

Abstract
Background
The HER3 receptor functions as a major cause of drug resistance in cancer treatment. It is believed that therapeutic targeting of HER3 is required to improve patient outcomes. It is not clear whether a novel strategy with two functional cooperative miRNAs would effectively inhibit erbB3 expression and potentiate the anti-proliferative/anti-survival effects of a HER2-targeted therapy (trastuzumab) and chemotherapy (paclitaxel) on HER2-overexpressing breast cancer cells.
Results
Combination of miR-125a and miR-205, as compared to either miRNA alone, potently inhibited expression of HER3 in HER2-overexpressing breast cancer BT474 cells. Co-expression of the two miRNAs not only reduced the levels of phosphorylated erbB3 (P-erbB3), Akt (P-Akt), and Src (P-Src), it also inhibited cell proliferation and increased cells at G1 phase. A multi-miRNA lentiviral vector - the cluster of miR-125a and miR-205 - was constructed to simultaneously express the two miRNAs in HER2-overexpressing breast cancer cells. Concurrent expression of miR-125a and miR-205 via the miRNA cluster transfection significantly enhanced trastuzumab-mediated growth inhibition and cell cycle G1 arrest in BT474 cells and markedly increased paclitaxel-induced apoptosis in another HER2-overexpressing breast cancer cell line HCC1954.
Conclusions
Here, we showed that functional cooperative miRNAs effectively suppressed erbB3 expression. This novel approach targeting of HER3 was able to enhance the therapeutic efficacy of trastuzumab and paclitaxel against HER2-overexpressing breast cancer.
Electronic supplementary material
The online version of this article (10.1186/s12575-018-0081-x) contains supplementary material, which is available to authorized users.
Keywords: HER3, miRNA-replacement therapy, Functional cooperation, HER2, Breast cancer
Background
The HER receptor tyrosine kinase (RTK) family, including the epidermal growth factor receptor (EGFR, also known as HER1/erbB1), HER2 (erbB2/neu), HER3 (erbB3), and HER4 (erbB4), is arguably the most important receptor family in the context of development and tumorigenesis [1, 2]. The HER3 receptor has no or much lower intrinsic kinase activity [3, 4]. It frequently co-expresses and interacts with another RTK in cancer cells to activate oncogenic signaling, such as PI-3 K/Akt pathway, MEK/MAPK pathway, and Src kinase [3, 5, 6]. Recent studies have identified oncogenic erbB3 gene mutations in colon and gastric cancers [7]; however, overexpression without gene alteration is still the major mechanism for HER3 to be associated with a worse survival in patients with a wide variety of solid tumors [8]. Indeed, elevated expression of HER3 has been shown to play a pivotal role in the development of HER2-overexpressing breast cancer [9, 10], castration-resistant prostate cancer (CRPC) [11], and ovarian cancer [12, 13]. Studies on the underlying mechanisms indicate that one of the major functions of HER3 signaling is to cause treatment failure in human cancers [14–16]. Especially in breast cancer, HER3 serves as a vital co-receptor of HER2, and its expression is a rate-limiting factor for HER2-induced breast cancer cell survival, proliferation, and progression [9, 10, 15]. We have shown that elevated expression of HER3 renders HER2-overexpressing breast cancer cells resistant to tamoxifen [17], HER2-targeted therapy (trastuzumab/Herceptin and lapatinib) [18, 19], and the chemotherapeutic agent paclitaxel [20]. It is believed that inhibition of HER3 signaling is required to overcome drug resistance and effectively treat the breast cancer patients with HER2-overexpressing tumors.
Although both HER2 and HER3 receptors play pivotal roles in breast tumorigenesis, only HER2-targeted therapy has been clinically used in the treatment of HER2-overexpressing breast cancer. To date, no HER3-targeted therapy has been approved for cancer treatment. Because of its lack of or low kinase activity [3, 4], targeting of HER3 with a blocking antibody (Ab) is the only strategy under preclinical studies [21, 22] and clinical investigations. We have shown that the fully human anti-HER3 monoclonal Ab MM-121 (Merrimack Pharmaceuticals, Inc., Cambridge, MA, USA), inhibiting ligand-dependent activation of HER3 [21, 22], is able to abrogate drug resistance and significantly enhance the antitumor activity of trastuzumab and paclitaxel against HER2-overexpressing breast cancer in vitro and in vivo [16, 23, 24]. Moreover, our recent findings support the notion that inhibition/downregulation of HER3 could be achieved by the class I HDAC inhibitor (HDACi) entinostat (or SNDX-275, MS-275) or the functional cooperative miRNAs [25–27]. While the mechanism of action of the miRNAs is different from the anti-HER3 blocking Abs, this novel approach aims to reduce the protein levels of HER3 rather than just inhibit its signaling, which may eliminate the chance for tumor cells to develop resistance after initial response.
In the current study, we have focused on studying the inhibitory effect of co-expression of miR-125a and miR-205 on erbB3 expression. We have also investigated whether the newly identified miRNA-based strategy, targeting of erbB3 with two functional cooperative miRNAs, can significantly enhance the anti-proliferative/anti-survival effects of trastuzumab and paclitaxel on HER2-overexpressing breast cancer cells.
Methods
Reagents and Antibodies
The restriction endonucleases XbaI and NotI and T4 DNA ligase were purchased from New England Biolabs, Inc. (Ipswich, MA, USA). Paclitaxel was from LC Laboratories (Woburn, MA, USA). Trastuzumab (Herceptin) and Ado-trastuzumab emtansine (T-DM1, also known as Kadcyla) were obtained from University of Colorado Hospital pharmacy. Antibodies used for western blot assays were as follows: erbB3 (Ab7) (LabVision Corp. Fremont, CA, USA); P-erbB3, caspase-8 (1C12), and caspase-3 (8G10), P-MAPK, MAPK, P-Akt (S473), Akt, P-Src(Y416), Src, PARP (Cell Signaling Technology, Inc., Beverly, MA, USA); E2F1, Cyclin D1, p27kip1 (Santa Cruz Biotechnology, Inc. Dallas, TX, USA); and β-actin (Sigma Co., St. Louis, MO, USA). All other reagents were purchased from Sigma Co. unless otherwise specified.
