

Abstract
Efavirenz (EFV), an antiretroviral that interacts clinically with co-administered drugs via activation of the pregnane X receptor (PXR), is extensively metabolized by the cytochromes P450. We tested whether its primary metabolite, 8-hydroxyEFV (8-OHEFV) can activate PXR and potentially contribute to PXR-mediated drug-drug interactions attributed to EFV. Luciferase reporter assays revealed that despite only differing from EFV by an oxygen atom, 8-OHEFV did not activate PXR. Corroborating this, treatment with EFV for 72 h elevated the mRNA abundance of the PXR target gene, Cyp3a11, an approximate 28-fold in primary hepatocytes isolated from PXR-humanized mice while treatment with 8-OHEFV did not result in a change in Cyp3a11 mRNA levels. FRET-based competitive binding assays and isothermal calorimetry demonstrated that even with the lack of ability to activate PXR, 8-OHEFV displays an affinity for PXR (IC50 12.1 µM; KD 7.9 µM) nearly identical to that of EFV (IC50 18.7 µM; KD 12.5 µM). The use of 16 EFV analogs suggest that other discreet changes to the EFV structure beyond the 8-position are well-tolerated. Molecular docking simulations implicate an 8-OHEFV binding mode that may underlie its divergence in PXR activation from EFV.
Keywords: nuclear receptor, ligand-binding, drug metabolism, cytochrome P450, molecular docking
Entry for the Table of Contents
Efavirenz interacts clinically with co-administered drugs via activation of the pregnane X receptor and is extensively metabolized by the cytochromes P450. Its primary metabolite, 8-hydroxyEFV (8-OHEFV), circulates in a near 1:1 ratio with EFV following chronic EFV use. We determined that 8-OHEFV does not activate PXR and thereby does not likely contribute to PXR-mediated drug-drug interactions attributed to EFV. Although it lacks the ability to activate PXR, 8-OHEFV appears to bind PXR with an affinity nearly identical to EFV. Molecular docking indicates that 8-OHEFV exhibits a binding mode distinct from EFV, which may underlie the divergence in PXR activation.
Introduction
The pregnane X receptor (PXR; NR1I2) is a critical regulator of an evolved defense mechanism against foreign substances[1]. Thus, as a xenobiotic sensor, PXR can be activated by a broad array of small-molecules. To date, large-scale structure-activity analyses have been performed in an attempt to identify general trends that confer specificity of PXR ligand recognition, activation and antagonism using drug libraries of structurally diverse compounds[2]; on the other hand, the impact of subtle molecular differences, including those resulting from drug metabolism, on binding to PXR and PXR activation has been explored to a lesser extent.
Similar to other ligand-activated nuclear receptors, PXR is organized in a modular structure and exhibits two highly conserved regions: a centralized DNA-binding domain containing two zinc fingers and a C-terminal ligand-binding domain (LBD)[3]. Activation of PXR has been demonstrated to modulate the expression of genes involved in drug metabolism and transport as means of detoxifying and eliminating xenobiotics from the body[1]. Members of the cytochrome P450 (CYP) 3A subfamily (CYP3A) including human CYP3A4 and murine Cyp3a11 are prototypic PXR target genes across species[1]. Consequently, PXR-mediated induction of the cytochromes P450 is the basis for a majority of drug-drug interactions in which a drug enhances the metabolism of a co-administered therapeutic agent, leading to decreased efficacy or increased toxicity[1, 4].
The non-nucleoside reverse transcriptase inhibitor efavirenz (EFV; Figure 1) is a cornerstone of HIV therapy, and in fact is designated by the World Health Organization (www.who.int) as an essential medicine; however, EFV is also a PXR agonist[5] and as a result exhibits a high incidence rate of drug-drug interactions upon co-administration with other P450 substrates, including anti-hepatitis C viral agents, antifungals and oral contraceptives[6]. Further, EFV autoinduces the formation of its primary cytochrome P450-dependent metabolite, 8-hydroxyEFV (8-OHEFV). This manifests itself clinically as an observed decrease in the ratio of EFV to 8-OHEFV following long-term treatment, resulting in EFV and 8-OHEFV circulating at a near equimolar concentration in plasma[7]; however, it is currently unclear whether 8-OHEFV itself can also bind to and activate PXR and potentially participate in the autoinduction attributed to EFV and the PXR-mediated drug-drug interactions associated with EFV. In fact, in general, the impact of metabolites of PXR-activating drugs on PXR activity remains largely uncharacterized. With this in mind, in the present study, we tested whether 8-OHEFV can activate PXR. In doing so, we also aimed to elucidate the relationship between ligand chemical structure and PXR activation using a panel of analogs based on the EFV (Figure 1) scaffold. Our data reveal that 8-OHEFV does not activate PXR and molecular docking simulations suggest that this may be due to EFV and 8-OHEFV binding to PXR in two distinct modes. The predicted binding mode for EFV is consistent with its activating properties, including van der Waals contacts with residues of the activation function 2 (AF2) helix, that would result in the stabilization of the activation PXR function. On the other hand, 8-OHEFV is predicted to adopt a binding mode wherein there is no engagement between the ligand and the AF2 helix. These data suggest that in contrast to EFV, 8-OHEFV does not activate PXR and therefore, does not contribute as an inducer to EFV-mediated drug-drug interactions that result from PXR activation.
Figure 1.

Chemical structures of EFV, 8-OHEFV, and 16 EFV analogs. Structural divergences from EFV are highlighted in red.
Results
Activation of PXR by EFV versus 8-OHEFV.
Using a luciferase reporter assay, we tested for the direct activation of PXR by EFV and 8-OHEFV. Two regulatory regions of CYP3A4 have been previously identified to contain PXR response elements critical for facilitating transcriptional activation by xenobiotics, including a proximal promoter region from bases −362 to +53 and a more distal region spanning bases −7836 to −7208 relative to the transcription start site[8]. As such, human hepatocellular carcinoma HepG2 cells were transfected with the plasmid pGL3-basic-CYP3A4–362(7836/7208ins)-luc, containing a firefly luciferase gene under the control of the PXR-activated regulatory elements in order to assay for PXR transcriptional activity[9]. In agreement with previously published data, we observe activation of PXR by EFV (10 µM; an approximate 4.3-fold increase in luciferase activity) (Figure 2)[10]; however, treatment with 8-OHEFV did not result in activation of PXR. RIF, a prototypic activator of human PXR employed here as a positive control, yielded the greatest increase in firefly luciferase activity of 9.5-fold relative to empty vector drug treatment (Figure 2). PCN was used here as a negative control for human PXR activation and as expected, treatment with this compound had no effect on luciferase reporter activity.
Figure 2.

Comparative effects of EFV, 8-OHEFV and EFV analogs (10 μM) on the activation of human PXR in transiently transfected HepG2 cells. Firefly and R. reniformis luciferase activities were quantified using Dual-Glo luciferase reporter assays (described in Experimental Section). Firefly luciferase activity was normalized against that of R. reniformis. All data are expressed relative to DMSO control as mean fold change (± S.D.). Experiments were performed in quadruplicate. Statistical significance, as determined using an unpaired t-test, from empty the vector drug treatments is indicated as **, p ≤ 0.01 and ***, p ≤ 0.001.
To test whether changes to the EFV structure beyond an oxygen insertion at the 8-position affected activation of PXR, we employed a panel of 16 EFV analogs (Figure 1). As shown in Figure 2, the two largest EFV analogs, 6 and 7, exhibited the greatest impact on PXR as firefly luciferase activity was increased 6.7- and 6.9-fold, respectively, in the presence of these compounds. Interestingly, 5, which is an enantiomer of 6, exhibited only an approximate 1.9-fold increase in luciferase activity. With the exception of the smallest analog, 10, all of the analogs tested stimulated PXR transcriptional responses that were elevated significantly over empty vector controls.
