

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) have a variety of potential indications that include management of pain and inflammation as well as chemoprevention and/or treatment of cancer. Furthermore, a specific form of ibuprofen, dexibuprofen or the S-(+) form, shows interesting neurological activities and has been proposed for the treatment of Alzheimer’s disease. In a continuation of our work probing the anticancer activity of small sulindac libraries, we have prepared and screened a small diversity library of α-methyl substituted sulindac amides in the profen class. Several compounds of this series displayed promising activity compared with a lead sulindac analog.
Graphical abstract
The non-steroidal anti-inflammatory drugs (NSAIDs) are widely used for the treatment of minor pain and chronic inflammatory diseases such as rheumatoid arthritis. A number of these drugs possess antipyretic activity in addition to having analgesic and anti-inflammatory effects, and thus have use in the treatment of fever. These effects are attributed to the ability of the NSAIDs to inhibit the cyclooxygenases (COX), which convert arachidonic acid to prostaglandins (PGs) [1]. Three distinct COX isozymes have been characterized; COX-1 is responsible for the regulation of prostaglandin biosynthesis in normal tissues and serves an important role in gastric cytoprotection and renal homeostasis, COX-2 is an inducible enzyme important for acute inflammatory responses and pyrexia in the body, while COX-3 currently has no established role in humans. Evidence is mounting that the NSAIDs may play a role in the treatment of patients with familial adenomatous polyposis and for the chemoprevention of colorectal cancer [2,3]. Experimental data as well as epidemiological and clinical studies suggest that the regular use of NSAIDs in a chemoprevention regimen can reduce the incidence of colorectal cancer by approximately 30–50% [4,5]. However, upper gastrointestinal, renal, or cardiovascular side effects resulting from COX inhibition limit the utility of NSAIDs for prevention regimens as they typically require high dosages and chronic administration [6–10]. It is now clear that NSAIDs demonstrate a variety of activities beyond COX inhibition and their effects on tumor cells may be a result of multifarious activities [11]. While NSAIDs are believed to exhibit their anticancer properties through inhibition of COX-2 that is overexpressed in various tumor cells, several COX-2 independent mechanisms have also been suggested for the chemopreventive and antineoplastic properties of NSAIDs. Other activities include activation of apoptosis, inhibition of angiogenesis, modulation of the adaptive immune system or direct inhibition of cancer cell growth by blocking signal transduction pathways responsible for cell proliferation [12–17].
As a member of the NSAIDs, sulindac has been shown to dramatically induce regression of adenomas in familial adenomatous polyposis (FAP) patients, prevent recurrence of adenomas [18,19] and reduce the risk of colon cancer and prostate cancer [20–22]. As such, it has been studied extensively and is clinically used as a chemopreventive agent [13]. Sulindac is considered a prodrug that is reductively metabolized in vivo to the more active sulfide as well as oxidized to the more hydrophilic and less active sulfone (see Figure 1). While sulindac contains a chiral sulfoxide group that reduces lipophilicity of the scaffold and improves solubility of the drug, the commercial compound is racemic, and the reversible cycling between the methyl sulfide and the methyl sulfoxide would scramble any chirality making the study of the effects of chirality at this center difficult in an in vivo setting. Oxidation to the sulfone is irreversible and the more hydrophilic product is considerably less active as a COX inhibitor.
Figure 1.
In vivo metabolic cycling (oxidation/reduction) of the sulindac sulfoxide moiety.
The biological mechanism of the antineoplastic effect of the sulindac metabolites appears to involve the selective induction of apoptosis as demonstrated in human breast, lung, prostate and colon cancer cell lines [23–26]. Our earlier studies suggested that a relatively simple alteration to sulindac in the form of sulindac sulfide amide (SSA) (Figure 2) can virtually abolish COX-related activity and toxicity while enhancing anticancer activity in vitro and maintaining similar in vivo xenograft activity [27] in a chemoprevention protocol. It is notable that the metabolic oxidation-reduction cycling demonstrated for sulindac (Figure 1) has also been shown to happen for the sulindac analog SSA yielding both the sulfoxide and sulfone metabolites in vivo [27].
Figure 2.
Overview of our structural modifications relative to our lead agent SSA in order to study the SAR of this class
Figure 2 shows our lead agent SSA and modifications reported herein to develop a broader SAR for the sulindac amides exemplified by SSA.
