

Abstract
Allergen specific immunotherapy has been shown to be the only effective treatment for long- lasting clinical benefit to IgE-mediated allergic diseases, but a fewer than 5% of patients choose the treatment because of inconvenience and a high risk of anaphylaxis. Recently, epicutaneous allergen-specific immunotherapy (EPIT) has proven effective, yet with limitations owing to strong skin reactions. We demonstrate here safer and faster EPIT, named μEPIT, by delivering powdered allergen and adjuvants into many micropores in the epidermis. We fabricated a microarray patch fractionally coated with a powder mixture of ovalbumin (OVA) model allergen, CpG, and 1,25-dihydroxyvitamin D3 (VD3). Topical application of the patch onto laser- microperforated skin resulted in a high level of epidermal delivery while greatly minimizing allergen leakage into circulation system as compared to current subcutaneous immunotherapy (SCIT). Moreover, only three times of μEPIT over two weeks could sufficiently inhibit allergen- specific IgE responses in mice suffering OVA-induced airway hyperresponsivness (AHR), which was unattainable by eight times of SCIT over three weeks. Mechanistically, μEPIT preferably enhanced IgG2a production suggesting TH1-biased immune responses and induced a high level of T-regulatory (Treg) cells against repeated allergen sensitization. The immune tolerance was confirmed by marked reduction in airway wall thickness as well as eosinophil and neutrophil infiltration into the respiratory airway. The μEPIT represents a novel and painless technology to treat IgE-mediated allergic diseases with little local skin reaction and a minimal risk of anaphylaxis.
Keywords: allergic asthma, adjuvant, epicutaneous immunotherapy (EPIT), immunoglobulin E (IgE), laser and micro-fractional epicutaneous (μEP) delivery
Graphical Abstract
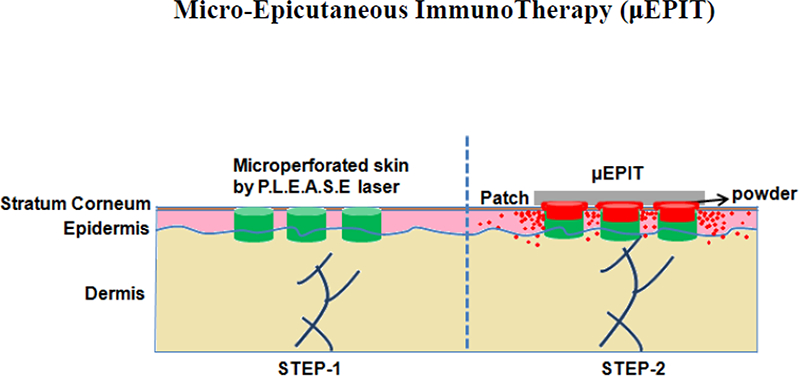
Introduction
Allergic asthma is frequent among atopic patients and caused by inhaled harmless environmental antigens obstructing airways of the lung along with elevated specific-immunoglobulin E (IgE) responses [1]. The prevalence of asthma is estimated to affect more than 300 million people globally, in particular, children [1] or ~25 million people in the United States alone [1]. To date, allergen-specific immunotherapy (AIT) represents the only curative treatment for type-1 allergic asthma, besides allergen avoidance and management practices [2–4]. In clinics, subcutaneous (SCIT) and sublingual (SLIT) immunotherapies have been employed to induce immunological tolerance and treat allergic patients [2–4]. However, SCIT and SLIT are only moderately effective, with great safety concerns in association with SCIT owing to a potential risk of severe systemic and local reactions [5,6]. Apart from the low efficacy and high risk, the treatment requires 50~70 visits to clinics/hospitals for repetitive allergen administration over 3–5 years [5,6]. Pharmacological treatments in case of emergency visits include administration of antihistamines, leukotriene modifiers and/or corticosteroids either systemically or using inhalers to suppress the ongoing allergic reaction, but extensive use of these drugs might cause severe side effects [7,8]. Therefore, tailor-made treatment choice for each patient is being considered to provide extended protection against allergic asthma [8]. Recently, epicutaneous allergen-specific immunotherapy (EPIT) is developed as a potential alternative, in which allergens are introduced into non-vascularized epidermis targeting potent antigen presenting cells in the epidermis for efficient and safe treatments [5,6,9–11].
However, epidermal delivery of allergen via intact skin faces numerous challenges as the skin is impermeable for large molecules, whereas breaking skin barrier often triggers unwanted TH2 immune responses [11]. Minimal disruption of the skin in humans was initially explored to deliver allergen into the epidermis by tape-stripping followed by application of allergen patches [5,6,10,11]. However, a large area of skin disruption as well as a long duration of allergen-patch application due to poor allergen delivery elicited strong local skin reactions during EPIT and weak immune tolerance in humans [11]. Thus, there is a need for improved epidermal allergen delivery.
We, along with others, have used ablative micro-fractional laser (AFL) to generate an array of self-renewable microchannels (MCs) in the skin for vaccine delivery and to elicit strong Th1 immune responses [12–15]. AFL is a mature cosmetic technology for skin resurfacing and has demonstrated a long record of safety in the clinics [12–15]. Several studies showed that application of patch that had been soaked in allergen-adjuvant solution to AFL-treated skin enhanced the efficacy of EPIT in mice of airway hyper-responsiveness (AHR), but the delivery efficacy was low [13–15]. Also, preservatives might be required to avoid microbial contamination during the prolonged period of patch application. In contrast, we delivered powdered substances into the epidermis via micro-perforated skin at a delivery efficacy of about 80% in less than 1 hr, without incurring overt skin reactogenicity [12]. In the present study we employed this needle-free, painless technology to deliver powdered allergen and to evaluate various adjuvants for their ability to induce immune tolerance. We found that micro-fractional delivery of powdered allergen plus 1,25-dihydroxyvitamin D3 (VD3) and CpG-ODN (TLR-9 agonist) into the epidermis, called here μEPIT, sufficiently suppressed allergen-specific IgE responses as a result of increased T regulatory (Treg) cells and IgG2a in AHR mice. The μEPIT results in substantial reduction of times of treatment with safer and more effective profiles than conventional SCIT, conferring great promise to be a new therapeutic intervention to treat IgE- mediated allergic diseases with a high patients’ compliance.
2. Materials and methods
2.1. Animals
Female BALB/c mice at 5–6 weeks of age were obtained from Charles River Laboratory, Wilmington, USA. The mice were housed under specific pathogen-free conditions at Massachusetts General Hospital (MGH) animal facility. The animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at MGH, Boston.
