

Abstract
Introduction:
Ibrutinib is the first BTK inhibitor to show efficacy in chronic lymphocytic leukemia (CLL) and is also the first BTK inhibitor to which patients have developed resistance. Mutations in BTK and PLCG2 are found in ≈80% of CLL patients with acquired resistance to ibrutinib, but it remains unclear if these mutations are merely associated with disease relapse or directly cause it.
Areas covered:
Unique properties of both CLL and ibrutinib that complicate attempts to definitively conclude whether BTK/PLCG2 mutations are passengers or drivers of ibrutinib-resistant disease are reviewed. Characteristics of mutations that drive drug resistance are summarized and whether BTK/PLCG2 mutations possess these is discussed. These characteristics include (1) identification in multiple patients with acquired resistance, (2) in vitro validation of drug-resistant properties, (3) mutual exclusivity with one another, (4) increasing frequency over time on drug, and (5) high frequency at the time and site of clinical relapse.
Expert Commentary:
While BTK/PLCG2 mutations have characteristics suggesting that they can drive ibrutinib resistance, this conclusion remains formally unproven until specific inhibition of such mutations is shown to cause regression of ibrutinib-resistant CLL. Data suggest that alternative mechanisms of resistance do exist in some patients.
Keywords: BTK inhibitors, chronic lymphocytic leukemia, ibrutinib, PLCG2
1. Introduction
Ibrutinib is a covalent, irreversible inhibitor of Bruton Tyrosine Kinase (BTK) that is remarkably effective for the treatment of chronic lymphocytic leukemia (CLL). Overall response rates are approximately 70–90%, even in patients with high risk features such del(17p) and unmutated IGHV [1, 2, 3, 4]. Ibrutinib significantly prolonged overall survival in phase 3 trials in both the relapsed/refractory and front-line settings versus ofatumumab and chlorambucil, respectively [1–3]. Its outstanding efficacy is presumed to stem from its ability to inhibit signal transduction through the B Cell Receptor (BCR) signaling pathway (Figure 1), a pathway thought to be critical for the proliferation and survival of CLL cells.
Figure 1:
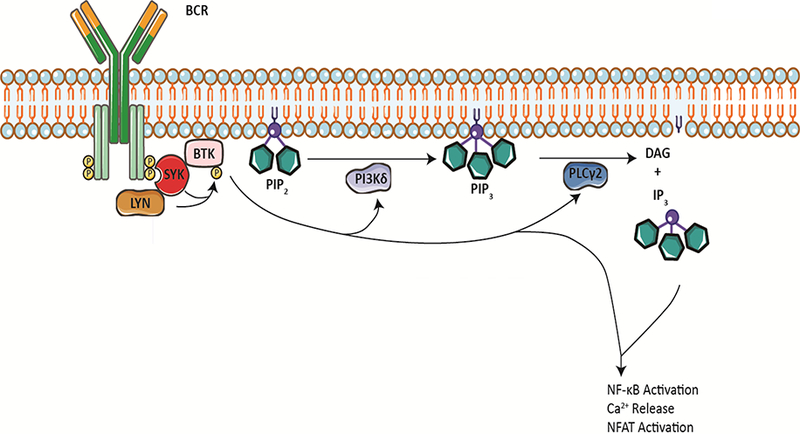
BTK is engaged downstream of the constitutively activated B cell receptor (BCR) in CLL. This leads to activation of phosphatidylinositol-3-kinase delta (PI3K?) and PLC?2 with subsequent hydrolysis of phosphatidylinositol-3-phosphate (PIP3) into diacylglycerol (DAG) and inositol-3-phosphate (IP3). Events downstream of activated BTK and PLC?2 include, but are not limited to, intracellular Ca2+ release as well as NF-?B and NFAT pathway activation.