To corroborate these findings, we examined the modulation of PXR target gene expression by EFV, 8-OHEFV and a subset of EFV analogs using primary hepatocytes isolated from PXR-humanized and PXR-null mice. In the case of EFV, Cyp3a11 mRNA levels were increased by an approximate 28-fold in the PXR-humanized hepatocytes following 72 h of treatment (Figure 3). This observed effect was PXR-dependent as EFV exhibited no impact on Cyp3a11 mRNA abundance in the PXR-null mice. Similar to our findings using the luciferase reporter assay, treatment of PXR-humanized mouse hepatocytes with 8-OHEFV did not result in modulation of Cyp3a11 mRNA levels (Figure 3). We employed EFV analogs 7 and 8 in these hepatocyte studies since analog 7, which departs significantly from EFV structure, was among the most active in the PXR luciferase reporter assay, whereas analog 8 is structurally similar to EFV and exhibited a similar response to EFV in the reporter assay experiments. Increased abundance of Cyp3a11 mRNA was observed for both analog 7 and 8 in the hepatocytes isolated from PXR-humanized mice. Consistent with the luciferase reporter assay results, the increase observed for 7 (37-fold) was greater than that observed for 8 (10-fold). Also, in agreement with the reporter assay results, we found that there was no change in the Cyp3a11 mRNA levels in PXR-humanized hepatocytes following treatment with the smallest analog 10.
Figure 3.

Impact of EFV, 8-OHEFV as well as analogs 7, 8 and 10 on PXR target gene Cyp3a11 mRNA expression in primary mouse hepatocytes isolated from PXR-humanized and PXR-null transgenic mice. Cyp3A11 and GAPDH mRNA levels were measured using real-time quantitative PCR 72 h post treatment with either 0.1% DMSO or 10μM drug. Cyp3A11 mRNA levels were normalized to GAPDH mRNA levels. All data are expressed as mean fold change from DMSO control for four independent experiments ± S.D. Statistical significance, as determined using an unpaired t-test, from the PXR-null mouse drug treatments is indicated as *, p ≤ 0.05, and **, p ≤ 0.01
TR-FRET analyses indicate binding of EFV, 8-OHEFV, and EFV analogs to the ligand binding pocket of PXR
In order to determine whether differences in binding affinity to PXR played a causal role in the divergence in the activation by EFV and 8-OHEFV, we examined the solution-binding properties of each of these compounds in addition to the EFV analogs using a competitive binding assay. This assay measures the displacement of the tight binding, fluorescent fluormone ligand from the xenobiotic-binding pocket of a terbium-labeled, antibody-PXR complex. Excitation of the fluormone-bound, PXR adduct at 332 nm results in fluorescence resonance energy transfer (FRET) between the terbium donor and fluormone, yielding an emission signal at 515 nm. Fluormone displacement by competing ligands results in decreases in the FRET signal and 515 nm/485 nm ratio. Titration with the positive control, SR12813, produced a dose-dependent decrease in the 515 nm/485 nm ratio (Figure 4) and an IC50 value of 140 nM, which is in agreement with previously published studies[11]. Figure 4 shows the titration curves for a representative subset of examined analogs, whereas all determined IC50 values are reported in Table 1. EFV exhibited an IC50 of 18.7 µM. The IC50 values for remaining EFV analogs that demonstrated activation of PXR ranged from 1.9 µM (5) to 77.8 µM (4). Analog 10, which did not activate PXR based on our luciferase reporter assay results also did not exhibit measurable binding to PXR. In contrast, despite its inability to activate PXR, 8-OHEFV appeared to interact with PXR with an IC50 value (12.14 µM) comparable to that observed for EFV.
Figure 4.

Competitive binding assays of EFV, 8-OHEFV and EFV analogs. Time-Resolved FRET signal intensity (515nm/485nm ratio) derives from the Fluormone●PXR complex. As the inhibitor concentration increases, Fluormone is displaced from the PXR-LBD resulting in a decrease in the FRET signal intensity. TR-FRET signal intensity (515nm/485nm) was plotted as a function of the inhibitor concentration. All data were normalized against the DMSO (1%) controls. Each data point represents the mean of four measurements (± S.D.). Details of the TR-FRET assay are described in the Experimental Section. Dose-response curves are shown for EFV, 8-OHEFV, analogs 5, 6, 10, 15 as well as the positive control SR12813, for simplicity and clarity.
Table 1.
IC50 values for EFV, 8-OHEFV, and EFV analogs.
| Compound | IC50 (μM)[a] |
|---|---|
| EFV | 18.7 ±5.8 |
| 8-OHEFV | 12.1 ±2.8 |
| 1 | 25.4 ± 22.6 |
| 2 | 20.5 ± 10.7 |
| 3 | 5.6 ±2.7 |
| 4 | 77.8 ±86.1 |
| 5 | 1.9 ±1.1 |
| 6 | 4.5 ±2.6 |
| 7 | 6.2 ±4.9 |
| 8 | 10.2 ±3.3 |
| 9 | 39.3 ± 24.1 |
| 10[b] | - |
| 11 | 3.9 ±3.6 |
| 12 | 9.4 ± 2.2 |
| 13 | 12.1 ±4.6 |
| 14 | 8.4 ±4.0 |
| 15 | 5.6 ±4.1 |
| 16 | 2.5 ±1.3 |
| SR2813 | 0.15 ±0.02 |
| PCN[b] | - |
The errors in the IC50 values were determined from the data fit to equation (1). For more details, refer to the experimental section.
IC50 values not determined.
Thermodynamic studies of EFV, 8-OHEFV and EFV analogs interaction with the PXR-LBD
Isothermal titration calorimetry (ITC) was also employed to directly measure binding of EFV, 8-OHEFV (Figure 5) and EFV analogs 4, 5, 6, 7, 8, 10, 15, and 16 to the PXR-LBD. As observed for the competitive TR-FRET binding assay, the PXR agonist SR12813 displayed tight binding to PXR with a KD of 186.06 nM. Association of SR12813 was accompanied by a large favorable enthalpy change (ΔHBIND = −9.2 kcal/mol) and a small, unfavorable binding entropy (TΔSBIND = −0.2 kcal/mol). The data were well described by a single-site binding model with an N value of 1.0. This is consistent with X-ray structures of SR12813•PXR complexes, all of which show 1:1 ligand-to-protein stoichiometry. Whereas ITC studies using SR12813 were relatively straightforward, analyses of PXR interactions with EFV and EFV-related analogs proved to be less so due to low binding enthalpies and poor ligand solution properties. However, using this approach, we were able to directly examine the binding thermodynamics for EFV and 8-OHEFV as well as the EFV analogs, 4, 8 and 15 (Figures 5A and 5B). Like SR12813, the data sets showed fits to the single site binding model, with N values of approximately 1.0 (Figure 5A). The KD values determined using the ITC method are commensurate with the IC50 values obtained by the competitive FRET assay. Indeed, the ITC results revealed similar binding affinities for EFV and 8-OHEFV of 12.5 µM and 7.9 µM, respectively. Consistent with the luciferase reporter assay and TR-FRET competitive binding studies, analog 10 exhibited very weak binding with a KD value greater than 400 µM. In contrast to SR12813, the thermodynamic profiles for EFV and the other examined analogs indicate that binding is associated with notably low enthalpy changes and are largely entropy driven.