2.2. Fabrication of powder microarray patches
Powder microarray patches were prepared following a published procedure [12]. In brief, a 1 cm2 plastic membrane was illuminated by P.L.E.A.S.E.® (Precise Laser EpidermAl SystEm, a generous gift of Pantec Biosolution AG) laser to generate 75 micropores per cm2 each in a size of 50–75 μm in base diameter and 20–30 μm in depth in the following parameters: pulse width - 50 μs, RepRate - 500 Hz, pulse per pore - 2, Array size - 10 mm, density - 4%, fluence - 2.8 J/cm2 and power 0.7 W. The pore depth is in lines with previous publication showing low energy application (4.53 J per cm2) in porcine skin [16]. Next, this plastic membrane was attached to an adhesive tape (3M center, MN, USA), followed by firm pressure of finely lyophilized powder ovalbumin (OVA) or OVA mixed with indicated adjuvant into each microholes. Excess powder on the surface was carefully removed to obtain powder-coated patch in a same pattern as the one generated in the skin using the same laser treatment. For μEPIT, 50 μg OVA was mixed with 5 μg CpG-ODN either alone or in combination with l0ng 1,25-dihydroxyvitamin D3 (VD3) or 5 μg rapamycin (Rapa) prior lyophilization and coating.
2.3. Micro-fractional epicutaneous delivery with reduced systemic exposure
To verify reduced systemic exposure following micro-fractional epicutaneous (μEP) delivery, 5 μg FITC powder (fluorescein isothiocyanate 1, Sigma-Aldrich, MO, USA) was coated onto a microarray patch as described above. The patch was topically applied to the shaved dorsal skin that had been micro-perforated with P.L.E.A.S.E.® laser to generate 75 pores per cm2 as above. Intradermal (ID) or subcutaneous (SC) injection of 10 μ1 FITC (5 μg) in PBS was also conducted in the similarly shaved dorsal skin in separate groups of mice for comparison. The fluorescence intensity was quantified using spectrofluorometer in blood plasma collected at indicated times after FITC delivery into the skin by various procedures.
2.4. Laser-facilitated μEPIT
Balb/c mice were sensitized to OVA (Sigma Aldrich) by intraperitoneal injection on days 0 and 7 of 10 μg OVA emulsified in 1 mg alum adjuvant (Thermo Fischer Scientific Inc., MA, USA). The OVA sensitized mice were treated with OVA adjuvantated by CpG and VD3 delivered by either μEP or SC injection. Specifically, the mouse skin was first microperforated with P.L.E.A.S.E.® laser using the same laser parameters above. A microarray patch coated with a mixture of OVA, VD3, and CpG powder was applied onto the laser-treated skin for 2 hours. Control mice received the same amount of OVA, CpG and VD3 in 50 μ1 PBS subcutaneously to evaluate Treg cell responses. In addition to CpG and VD3 duo, various adjuvants in place of either CpG or VD3 in separate μEPIT-treated groups were evaluated to determine the most effective μEPIT against IgE-mediated allergy. μEPIT was repeated three times each at an interval of one week, while eight SCIT treatments were given with 0.5, 1, 2, 4, 8, 16, 32, or 50 μg OVA in 50 μ1 for every three days over the course of three weeks. Control mice were treated with the same patch containing OVA, CpG and VD3 in sham light-treated skin. One week after the final treatment, the mice were challenged three times by intranasal (i.n.) administration of 50 μ1 1% OVA in PBS. After 2h of final intranasal challenge, the mice were sacrificed and analyzed for IgE-mediated allergy.
2.5. Assessment of OVA-specific antibodies
Mouse blood of 50 μ1 was collected from tail-vein at different times after final sensitization, treatment, and challenge. OVA-specific antibodies (IgE, IgG, IgGl and IgG2a) were quantified by ELISA. In brief, 10 μg OVA per ml in bicarbonate buffer, pH 9.6 was added to each well of the 96-well ELISA plate (Coming Life sciences, NY, USA) and incubated overnight at 4°C. After washing, mouse serum was gradient diluted and incubated for 2 hours at room temperature. HRP-labeled anti-mouse secondary antibodies against IgE, IgG, IgGl or IgG2a (all from Southern Biotech, AL, USA) were then added to each well and incubated for an hour. The assay was developed by o-phenylenediamine dihydrochloride-H2O2 substrate solution (Sigma-Aldrich) followed by measurement of the colored reaction at 490 nm using microplate ELISA reader (SpectraMax®, Molecular devices, CA, USA).
2.6. Allergic responses in bronchoalveolar lavages and airway wall thickness
Bronchoalveolar lavage fluid (BALF) was collected as described [17]. In brief, 1ml saline was instilled by syringe into lungs and sucked and the in-out procedure was repeated three times to collect BALF. The resultant BALF fluid was centrifuged at 100 × g for 10 min at 4°C. The cell pellet was suspended in saline for counting eosinophils and neutrophils under a microscopy after Wright staining. The cells were counted from twenty randomly selected fields in a sample-blind fashion and expressed in 104 cells per mL of BALF fluid. Lung tissues were fixed in 10% phosphate buffer/formalin, embedded, cut at 4μm tissue sections and stained with H&E and Wright-Giemsa. The sections were analyzed by nanozoomer slide scanner (Hamamatsu photonics, Japan). Airway wall thickness and the infiltration of eosinophils and neutrophils in the lung tissue sections were determined in twenty randomly selected views from each group by using nanozoomer software (Hamamatsu photonics, Japan).
2.7. OVA-specific Treg cells
To determine OVA-specific Treg cells, spleen was pressed through a 70 μm cell strainer (BD Biosciences, CA, USA) to prepare single cell suspension followed by treatment with ammonium- chloride-potassium lysing buffer to remove red blood cells. Lymphocytes were then stimulated with 0.1 mg/ml OVA for 16 hours at 37 °C with 1 μg/ml Golgin-plug (eBioscience, CA, USA) in the culture for the final 5 hr of incubation. The stimulated cells were then harvested and stained for surface markers with anti-mouse CD4 (GK 1.5) and CD25 (PC 61) antibodies (Biolegend, CA, USA). After surface marker staining, the cells were fixed, permeabilized (permeabilization buffer, eBioscience), and stained with PE anti-mouse FoxP3 antibody (MF 14; Biolegend). The stained cells were quantified on FACS Aria (BD Biosciences) and the results were analyzed by FlowJo software (version 7.6.5; OR, USA).