There are several broad classes of NSAIDs including the salicylates (e.g., aspirin) and the acetates or 2-aryl acetic acids (e.g., indomethacin). Sulindac belongs to the NSAID acetic acid class and is considered an indene-3-acetic acid. Addition of an α-Me group as indicated in Figure 2 would introduce a chiral center transforming the scaffold into the NSAID profen class or a 2-aryl propionic acid (e.g., ibuprofen – Advil® and naproxen – Aleve®). Most NSAID propionic acids, including ibuprofen, are sold as racemic mixtures. However, naproxen is available commercially as the S-isomer prepared by precipitation of an insoluble salt via the Pope-Peach method of chiral resolution. The impact of chirality at the α-Me position has been extensively studied for ibuprofen. Dexibuprofen, or (S)-(+)-ibuprofen, has been analyzed for toxicity and side effects, uptake, and neurological activity versus racemic ibuprofen. In fact, dexibuprofen has demonstrated varied and improved effects for Alzheimer’s disease [28]. Hence, we were interested in how introduction of an α-Me group would impact activity of SSA analogs against three common cancer cell lines from colon, breast, and prostate cancers that are standard cell lines used for preliminary chemoprevention and anticancer screening. We initiated a diversity program involving preparation of a sulindac profen core (α-Me sulindac) as shown in Figure 2 followed by diversification at both the amide position and the indene aryl group in order to study the structure-activity relationships in the profen amide series of sulindac. Herein, we present the preparation and preliminary screening of a series of novel sulindac amide derivatives containing a methyl group at the α-position with various alterations in the amide and aryl linkers. Our lead agent, SSA (Figure 2) was used as a standard control compound for comparison, as it shows improved activity relative to the clinical NSAID sulindac against colon cancer cells in vitro as well as good activity in vivo in a murine chemoprevention model of colon cancer [27].
Preparation of α-methyl sulindac amides 3–59 started with esterification of commercially available sulindac sulfide (to form 3–29), (+/−)-sulindac (to prepare 30–56), or a sulindac 3,4,5-trimethoxyphenyl analog (to afford 57–59) in the presence of MeOH/thionyl chloride. The corresponding methyl esters were formed in 90–96% yields. The introduction of the key α-methyl group was carried out using LDA and CH3I at −78 °C to furnish racemic or diastereomeric α-methyl sulindac ester derivatives in 73–93% yields [29]. The α-methyl esters were subsequently hydrolyzed to give the corresponding acids 2 in quantitative yields (Scheme 1). Finally, coupling of the α-methyl sulindac analogs 2 with various amines using HATU [30] as the coupling agent afforded compounds 3–59 in satisfactory yields as diastereomeric or racemic mixtures. When acid stereoisomers 2 were treated with a chiral amine, an inseparable mixture of diastereomeric amides was formed. However, separation of diastereomers was achievable in one case for the sulindac sulfide amide of L-histidine methylester (5 and epi-5 in Table 1), although relative and absolute configurations of those epimers were not determined.