Figure 5.

Thermodynamic analysis of binding of EFV, 8-OHEFV and selected EFV analogs to PXR-LBD-SRC1p construct. (A) ITC thermograms and corresponding binding isotherms for EFV (left) and 8-OHEFV (right). (B) Thermodynamic signatures of binding for EFV, 8-OHEFV, 4, 8 and 15. Errors in ΔHo and ΔSo were obtained when the data were fit to a single-site binding model using Microcal Origin software (Malvern). Errors in ΔGo were obtained through error propagation using the equation ΔGo = -RTlnKD and the error in KD.
To overcome the challenges posed by the low binding heats and poor solubility of the remaining EFV analogs, we employed a competitive ITC approach. In these studies, PXR was titrated with SR12813 in the presence of several EFV analogs of interest including 5, 6, 7 and 16, all of which are insoluble at concentrations higher than 400 μM. As observed for the direct titrations, the competitive ITC approach afforded KD values that are consistent with the FRET-based IC50 values (Table 2).
Table 2.
Thermodynamic parameters and dissociation constants of ligand binding to hPXR.
| Compound | Kd (μM) | ΔH° (kcal/mol) | -TΔS° (kcal/mol•K) | ΔG° (kcal/mol) |
|---|---|---|---|---|
| EFV[a] | 12.5 ±3.3 | −2.1 ±0.3 | −4.9 ±0.4 | −6.9 ± 0.2 |
| 8-OHEFV[a] | 7.9 ±7.7 | −3.1 ±1.2 | −3.8 ± 1.5 | −6.9 ± 0.3 |
| 4[a] | 78.7 ± 15.3 | −1.1 ±0.1 | −4.5 ±0.2 | −5.6 ±0.1 |
| 5[b] | 14.5 ±4.3 | − | − | −6.6 ±0.1 |
| 6[b] | 9.5 ±2.7 | − | − | −6.8 ± 0.2 |
| 7[b] | 4.2 ±1.1 | − | − | −7.3 ± 0.2 |
| 8[a] | 14.9 ±2.6 | −1.8 ±0.1 | −4.8 ±0.3 | −6.6 ± 0.1 |
| 10[b] | >400 | − | − | − |
| 15[a] | 10.9 ± 1.8 | −1.6 ±0.2 | −5.1 ±0.2 | −6.8 ±0.1 |
| 16[b] | 10.5 ±1.5 | − | − | −6.8 ± 0.2 |
| SR12813[a] | 0.28 ± 0.02 | −9.2 ±0.1 | 0.2 ±0.1 | −8.9 ±0.1 |
ΔGo values were obtained from the dissociation constants using the equation ΔGo = -RTlnKD. Errors in KD, ΔHo, and ΔSo were obtained by fitting the data to a single-site binding model using Microcal Origin software (Malvern). Errors in ΔGo were obtained through error propagation using the above equation and the error in KD.
Binding affinities for these compounds were determined by using the competitive inhibition ITC method (see Experimental Section). Errors in the KD values of the EFV analog were also determined through error propagation using equation (2) and the errors in the KD, APP of the parent ligand, SR12813.
Molecular docking of EFV, 8-OHEFV and EFV analogs to the ligand-binding pocket of PXR
A docking approach was used to further address the details of recognition of the EFV-related ligands by PXR. Of particular interest were the binding modes adopted by EFV and 8-OHEFV. To assess the correspondence between docking and experimental data, the binding modes predicted for SR12813 and hyperforin (HYF), both well-studied PXR agonists, were compared to those revealed by X-ray crystallography. Docking simulations of SR12813 binding predicted two low energy poses, the two lowest of which closely reproduced the pair of binding modes revealed in known X-ray structures. One is shown in Figure 6A. Relative to the ligand heavy atoms of the experimentally observed SR12813 binding modes, those of the docked conformations exhibit root-mean-square displacements (RMSD) of 2.62 (Figure 6A) and 2.50 Å. In the case of HYF, the lowest energy docking pose also recapitulated the experimentally observed ligand-binding mode. In this case, we observed an RMSD of 3.93 Å between the ligand heavy atoms of the crystallographically determined and predicted HYF binding modes. Whereas our docking protocol included a rigid PXR receptor, we explored the flexibility of the PXR pocket in subsequent energy minimizations of the selected, low energy docking poses. The observed results were commensurate with many of the side chain movements revealed by comparisons of the known ligand bound PXR structures, including those noted for M243, Q285, M323, H407 and L411.
Figure 6.

Molecular docking reproduces the structural aspects of SR12813 binding to PXR-LBD. A) Relative ligand positions for the observed (yellow) and the docked (grey) for the SR12813 conformations. RMSD values are reported in the text. B) Predicted binding mode of EFV (pink) relative to SR12813 (pale yellow) in the PXR pocket. Oxygen, nitrogen, sulfur, fluorine, and chlorine atoms are colored red, dark blue, orange, light blue, and green, respectively.
Docking simulations of EFV binding to PXR predicted two low energy docking poses. This is surprising given the small size of EFV (volume = 326 Å3) relative to that of the PXR pocket (volume ≈ 1400 Å3). Only one of the poses is consistent with the activation properties observed for EFV. This pose is shown in Figure 7A. Compared to SR12813, EFV is predicted to make only a fraction of the contacts, which is consistent with the marked difference in ligand size (SR12813 volume = 823 Å3) and observed binding affinities (100-fold). Indeed, SR12813 has been shown to interact with 15 residues; however, EFV is predicted to engage with 10 residues. The EFV docking pose places the ligand between two overlapping binding hotspots (regions with a high propensity for ligand binding) identified by X-ray analyses and computational solvent mapping studies[12]. These include M243, A244, S247, H407 and L411 (hotspot 1) as well as F281, H407 and L411 (hotspot 2). Importantly, additional predicted contacts M425 and F429 include residues of the activation function (AF2) helix. PXR pocket residues engage with the EFV ring systems on both faces. These include an edge-to-face contact with F281 on one side and hydrophobic interactions with M243 on the other. The AF2 helix residues are predicted to interact extensively with the EFV cyclopropylethynyl substituent. The docking pose also shows a hydrogen-bonding interaction between the oxazinone ring carbonyl group and S247, which is found to participate similarly in a number of crystallographic studies of ligand-bound PXR complexes[13]. Additional predicted hydrophobic contacts include, A244 and M425, both of which interact with the perimeter of the EFV ring system.
Figure 7.

Molecular docking simulations of EFV, 8-OHEFV, and the EFV analogs 7 and 10. Predicted contacts made by (A) EFV (pink), (B) 8-OHEFV (blue), (C) analog 7 (light green) and (D) analog 10 (orange) in the PXR pocket. Residues within 5Å of each ligand shown are labelled. The PXR pocket residues are depicted as white sticks with spheres at the Cα positions. The AF2 helix is depicted in the cartoon representation and shown for the sake of clarity and orientation purposes. Hydrogen-bonding contacts are indicated by the yellow dashed lines.