2.8. qRT-PCR
Total RNA was extracted from mouse skin at the site of antigen/adjuvant delivery using TRIzol® reagent (Thermo Fischer Scientific Inc.) and reverse transcribed into cDNA by TaqMans reverse transcriptase (Applied Biosystems, NY, USA). qRT-PCR was performed with the Fast Start DNA Master SYBR Green I and LightCycler 1.5 (Roche Diagnostics, IN, USA) in accordance to the manufacturer’s protocol. Oligonucleotide primers were forward, TGACGTCACTGGAGTTGTACGG and reverse, GGTTCATGTCATGGATGGTGC for TGF- β; forward, GGAAGCACGGCAGCAGAATA and reverse, AACTT GAGGGAGAAGTAGGAATGG for IL-12; forward, ACAGGAGAAGGGACGCCAT and reverse, GAAGCCCTACAGACGAGCTCA for IL-4; forward, GCTCTTACTGACTGGCATGAG and reverse, CGCAGCTCTAGGAGCATGTG for LL-10, and forward, GGTCACAGCCAGTCCTCTTAC and reverse, TCAAAGTGCCAGTGAACCCC IL-18. The primers were synthesized by MGH DNA core facility. The expression levels of target gene mRNA was defined relative to the expression of the reference gene GAPDH.
2.9. Statistics
One-way ANOVA followed by Turkey’s multiple comparison tests was used to analyze the differences among multiple groups. The significance of differences between two individual groups was analyzed by Mann-Whitney U t-test. All statistical analyses were performed using Prism GraphPad 5.0 (CA, USA). P value less than 0.05 was considered significant.
Results
3.1. Microfractional delivery of powdered allergens into the epidermis
We recently reported that microfractional delivery of powdered antigens via laser- microperforated skin greatly enhanced efficacy of epicutaneous vaccine-adjuvant delivery without incurring any skin lesion [12]. To extend this approach to EPIT, an array of 75 micropores per cm2 each in a size of~50–75 pm in base diameter and 20–30 pm in depth were generated in the skin using a laser device named P.L.E.A.S.E at two laser pulses and fluence of 2.8 J per cm2 (Figure 1A). Meanwhile, a patch was coated with powdered ovalbumin (OVA) in a pattern mirroring the array of the micropores in the skin (Figure 1B) as previously described. The patch was topically applied onto laser-treated skin in precise alignment with the micropores in the skin (Figure 1C). Most of OVA dots disappeared from the patch with only trace resident OVA on the patch after 2 hr application on the skin (Figure 1B, right), because OVA entered the skin sufficiently. OVA entry of these micropores was corroborated using fluorescently labeled OVA that filled each micropore fully after microfractional epicutaneous (μEP) delivery (Figure 1C, right). Interestingly, on a high magnification, OVA-alexa647 was found to be diffused predominantly into the epidermis via the top circumference of each cylindrical micropore (Fig. 1D, left). There was little OVA-fluorescence in the center or at the bottom of each micropore seen in a 3D image of the micropore, clearly suggesting primarily epidermic, rather than dermic, penetration of OVA-alexa647 (Fig. 1D, right), even though the micropore channeled into the dermis.
Figure 1.
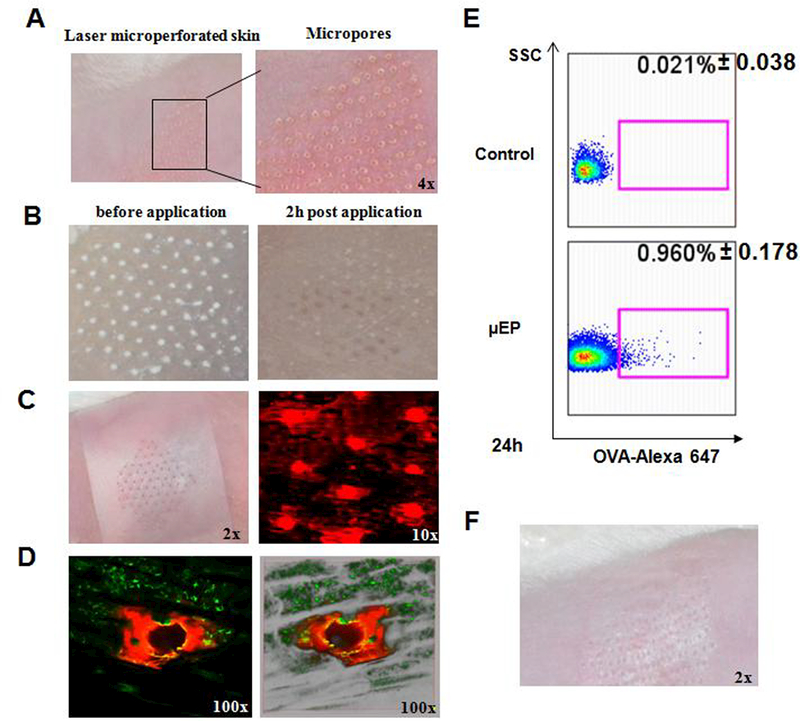
μEP delivery and skin reaction. A. Representative photographs of mouse skin after treatment with P.L.E.A.S.E laser device. B. Powdered OVA microarray patch before (left) and after (right) applied to laser microperforated skin for 2 hr. C. Topical application of powdered OVA microarray patch onto laser-treated skin (left). Microfractional epicutaneous (μEP) delivery of OVA-alex647 into the micropores generated by laser treatment (right). n=4. D. Enlarged microscopic (left) and 3D (right) images of one representative micropore. Note: OVA-Alexa- 647 is diffused into the epidermis via the top circumference of the micropore. E. Representative flow cytometry plots show cells carrying OVA-alex647 in the inguinal lymph node at 24h after μEP delivery compared to controls. n=4. F. A representative photograph captured after 72h of patch application displays little skin reaction. Magnifications (x) are indicated in individual panels.
The high efficacy delivery resulted in ~1% of cells carrying OVA-alexa647 in the draining inguinal lymph node of the mice, as measured by flow cytometry after 24h of patch application, which represented an 45-fold increase compared to controls in which the similar OVA patch was applied onto sham light-treated skin (Figure 1E). When the antigen was delivered in a microfractional fashion, we observed no inflammation or any adverse events in the skin during the treatment (Figure 1F). Apart from little local reaction, the microfractional delivery was much safer than subcutaneous administration in terms of systemic reaction. As shown in Figure 2, when FITC was administered by the μEP, intradermal (ID) or subcutaneous (SC) route, FITC entered the circulation via SC injection at 5~6 times greater than ID or μEP administration. The powdered form also significantly delayed OVA entrance of the circulation by hrs as compared to ID injection of OVA solution. Delayed and minimal entry of allergens into the circulation warrants the safety of this microfractional EPIT or μEPIT, because allergen entrance of the circulation is a prerequisite for triggering anaphylaxis.
Figure 2.