Cells and Cell Culture
Human breast cancer cell lines SKBR3, BT474 and HCC1954 were obtained from the American Type Culture Collection (Manassas, VA, USA). The identity of all cell lines was confirmed with DNA profiling by our Cancer Center’s DNA Sequencing & Analysis Core facility. Cell lines were free of mycoplasma contamination, determined by the MycoAlert™ Mycoplasma Detection Kit (Lonza Group Ltd., Basel, Switzerland) every 3 months. All cell lines were maintained in DMEM/F-12 (1:1) medium containing 10% FBS, and cultured in a 37 °C humidified atmosphere containing 95% air and 5% CO2 and split twice a week.
Lentiviral Vectors, Virus Production and Cell Infection
Lentiviral vector pCDH-CMV-MCS-EF1-Puro and pCDH-miR-125b were kindly provided by Dr. Paul Jedlicka (Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, CO, USA). Other miRNA expression vector for miR-125a or miR-205 was constructed by cloning the miRNA precursor into the multiple cloning site of pCDH-CMV-MCS-EF1-Puro. The primer sequences used to amplify the miRNA precursors by PCR were the following:
miR-125a primers
5′---GCTCTAGATGCTGTGTCTCTGTGGCTTC---3′
5′---ATAAGAATGCGGCCGCGAGGCGCTCAGAGTAGGTTG---3′
miR-205 primers
5′---GCTCTAGATATCTGGGTGGCTGTTTTGA---3′
5′---ATAAGAATGCGGCCGCGAGGCTTTTCAGTAGACAAGCAA--3′
Multi-miRNA expression Lentivector (cluster) was obtained from SBI system Bioscience LLC. (Palo Alto, CA, USA).
The lentiviral vector and lentivirus packaging plasmids pCMV-VSVG and pCMV-ΔA.9 were co-transfected into HEK293T cells with FuGene 6 (Roche Diagnostics Corp., Indianapolis, IN, USA). After 24 h, the culture media were replaced with fresh medium. The virus in conditioned medium were harvested in 3 consecutive days and filtered with low-protein binding filters (Millex-HV, 0.45-mm polyvinylidene difluoride; Millipore Corp., Billerica, MA, USA) before they were aliquoted and stored at − 80 °C freezer. Prior to infection, the lentivirus-containing media were thawed completely at room temperature, and mixed with a same amount of fresh medium containing polybrene (8 μg/ml). The culture media of the candidate breast cancer cells were then replaced with the lentivirus-containing media. After 24 h, the virus-infected cells were selected with puromycin (1 μg/ml) for 48 h, and then subjected to required experiments.
Analysis of miRNA Expression
Total RNA, including small RNA, was extracted and purified using the miRNeasy Mini Kit (QIAGEN Inc., Valencia, CA, USA) following the manufacturer’s instructions. For miRNA analysis, TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems Inc., Foster City, CA, USA) was first used to generate cDNA with the hairpin primers, which are specific to the mature miRNA and will not bind to the precursors. The expression levels of miR-125a, miR-125b, and miR-205 were then measured by real-time PCR using TaqMan MicroRNA Assays (assay ID: 002198, 000449, 000509, respectively; Applied Biosystems Inc.) according to the manufacturer’s protocol. RNU6B was used as an internal control to normalize all data using the TaqMan RNU6B Assay (assay ID: 001093; Applied Biosystems Inc.). RNU6B levels were unaffected by entinostat treatment. The relative miRNA levels were calculated using the comparative Ct method (ΔΔCt) as described previously [27].
Western Blot Analysis
Protein expression levels were determined by western blot analysis as described previously [19, 28]. Equal amounts of total cell lysates were boiled in Laemmli SDS-sample buffer, resolved by SDS-PAGE, transferred to nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA), and probed with the primary antibodies described in the figure legends. After the blots were incubated with horseradish peroxidase-labeled secondary antibody (Jackson Immuno Research Laboratories, West Grove, PA, USA), the signals were detected using the enhanced chemiluminescence reagents (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
Quantification of Apoptosis
An apoptotic ELISA kit (Roche Diagnostics Corp.) was used to quantitatively measure cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) as previously reported [19, 23]. This enzyme immunoassay was performed according to the manufacturer’s instructions.
Cell Proliferation Assay
The IncuCyte™ system (Essen BioScience, Inc., Ann Arbor, MI, USA) was used to kinetically monitor cell growth. It is an automated imaging platform providing real-time images and quantitative data generated throughout the entire cell culture process. Proliferation is successfully measured using an IncuCyte phase-only processing module as described previously [28]. Briefly, Cells were plated onto 96-well plates in 10%FBS DMEM/F12 medium or 0.5% FBS DMEM/F12 medium containing trastuzumab (20 μg/ml). The plate was placed into the IncuCyte™ system at 37 °C for 2–3 days. During this time of period, each well was repeatedly scanned and imaged at fixed time intervals (every 4 h). The data were analyzed by the IncuCyte software. Data reflects the means of three independent experiments.
Flow Cytometric Analysis of Cell Cycle
Flow cytometric analyses were performed to define cell cycle distribution for treated and untreated cells [19, 23]. Briefly, cells grown in 100-mm culture dishes were harvested and fixed with 70% ethanol. Cells were then stained for total DNA content with a solution containing 50 μg /ml propidium iodide and 100 μg/ml RNase I in PBS for 30 min at 37 °C. Cell cycle distribution was analyzed at the Flow Cytometry Core Facility of University of Colorado Cancer Center with a FAC Scan flow cytometer (BD Biosciences, San Jose, CA, USA).
Statistical Analysis
Statistical analyses of the experimental data were performed using a two-sided Student’s t test. Significance was set at the P < 0.05. All values are reported at the mean +/− SD from at least three independent experiments.