Interestingly, the docking studies predict a single major binding mode for 8-OHEFV. This compound is predicted to interact primarily with a binding-pocket hotspot defined by F288, W299 and Y306. This hotspot is distinct from described above and is located at the end of the pocket opposite that of the AF2 helix and the EFV docking site. The 8-OHEFV pose shows the ligand oxazinone ring system clamped on one face by V211 and on the other by M243. Further stabilization is provided by hydrophobic interactions between L209 and the edge of the 8-OHEFV ring system bearing the chlorine group. The 8-OHEFV trifluoromethyl and cyclopropylethynyl substituents engage with the F288-W299-Y306 hotspot. Importantly, the presence of the hydroxyl group at the 8-position and resulting change in ligand hydrogen-bonding properties apparently underlie the change in the binding modes predicted for 8-OHEFV relative to EFV. Indeed, we observe a new set of ligand-protein hydrogen bonds for the 8-OHEFV•PXR complex, which include the backbone carbonyl of L209 that forms a bridging interaction to both the 8-hydroxyl group and oxazinone ring nitrogen atom of the EFV metabolite (Figure 7B). Relative to the predicted EFV binding mode, 8-OHEFV displays 8Å shift of the ligand skeleton. The docking results do not reveal additional polar contacts between PXR and remaining 8-OHEFV ring heteroatoms. However, energy minimizations of the 8-OHEFV docking pose in the presence of explicit solvent suggest that the exposed ligand polar atoms would likely be stabilized by water molecules occupying the PXR pocket, completely satisfying the hydrogen-bonding potential of the 8-OHEFV.
The EFV analog, 7, displays a higher PXR activation level and binding affinity relative to EFV. To obtain insights into both enhanced properties, we also carried out docking simulations with 7. The docking pose obtained for this ligand is shown in Figure 7C. In agreement with its biochemical properties, docking results reveal a large interaction surface that combines pocket elements that dictate binding for both EFV and 8-OHEFV. Moreover, whereas predicted binding contributions include up to two hotspots for both EFV and 8-OHEFV, binding of 7 appears to involve four of the five identified hotspots. Similar to EFV and 8-OHEFV, docking suggests that the binding of analog 7 is dominated by hydrophobic contacts. Importantly, these include residues of the AF2 helix, namely M425 and F429. The docking results also reveal one hydrogen-bonding contact involving Q285 the amide carbonyl of 7. Finally, multiple low energy docking poses were predicted for the smallest EFV analog, 10, which, like 8-OHEFV, failed to activate PXR. Similar to 8-OHEFV, the docking results for 10 do not implicate ligand interactions with the AF2 helix (see Figure 7D).
In light of the abovementioned findings, we next tested whether 8-OHEFV could inhibit activation of PXR by RIF or EFV. To do so, primary human hepatocytes were incubated with either RIF or EFV in the presence or absence of 8-OHEFV, and CYP3A4 mRNA levels were measured. These studies revealed that co-incubation of 10 µM 8-OHEFV with 2 µM RIF results in an approximate 20% decrease in CYP3A4 mRNA induction as compared to 2 µM RIF alone (p=0.004). Interestingly, 8-OHEFV did not block activation of PXR by EFV in primary human hepatocytes at clinically relevant concentrations, or under conditions where the concentration of 8-OHEFV employed was 2-fold greater than that of EFV (data not shown).
Discussion
PXR is a ligand-activated transcription factor that is involved in the regulation of genes critical for maintaining cellular homeostasis within the liver and intestines[14]. In addition to its endogenous function(s), PXR exhibits broad substrate specificity and binds structurally diverse activators enabling the receptor to serve as a xenobiotic sensor. The coordinated actions of PXR’s DNA-binding domain and LBD function to eliminate foreign and potentially toxic substances from the body through modulation of the expression of genes that encode proteins involved in phase I and phase II drug metabolism as well as drug transport[1]. Moreover, PXR-mediated induction of drug metabolizing enzymes, including several cytochromes P450 that play major roles in the biotransformation of clinical relevant drugs, underlies a large portion of reported drug-drug interactions[15].
In this study, we employed a battery of approaches to gain insights into the interactions between EFV and PXR, and of particular interest, to test whether 8-OHEFV, the primary oxidative metabolite of EFV, is also a PXR agonist. EFV is extensively metabolized by cytochrome P450 enzymes to several metabolites including the major oxygenated metabolite, 8-OHEFV[16]. We have reported that this metabolite can be detected clinically not only in blood plasma, but also in seminal plasma and cerebrospinal fluid[17]. Of note, Ngaimisi et al found that in 129 male and female patients receiving 600 mg of EFV per day plasma concentrations of EFV were 10.61 ± 1.11 µM at 4 weeks and 10.33 ± 0.98 µM at 16 weeks. In addition, they observed 8-OHEFV concentrations of 6.51 ± 0.87 µM at 4 weeks and 6.84 ± 1.06 µM at 16 weeks[7]. As such, the abundance of this metabolite in circulation makes understanding its pharmacology of importance. Since 8-OHEFV only differs from EFV by an oxygen atom, we were intrigued to find that 8-OHEFV did not activate PXR in the luciferase reporter assays or stimulate any changes in Cyp3a11 mRNA abundance in humanized-PXR primary mouse hepatocytes. It was then unanticipated to observe that 8-OHEFV could competitively bind to the PXR-LBD and with an affinity comparable to EFV. Interestingly, a previous report by Gao et al. reported PXR activation may be attenuated with the addition of a single polar functional group, such as −OH or −NH2, to a known agonist[18]. Additional studies have also suggested the importance of hydroxyl groups in promoting antagonistic activity towards PXR[19]. Moreover, Lin et al. recently demonstrated that small structural changes in the scaffold of 1,4,5-substituted 1,2,3-triazole can convert analogs that act as PXR agonists to antagonists[20]. Thus, our work, when considered within the context of these published reports, raises the possibility that 8-OHEFV could be a PXR antagonist. In line with this, we found that when primary human hepatocytes are treated with RIF and 8-OHEFV at a near clinical concentration, a decrease in the induction of a PXR target gene was observed. On the other hand, we observed no antagonism of EFV by 8-OHEFV. At the molecular level, these results are consistent with 8-OHEFV and RIF being direct competitors of binding to the PXR pocket. Viewed in this light, our results also suggest that EFV and 8-OHEFV do not directly compete for the PXR ligand pocket. Our docking results support this notion. Indeed, the predicted binding modes for EFV and 8-OHEFV place both ligands in distinct, largely non-overlapping regions of the PXR pocket (see Figure 8). Due to the small size of both ligands and the flexibility of the PXR pocket, it may not be unreasonable to envision the simultaneous binding of EFV and 8-OHEFV. The simultaneous binding of two ligands by PXR was demonstrated recently by X-ray crystallography[21]. Future work studies will focus on testing this hypothesis. Nevertheless, the findings presented here suggest that 8-OHEFV does not activate PXR and therefore, does not likely contribute to EFV-mediated induction of drug metabolizing enzymes and transporters that is PXR-dependent.
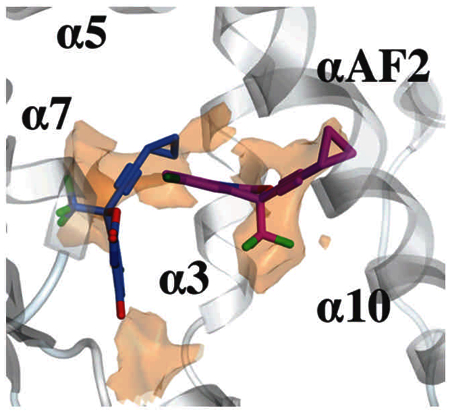
Figure 8: Identification of hydrophobic regions in the human PXR ligand-binding pocket.

The surface shown (orange) derives from an electrostatic features map that applies the Poison-Boltzmann Equation to predict preferred locations of hydrophobic as well as hydrogen-bonding donor and acceptor ligand groups (see the Materials and Methods section). EFV, 8-OHEFV, and 7 are depicted as purple, blue and green sticks, respectively. For clarity, only the hydrophobic locations are presented. The hydrophobic surface is contoured at 2.25 kcal/mol. The map was generated using the program MOE.