Despite this success, resistance to ibrutinib does occur, particularly in patients with high-risk disease biology (e.g. del(17p)) and heavily pretreated disease. In approximately 80% of patients, development of resistance is associated with mutations in BTK itself as well as in phospholipase Cγ2 (PLCG2) [5, 6, 7, 8]. These BTK mutations have been assumed to be the causative drivers of ibrutinib-resistant disease, as they affect the residue in BTK to which ibrutinib covalently binds and allow for continued signaling through the BCR signaling pathway in vitro even in the presence of ibrutinib [7, 8]. However, biochemical evidence that a particular BTK mutation renders the protein drug-resistant in vitro does not necessarily prove that such a mutation is responsible for the in vivo clinical phenotype of ibrutinib resistance and disease progression. In an individual patient, such mutations could hypothetically be bystanders associated with relapse, the exclusive and driving cause of relapse, or one of multiple co-occurring mechanisms causing relapse. Distinguishing between these possible explanations is important because the presumed efficacy of inhibitors targeting mutant BTK or PLCG2 will differ in each setting. Within this review, we examine evidence regarding whether BTK and PLCG2 mutations are necessary and sufficient for CLL relapse in patients who develop them while on ibrutinib therapy.
2. Reasons for discontinuation of ibrutinib therapy
Reported rates of ibrutinib discontinuation range from 28–51% after approximately three years of follow-up [5, 9]. Analyses of hundreds of ibrutinib-treated patients at various academic medical centers have identified three main reasons for ibrutinib discontinuation. First, a substantial portion of patients discontinue ibrutinib for reasons other than disease progression. These reasons include adverse events and drug intolerance (e.g. infection, atrial fibrillation, and gastrointestinal side effects) [4, 5, 9]. Age is a significant risk factor for discontinuations within this category [6], and these types of discontinuation more commonly occur in the initial years that a patient is on ibrutinib [5]. Thus, the fraction of discontinuation events that are attributable to reasons other than disease progression varies based on the patient population and length of follow-up, with frequencies ranging from 48% after 3.4 years of follow-up at OSU [5], to 63% after 3.2 years of follow-up at MD Anderson [9], to 75% in a multicenter retrospective study with 1.4 years of follow-up [4]. BTK and PLCG2 mutations have not been systematically studied in this population, and thus we will not focus any further on this group in this review.
A second portion of patients discontinue ibrutinib due to Richter’s transformation (RT) to a more aggressive lymphoma. Transformation tends to occur within the first two years and then the incidence appears to plateau. Reported rates are 5–7.3% of all patients after 17–24 months of ibrutinib [4, 5, 9, 10]. The frequency of BTK and PLCG2 mutations in RT is different than the frequency of such mutations in patients who have progressive CLL on ibrutinib without transformation. In the peripheral blood, BTK and PLCG2 mutations are found in about 80% of patients with progressive CLL (Table 1) but only about 40% of patients with RT [5, 11]. Likely due to the greater difficulty in obtaining transformed tissue, the molecular details of the Richter’s tissue remain largely unexplored to date. However, one study has reported that three out of six cases of RT displayed BTK mutations in the nodal tissue [11]. The different BTK/PLCG2 mutational frequency in RT suggests an underlying biology that is distinct from simple progression of CLL without transformation (discussed below). Given this distinct biology and the need for more comprehensive molecular analyses of RT, our discussion of BTK and PLCG2 mutations in this review will primarily pertain to progression of CLL without transformation.