Results
Lentiviral Expression Vectors of miR-125a and miR-205 have been Established
MiRNA-replacement therapy is actively explored as a novel strategy to treat human cancers [29–33]. While majority of the studies focus on the therapeutic potential of single miRNA, a new approach combining two miRNAs has been evaluated in lung cancer cells [34]. Our current studies aim to examine the hypothesis that functional cooperative miRNAs will effectively inhibit erbB3 expression in HER2-overexpressing breast cancer cells. Thus, we decided to test our innovative idea with ectopic expression of miR-125a and/or miR-205. We utilized the lentiviral vector pCDH-CMV-MCS-EF1-Puro (pCDH) to construct the expression vectors of miR-125a and miR-205 (Fig. 1a). First, genome DNA extracted from SKBR3 cells was used as a template to amplify the precursors of miR-125a and miR-205 via the PCR primers shown in Fig. 1b. The PCR products and pCHD vector digested with the restriction endonucleases XbaI and NotI were then electrophoretically analyzed (Fig. 1c). After linked by a T4 DNA ligase, the resulting sub-clone vectors (Fig. 1d) were transformed into the DH5α bacteria. The plasmids obtained from the bacteria colonies (9 of each miRNA expression vector) were analyzed by PCR assays via the primers shown in Fig. 1b to evaluate the presence of the precursor of miR-125a or miR-205 (Fig. 1e top). Finally, one of the positive clones of miR-125a- or miR-205-expressing vector was further verified via DNA sequencing analysis (Fig. 1e bottom). Thus, the lentiviral expression vector of miR-125a (pCDH-miR-125a) or miR-205 (pCDH-miR-205) with accurate sequence has been established. The three miRNA expression vectors (pCDH-miR-125b obtained from Dr. Paul Jedlicka at our Department, pCDH-miR-125a, and pCDH-miR-205) were then used in the following experiments.
Fig. 1.

The lentiviral vector expressing miR-125a or miR-205 was constructed. a Map of the lentiviral vector pCDH-CMV-MCS-EF1-Puro. b Primers used in PCR assays to amplify the precursor of miR-125a or miR-205. The capital letters in red and blue indicate the digested sites of Xba I and Not I, respectively. c Electrophoretogram of the PCR products and the lentiviral vector digested with Xba I and Not I. d Strategy to construct the lentiviral vector with miRNA expression. e Results of PCR and DNA sequencing analysis of positive colonies
Co-Expression of Two erbB3-Targeting miRNAs Shows Functional Cooperation and Potently Inhibits erbB3 Expression in Human Breast Cancer Cells
The human HER2-overexpressing breast cancer BT474 cells were seeded onto 6-well plate for overnight. The cells were then infected with the lentivirus containing pCDH-miR-125a, pCDH-miR-125b, or pCDH-miR-205 alone or both pCDH-miR-125a and pCDH-miR-205 or both pCDH-miR-125b and pCDH-miR-205 for 48 h (Fig. 2a) or 72 h (Fig. 2b). Taqman real-time PCR analyses revealed that the expression of each miRNA was specifically increased about 2–8 folds as compared to the empty vector (pCDH) control. Although all three miRNAs (miR-125a, miR-125b, and miR-205) have been shown to directly target the 3’UTR of erbB3 mRNA [35, 36], a clear reduction of HER3 protein levels was only observed with ectopic expression of miR-205, but not miR-125a or miR-125b. Interestingly, the combinations of miR-125a and miR-205 or miR-125b and miR-205 were able to profoundly reduce HER3 levels (Fig. 2c). It appeared that co-expression of miR-125a and miR-205 exhibited a more potent inhibitory effect on HER3. Collectively, these data suggested that the expression of miR-125a, miR-125b, and/or miR-205 could be specifically increased in breast cancer cells; and two miRNAs, especially miR-125a and miR-205 showed functional cooperation to suppress erbB3 expression in HER2-overexpressing breast cancer cells.
Fig. 2.
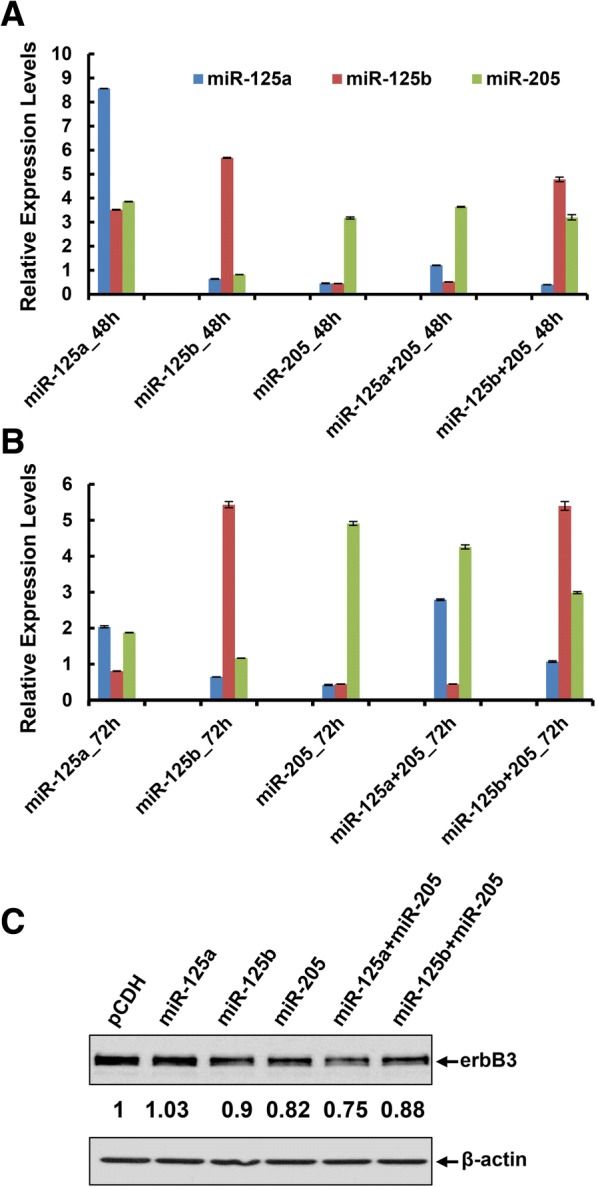
Ectopic expression of miR-125a, miR-125b, and/or miR-205 via transient transfection inhibited erbB3 expression in HER2-overexpressing breast cancer cells. BT474 cells were infected with the lentivirus containing pCDH-miR-125a, pCDH-miR-125b, or pCDH-miR-205 alone or both pCDH-miR-125a and pCDH-miR-205 or both pCDH-miR-125b and pCDH-miR-205 for 48 h (a) or 72 h (b). The expression levels of miR-125a, miR-125b, and miR-205 were measured by qRT-PCR using TaqMan miRNA assays. All results were normalized with the internal control RNU6B. Bars, SD. Data shows the representative of three independent experiments. c The expression levels of HER3 (erbB3) were detected by western blot analysis upon lentiviral infection for 72 h. β-actin was used as a loading control