In trying to understand how chemical structure can impact the ability of a ligand to activate PXR, EFV served as an ideal probe for this work since it has been demonstrated to activate PXR, is a clinically used drug that has been shown to be involved in drug-drug interactions, and has a chemical structure with several features that can be readily modified synthetically rendering it a useful tool for structure-activity studies. The EFV analogs employed here include a set of closely-related compounds displaying a broad range of modifications that facilitate understanding the ligand structural requirements for recognition and PXR agonism. The analogs range in size from 223 to 423 Da and alterations include targeting of the EFV oxazinone scaffold (analogs 8, 11–16) and cyclopropylethynyl substituent at the 4-position of the oxazinone ring (analogs 3, 4 and 13). Other departures from the parent EFV structure include opening of the oxazinone ring (analogs 1, 2, 5–7 and 10).
We found that modifications to the oxazinone ring result in minor effects on PXR activation. For example, the replacement of the oxazinone ring carbonyl moiety with a methylene or methyl group to yield analogs 8 and 11, respectively, results in induction of PXR activity levels that are similar (38% and 45%, respectively) to that found for EFV. Results obtained for analogs with heteroatom replacements at the endocyclic nitrogen and oxygen atoms also showed no significant effects on PXR activity relative to the parent EFV ligand. Interestingly, differential activation was observed for analogs 5 and 6, which are enantiomers, with analog 6 having a greater impact on PXR activity than analog 5. This result is not surprising as similar differences in PXR activation have been previously reported for enantiomeric compounds[22]. In this case, docking predicts two distinct binding modes for 5 and 6 with the latter nearly reproducing the interactions observed for 7 (data not shown), which displays a comparable activation level (see below). Finally, we find that an intact oxazinone ring is not required for PXR activation. Indeed, analogs 1 and 2, display small reductions in PXR activation (27% and 17%, respectively) relative to EFV. Although an intact oxazinone ring does not appear to be crucial for binding and transactivation, removal of the cyclopropylethynyl substituent to yield the smallest of the examined analogs, 10, nearly abolishes both activities.
We used a molecular docking approach to obtain insights into the basis of PXR binding and activation by EFV, 8-OHEFV and closely-related analogs. For EFV, docking reveals a binding mode that is consistent with its moderate binding affinity, thermodynamic profile and PXR activation. Relative to SR12813, hyperforin and other tight binding agonists, EFV engages in a significantly smaller number of receptor contacts and identified pocket hotspots. Moreover, entropy driven binding for EFV is consistent with the dominance of stabilizing hydrophobic interactions in the modeled EFV-PXR complex. Importantly, docking reveals a binding mode in which EFV forms direct van der Waals contacts with residues L411, M425 and F429 that are located on the AF2 helix. These interactions presumably stabilize the activated PXR form. Similar interactions are observed with analog 7 and other large analogs, including 6 and 9 (data not shown). Relative to EFV, these analogs promote higher levels of PXR activation. For 6 and 7, we observe higher affinity binding suggesting a more extensive network of ligand-pocket contacts. For 8-OHEFV, which failed to activate PXR, docking predicts a binding mode that places the ligand in a remote location relative to the AF2 helix. Despite being unable to engage with residues of the AF2 helix, 8-OHEFV binds PXR with an affinity similar to that of EFV. Like EFV, the binding of 8-OHEFV is entropy driven underscoring an important role for hydrophobic interactions. We also observe a slightly more favorable binding enthalpy for 8-OHEFV, which might be due to the formation of two hydrogen-bonding contacts with the ligand; the EFV pose suggest one such interaction. Analyses of the electrostatic properties of the PXR pocket reveals two regions of significant hydrophobicity (Figure 8). One is these regions is proximal to the AF2 helix. The other is situated at the other end of the pocket near the W299-F288-Y306 hotspot. Interestingly, the observed 8-OHEFV pose occupies the former, whereas EFV binding in the latter. The docking results also suggest that weak binding and failure to activate PXR for analog 10 likely results from the small number of contacts engaged by 10. Moreover, its tendency to maximize hydrogen-bonding contacts resulted in a number of nonproductive, isoenergetic docking poses, the majority of which showed interactions with the region of the PXR pocket occupied by 8-OHEFV. Importantly, none of the identified 10 poses revealed interactions with the AF2 helix. On the other hand, analog 7 is large enough to span the PXR and interact with residues defining both hydrophobic regions. The same is true for other known strong binders and activators of PXR. Importantly, the binding pose observed for analog 7 resembles that of another anti-HIV non-nucleotide reverse transcriptase inhibitor, PNU-142721 that has also been shown to be an PXR agonist[23].
In summary, we have demonstrated that unlike EFV, 8-OHEFV does not activate PXR. Through quantitative studies of PXR activation and binding using EFV, 8-OHEFV and a panel of small-molecule probes based on the EFV scaffold, we find that molecular size and, likely, hydrogen-bonding properties are key features in conferring PXR agonism for ligands displaying molecular weights of approximately 300 Da. In addition, it also appears that for ligands of this size, agonism and antagonism may be further determined by interactions with two hydrophobic mini-pockets that compose the PXR cavity. These findings can be leveraged to inform small-molecule drug development in an effort to aid in the prediction of xenobiotics that potentially activate PXR.
Experimental Section
Compounds
Pregnenolone-16α-carbonitrile (PCN; 5-Pregnen-3β-ol-20-one-16α-carbonitrile), rifampicin (RIF; 3-(4-methylpiperazinyliminomethyl)-rifamycin SV) and SR 12813 (tetraethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate) were purchased from Sigma-Aldrich (St. Louis, MO). EFV ((S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one) was obtained from the National Institutes of Health AIDS Reagent Program (Germantown, MD). 8-OHEFV ((S)-6-chloro-4-(cyclopropylethynyl)-8-hydroxy-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one) and EFV analogs: 1 ((R)-2-(2-amino-5-chlorophenyl)-4-cyclopropyl-1,1,1-trifluorobut-3-yn-2-ol), 2 ((S)-2-(2-amino-5-chlorophenyl)-4-cyclopropyl-1,1,1-trifluorobut-3-yn-2-ol), 3 ((S,E)-6-chloro-4-(2-cyclopropylvinyl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one), 4 (6-chloro-4-(pent-1-yn-1-yl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one), 5 ((R)-2-(5-chloro-2-((4-methoxybenzyl)amino) phenyl)-4-cyclopropyl-1,1,1-trifluorobut-3-yn-2-ol), 6 ((S)-2-(5-chloro-2-((4-methoxybenzyl)amino)phenyl)-4-cyclopropyl-1,1,1-trifluorobut-3-yn-2-ol), 7 (N-(4-chloro-2-(4-cyclopropyl-1,1,1-trifluoro-2-hydroxybut-3-yn-2-yl)phenyl)-4-methoxybenzamide), 8 ((S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazine), 9 ((S)-6-chloro-4-(cyclopropylethynyl)-2-(4-methoxyphenyl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazine), 10 (1-(2-amino-5-chlorophenyl)-2,2,2-trifluoroethan-1-one), 11 (6-chloro-4-(cyclopropylethynyl)-2-methyl-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazine), 12 ((S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-3,4-dihydroquinazolin-2(1H)-one), 13 ((S,E)-6-chloro-4-(2-cyclopropylvinyl)-4-(trifluoromethyl)-3,4-dihydroquinazolin-2(1H)-one), 14 (6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-3,4-dihydroquinolin-2(1H)-one), 15 (6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-4H-benzo[d][1,3]dioxin-2-one) and 16 (7-chloro-1-(cyclopropylethynyl)-1-(trifluoromethyl)isochroman-3-one) were synthesized by Toronto Research Chemicals (Toronto, ON, Canada). EFV, 8-OHEFV, and analogs 1, 2, 3, 5, 6, 8, 9, 12, and 13 were obtained as the R or S enantiomers (as indicated); analogs 4, 7, 11, 14, 15 and 16 as racemic mixtures; and analogs 9 and 11 as mixtures of diastereomers.