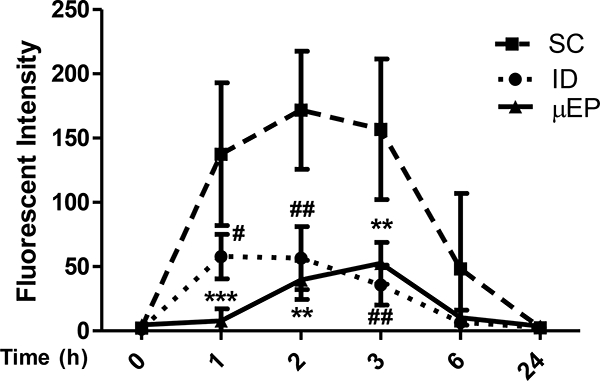
Reduced leakage of FITC administered via μEP or ID as compared to the SC route. Fluorescent intensity of FITC was measured in blood at indicated times following FITC administration. Data are shown as mean±SD (n=5). **p<0.001 for μEP vs SC; #p<0.05 and ##p<0.01 for ID vs SC.
3.2. Cytokine responses in the skin provoked by various adjuvants
Allergic asthma is an IgE-mediated Th2 immune response [1,2]. Adjuvants that induce Th1 and/or Treg cell immune response should bolster the efficacy of immunotherapy, shorten the period of treatment time, and reduce the number of hospital visits considerably [6,7]. To this end, we evaluated effects of various adjuvants either alone or in various combinations on cytokine production in the skin at 24 hours after ID immunization, using real-time RT-PCR. We found that CpG or VD3 alone induced little or a low amount of TGF-β, IL-10 or IL-18 in comparison with sham treatment (Figure 3, A, D and E), while CpG but not VD3 augmented IL-12 production significantly (Figure 3B). However, a combination of the two profoundly increased TGF-β expression by 4-fold and IL-10 by 1000-fold relative to CpG or VD3 alone, two primary cytokines for inducing Treg cells (Figure 3A and 3D) [2–4,18]. The duo adjuvant also robustly enhanced IL-12 and IL-18 production by 100~200-fold (Figure 3B) or 40-fold (Figure 3E) compared to sham controls, respectively, which together suppressed IL-4 and IgGl production while increasing IgG2 production as clearly demonstrated by previous investigations [15,19]. The robust increases in the production of TGF-β, IL-10, IL-12 and IL-18 were accompanied with only a small increment in IL-4 production in the skin measured in parallel (Figure. 3C). Monophosphoryl lipid A (MPL) either alone or combined with VD3 or CpG+VD3 was far lesser effective than CpG+VD3 in enhancing TGF-β production (Figure 3A). MPL alone or MPL plus CpG and VD3 did not induce IL-10 or IL-18 production either, although MPL+VD3 increased the production of these two cytokines stronger than or similarly as CpG+VD3 (Figure 3 D and E). CpG+MPL+VD3 also provoked strong IL-12 production (Figure 3 B). Other adjuvants RA (retinoic acid) and rapamycin (Rapa) were not nearly as effective as CpG combined with VD3 in our study although the two adjuvants have been reported for their anti-allergen effects in other studies [19,20]. We thus chose CpG and VD3 duo for subsequent investigations.
Figure 3.
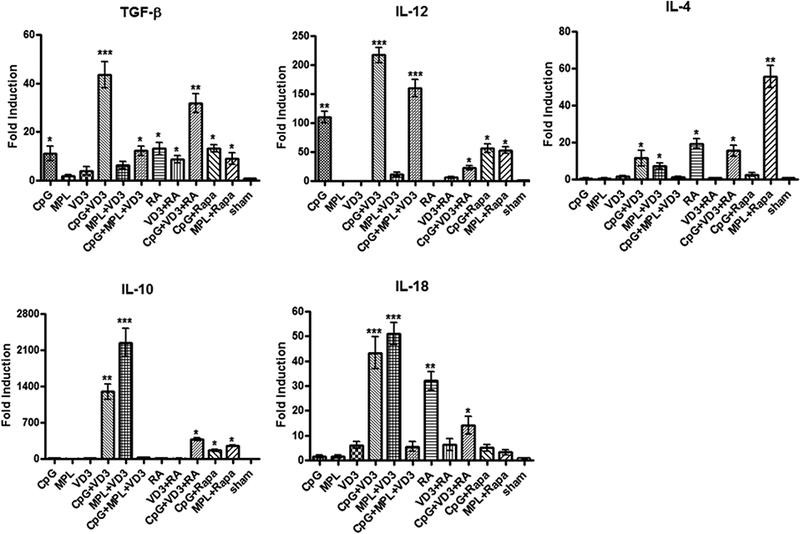
Cytokine responses induced by various adjuvants at immunization sites. Mice were ID injected with various adjuvants either alone or in various combinations. Skin tissue samples of 1 cm2 at the injection site were harvested 24h later to extract total RNA. Cytokine levels were quantified by real-time RT-PCR and are expressed as mean±SD of fold increases relative to GAPDH (n=6). *p<0.05; **p<0.01; and ***p<0.001, compared with sham treatment.
3.3. Anti-allergen immune responses induced by μEPIT
TGF-β and IL-10 are well known important to induce Treg cells [2–4,18] and thus CpG plus VD3 that stimulated high levels of TGF-β and IL-10 production in the skin were included in our subsequent studies. Strikingly, a single μEPIT treatment with OVA+CpG+VD3 elevated the proportion of CD4+CD25+ Treg cells in the spleen of OVA-sensitized mice by 7-fold compared to SCIT containing the same amount of CpG, VD3, and OVA (Figure 4C) as analyzed by Foxp3 expression (Figure 4B), confirming a high correlation between local TGF-β and IL-10 production and Treg cell development in the spleen. We subsequently treated OVA-sensitized mice by conventional SCIT using subcutaneous administration of increasing amounts of OVA alone every three days for a duration of 3 weeks with a total of 8 treatments, mimicking clinic treatment as depicted in Figure 5A. Alternatively, μEPIT containing 50 μg OVA, along with CpG, CpG+VD3 or CpG+ Rapamycin, were used to treat OVA-sensitized mice for 3 times each at an interval of one week (Fig. 5A). The sham group was treated with the same powder patch containing OVA, CpG, and VD3 applied onto sham light-treated skin. The subtypes of OVA- specific antibodies were determined in the mice two weeks after two sensitizations, 2 days after the final treatment, or 2 hr after the final sensitization, respectively. As shown in Figure 5B, after sensitizations, all mice produced similar levels of anti-OVA IgE at a titer about 1: 1500. Eight SCIT treatments failed to suppress IgE production in OVA-sensitized mice, nor did sham controls, or μEPIT with CpG alone (Figure 5A). On the contrary, 3 μEPITs with CpG + VD3 duo adjuvant resulted in a 4-fold decrease of IgE titers and the low IgE level was sustained after repeated intranasal challenges (Figure 5B). Rapamycin in place of VD3 gave rise to much less suppression on IgE response post-challenge (Fig. 5B). The anti-IgE response was correlated with the level of Treg cells in the spleen measured 2 hr after the final challenge (Figure 5F), but not with humoral immune responses in these sensitized mice (Figures C&D). In this regard, CpG alone, CpG plus rapamycin, and to a lesser degree, CpG+VD3, significantly increased the production of IgGl, a Th2 antibody, IgG2a, a Th1 antibody, or total IgG following the treatments as compared to SCIT or sham therapy and the level was not altered significantly post-challenges (Figure C&D). The result suggests that CpG + VD3 adjuvant used in μEPIT can greatly induce Treg cells and suppress OVA-specific IgE response.