Concurrent Expression of a Multi-miRNA Cluster Targeting of erbB3 Significantly Inhibits Proliferation of HER2-Overexpressing Breast Cancer Cells
We hypothesized that functional cooperative miRNAs targeting of erbB3 would be a powerful strategy to develop a miRNA-replacement therapy. However, delivering two distinct miRNAs at same time into a cancer cell may not be efficient and convenient. To better address our hypothesis, we then designed a multi-miRNA lentiviral vector simultaneously expressing both miR-125a and miR-205 and constructed the expression vector (Fig. 3a), which was designated as “miRNA cluster” in the following studies. Two HER2-overexpressing breast cancer cell lines SKBR3 and BT474 were used to test the efficacy of the miRNA cluster. The expression levels of both miR-125a and miR-205 were increased upon infection with the lentivirus containing the cluster (Fig. 3b bar graphs). Although the fold induction of miR-125a or miR-205 in cluster transfection was much less than that in single miRNA transfection, the miRNA cluster exhibited a much more potent activity than any one miRNA to downregulate HER3 in both SKBR3 and BT474 cells (Fig. 3b western blots). The data supported that two miRNAs expressed simultaneously via one vector, even at a very low expression level, could clearly inhibit erbB3 expression.
Fig. 3.
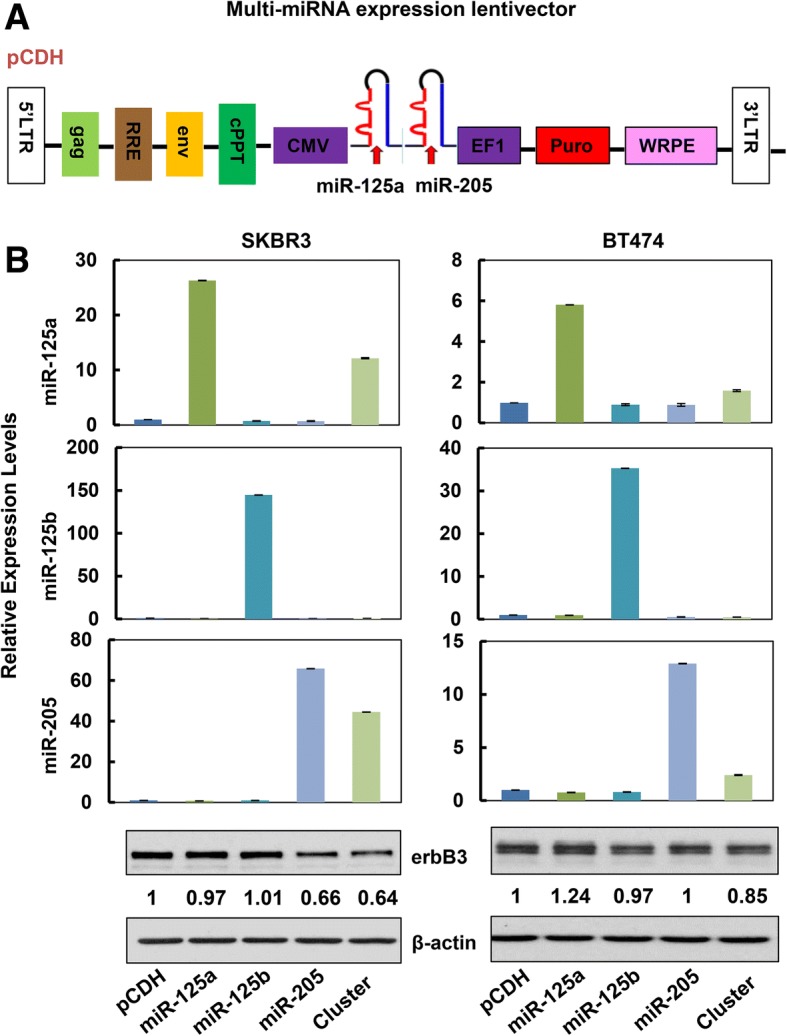
Co-expression of miR-125a and miR-205 via transient transfection with the multi-miRNA expression vector (cluster) exhibited potent activity to inhibit erbB3 expression in HER2-overexpressing breast cancer cells. a Map of the multi-miRNA lentivector expression vector (cluster). The precursors of miR-125a and miR-205 were linked together and driven by the same CMV promoter. b SKBR3 cells and BT474 cells were infected with the lentivirus containing pCDH-miR-125a, pCDH-miR-125b, or pCDH-miR-205 alone or the pCDH-miR-125a-miR-205 (cluster) for 48 h. The expression levels of miR-125a, miR-125b, and miR-205 were measured by qRT-PCR using TaqMan miRNA assays. All results were normalized with the internal control RNU6B. Bars, SD. Data shows the representative of three independent experiments. The expression levels of HER3 (erbB3) were detected by western blot analysis upon lentiviral infection for 72 h. β-actin was used as a loading control
We next examined the effects of ectopic expression of miR-125a, miR-205, or the cluster on proliferation of HER2-overexpressing breast cancer cells. The empty vector pCDH was used as a negative control. Upon infection of BT474 cells with the lentivirus containing pCDH-miR-125a, pCDH-miR-205, or pCDH-miRNA cluster, we obtained a number of cell clones from each lentiviral infection group upon puromycin selection. To avoid any potential artificial effects caused by induction of hundred- or thousand-fold of the miRNAs, we on purposely selected the cell clones that had increased expression levels of miR-125 and/or miR-205 within 10–20 folds as compared the empty vector control. Real-time PCR analyses showed that the expression of miR-125a was increased about 6-fold and the expression of miR-205 was enhanced about 16-fold in single miRNA transfected cell clones. In the cluster transfection, the expression of miR-125a and miR-205 was increased approximately 3- and 8-fold, respectively (Fig. 4a). These particular cell clones were designed as pCDH, miR125a, miR205, or cluster in the following experiments. While the miR125a and miR205 cell clones grew at a similar rate as the control (pCDH) cell clone, the cluster cell clone experienced a significant growth inhibition (Fig. 4b), although the fold induction of miR-125a and miR-205 was much less in the cluster than that in single miRNA transfected clones (Fig. 4a). Cell cycle analysis revealed that the cluster cell clone had increased percentage of cells at G1 phase and reduced cells at S phase (Fig. 4c). This might shed a light on the mechanism of growth inhibition-induced by the miRNA cluster. Furthermore, the expression of erbB3 and its phosphorylation (p-erbB3) as well as the downstream signaling kinases - phosphorylated Akt (p-Akt), MAPK (p-MAPK) and Src (p-Src) were also decreased in the cluster clone (Fig. 4d). Collectively, our data demonstrated that concomitant expression of the miRNA cluster targeting of erbB3 significantly inhibited proliferation of HER2-overexpressing breast cancer cells, likely via cell cycle G1 arrest as well as inactivation of the HER3 signaling pathways.