Isolation of primary mouse hepatocytes and drug treatments
All animals used in this study were exposed to a 12 h light, 12 h dark cycle to minimize stress exposure prior to hepatocyte isolations. Further, all animal related procedures described herein were approved by Johns Hopkins University’s Institutional Animal Care and Use Committee. Hepatocytes were isolated from 9 to 13-week-old male PXR-humanized and PXR-null mice bred on a C57BL/6J background (Taconic Biosciences; Hudson, NY). Primary mouse hepatocytes were isolated via collagenase perfusion as previously described [24]. Hepatocytes were plated in six-well collagen-coated plates (Corning, Tewksbury, MA) at a density of 2.4 × 105 cells per well. Cells were incubated overnight at 37°C and 5% CO2 prior to replacing the cell culture medium with fresh Williams E medium containing 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin-streptomycin for drug treatments. Hepatocytes were treated with either DMSO vehicle (0.1% final) or 10 µM drug for RNA isolation over a range of time points. For 48 h and 72 h drug treatments, cell culture medium was replaced every 24 h and hepatocytes were retreated with either DMSO or drug. A total of four independent experiments were performed for hepatocytes isolated from PXR-humanized and PXR-null mice.
Cryopreserved primary human hepatocyte culture and treatment
Cryoplateable primary human hepatocytes from one adult male (age 38) and two adult female (ages 34 and 39) donors were purchased from BioIVT (Baltimore, MD) and were thawed and plated according to BioIVT protocol onto 24-well collagen-coated plates (Corning, Tewksbury, MA). Cells had a viability of ≥85% at the time of plating, as determined by trypan blue exclusion. After plating, cells were incubated overnight at 37°C and 5% CO2 prior to replacing the cell culture medium with fresh InVitroGRO CP medium (BioIVT) supplemented with Torpedo Antibiotic Mix (BioIVT) containing the drug treatments outlined below. After that, medium and drug treatment was replaced every 24 hours. Hepatocytes were treated for 48 hours with either RIF (2 µM or 10 µM) or EFV (10 µM) in the presence or absence of 10 µM 8-OHEFV. DMSO-treated hepatocytes (0.1% DMSO final) were employed as vehicle controls. Statistical analysis was performed using a paired t-test.
RNA isolation and quantitative real-time PCR
RNA was prepared from primary mouse hepatocytes and cryopreserved human hepatocytes using TRIzol (Thermo Fisher Scientific) following the manufacturer’s instructions for cells grown in a monolayer. Total RNA (1 µg) was reverse transcribed using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). The following primers were used for quantitative real-time polymerase chain reaction (qPCR): mouse Cyp3a11 forward 5’-ATAGAGCTTTGCTGTCCCCC-3’ and reverse 5’-CGGCTTTCCTTCATTCTGTC-3’; mouse Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) forward 5’-GAAGGCCGGGGCCCACTTGA-3’ and reverse 5’-TCTCCAGGCGGCACGTCAGA-3’; human Cyp3a4 forward 5’-GGAAAAGTGTGGGGCTTTTATGATGG-3’ and reverse 5’-GCCTGTCTCTGCTTCCCGCC-3’; and human Gapdh forward 5’-CTTCTTTTGCGTCGCCAGCCGA-3’ and reverse 5’-CACGACGTACTCAGCGCCAGC-3’. A Gapdh PCR product was generated from mouse and human hepatocyte RNA using conventional PCR and ligated into CloneJET PCR cloning kit for subcloning (Thermo Fisher Scientific). Serial dilutions of the resulting Gapdh plasmids were used to generate a standard curve for the quantitation of mRNA levels using Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific). Gapdh served as the housekeeping gene for mRNA level normalization. The following qPCR cycling reactions were performed: denaturation of dsDNA for 10 min at 95°C proceeded by 40 cycles of denaturation for 15 sec at 95°C, annealing for 30 sec at 60°C, and extension for 30 sec at 72°C. Resulting Cyp3a11 or Cyp3a4 copy numbers were normalized to Gapdh for each treatment and data was reported as fold change from DMSO control for each time point.
Transient transfection and dual-luciferase reporter gene assay
HepG2 human hepatocellular carcinoma cells (American Type Culture Collection, Manassas, VA) were cultured in T-75 flasks in high glucose Dulbecco’s Modified Eagle Medium (DMEM; American Type Culture Collection) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. Cells were maintained at 37°C and 5% CO2 prior to subculturing for transfection. At approximately 80–90% confluency, HepG2 T-75 flasks were trypsinized and plated at 2 × 106 cells/mL into a new flask. After 24 h, culture medium was replaced with Opti-MEM I reduced serum medium (Thermo Fisher Scientific) and T-75 flasks were transfected with a master mix containing FuGENE 6 transfection reagent (3 µL/µg plasmid DNA per flask; Promega, Madison, WI), Opti-MEM I reduced serum medium (500µL per flask), pGL4.74 [hRluc/TK] internal control vector (500 ng per flask; Promega), pGL3-basic-CYP3A4–362(7836/7208ins)-luc reporter construct (2 µg per flask; Blue Heron Biotech, Bothell, WA), a receptor construct (6 µg pCMV6-entry-mPXR or 2 µg pCMV6-XL4-PXR per flask; OriGene Technologies, Rockville, MD) or an empty vector (6 µg pCMV6-entry or 2 µg pCMV6-XL4 per flask; OriGene Technologies). Cells were transfected for 24 h and then trypsinized, counted and plated into fresh DMEM at 1 × 105 cells/well into a 96-well white plate (Costar). Following 24 h plating (48 h post-transfection), cells were treated in triplicate with either 0.1% DMSO control or 10 µM drug prepared in DMEM for a total of 24 h. After treatment, cells were lysed and the firefly luciferase and R. reniformis luciferase activities were quantified using a Dual-Glo Luciferase Reporter Assay (Promega). Luminescence was measured directly in the 96-well plate using a Synergy HT plate reader (BioTek, Winooski, VT). Firefly luciferase activity was normalized by taking the ratio of firefly luminescence to that of R. reniformis and results were expressed as fold change from DMSO control. A total of four independent experiments were performed.
Competitive ligand binding assay
A LanthaScreen PXR Competitive Binding Assay (Thermo Fisher Scientific), which is based on the principle of time-resolved fluorescence resonance energy transfer (TR-FRET). The PXR-LBD (amino acids 111 to 434) construct containing an N-terminal glutathione S-transferase tag (10 nM) was incubated with varying concentrations of competitor ligands, namely SR12813 (positive control), PCN (negative control), EFV, 8-OHEFV and remaining EFV analogs in the presence of Fluormone PXR Green (PXR ligand) (40 nM) and a Tb-labeled anti-glutathione S-transferase antibody (10 nM) in a covered 384-well plate (Corning) for 1 hour at room temperature. The fluorescence intensities were then measured using a Tecan Safire 2 (Tecan Group Ltd., Männedorf, Switzerland) multiplate reader. The samples were excited at 332 nm and fluorescence intensities were measured at both 485 nm (Tb emission) and 515 nm (Fluormone emission). Data was expressed as the ratio of Fluormone (515 nm) over terbium (485 nm) emission as a function of log[inhibitor] for each ligand. Data were fit to the following equation to determine the half-maximal inhibitory concentration (IC50) values.
| (1) |
where Fmeas is the measured FRET signal, Fmax is the maximum FRET signal, Fmin is the minimum FRET signal, [L] is the ligand concentration, and N is the Hill coefficient.