Table 1:
Selected Sequencing Studies of Ibrutinib-Resistant CLL Patient Populations - Simple CLL Progression Only
| Number (%) of Patients with Indicated Mutations | Number of Ibrutinib-Resistant Patients with summed BTK/PLCG2 VAFs of:2 |
|||||||
|---|---|---|---|---|---|---|---|---|
| Study | Number of Patients1 |
BTK only | PLCG2 only | BTK and PLCG2 | No identified BTK or PLCG2 mutation |
<10% | 10–30% | >30% |
| Maddocks [6]3 | 11 | 7 (64%) | 2 (18%) | 2 (18%) | 0 (0%) | 2 (18%) | 2 (18%) | 7 (64%) |
| Woyach [5]3 | 35 | 25 (71%) | 1 (3%) | 3 (8.6%) | 6 (17%) | 5 (17%) | 7 (24%) | 17 (59%) |
| Ahn [10]4 | 10 | 2 (20%) | 1 (10%) | 5 (50%) | 2 (20%) | 3 (43%) | 0 (0%) | 4 (57%) |
| Kadri [11] | 3 | 1 (33%) | 0 (0%) | 0 (0%) | 2 (67%) | 0 (0%) | 0 (0%) | 1 (100%)5 |
| Burger [18] | 5 | 1 (20%) | 1 (20%) | 0 (0%) | 3 (60%) | Not reported | ||
| Totals | 64 | 36 (56%) | 5 (7.8%) | 10 (16%) | 13 (20%) | 10 (21%) | 9 (19%) | 29 60%) |
Number of patients reflects only those patients in the studies that had CLL simple progression and next generation sequencing of the BTK and PLCG2 loci.
Only patients that had detectable BTK and/or PLCG2 mutations are included in the percentage calculations.
Eleven patients in the Maddocks cohort are also in the Woyach cohort. These eleven patients have been removed from the statistics reported for the Woyach cohort to avoid double counting.
One subject with BTK and PLCG2 mutations did not have VAF testing; the denominator for VAF of the various frequencies is seven patients.
Cancer cell fraction, and not variant allele frequency, was reported.
The final common reason for ibrutinib discontinuation is CLL simple progression. To clearly define this term, by “CLL simple progression” we mean the appearance of relapsed disease according to iwCLL 2008 criteria [12] while on ibrutinib without transformation to other hematologic malignancies. As with the other reasons for ibrutinib discontinuation, the rates of CLL simple progression vary over time. The cumulative incidence of CLL simple progression was 5% at 2 years, 10.8% at 3 years, and 19.1% at 4 years in a recent study [5]. This nearly exponential pattern to the development of acquired resistance is characterized by very low rates of progression initially and much higher rates the longer that a patient is on therapy; similar kinetics have been seen in other cohorts of CLL patients with simple progression [9]. The majority of patients with this type of progression display BTK and/or PLCG2 mutations and will thus be the focus of our review. An aggregation of cases from five independently reported cohorts demonstrates that 51 out of 64 patients (80%) had mutations in either BTK and/or PLCG2 at the time of clinical relapse (Table 1). While these mutations are therefore associated with ibrutinib resistance, this does not necessarily prove that they are the cause of ibrutinib resistance. An alternative hypothesis -- that cells with such mutations are good markers for ibrutinib-resistant disease but do not necessarily drive the phenotype -- could also be consistent with the high rates of BTK and PLCG2 mutations found in patients with simple progression. Distinguishing between these hypotheses may be particularly difficult in CLL, as discussed below.
3. Correlation versus causation: unique aspects that make the distinction difficult
In certain other malignancies, there is less doubt about the causative role that specific mutations in driving oncogenes play in acquired resistance. For example, the T315I mutation in BCR-ABL is accepted as the driver of resistance to imatinib, dasatinib, and bosutinib in chronic myelogenous leukemia (CML) [13]. A corresponding mutation in the gatekeeper amino acid in EGFR, T790M, confers acquired resistance to erlotinib and gefitinib in EGFR-mutant lung adenocarcinoma [14]. Certain characteristics of both CLL and ibrutinib complicate the picture and make it more difficult to establish this link in the case of BTK and PLCG2 mutations.