Figure 4.
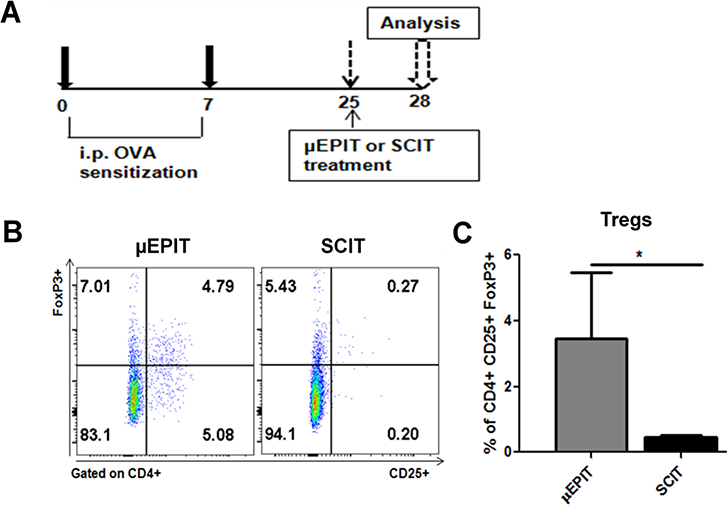
Strong induction of Treg cells by μEPIT. A. Depict of experimental design for OVA- sensitization and μEPIT. B. Representative flow cytometry profiles of FoxP3+ Treg cells in the spleen 72 hr after a single treatment of μEPIT or SCIT. C. A single microarray patch application induces FoxP3 Treg cells in mice sensitized with OVA. Flow cytometric data in B are summarized as mean percentages ±SD of CD25+Foxp3+ Treg cells on gated CD4+ cells (n=5). *p<0.05 compared to SCIT.
Figure 5.
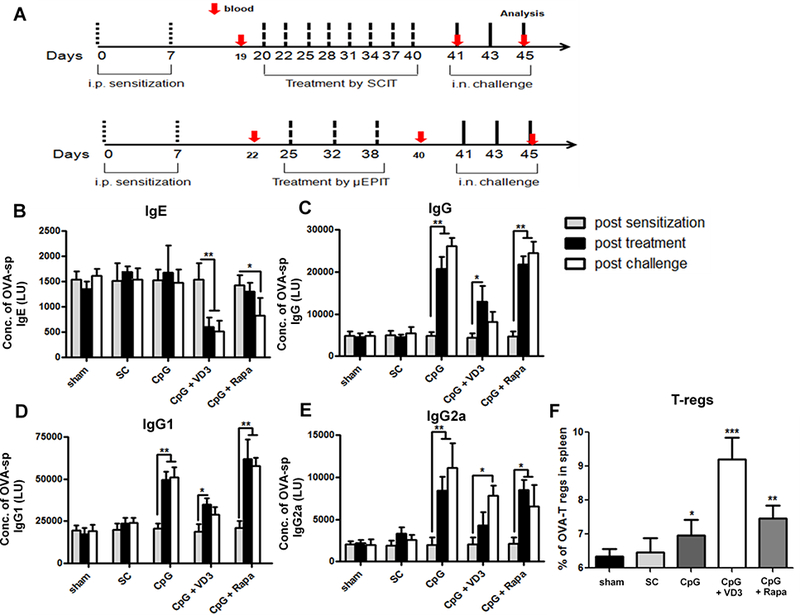
μEPIT induces Treg cells and suppresses OVA-specific IgE in AHR mice. A. Schedules of OVA sensitization, treatments, and challenges. B-E. μEPIT containing CpG and VD3 duo adjuvant suppresses allergen-specific IgE and modulates IgG, IgGl and IgG2a in AHR mice. The antibodies were measured in mouse sera collected after the final sensitization, treatment and challenge. Data are shown as mean±SD (n=8). *p<0.05, **p<0.01, individual treatment group compared between post sensitization and post treatment or post challenge. F. μEPIT induces OVA-specific Treg cells in spleen as determined by flow cytometry at the end of the experiment. *p<0.05, **p<0.01 and ***p< 0.001 for percentages of Treg cells compared to sham treatment as analyzed as Figure 4B.
3.4. μEPIT alleviates allergic responses induced by OVA challenge
After treatments, the mice were challenged three times by intranasal administering 1% OVA solution to determine immune tolerance in the mice. The mice were sacrificed 2hr after final challenge for histologic examination. Representative result of airway wall thickness of different groups were shown in Figure 6A, in which non-sensitized mice had an average airway wall thickness of 25 μm, whereas sham-treated mice showed an average thickness of about 75 μm owing to an allergic response to OVA challenge (Fig. 6A and B). The airway wall thickness was diminished slightly in mice treated with SCIT or μEPIT comprised of CpG alone, but was without statistical significance. On the contrary, the allergic response was profoundly suppressed by CpG and VD3 duo adjuvant in μEPIT, bringing about the airway morphology close to non-sensitized mice and significantly lowers than sham-treated mice (Fig. 6A and B). Under similar treatments, the airway wall thickness of CpG + rapamycin group was about 60 μm, which was significantly lower than the sham or SCIT group, but the treatment was much less sufficient when compared to CpG+VD3 group (Figure 6A and B). The result concludes that CpG+VD3 adjuvant is most effective to alleviate the allergic response against OVA compared to other adjuvant tested when delivered by μEPIT. Indeed, infiltrations of allergic eosinophils and neutrophils were reduced most and profoundly in the lung and bronchoalveolar lavage fluid (BALF) only in mice treated with CpG+VD3 duo in pEPIT (Figure 7 B&C). SCIT was totally ineffective in suppression of OVA allergic responses and pEPIT with CpG alone or CpG+rapamycin as adjuvant showed modest efficacy of anti-allergic responses. Representative histology results of cellular infiltration in different groups were shown in Figure 7A, where very few eosinophils or neutrophils were found in lung and BALF in mice receiving μEPIT with CpG+VD3 as adjuvant. On the contrary, as many as 10 times more eosinophils or neutrophils were seen in the sham and SCIT groups. The number of these inflammatory cells was significantly diminished by 30~50% in mice treated with μEPIT using CpG alone or CpG+rapamycin as adjuvant. Overall, these results were in good agreement with the airway wall thickness data (Figure 6).