Scheme 1.
Synthetic pathways to analogs 3–59. Reagents and conditions: (a) SOCl2, MeOH (b) LDA, MeI, THF, −78 oC (c) KOH, EtOH/H2O (d) HATU, DIEA, MeCN
Table 1.
Screening data for compounds SSA and 3–59.
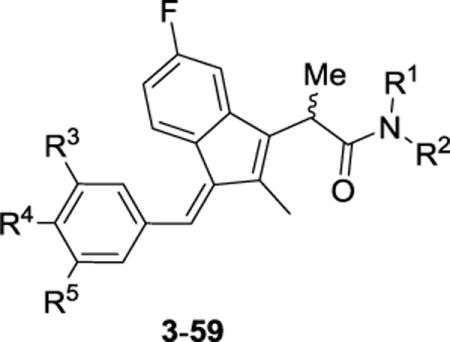
| |||||
|---|---|---|---|---|---|
| R3, R4 & R5 | Cmpds | R1 & R2 | CC50 (uM)
|
||
| HT29 | PC3 | MDA-MB-231 | |||
| SSA | 0.65±0.03 | 3.12±0.15 | 2.67±0.08 | ||
| 3 | R1 = H, R2 = benzyl | 22.88±2.78 | >50.00 | >50.00 | |
| 4 | R1 = H, R2 = furan-2-ylmethyl | 11.15±0.70 | 23.49±4.84 | 26.11±3.36 | |
| 5 | R1 = H, R2 = L-histidine methyl ester | 6.62±0.26 | 14.41±0.61 | 12.66±0.65 | |
| epi-5 | R1 = H, R2 = L-histidine methyl ester | 10.74±0.62 | 16.83±0.73 | 14.96±0.54 | |
| 6 | R1 = H, R2 = −CH2CH2N(CH3)2 | 1.80±0.04 | 5.07±0.45 | 3.88±0.52 | |
| 7 | R1 = H, R2 = −CH2CH2CH2N(CH3)2 | 2.58±0.05 | 7.24±0.49 | 6.42±0.18 | |
| 8 | R1 = H, R2 = −CH2CH2N(CH2CH3)2 | 2.03±0.06 | 6.21±0.37 | 5.38±0.19 | |
| 9 | R1 = H, R2 = −CH2CH2CH2N(CH2CH3)2 | 2.17±0.10 | 6.13±0.47 | 4.23±0.39 | |
| 10 | R1 = CH3, R2 = −CH2CH2N(CH3)2 | 1.43±0.05 | 5.55±0.28 | 3.66±0.58 | |
| 11 | R1 = CH3, R2 = −CH2CH2CH2N(CH3)2 | 2.12±0.08 | 5.50±0.52 | 4.11±0.36 | |
| 12 | R1 = CH2CH3, R2 = −CH2CH2N(CH2CH3)2 | 2.31±0.06 | 6.59±0.49 | 5.98±0.18 | |
| 13 | R1 = CH3, R2 = −CH2CH2N(CH2CH3)2 | 2.22±0.08 | 7.27±0.36 | 6.71±0.33 | |
| R3, R5 = H | 14 | R1 = CH2CH3, R2 = −CH2CH2N(CH3)2 | 1.13±0.03 | 3.54±0.24 | 3.18±0.12 |
| R4 = SCH3 | 15 | R1 = CH3, R2 = −CH2CH2N(CH3)H | 1.31±0.05 | 4.06±0.09 | 3.21±0.11 |
| 16 | R1 = CH3, R2 = −CH2CH2CH2N(CH3)H | 1.98±0.09 | 4.96±0.30 | 3.83±0.39 | |
| 17 | R1 = CH2CH3, R2 = −CH2CH2N(CH2CH3)H | 1.30±0.02 | 3.26±0.13 | 2.51±0.09 | |
| 18 | R1 = CH2CH3, R2 = −CH2CH2CH2N(CH2CH3)H | 2.01±0.07 | 4.60±0.20 | 3.97±0.46 | |
| 19 | R1 = CH(CH3)2, R2 = −CH2CH2N(CH(CH3)2)H | 1.50±0.05 | 3.91±0.14 | 3.83±0.36 | |
| 20 | R1 = CH(CH3)2, R2 = −CH2CH2CH2N(CH(CH3)2)H | 1.26±0.02 | 3.18±0.18 | 2.91±0.13 | |
| 21 |
|
2.03±0.09 | 5.68±0.45 | 5.00±0.28 | |
| 22 |
|
2.03±0.08 | 5.06±0.39 | 5.32±0.40 | |
| 23 |
|
2.22±0.08 | 5.91±0.39 | 5.32±0.46 | |
| 24 | R1, R2 = − (−CH2CH2−)2NCH2CH2N(CH3)2 | 2.02±0.09 | 5.98±0.40 | 3.77±0.53 | |
| 25 | R1, R2 = − (−CH2CH2−)2NCH2CH2CH2N(CH3)2 | 1.50±0.03 | 4.14±0.19 | 3.06±0.10 | |
| 26 | R1, R2 = − (−CH2CH2−)2N(pyridin-4-yl) | 2.20±0.08 | 4.68±0.20 | 3.95±0.55 | |
| 27 | R1, R2 = − (−CH2CH2−)2CH(pipyridin-1-yl) | 1.89±0.05 | 4.36±0.29 | 4.30±0.63 | |
| 28 |
|
1.10±0.05 | 4.24±0.28 | 3.49±0.11 | |
| 29 |
|
0.84±0.05 | 3.93±0.27 | 3.57±0.13 | |
|
| |||||
| 30 | R1 = H, R2 = benzyl | 17.19±1.24 | 15.70±0.89 | 26.00±2.56 | |
| 31 | R1 = H, R2 = furan-2-ylmethyl | >50.00 | >50.00 | >50.00 | |
| 32 | L-histidine methylester | >50.00 | >50.00 | >50.00 | |
| 33 | R1 = H, R2 = −CH2CH2N(CH3)2 | 12.69±1.43 | >50.00 | >50.00 | |
| 34 | R1 = H, R2 = −CH2CH2CH2N(CH3)2 | 22.46±3.02 | >50.00 | >50.00 | |
| 35 | R1 = H, R2 = −CH2CH2N(CH2CH3)2 | 11.14±1.40 | 39.96±4.83 | 36.56±8.17 | |
| 36 | R1 = H, R2 = −CH2CH2CH2N(CH2CH3)2 | 15.52±1.92 | >50.00 | >50.00 | |
| 37 | R1 = CH3, R2 = −CH2CH2N(CH3)2 | 11.85±1.41 | 44.55±4.46 | >50.00 | |
| 38 | R1 = CH3, R2 = −CH2CH2CH2N(CH3)2 | 16.62±2.28 | >50.00 | >50.00 | |