Protein cloning, expression and purification
For the solution-binding studies (ITC), we used a PXR construct (His6-tag PXR-LBD-L5-SRC1p), which contains an N-terminal His6-tag followed by the PXR ligand-binding domain (residues 130–434) fused to a peptide fragment of the SRC1 protein (residues 623–710) via penta-peptide (-GGSGG-) linker. The His6-tag PXR-LBD-L5-SRC1p construct was generated using a previously published protocol. In brief, sequence encoding the PXR ligand-binding domain was amplified from a plasmid containing the full-length PXR gene (GenBank ID: NM_003889) using the following primers:
Forward:
5’-CACCATGAAAAAAGGTCACCACCATCACCATCACGGTAGTGAACGGACAGGGACTCAGC-3’ and
Reverse:
5’-TTATGAGGGGCTACCCTCCTGTAAGAGCCGGTGTAGAATTTTATGCCGTTCTGTCAATGAGCTATGAGAAGAGCCACCAGAGCCACCGCTACCTGTGATGCCGAACAACTC-3’.
The resulting PCR product was then cloned into a pENTR/SD/D-TOPO vector using a TOPO cloning kit (Thermo Fisher Scientific). A subsequent LR clonase reaction was performed to transfer the DNA insert into the final destination vector, pDEST14, using an LR cloning kit (Thermo Fisher Scientific). PXR-LBD-L5-SRC1p was expressed in E. coli BL21 (DE3) cells. The cells were grown in Terrific Broth medium supplemented with 0.2% glucose and 100 µg/mL ampicillin at 37°C until the cell density reached an OD600 of 3–4. Following this, the fermentation temperature was lowered to 16°C and protein expression was induced with the addition of 0.2 mM isopropyl β-D-thiogalactopyranoside. Sixteen hours post induction, cells were pelleted by centrifugation and resuspended in lysis buffer (25 mM HEPES pH 7.9, 5% v/v glycerol, 150 mM sodium chloride, 1 mM tris(2-carboxyethyl)phosphine hydrochloride, 10 mM imidazole) containing an EDTA-free ProBlock Gold Bacterial 2D protease inhibitor cocktail (Gold Biotechnology, St. Louis, MO). The cells were then lysed using a microfluidizer (Watts Fluidair, Kittery, ME). Clarified lysates were then applied to a Ni2+-affinity column (HisTrap HP 5 mL; GE Healthcare, Marlborough, MA) pre-equilibrated with lysis buffer containing 30 mM imidazole. Lysis buffer supplemented with 250 mM imidazole was used for the elution step. The fractions containing PXR-LBD-L5-SRC1p were pooled and applied onto a desalting column (HiPrep 26/10; GE Healthcare) followed by a cation exchange column (HiTrap SP HP 5mL; GE Healthcare) pre-equilibrated with buffer A (25 mM HEPES pH 7.2, 5% v/v glycerol, 50 mM sodium chloride, 1mM tris(2-carboxyethyl)phosphine hydrochloride). PXR-LBD-L5-SRC1p was eluted using buffer A supplemented with 1 M NaCl. The purity of protein-containing fractions was judged by SDS-polyacrylamide gel electrophoresis and coomassie staining.
Isothermal titration calorimetry
For each ITC experiment, purified PXR-LBD-SRC1p was dialyzed overnight against 20 mM Tris-HCl pH 8.5, 200 mM sodium chloride, 1 mM tris(2-carboxyethyl)phosphine hydrochloride using Slide-A-Lyzer cassettes (20 kDa molecular weight cut-off) (Thermo Fisher Scientific). Following dialysis, the sample protein concentrations were determined spectrophotometrically (extinction coefficient at 280nm = 26930 M−1 cm−1; mol. wt. = 39130.03 Da). Experiments were performed at 25°C using a VPITC microcalorimeter (Malvern, Westborough, MA). Protein (2.1 mL at 40 µM) and ligand (500 µL at 0.4 mM) solutions were prepared in dialysis buffer supplemented with 0.05% Tween 20 and a final concentration of 5% DMSO. All titrations consisted of a total of 30 injections (a 5 µL injection, followed by 29, 10 µL injections) with constant stirring at 300 rpm. The data were fit to a single-site binding model using Microcal Origin software (Malvern). Data were further analyzed using the NITPIC and SEDPHAT program, to obtain the thermodynamic parameters of ligand binding to PXR[25]. A buffer-ligand titration was performed for each compound and subtracted as background from the protein-ligand titration prior to fitting.
Competitive isothermal titration calorimetry
The competitive ITC experiments were carried as described above. In these studies, we monitored the binding of the SR12813 ligand (70 μM) in the presence of competitor ligands (EFV analogs, 100–200 μM), which were present in the injection syringe and sample cell. The data were fit to a single-site binding model using Microcal Origin software (Malvern) to obtain the apparent ligand dissociation constant (KD,APP) for SR12813. The dissociation constant of the inhibitor (KI) was obtained using the following equation:
| (2) |
where KD,APP is the ligand dissociation constant for SR12813 determined in the presence of inhibitor, KD is the SR12813 dissociation constant in the absence of inhibitor, [I] is the inhibitor concentration, and Ki is the inhibitor dissociation constant. The KI values for analogs 5, 6, 7, 10, and 16 are given in Table 2.
Molecular docking simulations
Molecular docking simulations were performed using AutoDock 4.2[26]. Ligand structures were generated using ChemDrawProfessional 16.0 and minimized using the Builder tool in MOE[27] (Molecular Operating Environment, Chemical Computing Group Inc., 2016). The PXR receptor model (PDB Id: 1NRL) was prepared for docking using the MOE structure preparation tool. The model was then energy minimized using YASARA[28], which employs a simulated annealing protocol with explicit solvent and an optimized force field based on AMBER (YAMBER3). AutoDock was used to assign the PXR receptor model and the ligand atom partial charges. All ligands were docked into the PXR receptor using a 54 × 54 × 54 point grid with a spacing of 0.375 around the putative ligand binding pocket. The Lamarckian algorithm was used to search for low energy binding modes and the parameters were set to fifty iterations of genetic algorithm with a maximum of 2.5 × 107 energy evaluations per docked ligand. All final selected, low energy docking poses were energy minimized with explicit using YASARA.
Analysis of the PXR pocket electrostatic and hydrophobic features.
Electrostatic feature map applies the Poisson-Boltzmann Equation (PBE) to the prediction of electrostatically preferred locations of pocket or surface hydrophobic, H-bond acceptor and H-bond donor regions. Solving the PBE has been well studied[29]. In MOE, the PBE is solved by a finite difference multi-grid technique. Given a collection of receptor atoms with the i-th atom having 3D coordinates xi, partial charge qi, van der Waals radius Ri, and homogeneous van der Waals well depth Ei. The receptor charge density f is modeled as a sum of atom-centered Gaussian densities each of which has the form:
| (3) |
where a is a scale parameter (a = 0 results in a point charge) and ci is the normalization constant that integrates fi to qi. The MOE Surfaces and Maps application uses OPLS van der Waals radii for the receptor atoms and an a parameter of 0.4. Solving the PBE for the electrostatic potential, u, enables the generation of predictive maps. The hydrophobic map is an isocontour of the potential - (qO + qH) u + vC where vC is the van der Waals potential of a carbon atom, which are the regions of receptor contact and low preference of the ions. PXR was ionized and charged using Protonate 3D (MOE) and CHARMM27 atomic partial charges (MOE).