Figure 6.
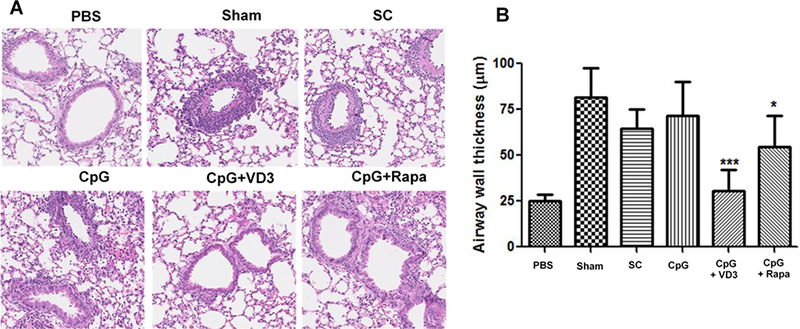
μEPIT reduces airway wall thickness. A. Representative histology shows the airway wall thickness after three challenges. PBS, non-sensitized control; Sham, sham light-treated skin; SC, subcutaneous immunotherapy; and μEPIT with OVA along with CpG, CpG+VD3 or CpG+rapamycin (the lower panel). B. Reduction of airway wall thickness by μEPIT comprised of CpG and VD3 duo adjuvant. The measurement of airway wall was performed from twenty randomly selected views from each group using nanozoomer software and data are shown as mean±SD (n=8). *p<0.05, **p<0.01 and ***p<0.001 compared to sham treatment.
Figure 7.
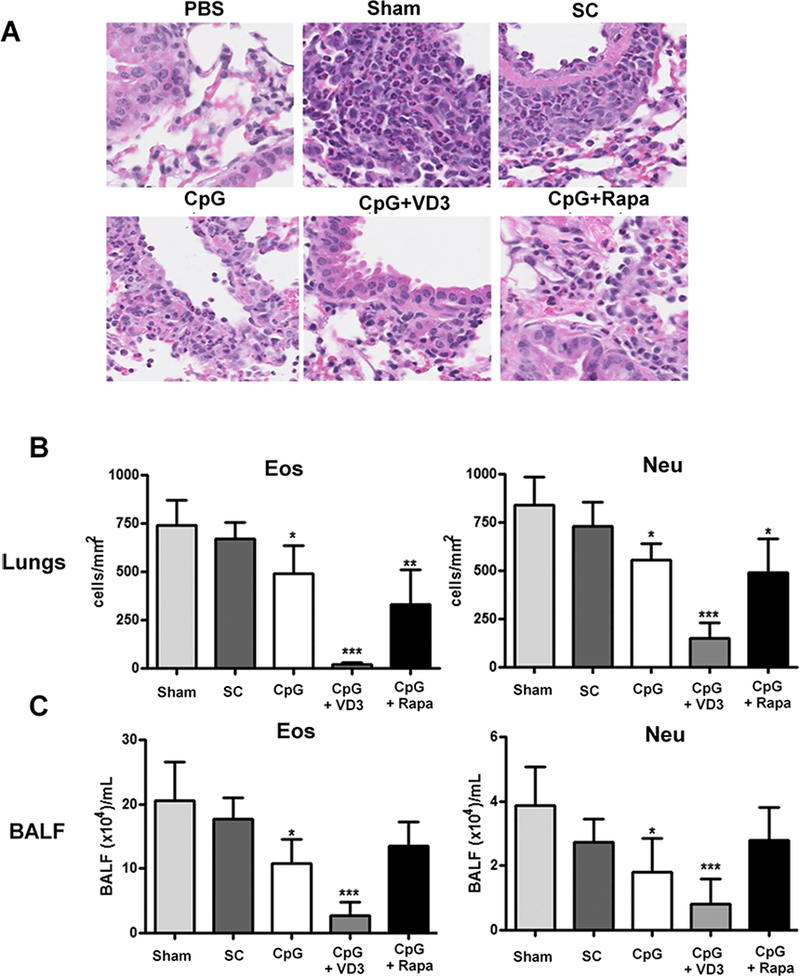
μEPIT inhibits infiltration of eosinophils and neutrophils into the lung and bronchoalveolar lavage fluid (BALF). A. Representative microscopic images from indicated groups show infiltration of eosinophils and neutrophils into the lung. B. Analysis of eosinophils and neutrophils in lung and BALF. Cells were counted and analyzed from twenty randomly selected views in each group using nanozoomer software and data are shown as mean±SD (n=8). *p<0.05, **p<0.01 and ***p<0.001 compared to sham treatment as above.
4. Discussion
We introduce here a novel terminology μEPIT that involves delivery of powdered allergen and adjuvant into many micropores in the epidermis for safer and more effective immunotherapy of IgE-mediated allergy. Although AFL was used to generate 75 pores/cm2in the skin each at a size of 50–75 μm in base diameter and 20–30 μm in depth in the study, the powdered allergen appeared to be primarily delivered into the epidermis via the top circumference of individual micropores. This is not surprising in that the powdered allergen must be first dissolved in situ by interstitial fluid at the contact between the epidermic layer and the powder as depicted in the graphical abstract. The powder allergen is then diffused horizontally into the epidermis along a descent concentration gradient, followed by vertical distribution from the epidermis into the dermis, rather than directly enters the micropores as previously thought [12,16]. Each of these micropores is so small that it can heal within days by fast-growing epithelial cells surrounding each micropore, provided that the distances between any micropores are sufficient [21]. The quick healing favors Th1 immune responses because a Th1 immune response is prominent at the early phase of skin repair, in general, followed by a TH2-skewed immune response in the late phase of the skin repair [23]. Hence, micro-fractional delivery of allergens not only minimizes skin lesion, but also averts Th2 immune response at the immunization site.