| 39 | R1 = CH2CH3, R2 = −CH2CH2N(CH2CH3)2 | 4.45±0.29 | 19.66±3.78 | 23.50±5.73 | |
| 40 | R1 = CH3, R2 = −CH2CH2N(CH2CH3)2 | 5.83±0.56 | 29.32±4.61 | >50.00 | |
| R3, R5 = H | 41 | R1 = CH2CH3, R2 = −CH2CH2N(CH3)2 | 9.51±1.39 | 28.13±2.28 | 31.96±8.21 |
| 42 | R1 = CH3, R2 = −CH2CH2N(CH3)H | 10.72±2.24 | 27.92±1.58 | >50.00 | |
| R4 = S(O)CH3 | 43 | R1 = CH3, R2 = −CH2CH2CH2N(CH3)H | 10.54±1.03 | 33.73±4.47 | 36.76±9.36 |
| 44 | R1 = CH2CH3, R2 = −CH2CH2N(CH2CH3)H | 9.99±0.80 | 26.26±2.82 | 27.87±4.95 | |
| 45 | R1 = CH2CH3, R2 = −CH2CH2CH2N(CH2CH3)H | 6.98±0.65 | 24.75±5.07 | 25.56±3.45 | |
| 46 | R1 = CH(CH3)2, R2 = −CH2CH2N(CH(CH3)2)H | 8.06±1.32 | 15.17±0.52 | 21.34±4.68 | |
| 47 | R1 = CH(CH3)2, R2 = −CH2CH2CH2N(CH(CH3)2)H | 7.13±1.15 | 15.67±2.08 | 23.00±4.35 | |
| 48 |
|
13.66±0.96 | 31.95±2.96 | 39.25±8.84 | |
| 49 |
|
9.67±1.53 | 36.26±2.84 | 39.29±11.30 | |
| 50 |
|
>50.00 | >50.00 | >50.00 | |
| 51 | R1, R2 = −(−CH2CH2−)2NCH2CH2N(CH3)2 | 38.43±6.41 | 49.96±6.47 | >50.00 | |
| 52 | R1, R2 = −(−CH2CH2−)2NCH2CH2CH2N(CH3)2 | 31.96±3.07 | >50.00 | >50.00 | |
| 53 | R1, R2 = − (−CH2CH2−)2N(pyridin-4-yl) | 4.99±0.29 | 10.50±1.3 | 19.62±5.35 | |
| 54 | R1, R2 = − (−CH2CH2−)2CH(pipyridin-1-yl) | 48.42±6.92 | >50.00 | >50.00 | |
| 55 |
|
9.35±0.60 | 41.50±3.71 | 24.87±4.33 | |
| 56 |
|
7.82±0.71 | 24.36±2.32 | 32.00±3.93 | |
|
| |||||
| 57 | R1 = H, R2 = benzyl | 2.77±1.33 | 4.92±1.64 | 3.81±1.78 | |
| R3, R4 & R5 = OCH3 | 58 | R1 = H, R2 = furan-2-ylmethyl | 2.88±0.86 | 3.88±0.76 | 5.51±2.05 |
| 59 | R1, R2 = − (−CH2CH2−)2NCH2CH2CH2N(CH3)2 | 3.69±0.08 | 9.61±0.62 | 7.34±0.23 | |
The N,N-dimethylaminoethyl amide derivatives 15 and 42 were treated with Boc-L-valine or Boc-L-proline in the presence of HATU [30] to give the BOC-protected amides 60–63. Removal of the BOC group with 1 N HCl yielded the target compounds in good yields (Scheme 2). Sulindac analogs 3–63 were screened against three cancer cell lines—prostate, colon, and breast—using a quantitative high-throughput screen (qHTS) format according to the method of Mathew et al [31].
Scheme 2.
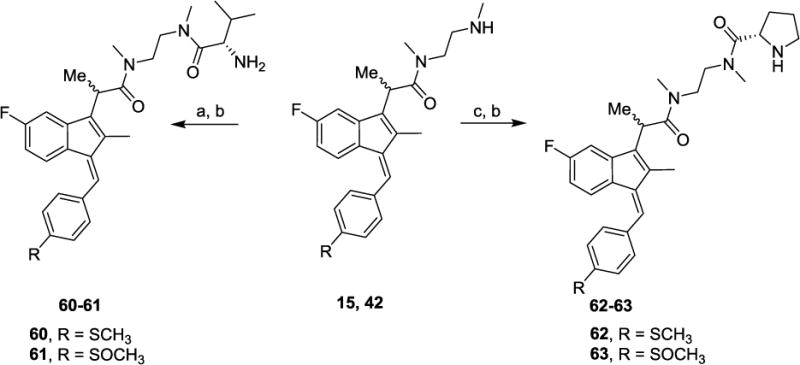
Synthetic pathways to analogs 60–63. Reagents and conditions: (a) Boc-L-valine, HATU, DIEA, MeCN (b) 1 N HCl (c) Boc-L-proline, HATU, DIEA, MeCN
Additional screening of selected compounds was performed at St. Jude Children’s Research Hospital against a panel of cancer cell lines, which consisted of CPC300 cells derived from a mouse model of choroid plexus carcinoma (CPC), where the mouse model has the genetic background Trp53LoxP-RBLoxP-PtenLoxP. In addition, four acute lymphoblastic leukemia (ALL) cell lines and one lymphoma line were included as follows: a. a cell line established from the peripheral blood of a 12-year old girl with ALL at relapse in 1977 that carries a near haploid karyotype (NALM16 cells); b. RAJI: Burkitt’s lymphoma, with FAB L3 (RAJI cells); c. acute T cell leukemia Jurkat e6–1 cells (JURKT cells); d. a precursor B-cell ALL patient-derived cell line expressing only wild-type MLL and wild-type AF4 (REH cells); e. a cell line (697) established from bone marrow cells obtained from children with ALL in relapse.