Acknowledgements
This work was supported by the National Institutes of Health National Institute of General Medical Sciences (R01 GM103853, awarded to NNB), a PhRMA Foundation Predoctoral Fellowship in Pharmacology (awarded to JML), and a National Science Foundation Predoctoral Fellowship (awarded to CJSH).
References:
- [1].Kliewer SA, Goodwin B, Willson TM, Endocr. Rev 2002, 23(5), 687–702. [DOI] [PubMed] [Google Scholar]
- [2].a) Shukla SJ, Sakamuru S, Huang R, Moeller TA, Shinn P, Vanleer D, Auld DS, Austin CP, Xia M, Drug Metab. Dispos 2011, 39(1), 151–159; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao L, Nickbarg E, Wang W, Thomas A, Ziebell M, Prosise WW, Lesburg CA, Taremi SS, Gerlach VL, Le HV, Cheng KC, Biochem. Pharmacol 2011, 81(5), 669–679; [DOI] [PubMed] [Google Scholar]; c) Ratajewski M, Grzelak I, Wisniewska K, Ryba K, Gorzkiewicz M, Walczak-Drzewiecka A, Hoffmann M, Dastych J, Toxicol. Lett 2015, 232(1), 193–202; [DOI] [PubMed] [Google Scholar]; d) Lin WW, Goktug AN, Wu J, Currier DG, Chen TS, Assay Drug Dev. Technol 2017, 15(8), 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Germain P, Staels B, Dacquet C, Spedding M, Laudet V, Pharmacol. Rev 2006, 58(4), 685–704. [DOI] [PubMed] [Google Scholar]
- [4].a) Palleria C, Di Paolo A, Giofre C, Caglioti C, Leuzzi G, Siniscalchi A, De Sarro G, Gallelli L, J. Res. Med. Sci 2013, 18(7), 601–610; [PMC free article] [PubMed] [Google Scholar]; b) Evans RM, Mol. Endocrinol 2005, 19(6), 1429–1438. [DOI] [PubMed] [Google Scholar]
- [5].Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H, Pharmacol J. Exp. Ther 2007, 320(1), 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Vadlapatla RK, Patel M, Paturi DK, Pal D, Mitra AK, Expert Opin. Drug Metab. Toxicol 2014, 10(4), 561–580; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tittle V, Bull L, Boffito M, Nwokolo N, Clin. Pharmacokinet 2015, 54(1), 23–34; [DOI] [PubMed] [Google Scholar]; c) Khatri A, Dutta S, Dunbar M, Podsadecki T, Trinh R, Awni W, Menon R, Antimicrob. Agents Chemother 2016, 60(5), 2965–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ngaimisi E, Mugusi S, Minzi OM, Sasi P, Riedel KD, Suda A, Ueda N, Janabi M, Mugusi F, Haefeli WE, Burhenne J, Aklillu E, Clin. Pharmacol. Ther 2010, 88(5), 676–684. [DOI] [PubMed] [Google Scholar]
- [8].Goodwin B, Hodgson E, Liddle C, Mol. Pharmacol 1999, 56(6), 1329–1339. [DOI] [PubMed] [Google Scholar]
- [9].Chang TK, Waxman DJ, Methods Enzymol 2005, 400, 588–598. [DOI] [PubMed] [Google Scholar]
- [10].Sharma D, Lau AJ, Sherman MA, Chang TK, Biochem. Pharmacol 2013, 85(11), 1700–1711. [DOI] [PubMed] [Google Scholar]
- [11].Chen Y, Tang Y, Robbins GT, Nie D, J. Pharmacol. Exp. Ther 2010, 334(3), 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S, Biochemistry 2009, 48(48), 11572–11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR, Science 2001, 292(5525), 2329–2333. [DOI] [PubMed] [Google Scholar]
- [14].Aranda A, Pascual A, Physiol. Rev 2001, 81(3), 1269–1304. [DOI] [PubMed] [Google Scholar]
- [15].Rendic S, Guengerich FP, Chem. Res. Toxicol 2015, 28(1), 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z, J. Pharmacol. Exp. Ther 2003, 306(1), 287–300. [DOI] [PubMed] [Google Scholar]
- [17].Avery LB, VanAusdall JL, Hendrix CW, Bumpus NN, Drug Metab. Dispos 2013, 41(2), 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gao YD, Olson SH, Balkovec JM, Zhu Y, Royo I, Yabut J, Evers R, Tan EY, Tang W, Hartley DP, Mosley RT, Xenobiotica 2007, 37(2), 124–138. [DOI] [PubMed] [Google Scholar]
- [19].a) Hodnik Z, Peterlin Masic L, Tomasic T, Smodis D, D’Amore C, Fiorucci S, Kikelj D, J. Med. Chem 2014, 57(11), 4819–4833; [DOI] [PubMed] [Google Scholar]; b) Lau AJ, Chang TK, Pharmacol. Res 2015, 100, 64–72. [DOI] [PubMed] [Google Scholar]
- [20].Lin WW, Wang YM, Chai SC, Lv LL, Zheng J, Wu J, Zhang QJ, Wang YD, Griffin PR, Chen TS, Nat. Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Delfosse V, Dendele B, Huet T, Grimaldi M, Boulahtouf A, Gerbal-Chaloin S, Beucher B, Roecklin D, Muller C, Rahmani R, Cavailles V, Daujat-Chavanieu M, Vivat V, Pascussi JM, Balaguer P, Bourguet W, Nat. Commun 2015, 6, 8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Mu Y, Stephenson CRJ, Kendall C, Saini SPS, Toma D, Ren SR, Cai HB, Strom SC, Day BW, Wipf P, Xie W, Mol. Pharmacol 2005, 68(2), 403–413; [DOI] [PubMed] [Google Scholar]; b) Rulcova A, Prokopova I, Krausova L, Bitman M, Vrzal R, Dvorak Z, Blahos J, Pavek P, J. Thromb. Haemost 2010, 8(12), 2708–2717. [DOI] [PubMed] [Google Scholar]
- [23].Cheng Y, Redinbo MR, Protein Sci 2011, 20(10), 1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee P, Peng H, Gelbart T, Beutler E, Proc. Natl. Acad. Sci. U. S. A 2004, 101(25), 9263–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Houtman JCD, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, Schuck P, Protein Sci 2007, 16(1), 30–42; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keller S, Vargas C, Zhao HY, Piszczek G, Brautigam CA, Schuck P, Anal. Chem 2012, 84(11), 5066–5073; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scheuermann TH, Brautigam CA, Methods 2015, 76, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, J. Comput. Chem 2009, 30(16), 2785–2791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ, J. Comput. Chem 1998, 19(14), 1639–1662. [Google Scholar]
- [27].S. S. W. Chemical Computing Group ULC, Suite #910, Montreal, QC, Canada, H3A 2R7, Molecular Operating Environment (MOE), 201308, 2017. [Google Scholar]
- [28].Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G, Proteins-Structure Function and Bioinformatics 2004, 57(4), 678–683. [DOI] [PubMed] [Google Scholar]
- [29].Gilson MK, Sharp KA, Honig BH, J. Comput. Chem 1988, 9(4), 327–335. [Google Scholar]