, the promoter activity was greatly reduced compared to the K6 wild type, albeit not abolished. Although we cannot completely rule out spacing effects in these deletions, because the three deletions are of different sizes and therefore change spacing differently, it is more likely that these sites, which were found to bind nuclear proteins independently of the TATA box, are necessary for full K6 promoter activity. Deletion of both site A and site B did not decrease the promoter activity more than deletion of just one site.
Deletion of the PAL binding site as well as of the AP-1 site also led to a dramatic decrease in CAT activity. Therefore the PAL binding proteins and the AP-1 proto-oncoproteins are also important in K6 gene transcription.
Discussion
In the promoter of the human K6 gene, three regions of DNA–protein interaction can be demonstrated, each with characteristic functional and structural properties. The distal region is an AP-1 site that interacts with fos and jun proto-oncoproteins. The middle one is a large palindromic sequence that interacts symmetrically with the cognate binding protein. The proximal region is directly upstream from the TATA box and independently binds two proteins at adjacent sites.
The palindromic site is required for K6 promoter activity. The corresponding protein apparently binds symmetrically as a dimer, but either arm of the palindrome is sufficient to compete with the binding. Thus each monomer can bind DNA independently. The palindromic binding site exhibits different protein binding patterns in HeLa cells and in HEK. Whether the additional band observed with HeLa nuclear extract has any transcriptional function is still unknown, but it may be responsible for the differential expression of the K6 gene in HEK and in HeLa cells.
Similar differences are observed in the TATA box-proximal region. Again, the function of the additional binding proteins present in HeLa nuclei are unknown, but they may be responsible for the lack of K6 expression in HeLa cells. The two protein binding sites in the proximal region, together with the TATA box, form a very tight cluster of protein recognition elements This suggests that the corresponding proteins interact when bound to DNA, but this interaction does not function simply at the level of protein binding to DNA, i.e., the binding per se of each protein is neither synergistic with nor antagonistic to the binding of the others.
The binding patterns of the keratinocyte and HeLa proteins are identical at the AP-1 site, although we cannot exclude the possibility that different members of the fos and jun families are present in the two cell types. This binding demonstrates the presence of functional AP-1 proteins in epidermal keratinocytes. Activation of AP-1 is often associated with other regulatory elements in the same gene (Nicholson et al., 1990; Schüle et al., 1990a,b; Zhang and Young, 1991); however, we have not detected binding sites for other nuclear proteins in the immediate vicinity of the AP-1 site.
The presence of the AP-1 site is especially important, because the AP-1 binding proteins are elevated during spontaneous or induced malignancies. The level of c-fos mRNA expression is high in human epidermis, higher than in most adult tissues (Basset-Séguin et al., 1990), especially in the granular layer (Fisher et al., 1991). Levels of c-jun protein and its mRNA have not been as extensively studied. AP-1 was originally identified as a TPA-responsive element, and indeed tumor promoters, such as TPA further increase c-fos expression when applied to mouse skin (Rose-John et al., 1988; Dotto et al., 1986; Angel et al., 1987; Lee et al., 1987). Treatment of skin with tumor promoters induces formation of papillomas, premalignant benign hyperproliferative lesions (Nischtet al., 1988; Roop et al., 1988). In addition to TPA treatment, a second oncogenic event is required for progression to frank carcinomas (Greenhalgh et al., 1990). Among the pleiotropic effects of TPA, concomitant induction of the expression of K6 keratin has been observed (Schweitzer and Winter, 1982; Finch et al., 1991). The induction of K6 in premalignant tissues may be a consequence of activation of the AP-1 site by fos and jun.
However, K6 keratin can also be expressed in noncarcinogenic hyperproliferative diseases such as psoriasis or wound healing. In these cases, which are never associated with malignancies, it has been shown that fos is not induced and may be even suppressed (Elder et al., 1990; Basset-Séguin et al. 1991). The AP-1 site of the K6 gene is therefore not directly implicated. The level of TGFα in hyperproliferative diseases, especially in psoriatic plaques, is elevated (Coffey et al., 1987). Thus, expression of the K6 gene in nonmalignant hyperproliferative diseases may involve the action of the EGF-RE in the K6 promoter that we characterized previously (Jiang et al., 1993).