Screening results for all reported compounds against prostate, colon and breast cancer cell lines are summarized in Tables 1 and 2. The incorporation of a methyl group at the α-position resulted in significant changes in the anticancer activity. To further explore SAR, modifications were also made at the acetamide linker and benzylidene ring. Compounds 3–29 have a 4-methylthiobenzylidene ring at the C-1 position and an α-methyl group at the C-3 position while varying the amide. These compounds are racemic mixtures except in the cases noted below where a chiral amine was used to form the terminal amide. All these compounds showed significant activity against cancer but were less potent than the parent SSA. Compounds 3 and 4, with a benzyl ring or furyl ring at the acetamide linker, did not show significant activity against the three cancer cell lines. In the case of 5, both epimers were separately screened against the cancer cells. Notably, stereoisomer 5 showed similar activity to epi-5. The compounds with an ethylenediamino or a propylenediamino acetamide linker at the C-3 position (6–20) displayed excellent activity against HT29, PC3 and MDA-MB-231 cells with CC50 values ranging from 1.13–7.27 μM. The compounds (21–27) having heterocyclic amides showed significant activity against all three cancer cell lines similar to acyclic amide analogs 6–20. As mixtures of diastereomers, compounds 28 and 29 demonstrated excellent inhibitory activity against all three cancer cell lines, but separation would be necessary to demonstrate differential isomer activity.
Table 2.
Screening data compounds 60–63.
| Compounds | CC50 (μM)
|
||
|---|---|---|---|
| HT29 | PC3 | MDA-MB-231 | |
| 60 | 2.22±0.10 | 4.83±0.24 | 3.86±0.24 |
| 61 | 26.04±2.07 | 28.84±1.52 | 47.57±19.45 |
| 62 | 2.06±0.13 | 5.21±0.31 | 3.99±0.24 |
| 63 | 20.05±1.37 | 27.89±1.86 | >50.00 |
We also explored the SAR of selected acetamide modifications in the 4-methylsulfinylbenzylidene series (30–56). Due to the chirality of the sulfoxide and the newly installed α-methyl groups, these compounds were produced as inseparable mixtures of diastereomers. Several compounds in this group were found to inhibit the cancer cell lines, but all were, in general, 10–15 fold less active than corresponding thiobenzylidene analogs, suggesting that the 4-methylsulfinyl substitution is not favorable. For three selected acetamides, we explored the 3,4,5-trimethoxy substituted benzylidene (57–59) as racemic mixtures. Compounds 57 and 58 showed better activity than their corresponding 4-methylthiobenzylidene (3, 4) or 4-methylsulfinylbenzylidene (28, 29) analogs. In contrast, compound 59 was less active than its corresponding 4-methylthiobenzylidene analog 25, but more active than its 4-methylsulfinylbenzylidene analog 52.
Intestinal cells, cancer cells, and other cell types are known to have specific amino acid (AA) uptake mechanisms that have been utilized to increase drug uptake upon AA conjugation. For example, conjugation of the active HSV agents acyclovir and penciclovir as their valyl esters through the AA-CO2H group results in blood levels 3–5 times that of the parent drug due to specific valine uptake mechanisms through the intestinal epithelium [32, 33]. Hence, a small diamide library was prepared by coupling L-valine and L-proline with compounds 15 and 42 (Scheme 2) to create potential candidates for improved uptake in vivo. Table 2 lists the anticancer activity of the inseparable diastereomeric diamides 60–63. The 4-methylthiobenzylidene analogs 60 and 62 displayed comparable activity to the parent comparison 15, and the methylsulfinyl analogs 61 and 63 showed a similar decrease in activity relative to the parent 42. Since 15 has comparable activity to our lead control agent SSA, both 60 and 62 may be candidates for assessment of the effects of AA (specifically valyl or prolyl) conjugation on gastric uptake and bioavailability.
Anticancer activities of selected compounds have been evaluated against an additional panel of cancer cell lines at St. Jude Children’s Research Hospital [four acute lymphoblastic leukemia (ALL) cell lines, one lymphoma line, and a cell line derived from a mouse model of choroid plexus carcinoma (CPC300)], namely, 6–29, 57, 59, 60 and 62 (see methods in [34]). To address possible toxicity concerns associated with lipophilic basic amine scaffolds, the series 6–29 and 59–62 were also evaluated in a BJ cytotoxicity assay (description provided in Supplemental Materials). All screened compounds showed EC50 > 7.57 μM, suggesting no overt cytotoxicity for any of these compounds. Compounds showing activity against cell lines in the panel are listed in Table 3, including compound 14 with sub-nanomolar and 20 and 24 with low micromolar inhibition against a leukemia cell line. Racemic 57 is a promising lead for further development, since it shows good inhibitory activity against five acute lymphoblastic leukemia cell lines used on this panel (Table 3), while also potently inhibiting prostate, colon and breast cancer cell lines (Table 1). In comparison, lead compound sulindac sulfide amide (SSA) showed no activity against any of the cancer cell lines in this panel.