Fig. 4.

Concurrent expression of miR-125a and miR-205 via transfection with the miRNA cluster significantly inhibited proliferation, induced cell cycle G1 arrest, and inactivated HER3 signaling in BT474 cells. a The stable sub-clones of BT474 cells transfected with empty vector (pCDH), pCDH-miR-125a, or pCDH-miR-205 alone or pCDH-miR-125a-miR-205 (cluster) were selected. The relative expression levels of miR-125a and miR-205 were measured by qRT-PCR using TaqMan miRNA assays. In the cluster cell clone, both miR-125a and miR-205 were upregulated. b Proliferation of each cell clone was examined via analysis of the IncuCyte system. Bars, SD. Data shows the representative of three independent experiments. c Cell cycle progression was analyzed by flow cytometry. d The stable sub-clones were collected and subjected to western blot analyses with the specific antibodies directed against p-erbB3, HER3 (erbB3), p-Src, Src, p-Akt, Akt, p-MAPK, MAPK, or β-actin
The miRNA Cluster Targeting of erbB3 Enhances Anti-Proliferative/Anti-Survival Effects of Trastuzumab and Paclitaxel on HER2-Overexpressing Breast Cancer Cells
Our recent studies showed that activation of HER3 signaling and/or elevated expression of HER3 resulted in therapeutic resistance to trastuzumab and paclitaxel in HER2-overexpressing breast cancer cells [18, 20]. Thus, we first investigated whether inhibition of erbB3 expression with the miRNA cluster could enhance the anti-proliferative effects of trastuzumab on HER2-overexpressing breast cancer cells. The pCDH, miR125a, miR205, and cluster cell clones described above were treated with trastuzumab (20 μg/ml) for 24 h. Cell cycle analysis showed a remarkable G1 arrest and S phase reduction in the cluster clone than the single miRNA clone or the vector control (Fig. 5a). Cell proliferation was more significantly inhibited in the cluster clone as compared to that in miR125a or miR205 clone or the vector control clone (Fig. 5b). Western blots evaluating the molecular markers critical for G1-S transition found a clear reduction of E2F1 and a striking upregulation of p27kip1 in the cluster clone-treated by trastuzumab (Fig. 5c). Similar results were also obtained with the BT474 cell clones treated by another HER2-targeted therapy, T-DM1, i.e. the cluster cell clone compared with the single miRNA clone or the vector control was more sensitive to T-DM1-induced growth inhibition (Additional file 1: Figure S1). We next tested the antitumor activity of paclitaxel using another HER2-overexpressing breast cancer cell line HCC1954 upon infection with the lentivirus containing pCDH-miR-125a, pCDH-miR-205, or pCDH-miRNA cluster. Same process was performed to select proper cell clones of each miRNA transfection. The expression levels of miR-125a and miR-205 were upregulated by approximately 12-fold and 1.6-fold, respectively in the single miRNA transfected clones, whereas about 9-fold and 1.5-fold increase of miR-125a and miR-205, respectively was observed in the cluster-transfected clone (Fig. 6a & b). The proliferation of HCC1954 cells was significantly inhibited in the cluster cell clone as compared to the cell clones with single miRNA transfection or vector control (Fig. 6c), which was in agreement with our experimental data obtained with BT474 cells (Fig. 4b). The expression of HER3, p-HER3, and p-Akt was also decreased in the cluster cell clone, consistent with our observation in BT474 cells (Fig. 4d). The HCC1954 cell clones were then challenged by paclitaxel (10 nmol/L) for 24 h. An apoptotic ELISA revealed significantly more cells undergoing apoptosis in the cluster cell clone (Fig. 6e). Western blot assays also detected more PARP cleavage, a hallmark of apoptosis, and increased cleaved caspase-8 and caspase-3 (Fig. 6f). Taken together, our data indicated that the miRNA cluster (miR-125a and miR-205) targeting of erbB3 significantly enhanced anti-proliferative/anti-survival effects of trastuzumab and paclitaxel on HER2-overexpressing breast cancer cells.
Fig. 5.

Co-expression of miR-125a and miR-205 via cluster transfection profoundly enhanced trastuzumab-induced growth inhibition and cell cycle G1 arrest in BT474 cells. The stable sub-clones of BT474 cells treated with trastuzumab (20 μg/ml) for 48 h were collected and subjected to cell cycle analysis by flow cytometry (a) and cell proliferation (the IncuCyte system) (b). c The stable sub-clones of BT474 cells treated with trastuzumab (20 μg/ml) for 24 h were collected and subjected to western blot analysis with the specific antibodies directed against E2F1, Cyclin D1, p27kip1 (p27), or β-actin
Fig. 6.