One of the key differences between this μEPIT and current microneedle-based epidermic delivery is that powdered allergen and adjuvant, rather than liquid forms of these molecules, are employed for EPIT, which presents several advantages. Firstly, powdered antigens are more stable against degradation and chemical modification. The powder patch can be readily stored, transported, and used directly without a need of reconstitution, thereby eliminating programmatic errors in association with reconstitution either in wrong diluents or injection in wrong syringes. Secondly, unlike dissolvable microneedle arrays, no any additives, excipients, or specific formulations are required to preserve the immunogenicity of the allergen and adjuvant. Today, high quality allergen extracts are lyophilized and transported for specific immunotherapy needs in clinics. Various technologies are also available to transform liquid immunotherapeutic agents into dry forms, like freeze-drying, spray-drying, and spray-freeze-drying. The powder patch is readily fabricated, requiring no complex manufacturing and can be conveniently expanded into large-scale Good Manufacturing Practice (GMP) production. Thirdly, powdered allergen, after being dissolved in situ by interstitial fluids, entered the epidermis and the dermis, by which the allergen constantly stimulated immune system for days [21], simulating multiple doses of immunotherapy in favor of immune tolerance, while profoundly diminishing the risk of anaphylaxis. As shown in Figure 2, in contrast to SC, μEP gives rise to a minimal penetration of FITC that is a small molecule, into the circulation compared to subcutaneous injection. It is also an hrs’ delay relatively to ID injection. It may be worthwhile to point out that allergens are usually much larger than FITC and the leakage could be further reduced. Finally, the μEPIT is needle-free, painless, and can be potentially performed at home, which can greatly improve patients’ compliance.
Allergy is an IgE-mediated Th2 type immune response. Adjuvants inducing Tregs or TH1 immune response are effective in treating IgE-mediated allergic diseases in humans and mice [6,7,15,24]. Therefore, we examined whether different adjuvants either alone or in various combinations, could enhance immune-modulation locally and systemically. These are small molecules with well-known chemical structures and mechanism underlying its adjuvanicity. Among the adjuvants tested, our investigation demonstrated that a combination of CpG and VD3 vigorously increased the production of immune-modulating cytokines like TGF-β and IL-10 as well as THl-type cytokines like IL-12 locally, concomitant with a high amount of Treg cells produced in the spleen (Fig. 3A and D). The effect of VD3 (calcipotriol) may be ascribed to its ability to induce immune tolerogenic dendritic cells and Treg cells [25,26]. In addition, it also inhibits activated antigen-specific T cell response and IgE expression in B-cells [26,27]. CpG as an adjuvant showed potential benefit to alleviate allergy symptoms by inducing TH1 response [7,15,23]. The finding that VD3 and CpG can greatly induce a high level of Treg cells and TH1 immune response is in good agreement with the well-described effects of VD3 and CpG in induction of Treg cells and TH1 immune response [13–15,24,29,30].
To date, SCIT is widely considered as a ‘gold standard’ protocol compared to SLIT for treating IgE-mediated allergies, but patients experience significant systemic and local side effects during the therapy, apart from dozens of hospital visits during a long period of time [30–32]. Strikingly, a single μEPIT increased the frequency of Foxp3 Treg cells by 7-fold over SCIT. Three μEPIT treatments for a duration of two weeks showed much better outcomes than eight SCIT treatments over three weeks (Figure 5–7), evidenced by substantial reduction of allergen-specific IgE, airway wall thickness, and infiltration of inflammatory eosinophils and neutrophils in the respiratory system in AHR mice. In addition, IgG2a response against OVA was significantly enhanced suggestive of a TH1-dominated response. Our findings confirm and extend previous observations showing that EPIT promotes long-term immune tolerance by inducing allergen-specific Tregs in both humans and mice [15,24,31–33], suggesting that μEPIT is safer, faster and more effective in treating IgE-mediated allergy than SCIT.
In summary, we have fabricated a powdered allergen microarray patch that gave rise to efficient epidermic delivery of allergens when applied onto laser-micropeforated skin. The powdered allergens applied onto the skin are largely restricted within the epidermis for a prolonged period of time as compared to subcutaneous injection, thereby preventing anaphylaxis. Moreover, this needle-free μEPIT greatly improves the efficacy of immunotherapy of IgE-mediated type-1 allergy with minimal or no pain, and can potentially reduce the number of hospital visits, shorten the treatment period, and increase patients’ compliance.
5. Conclusion
The present investigation shows effectiveness of μEPIT to treat IgE-mediated allergy and to reduce the number of immunotherapy from eight times to 3 times and a duration from three weeks to two weeks, yet with significantly better outcome. The μEPIT enhances Treg cells against administered allergens and suppresses IgE response, whereas provoking little local reaction at the site of patch application and greatly minimizing the risk of anaphylaxis when compared to SCIT. The μEPIT shows great promise to treat IgE-mediated type-1 allergy and merits further investigation in larger animals or humans.
Acknowledgement
We thank the Pantec Biosolutions AG, Liechtenstein for the gift of P L E A S E. Laser device and the Photopathology Core, Wellman center for their excellent technical support in obtaining tissue sections and flow cytometry analyses. This work is supported by the National Institute of Health grants AIl 13458–01 and AI089779 and department fund to M.X.W.
Footnotes
Conflict of Interest
Authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.National Institute of Allergy and Infectious Disease. Asthma. http://www.niaid.nih.gov/topics/asthma/Pages/default.aspx. (updated October 1 2015) [Google Scholar]
- 2.Akdis CA, Akdis M, Mechanisms of allergen-specific immunotherapy and immune tolerance to allergens, World Allergy Organ. J. 8 (2015) 17–015-0063–2. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casale TB, Stokes JR, Immunotherapy: what lies beyond, J. Allergy Clin. Immunol. 133 (2014) 612–9: quiz 620. [DOI] [PubMed] [Google Scholar]
- 4.Jutel M, Agache I, Bonini S, Burks AW, Calderon M, Canonica W, Cox L, Demoly P, Frew AJ, O’Hehir R, Kleine-Tebbe J, Muraro A, Lack G, Larenas D, Levin M, Nelson H, Pawankar R, Pfaar O, van Ree R, Sampson H, Du Toit G, Werfel T, Gerth van Wijk R, Zhang L, Akdis CA, International consensus on allergy immunotherapy, J. Allergy Clin. Immunol. (2015). [DOI] [PubMed] [Google Scholar]