Table 3.
Cancer cell line screening data. Confidence intervals are shown in parentheses; NA: not active; ND: undetermined due to questionable curve fit.
| Cmpds | EC50 (μM)
|
||||||
|---|---|---|---|---|---|---|---|
| CPC300 | NALM16 | RAJI | JURKT | REH | 697 | BJ | |
| 6 | NA | 76 (28–73) | NA | NA | NA | NA | >7.57 |
| 8 | NA | NA | NA | NA | 14 (13–26) | NA | >7.57 |
| 9 | NA | NA | NA | NA | NA | NA | >7.57 |
| 11 | NA | NA | NA | NA | 38 (30–48) | NA | >7.57 |
| 14 | NA | NA | <0.00038 | NA | 10 (9.6–12) | NA | >7.57 |
| 19 | NA | NA | NA | NA | NA | NA | >7.57 |
| 20 | NA | 1.5 (0.58–1.8) | NA | NA | NA | NA | >7.57 |
| 21 | NA | NA | NA | 76 (16–66) | 76 (19–72) | 76 (28–73) | >7.57 |
| 23 | NA | NA | 45 (19–73) | NA | 26 (19–39) | NA | >7.57 |
| 24 | NA | 2.1 (1.2–2.2) | NA | NA | NA | NA | >7.57 |
| 26 | NA | NA | NA | NA | NA | 76 (58–75) | >7.57 |
| 29 | NA | NA | ND | NA | 11 (10–13) | NA | >7 .57 |
| 57 | NA | 8 (7.9–8) | 13 (13–13) | 7.7 (7.4–7.9) | 7 (6.9–7.1) | 6.1 (6–6.1) | >7.57 |
As a critical step in further selection of candidates for development, metabolic profiling is crucial to determine likely in vivo stability. Such information can be useful in moving candidates into various anticancer animal models. As such, we assessed the effects of the various acetamide linker modifications on metabolic vulnerability utilizing the robust CYP3A4 metabolism model implemented in StarDrop (from Optibrium Ltd.) to predict metabolic sites that may contribute to the metabolism of various analogs. For these computations, we selected compounds with low micromolar potencies against HT29, PC3 and MDA-MB-231 cancer cell lines comparable to SSA that also retained the 4-methylthiobenzylidene group at the C-1 position of the indene moiety. This allowed for a focused comparison of the metabolic stability of the altered linker region with respect to SSA. Although the N,N-dimethylaminoethyl group in the acetamide linker of SSA contains two labile sites (Figure S-1), we identified a number of analogs that contain linkers devoid of predicted labile sites, namely, compounds 22, 23, 26, 27, 60 and 62 as shown in Supplemental Figures S-2 through S-7, respectively. We also identified analogs 21, 24, 28 and 29 as compounds with reduced predicted metabolic lability in the linker region compared to SSA. These results suggest that metabolically labile sites in the linker region of SSA may be removed through cyclization or substitution of the basic amine moiety leading to analogs with reduced vulnerability to metabolic transformation in this region. This information may be useful in selecting particular compounds in this series for in vivo evaluation or in generation of additional synthetic analogs to reduce metabolic liabilities.
In conclusion, we identified a series of α-methyl sulindac amides with good anticancer activity compared to the lead agent SSA. 4-Methylthiobenzylidene analogs (3–29) displayed better activity relative to 4-methylsulfinylbenzylidene or 3,4,5-trimethoxybenzylidene analogs (30–59). A number of new analogs with comparable activity to the lead SSA were identified. In general, α-methylation of the sulindac amide scaffold results in only slightly diminished activity relative to the parent unmethylated analogs suggesting that alterations at this position have limited impact on overall potency, although potentially leading to distinct chemotypes with interesting activity profiles. For example, racemic compound 57 is a neutral analog at physiological pH and is a reasonably potent inhibitor in the three cancer cell lines screened as well as being active against four acute lymphoblastic leukemia cell lines and one lymphoma line (while showing no toxicity against a normal human foreskin fibroblast BJ cell line). Future studies relating to the mechanism of action, cyclo-oxygenase inhibition and selectivity of these derivatives may further elucidate the potential value of this substitution relative to known drugs that contain an α-methyl function. For example, the profen class of NSAIDs is generally a racemic mixture at the α-methyl position. When enantiomers are separated (e.g., in flurbiprofen), the two analogs exhibit altered target activities and differential toxicity profiles, once again demonstrating the variety of target activities of the broad NSAID class [35]. In fact, the R-enantiomer of flurbiprofen, like dexibuprofen, has been examined for the treatment of early stage Alzheimer’s disease [28, 35]. For the more promising analogs reported herein, future work will entail preparation and analysis of individual stereoisomers and more detailed profiling of toxicity and mechanisms of activity in an effort to better assess the potential role of α-methylation in NSAID SAR, not only as it relates to cancer chemoprevention, but also other possible indications.