The miRNA cluster transfection significantly increased paclitaxel-mediated cytotoxicity effects on HCC1954 breast cancer cells. a & b The stable sub-clones of HCC1954 cells transfected with empty vector (pCDH), pCDH-miR-125a, or pCDH-miR-205 alone or pCDH-miR-125a-miR-205 (cluster) were selected. The relative expression levels of miR-125a and miR-205 were measured by qRT-PCR using TaqMan miRNA assays. c Proliferation of the sub-clones was measured by analysis of the IncuCyte system. d The stable sub-clones were collected and subjected to western blot analyses of p-erbB3, HER3 (erbB3), p-Akt, Akt, or β-actin. e & f Each sub-clone treated with paclitaxel (10 nmol/L) for 24 h were collected and subjected to apoptotic-ELISA (e) or western blot analyses with specific antibodies directed against (F-PARP, full length PARP; C-PARP, cleaved PARP), caspase-8 (F-Casp-8, full length caspase-8; C-Casp-8, cleaved caspase-8), caspase-3 (F-Casp-3, full length caspase-3; C-Casp-3, cleaved caspase-3), or β-actin (f). Bars, SD. Data shows the representative of three independent experiments
Discussion
Mounting evidence implicates that the major function of the HER3 receptor in human cancers is to cause treatment failure [14–16]. Effective inhibition of HER3 is thought to be required to overcome drug resistance, enhance therapeutic efficacy, and improve outcomes of cancer patients. Among the anti-HER3 monoclonal Abs actively under preclinical investigations and clinical testing [21, 37–39], two of them - MM-121 (or seribantumab, Merrimack Pharmaceuticals, Inc.) and patritumab (Daiichi-Sankyo Co. Ltd., Tokyo, Japan) - have shown encouraging clinic benefits in patients with non-small cell lung cancer [40, 41]. Thus, it is not surprising that the U.S. Food and Drug Administration (FDA) has recently (10/30/2017) granted orphan drug designation to MM-121 for the treatment of heregulin, which is the ligand for HER3, positive non-small cell lung cancer (http://investors.merrimack.com/node/11346). In an attempt to identify novel therapeutics and approaches inhibiting HER3 receptor, we discovered that the HDACi entinostat selectively inhibited HER3 expression in HER2-overexpressing breast cancer cells [25]. Further studies revealed that entinostat induced expression of miR-125a, miR-125b, and miR-205, all of which were reported to directly target the 3’UTR of erbB3 mRNA [35, 36], and the three miRNAs acted in concert to inhibit HER3 protein translation in HER2-overexpressing breast cancer cells [27]. These exciting data support the hypothesis that effective targeting of HER3 could also be achieved by the HDACi entinostat or functional cooperative miR-125a, miR-125b, and miR-205. Because this novel approach aims to reduce HER3 protein levels, not just inhibits HER3 signaling, it has potential to eliminate the chance for tumor cells to develop resistance after initial response. To test this hypothesis, we decided to examine our innovative idea with miR-125a and miR-205, and to determine whether co-expression of the two miRNAs will exert functional cooperation to inhibit erbB3 expression in HER2-overexpressing breast cancer cells. We did not chose miR-125b in our studies, because miR-125b has been shown to behave as an oncomir under certain circumstances [42, 43]. In contrast, both miR-125a and miR-205 are consistently found to act as tumor suppressors in breast cancers [44, 45]. Indeed, our data showed that co-expression of miR-125a and miR-205 was more potent than either miRNA alone to inhibit erbB3 expression, decrease the levels of p-HER3, p-Src, p-Akt, and E2F1, and induce expression of p27kip1 in HER2-overexpressing breast cancer cells. It is worth mentioning that entinostat seems to be more powerful than our approach to repress erbB3 expression. However, the HDACi clearly shows additional side effects. In our study, since we on purposely increased expression of each miRNA at a limited amount, each single miRNA did not show much effect on erbB3 expression. However, the combination of two miRNAs exhibited a cooperative effect on inhibiting erbB3. We believe this innovative approach would be superior to entinostat in reducing the off-target effects.
MiRNA-based therapy is actively explored as a new strategy to treat human diseases, including cancer [46]. A locked nucleic acid (LNA)-modified miR-122 antagonist, named miravirsen/ SPC3649 (Santaris Pharma A/S, Copenhagen, Denmark), is the first miRNA reached clinical testing for the treatment of hepatitis C. It is currently under phase II clinical trial in patients with hepatitis C (http://clinicaltrials.gov/ct2/results?term=mir122&Search=Search). Because of their regulatory potential on the entire signaling networks within the cells and involvement in cancer development and progression, miRNAs have also emerged as promising molecular targets for the treatment of human cancers [29, 30, 33, 47]. Recent studies in this area drive the development of miRNAs as cancer therapeutics moving quickly from bench to clinical application [46, 48, 49]. In May 2013, a clinical trial using miR-34 mimics (trade name: MRX34, Mirna Therapeutics, Austin, TX) as a replacement therapy (ClinicalTrials.gov Identifier: NCT01829971) was initiated. This was the first and only miRNA replacement therapy carried out under clinical trials in multiple human cancers up until now [32, 50]. Unfortunately, the company (Mirna Therapeutics, Inc.) voluntarily halted patient enrollment and closed the phase 1 study of MRX34 on September 20, 2016, following multiple immune-related severe adverse events (SAE) observed in patients dosed with MRX34 over the course of the trial. It was hoped that further analysis of its full preclinical and clinical data set would provide useful information on the future development of MRX34 as a cancer therapeutics (https://www.bizjournals.com/austin/news/2016/09/21/austin-drug-company-halts-clinical-studies-after.html).