We propose a model in which there are at least two independent pathways for induction of the K6 keratin. The one relevant in carcinogenesis may involve the AP-1 site and the fos and jun proteins; the other, relevant in non-malignant diseases, may involve the EGF-RE and its binding protein.
Acknowledgments
Our special thanks to Mike Levine, whose work contributed to every single experiment described. We also thank E. Zifffor the gift of fos-specific antibody.
Our research was supported by National Institutes of Health grants AR30682 and AR39176, the New York University Skin Disease Research Center Grant AR39749, and the R. L. Baer Foundation. M. Blumenberg is a recipient of the Irma T. Hirschl Career Scientist Award.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Françoise Bernerd is currently at CIRD-GALDERMA, Sophia-Antipolis, 06565 Valbonne, France.
References
- Angel P., Imagawa M., Chiu R., Stein B., Imbra R. J., Rahmsdorf H. J., Jonat C., Herrlich P., and Karin M. (1987), Cell 49, 729–739. [DOI] [PubMed] [Google Scholar]
- Basset-Séguin N., Escot C., Blanchard J. M., Kerai C., Verrier B., Mion H., and Guilhou J. J. (1990), J Invest Dermatol 64, 418–422. [DOI] [PubMed] [Google Scholar]
- Basset-Séguin N., Escot C., Moles J. P., Blanchard J. M., Kerai C., and Guilhou J. J. (1991), J Invest Dermatol 97, 672–678. [DOI] [PubMed] [Google Scholar]
- Blessing M., Jorcano J. L., and Franke W. W. (1989), EMBO J 8, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. and Okayama H. (1987), Mol Cell Biol 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey R. J. Jr., Derynck R., Wilcox J. N., Bringman T. S., Goustin A. S., Moses H. L., and Pittelkow M. R. (1987), Nature 328, 817–820. [DOI] [PubMed] [Google Scholar]
- Dignam J. D., Martin P. L., Shastry B. S., and Roeder R. G. (1983), Methods Enzymol 101, 582–598. [DOI] [PubMed] [Google Scholar]
- Dotto G. P., Gilman M. Z., Maruymama M., and Weinberg R. (1986), EMBO J 5, 2853–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder J. T., Tavakkol A., Klein S. B., Zeigler M. E., Wicha M., and Voorhees J. J. (1990), J Invest Dermatol 94, 19–25. [DOI] [PubMed] [Google Scholar]
- Faisst S. and Meyer S. (1992), Nucleic Acids Res 20, 3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch J., Andrews K., Krieg P., Furstenberger G., Slaga T., Ootsuyama A., Tannoka H., and Bosden G. T. (1991), Carcinogenesis 12, 1519–1522. [DOI] [PubMed] [Google Scholar]
- Fisher C., Byers M. R., Iadarola M. J., and Powers E. A. (1991), Development 111, 253–258. [DOI] [PubMed] [Google Scholar]
- Franza B. R. Jr., Rauscher F. J., Josephs S. F., and Curran T. (1988), Science 239, 1150–1153. [DOI] [PubMed] [Google Scholar]
- Greenhalgh D. A., Welty D. J., Player A., and Yuspa S. H. (1990), Proc Natl Acad Sci USA 87, 643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid H. W., Moll I., and Franke W. W. (1988), Differentiation 37, 137–157. [DOI] [PubMed] [Google Scholar]
- Jiang C.-K., Epstein H. S., Tomic M., Freedberg I. M., and Blumenberg M. (1990), Nucleic Acids Res 18, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.-K., Epstein H. S., Tomic M., Freedberg I. M., and Blumenberg M. (1991), J Invest Dermatol 96, 162–167. [DOI] [PubMed] [Google Scholar]
- Jiang C.-K., Magnaldo T., Ohtsuki M., Freedberg I. M., Bernerd F., and Blumenberg M. (1993), Proc Natl Acad Sci USA 90, 6786–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. and Ziff E. (1988), Nature 336, 646–656. [DOI] [PubMed] [Google Scholar]