Supplementary Material
Anticancer activities of 60 new α-Me analogs of sulindac sulfide amide are reported.
Several compounds (6–29 and 60) show comparable inhibition of prostate, colon, breast cancer.
Addition of an α-methyl group to the lead scaffold does not dramatically alter activity.
Several analogs (6, 8, 11, 14, 20, 21, 24, 29, 57) show activity against several other cell lines.
Separated isomers, 5 and epi-5, show similar activities.
Acknowledgments
This work was supported by grants from the National Institutes of Health NCI 1R01CA131378 (RCR PI) and DOD Era of Hope award W81XWH-07-1-0463 (RCR PI). We thank Lucile White, Lynn Rasmussen and Melinda Ingram of Southern Research for HTS support services. Additionally, Judith V. Hobrath of the Drug Discovery Unit in the College of Life Sciences at the University of Dundee gave critical input into manuscript preparation and provided information relative to metabolism of the sulindac analogs presented in this work. We would also like to acknowledge contributions by the High Throughput Biology Center in the Department of Chemical Biology and Therapeutics at St. Jude Children’s Research Hospital in Memphis, TN for BJ cell line data and additional anticancer cell line screens. The work at St. Jude was supported by American Lebanese Syrian Associated Charities (ALSAC).
Abbreviations
- NSAID
non-steroidal anti-inflammatory drug
- COX
cyclooxygenase
- PG
prostaglandin
- FAP
familial adenomatous polyposis
- SSA
sulindac sulfide amide
- SAR
structure-activity relationship
- qHTS
quantitative high-throughput screen
- CPC
choroid plexus carcinoma
- ALL
acute lymphoblastic leukemia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary materials.
Supplementary materials contain metabolic site predictions and additional experimental details.
References
- 1.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 2.Reeder MK, Pamakcu R, Weinstein IB, Hoffman K, Thompson WJ. In: Cancer chemoprevention, Promising cancer chemoprevention agents. Hawk ET, Kelloff GJ, Sigman CC, editors. Totowa (NJ): Humana Press Inc; 2004. p. 401. [Google Scholar]
- 3.Soh JW, Weinstein IB. Role of COX-independent targets of NSAIDs and related compounds in cancer prevention and treatment. Prog Exp Tumor Res. 2003;37:261–285. doi: 10.1159/000071377. [DOI] [PubMed] [Google Scholar]
- 4.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 5.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3:166–174. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 6.Vane JR, Botting RM. Mechanism of action of antiinflammatory drugs. Int J Tissue React. 1998;20:3–15. [PubMed] [Google Scholar]
- 7.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem Pharmacol. 2002;63:817–821. doi: 10.1016/s0006-2952(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 9.Cannon CP, Cannon PJ. COX-2 inhibitors and cardiovascular risk. Science. 2012;336:1386–1387. doi: 10.1126/science.1224398. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra154. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricchi P, Zarrilli R, Di Palma A, Acquaviva AM. Nonsteroidal anti-inflammatory drugs in colorectal cancer: from prevention to therapy. Br J Cancer. 2003;88:803–807. doi: 10.1038/sj.bjc.6600829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alberts DS, Hixson L, Ahnen D, Bogert C, Einspahr J, Paranka N, Brendel K, Gross PH, Pamukco R, Burt RW. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J Cell Biochem. 1995;59:18–23. doi: 10.1002/jcb.240590804. [DOI] [PubMed] [Google Scholar]
- 13.Piazza GA, Keeton AB, Tinsley HN, Whitt JD, Gary BD, Mathew B, Singh R, Grizzle WE, Reynolds RC. NSAIDs: old drugs reveal new anticancer targets. Pharmaceuticals. 2010;3:1652–1667. doi: 10.3390/ph3051652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin T, Wang G, Ye T, Wang Y. Sulindac, a non-steroidal anti-inflammatory drug, mediates breast cancer inhibition as an immune modulator. Scientific Reports. 2016;6:19534–41. doi: 10.1038/srep19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochemical Pharmacology. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 16.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3:1679–1683. [PubMed] [Google Scholar]
- 17.Liggett JL, Zhang X, Eling TE, Baek SJ. Anti-tumor activity of non-steroidal anti-inflammatory drugs: Cyclooxygenase-independent targets. Cancer Letters. 2014;346:217–224. doi: 10.1016/j.canlet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoner GD, Budd GT, Ganapathi R, DeYoung B, Kresty LA, Nitert M, Fryer B, Church JM, Provencher K, Pamukcu R, Piazza G, Hawk E, Kelloff G, Elson P, van Stolk RU. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv Exp Med Biol. 1999;470:45–53. doi: 10.1007/978-1-4615-4149-3_5. [DOI] [PubMed] [Google Scholar]