While majority of the basic research and clinical evaluations focusing on the therapeutic potential of one miRNA, a new approach combining two miRNAs has just been studied in lung cancer cells [34]. Considering this innovative idea, our research focused on determining the functional cooperation of miR-125a and miR-205 in suppressing erbB3 expression. We on purposely selected the cell clones with an increased expression of miR-125a and/or miR-205 around 10-fold as compared to the cells transfected with the empty vector control (Figs. 2, 3, 4 and 6). With this limited elevation of miRNA expression levels, single miRNA (miR-125a or miR-205) enhancement had minor effect on erbB3 expression, whereas co-expression of the two miRNAs significantly inhibited erbB3 (Figs. 2c, 3b, & 4d). This approach would reduce the non-specific targeting of each miRNA, supporting the notion that “sister” miRNAs, which have common targets [51], may exert synergistic activity to only repress their common targets. Moreover, ectopic expression of both miR-125a and miR-205, as compared to either miRNA alone, not only inhibited proliferation, it also significantly enhanced trastuzumab-mediated growth inhibition and cell cycle G1 arrest and paclitaxel-induced apoptosis in HER2-overexpressing breast cancer cells. Thus, concurrent expression of “sister” miRNAs may be a more promising miRNA-replacement therapy for cancer treatment.
Conclusions
In the current study, we demonstrate that co-expression of miR-125a and miR-205 is more potent than that of single miRNA to inhibit cell proliferation and enhance trastuzumab- and paclitaxel-mediated anti-proliferative/anti-survival effects on HER2-overexpressing breast cancer cells. Our data support further investigations developing the miRNA (miR-125a/miR-205) cluster as a novel, effective HER3-targeted therapy to enhance the efficacy of trastuzumab and paclitaxel against HER2-overexpressing breast cancer.
Additional file
Figure S1. Transfection with the multi-miRNA (cluster) lentiviral vector significantly enhanced T-DM1-induced growth inhibitory effects on BT474 cells. The stable clones of BT474 cells transfected with empty vector (pCDH), pCDH-miR-125a, or pCDH-miR-205 alone or pCDH-miR-125a-miR-205 (cluster) were seeded onto 96-well plates. After 24 hrs, the 96-well plates were then placed into the IncuCyte system to measure cell proliferation in a real time. Data show growth curves of the clones in comparison with their responses to the treatment of T-DM1 (10 μg/ml) in the indicated time period. (TIF 2615 kb)
Acknowledgements
The authors are grateful to Ms. Lisa Litzenberger (Department of Pathology, University of Colorado Anschutz Medical Campus) for her excellent art preparation.
Funding
This work was supported in part by a grant from the National Institutes of Health (NIH), USA (1 R01CA201011 to BL).
Abbreviations
- Ab
Antibody
- CPRC
Castration-resistant prostate cancer
- EGFR
Epidermal growth factor receptor
- ELISA
Enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- HDAC
Histone deacetylase
- HDACi
HDAC inhibitor
- HER
Human epidermal growth factor receptor
- LNA
Locked nucleic acid
- MAPK
Mitogen-activated protein kinase
- MEK
MAPK kinase
- miRNA
microRNA
- PARP
Poly(ADP-ribose) polymerase
- PI-3 K
Phosphoinositide 3-kinase
- RTK
Receptor tyrosine kinase
Authors’ Contributions
The authors’ contributions to this work are reflected in the order shown, with the exception of ZH and BL who supervised the research and finalized the report. HL and JH performed the experiments and data analysis. HL and BL drafted the manuscript. HL, ZH, and BL conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 2.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/S0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 4.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16:1373–1383. doi: 10.1158/1078-0432.CCR-09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mujoo K, Choi BK, Huang Z, Zhang N, An Z. Regulation of ERBB3/HER3 signaling in cancer. Oncotarget. 2014;5:10222–10236. doi: 10.18632/oncotarget.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 overexpression and survival in solid tumors: a meta-analysis. J Natl Cancer Inst. 2013;105:266–273. doi: 10.1093/jnci/djs501. [DOI] [PubMed] [Google Scholar]
- 9.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 11.Jathal MK, Chen L, Mudryj M, Ghosh PM. Targeting ErbB3: the new RTK(id) on the prostate cancer block. Immunol Endocr Metab Agents Med Chem. 2011;11:131–149. doi: 10.2174/187152211795495643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills GB, Yarden Y. The rebirth of a phoenix: ovarian cancers are addicted to ErbB-3. Cancer Cell. 2010;17:217–218. doi: 10.1016/j.ccr.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 13.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, Moustafa Z, Thomas RK, Greulich H, Schinzel A, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010;17:298–310. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amin DN, Campbell MR, Moasser MM. The role of HER3, the unpretentious member of the HER family, in cancer biology and cancer therapeutics. Semin Cell Dev Biol. 2010;21:944–950. doi: 10.1016/j.semcdb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee Y, Ma J, Lyu H, Huang J, Kim A, Liu B. Role of erbB3 receptors in cancer therapeutic resistance. Acta Biochim Biophys Sin. 2014;46:190–198. doi: 10.1093/abbs/gmt150. [DOI] [PubMed] [Google Scholar]
- 16.Ma J, Lyu H, Huang J, Liu B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol Cancer. 2014;13:105. doi: 10.1186/1476-4598-13-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007;120:1874–1882. doi: 10.1002/ijc.22423. [DOI] [PubMed] [Google Scholar]
- 18.Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, Esteva FJ, Liu B. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70:1204–1214. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 19.Lyu H, Yang XH, Edgerton SM, Thor AD, Wu X, He Z, Liu B. The erbB3- and IGF-1 receptor-initiated signaling pathways exhibit distinct effects on lapatinib sensitivity against trastuzumab-resistant breast cancer cells. Oncotarget. 2016;7:2921–2935. doi: 10.18632/oncotarget.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Huang X, Lee CK, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of survivin. Oncogene. 2010;29:4225–4236. doi: 10.1038/onc.2010.180. [DOI] [PubMed] [Google Scholar]