- 5.Senti G, Graf N, Haug S, Ruedi N, von Moos S, Sonderegger T, et al. Epicutaneous allergen administration as a novel method of allergen-specific immunotherapy, J Allergy Clin Immunol, 124 (2009)997–1002. [DOI] [PubMed] [Google Scholar]
- 6.Senti G, von Moos S, Kündig TM. Epicutaneous Immunotherapy for Aeroallergen and Food Allergy. Curr Treat Options Allergy, 1 (2013) 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer RN, Manohar M, Singh AM, Jay DC, Nadeau KC, The future of biologics: applications for food allergy, J. Allergy Clin. Immunol. 135 (2015) 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price D, Harrow B, Small M, Pike J, Higgins V. Establishing the relationship of inhaler satisfaction, treatment adherence, and patient outcomes: a prospective, real-world, cross-sectional survey of US adult asthma patients and physicians. World Allergy Organ J. 8 (2015) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupont C, Kalach N, Soulaines P, Legoue-Morillon S, Piloquet H, Benhamou PH, Cow’s milk epicutaneous immunotherapy in children: a pilot trial of safety, acceptability, and impact on allergic reactivity, J. Allergy Clin. Immunol. 125 (2010) 1165–1167. [DOI] [PubMed] [Google Scholar]
- 10.Senti G, von Moos S, Tay F, Graf N, Sonderegger T, Johansen P, et al. Epicutaneous allergen- specific immunotherapy ameliorates grass pollen-induced rhinoconjunctivitis: a double-blind, placebo- controlled dose escalation study, J Allergy Clin Immunol. 129 (2012) 128–135 [DOI] [PubMed] [Google Scholar]
- 11.Senti G, von Moos S, Tay F, Graf N, Johansen P, Kündig TM. Determinants of efficacy and safety in epicutaneous allergen immunotherapy: summary of three clinical trials, Allergy. 70 (2015):707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Kositratna G, Zhou C, Manstein D, Wu MX. Micro-fractional epidermal powder delivery for improved skin vaccination, J Control Release. 192 (2014) 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss R, Hessenberger M, Kitzmüller S, Bach D, Weinberger EE, Krautgärtner WD, Hauser- Kronberger C, Malissen B, Boehler C, Kalia YN, Thalhamer J, Scheiblhofer S. Transcutaneous vaccination via laser microporation, J Control Release. 162 (2012) 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bach D, Weiss R, Hessenberger M, Kitzmueller S, Weinberger EE, Krautgartner WD, Hauser- Kronberger C, Boehler C, Thalhamer J, Scheiblhofer S. Transcutaneous immunotherapy via laser-generated micropores efficiently alleviates allergic asthma in Phi p 5-sensitized mice, Allergy. 67 (2012) 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hessenberger M, Weiss R, Weinberger EE, Boehler C, Thalhamer J, Scheiblhofer S. Transcutaneous delivery of CpG-adjuvanted allergen via laser-generated micropores. Expert Opin Drug Deliv. 10 (2013) 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachhav YG, Summer S, Heinrich A, Bragagna T, Böhler C, Kalia YN. Effect of controlled laser microporation on drug transport kinetics into and across the skin. J Control Release. 2010. August 17;146(l):31–36. [DOI] [PubMed] [Google Scholar]
- 17.Maxeiner JH, Karwot R, Hausding M, Sauer KA, Scholtes P, Finotto S. A method to enable the investigation of murine bronchial immune cells, their cytokines and mediators. Nat Protoc. 2007;2(1): 105–12. [DOI] [PubMed] [Google Scholar]
- 18.Akdis Cezmi A., Akdis Mübeccel. Mechanisms of immune tolerance to allergens: role of IL-10 and Tregs, J Clin Invest. 124 (2014) 4678–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goswami S, Angkasekwinai P, Shan M, Greenlee KJ, Barranco WT, olikepahad S et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009. May; 10(5):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mushaben EM, Kramer EL, Brandt EB, Khurana Hershey GK, Le Cras TD. Rapamycin attenuates airway hyperreactivity, goblet cells, and IgE in experimental allergic asthma. J Immunol. 2011. December 1; 187(11): 5756–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Li B, Wu MX. Effective and lesion-free cutaneous influenza vaccination. Proc Natl Acad Sci USA. 2015. April 21;112(16):5005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YC, Park JH, Prausnitz MR (2012) Microneedles for drug and vaccine delivery. Adv Drug Deliv Rev 64(14): 1547–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rani M, Zhang Q, Schwacha MG (2014) Burn wound γδ T-cells support a Th2 and Thl7 immune response. J Burn Care Res 35(l):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011. April; 10(4):499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolic T, Roep BO. Regulatory multitasking of tolerogenic dendritic cells - lessons taken from vitamin d3-treated tolerogenic dendritic cells. Front Immunol. 2013. 14;4:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang SW1, Kim SH, Lee N, Lee WW, Hwang KA, Shin MS, Lee SH, Kim WU, Kang I 1,25- Dihyroxyvitamin D3 Promotes FOXP3 Expression via Binding to Vitamin D Response Elements in Its Conserved Noncoding Sequence Region. J Immunol. 2012. June l;188(ll):5276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heine G, Tabeling C, Hartmann B, González Calera CR, Kuhl AA, Lindner J, Radbruch A, itzenrath M, Worm M. 25-hydroxvitamin D3 promotes the long-term effect of specific immunotherapy in a murine allergy model. J Immunol. 2014. August 1; 193(3): 1017–23. [DOI] [PubMed] [Google Scholar]
- 28.Milovanovic M, Heine G, Hallatschek W, Opitz B, Radbruch A, Worm M. Vitamin D receptor binds to the ε germline gene promoter and exhibits transrepressive activity. J Allergy Clin Immunol. 2010. November;126(5): 1016–23, 1023.el-4. [DOI] [PubMed] [Google Scholar]
- 29.Frew AJ. Allergen immunotherapy. J Allergy Clin Immunol. 2010. February;125(2 Suppl 2):S306–13. [DOI] [PubMed] [Google Scholar]
- 30.Cox L, Nelson H, Lockey R, Calabria C, Chacko T, Finegold I, Nelson M, Weber R, Bernstein DI, Blessing-Moore J, Khan DA, Lang DM, Nicklas RA, Oppenheimer J, Portnoy JM, Randolph C, Schuller DE, Spector SL, Tilles S, Wallace D. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol. 2011. January;127(1 Suppl):S1–55. [DOI] [PubMed] [Google Scholar]
- 31.von Moos S, Johansen P, Tay F, Graf N, Kundig TM, Senti G, Comparing safety of abrasion and tape-stripping as skin preparation in allergen-specific epicutaneous immunotherapy, J. Allergy Clin. Immunol 134 (2014) 965–7.e4. [DOI] [PubMed] [Google Scholar]
- 32.Mondoulet L, Dioszeghy V, Puteaux E, Ligouis M, Dhelft V, Plaquet C, Dupont C, Benhamou PH, Specific epicutaneous immunotherapy prevents sensitization to new allergens in a murine model, J. Allergy Clin. Immunol 135 (2015) 1546–57.e4. [DOI] [PubMed] [Google Scholar]
- 33.Radulovic S, Jacobson MR, Durham SR, Nouri-Aria KT. Grass pollen immunotherapy induces Foxp3-expressing CD4+ CD25+ cells in the nasal mucosa. J Allergy Clin Immunol. 2008. June;121(6):1467–72, 1472.e1 [DOI] [PubMed] [Google Scholar]