- Leask A., Byrne C., and Fuchs E. (1991), Proc Natl Acad Sci USA 88, 7948–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-E., Dhadly M. S., Temizer D. H., Clifford J. A., Yoshizumi M., and Quertermous T. (1991), J Biol Chem 266, 19034–19039. [PubMed] [Google Scholar]
- Lee W., Mitchell P., and Tjian R. (1987), Cell 49, 741–752. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F., and Sambrook J. (1982), Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Moll R., Franke W. W., Schiller D. L., Geiger B., and Krepler R. (1992), Cell 31, 11–24. [DOI] [PubMed] [Google Scholar]
- Nakajima N., Horikoshi M., and Roeder R. G. (1988), Mol Cell Biol 8, 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson R. C., Mader S., Nagpal S., Leid M., Rochette-Egly C., and Chambon P. (1990), EMBO J 9, 4443–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischt R., Roop D. R., Mehrel T., Yuspa S. H., Rentrop M., Winter H., and Schweizer J. (1988), Mol Carcinogenesis 1, 96–108. [DOI] [PubMed] [Google Scholar]
- Rauscher F. J., Sambucetti L. C., Curran T., Distel R. J., and Spiegelman B. M. (1988), Cell 52, 471–480. [DOI] [PubMed] [Google Scholar]
- Rentrop M., Knapp B., Winter H., and Schweizer J. (1986), J Cell Biol 103, 2583–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roop D. R., Huitfeldt H., Kilkenny A., and Yuspa S. H. (1987), Differentiation 35, 143–150. [DOI] [PubMed] [Google Scholar]
- Roop D. R., Krieg T. M., Mehrel T., Cheng C. K., and Yuspa S. H. (1988), Cancer Res 48, 3245–3252. [PubMed] [Google Scholar]
- Rose-John S., Furstenberger G., Krieg P., Besemfelder E., Rincke G., and Marks F. (1988), Carcinogenesis 9, 831–835. [DOI] [PubMed] [Google Scholar]
- Schermer A., Jester J. V., Hardy C., Milano D., and Sun T.-T. (1989), Differentiation 42, 103–110. [DOI] [PubMed] [Google Scholar]
- Schüle R., Rangarajan P., Kliewer S., Ransone L. J., Bolado J., Yang N., Verma I. M., and Evans R. M. (1990a), Cell 62, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Schüle R., Umensono K., Mangelsdorf D. J., Bolado J., Pike J. W., and Evans R. M. (1990b), Cell 61, 497–504. [DOI] [PubMed] [Google Scholar]
- Schweizer J., Kinjo M., Furstenberger G., and Winter H. (1984), Cell 37, 159–170. [DOI] [PubMed] [Google Scholar]
- Schwiezer J. and Winter H. (1982), Cancer Res 42, 1517–1529. [PubMed] [Google Scholar]
- Silverberg E. and Lubera J. A. (1989), CA Cancer J Clin 39, 3–20. [DOI] [PubMed] [Google Scholar]
- Smith G. H., Mehrel T., and Roop D. R. (1990), Cell Growth Differ 2, 161–170. [PubMed] [Google Scholar]
- Surya B., Yu J., Manabe M., and Sun T.-T. (1990), J Cell Sci 97, 419–432. [DOI] [PubMed] [Google Scholar]
- Tomic M., Jiang C.-K., Epstein H. S., Freedberg I. M., Samuels H. H., and Blumenberg M. (1990), Cell Regul 1, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner A. L., Eichman M. J., and Fuchs E. (1985), Proc Nat Acad Sci USA 82, 4683–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosatka R. J., Hermanowski-Vosatka A., Metz R., and Ziff E. B. (1989), J Cell Physiol 138, 493–502. [DOI] [PubMed] [Google Scholar]
- Weiss R. A., Eichner R., and Sun T.-T. (1984), J Cell Biol 98, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. B., Weinberg W., Johnson R., Yuspa S., and Levine A. J. (1990), Cell 61, 1315–1327. [DOI] [PubMed] [Google Scholar]
- Yuspa S. H., Kilkenny A., Cheng C., Roop D., Hennings H., Kruszewski F., Lee E., Strickland J., and Greenhalgh D. A. (1991), Environ Health Perspect 93, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. and Young A. P. (1991), J Biol Chem 266, 24332–24338. [PubMed] [Google Scholar]