- 21.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, Linggi B, Kalra A, Paragas V, Bukhalid R, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, Liu B. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12:134. doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, Tan J, Edgerton SM, Thor AD, Lee CK, Liu B. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013;15:R101. doi: 10.1186/bcr3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Gao L, Wang S, Lee CK, Ordentlich P, Liu B. HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009;69:8403–8411. doi: 10.1158/0008-5472.CAN-09-2146. [DOI] [PubMed] [Google Scholar]
- 26.Huang X, Wang S, Lee CK, Yang X, Liu B. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 2011;307:72–79. doi: 10.1016/j.canlet.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, Liu B. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013;4:e556. doi: 10.1038/cddis.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyu H, Huang J, Edgerton SM, Thor AD, He Z, Liu B. Increased erbB3 promotes erbB2/neu-driven mammary tumor proliferation and co-targeting of erbB2/erbB3 receptors exhibits potent inhibitory effects on breast cancer cells. Int J Clin Exp Pathol. 2015;8:6143–6156. [PMC free article] [PubMed] [Google Scholar]
- 29.Bader AG, Brown D, Winkler M. The promise of microRNA replacement therapy. Cancer Res. 2010;70:7027–7030. doi: 10.1158/0008-5472.CAN-10-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho WC. MicroRNAs as therapeutic targets and their potential applications in cancer therapy. Expert Opin Ther Targets. 2012;16:747–759. doi: 10.1517/14728222.2012.696102. [DOI] [PubMed] [Google Scholar]
- 31.Thorsen SB, Obad S, Jensen NF, Stenvang J, Kauppinen S. The therapeutic potential of microRNAs in cancer. Cancer J. 2012;18:275–284. doi: 10.1097/PPO.0b013e318258b5d6. [DOI] [PubMed] [Google Scholar]
- 32.Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, Zarone MR, Gulla A, Tagliaferri P, Tassone P, Caraglia M. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014;3:e194. doi: 10.1038/mtna.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taipaleenmaki H, Browne G, Akech J, Zustin J, van Wijnen AJ, Stein JL, Hesse E, Stein GS, Lian JB. Targeting of Runx2 by miR-135 and miR-203 impairs progression of breast cancer and metastatic bone disease. Cancer Res. 2015;75:1433–1444. doi: 10.1158/0008-5472.CAN-14-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, Dysart S, Chen X, Bader AG, Slack FJ. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene. 2015;34:3547–3555. doi: 10.1038/onc.2014.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iorio MV, Casalini P, Piovan C, Di Leva G, Merlo A, Triulzi T, Menard S, Croce CM, Tagliabue E. microRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009;69:2195–2200. doi: 10.1158/0008-5472.CAN-08-2920. [DOI] [PubMed] [Google Scholar]
- 36.Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282:1479–1486. doi: 10.1074/jbc.M609383200. [DOI] [PubMed] [Google Scholar]
- 37.Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S, Sineshchekova O, Saxena P, Sutton CR, Chen D, et al. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res. 2013;73:6024–6035. doi: 10.1158/0008-5472.CAN-13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LoRusso P, Janne PA, Oliveira M, Rizvi N, Malburg L, Keedy V, Yee L, Copigneaux C, Hettmann T, Wu CY, et al. Phase I study of U3-1287, a fully human anti-HER3 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:3078–3087. doi: 10.1158/1078-0432.CCR-12-3051. [DOI] [PubMed] [Google Scholar]
- 39.Mirschberger C, Schiller CB, Schraml M, Dimoudis N, Friess T, Gerdes CA, Reiff U, Lifke V, Hoelzlwimmer G, Kolm I, et al. RG7116, a therapeutic antibody that binds the inactive HER3 receptor and is optimized for immune effector activation. Cancer Res. 2013;73:5183–5194. doi: 10.1158/0008-5472.CAN-13-0099. [DOI] [PubMed] [Google Scholar]
- 40.Schoeberl B, Kudla A, Masson K, Kalra A, Curley M, Finn G, Pace E, Harms B, Kim J, Kearns J, et al. Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121) NPJ Syst Biol Appl. 2017;3:16034. doi: 10.1038/npjsba.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yonesaka K, Hirotani K, Kawakami H, Takeda M, Kaneda H, Sakai K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene. 2016;35:878–886. doi: 10.1038/onc.2015.142. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z, Liu H, Desai S, Schmitt DC, Zhou M, Khong HT, Klos KS, McClellan S, Fodstad O, Tan M. miR-125b functions as a key mediator for snail-induced stem cell propagation and chemoresistance. J Bio Chem. 2013;288:4334–4345. doi: 10.1074/jbc.M112.419168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Tan G, Dong L, Cheng L, Li K, Wang Z, Luo H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS One. 2012;7:e34210. doi: 10.1371/journal.pone.0034210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12:201. doi: 10.1186/bcr2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu H, Mo YY. Targeting miR-205 in breast cancer. Expert Opin Ther Targets. 2009;13:1439–1448. doi: 10.1517/14728220903338777. [DOI] [PubMed] [Google Scholar]
- 46.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 47.Nana-Sinkam SP, Croce CM. Clinical applications for microRNAs in cancer. Clin Pharmacol Ther. 2013;93:98–104. doi: 10.1038/clpt.2012.192. [DOI] [PubMed] [Google Scholar]
- 48.Catela Ivkovic T, Voss G, Cornella H, Ceder Y. microRNAs as cancer therapeutics: a step closer to clinical application. Cancer Lett. 2017;407:113–122. doi: 10.1016/j.canlet.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 2017;8:132–143. doi: 10.1016/j.omtn.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams BD, Parsons C, Slack FJ. The tumor-suppressive and potential therapeutic functions of miR-34a in epithelial carcinomas. Expert Opin Ther Targets. 2016;20:737–753. doi: 10.1517/14728222.2016.1114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wahdan-Alaswad R, Liu B. “Sister” miRNAs in cancers. Cell Cycle. 2013;12:3703–3704. doi: 10.4161/cc.26875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Transfection with the multi-miRNA (cluster) lentiviral vector significantly enhanced T-DM1-induced growth inhibitory effects on BT474 cells. The stable clones of BT474 cells transfected with empty vector (pCDH), pCDH-miR-125a, or pCDH-miR-205 alone or pCDH-miR-125a-miR-205 (cluster) were seeded onto 96-well plates. After 24 hrs, the 96-well plates were then placed into the IncuCyte system to measure cell proliferation in a real time. Data show growth curves of the clones in comparison with their responses to the treatment of T-DM1 (10 μg/ml) in the indicated time period. (TIF 2615 kb)