- 19.Keller JJ, Offerhaus GJ, Polak M, Goodman SN, Zahurak ML, Hylind LM, Hamilton SR, Giardiello FM. Rectal epithelial apoptosis in familial adenomatous polyposis patients treated with sulindac. Gut. 1999;45:822–828. doi: 10.1136/gut.45.6.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal B, Rao CV, Bhendwal S, Ramey WR, Shirin H, Reddy BS, Holt PR. Lovastatin augments sulindac-induced apoptosis in colon cancer cells and potentiates chemopreventive effects of sulindac. Gastroenterology. 1999;117:838–847. doi: 10.1016/s0016-5085(99)70342-2. [DOI] [PubMed] [Google Scholar]
- 21.Rahman MA, Dhar DK, Masunaga R, Yamanoi A, Kohno H, Nagasue N. Sulindac and exisulind exhibit a significant antiproliferative effect and induce apoptosis in human hepatocellular carcinoma cell lines. Cancer Res. 2000;60:2085–2089. [PubMed] [Google Scholar]
- 22.Du J, Guo Y, Bao Y, Xing M, Mahmoud AM, Che Z, Chen Z, Yang W. Anticancer activities of sulindac in prostate cancer cells associated with c-Jun NH2-terminal kinase 1/β-catenin signaling. Oncology Letters. 2014;8:313–316. doi: 10.3892/ol.2014.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH, Brendel K, Burt RW, Alberts DS, Pamukcu R, Ahnen DJ. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–3116. [PubMed] [Google Scholar]
- 24.Han EK, Arber N, Yamamoto H, Lim JT, Delohery T, Pamukcu R, Piazza GA, Xing WQ, Weinstein IB. Effects of sulindac and its metabolites on growth and apoptosis in human mammary epithelial and breast carcinoma cell lines. Breast Cancer Res Treat. 1998;48:195–203. doi: 10.1023/a:1005924730450. [DOI] [PubMed] [Google Scholar]
- 25.Lim JT, Piazza GA, Han EK, Delohery TM, Li H, Finn TS, Buttyan R, Yamamoto H, Sperl GJ, Brendel K, Gross PH, Pamukcu R, Weinstein IB. Sulindac derivatives inhibit growth and induce apoptosis in human prostate cancer cell lines. Biochem Pharmacol. 1999;58:1097–1107. doi: 10.1016/s0006-2952(99)00200-2. [DOI] [PubMed] [Google Scholar]
- 26.Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA, Jr, Chou T-C. Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res. 1999;59:6178–6184. [PubMed] [Google Scholar]
- 27.Piazza GA, Keeton AB, Tinsley HN, Gary BD, Whitt JD, Mathew B, Thaiparambil J, Coward L, Gorman G, Li Y, Sani B, Hobrath JV, Maxuitenko YY, Reynolds RC. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev Res (Phila) 2009;2:572–580. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ettcheto M, Sánchez-López E, Pons L, Busquets O, Olloquequi J, Beas-Zarate C, Pallas M, Garcia ML, Auladell C, Folch J, Camins A. Dexibuprofen prevents neurodegeneration and cognitive decline in APPswe/PS1dE9 through multiple signaling pathways. Redox Biol. 2017;13:345–352. doi: 10.1016/j.redox.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyamoto K, Tsuchiya S, Ohta H. Stereochemistry of Enzyme-Catalyzed Decarboxylation of Alpha-Methyl-Alpha-Phenylmalonic Acid. Journal of the American Chemical Society. 1992;114:6256–6257. [Google Scholar]
- 30.Carpino LA. 1-Hydroxy-7-Azabenzotriazole - an Efficient Peptide Coupling Additive. Journal of the American Chemical Society. 1993;115:4397–4398. [Google Scholar]
- 31.Mathew B, Hobrath JV, Lu W, Li Y, Reynolds RC. Synthesis and preliminary assessment of the anticancer and Wnt/β-catenin inhibitory activity of small amide libraries of fenamates and profens. Medicinal Chemistry Research. 2017;26:3038–3045. doi: 10.1007/s00044-017-2001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DK, Lee N, Kim YW, Chang K, Im G-J, Choi W-S, Kim KH. Synthesis and evaluation of amino acid esters of 6-deoxypenciclovir as potential prodrugs of penciclovir. Bioorg Med Chem. 1999;7:419–424. doi: 10.1016/s0968-0896(98)00235-1. [DOI] [PubMed] [Google Scholar]
- 33.Han HK, Oh DM, Amidon GL. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharm Res. 1998;15:1382–1386. doi: 10.1023/a:1011945420235. [DOI] [PubMed] [Google Scholar]
- 34.Mathew B, Hobrath JV, Connelly MC, Guy RK, Reynolds RC. Bioorganic & Medicinal Chemistry Letters. 2017;27:4614. doi: 10.1016/j.bmcl.2017.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith KDB, Paylor R, Pautler RG. R-Flurbiprofen Improves Axonal Transport in the Tg2576 Mouse Model of Alzheimer’s Disease as Determined by MEMRI. Magn Reson Med. 2011;65:1423–1429. doi: 10.1002/mrm.22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.