First, there are no sine qua non truncal mutations in CLL. In other words, CLL has no driving somatic mutations that meet the following criteria: (1) present within all or almost all patients with CLL; and (2) present in all evolving tumor subclones of a given patient, suggesting that they were present in the initiating neoplastic cell. This is in contrast to typical CML, diagnosis of which requires the presence of the BCR-ABL fusion gene [15], and also in contrast to EGFR-mutant lung adenocarcinomas, which by definition carry an activating mutation in EGFR. In these diagnoses, since the truncal oncogenic mutation was presumably a key initiating event and the cancer cells all thus depend on the activated oncogene for survival, the driver of resistance to a drug targeting that oncogene will logically be an acquired mutation within the oncogene itself. Rather than dependence on a single oncogene, CLL cells are thought to have dependence on a signal transduction pathway, the BCR signaling pathway [16]. In CLL cells, tonic and activated signaling through the BCR activates kinases, such as BTK, that lead to upregulation of the activity of transcription factors (e.g. NF-κΒ and NFAT) that ultimately promote cell survival and proliferation (Figure 1). In vitro, loss of signaling through this pathway (as demonstrated through the use of kinase inhibitors including ibrutinib) disrupts pro-survival signals from microenvironmental stimuli, resulting in decreased CLL cell proliferation and modest increases in CLL cell apoptosis [17]. A priori, it is unclear how the development of resistance may differ in neoplastic cells that are addicted to oncogenic pathways (such as the BCR pathway) rather than individual oncogenes. While drug-resistant mutants in key proteins of the pathway are one explanation for how resistance could develop in this setting, augmentation of parallel pathways could also cause resistance. Dependence on a pathway, rather than a single gene, likely expands the number of routes to resistance.
Second, CLL is generally an indolent disease. The initial development of disease and the subsequent development of drug resistance take place over months to years. Since the time between the first exposure to ibrutinib and the subsequent clinical phenotype of drug resistance is somewhat lengthy, it is more difficult to definitively link the two than if the time between them were very short. That being said, the kinetics of the development of drug resistance in CLL and chronic phase CML are quite similar, suggesting that conclusions from imatinib resistance in CML (where drug-resistant point mutations are common) may inform conclusions about ibrutinib resistance in CLL. For example, the growth rate of ibrutinib-resistant CLL clones was estimated to be 1.5–1.9% per day [18], while the growth rate of imatinib-resistant CML clones was estimated at 2% per day [19].
A third complicating factor when studying ibrutinib resistance in CLL is that the site of resistance is heterogeneous among patients and not always biopsied. Since samples of neoplastic tissue are easily obtained by venipuncture, peripheral blood samples remain the foundation of studies of the genomics of simple progression [5, 6]. However, simple progression does not always occur solely in the blood. Some patients have isolated lymphadenopathy, some patients have isolated lymphocytosis, and some patients have both [5, 6]. Focusing only on mutations found in the peripheral blood ignores the fact that resistance is polyclonal [5, 10, 18] and resistant clones - particularly those that drive the resistant phenotype of progressive lymphadenopathy while on ibrutinib -- may be separated in space from the clones that are ultimately obtained and analyzed.
A fourth complicating factor is ibrutinib’s unique status as an irreversible kinase inhibitor. Ibrutinib, osimertinib, afatinib, and neratinib are the only FDA-approved irreversible kinase inhibitors, targeting BTK, EGFR, EGFR/HER2, and HER2, respectively [20]. The mechanisms by which resistance develops to these irreversible kinase inhibitors may be conceptually distinct from the mechanisms by which resistance develops to reversible kinase inhibitors [21]. Drug-resistant mutants to reversible kinase inhibitors are often characterized by their effects on IC50 values, the amount of drug needed to achieve 50% inhibition in a given biochemical or cellular assay. This IC50 is arbitrary when applied to irreversible inhibitors, as it depends on both the drug’s effect on the enzyme and the time that the target is exposed to drug. With irreversible inhibitors, even mutants that react more slowly with the inhibitor will become fully inactivated given long enough contact with the drug [21]. Therefore, knowledge regarding resistance to reversible kinase inhibitors may not be applicable to ibrutinib in CLL. For example, one of the biochemical effects of the BTK C481S mutation is conversion of ibrutinib from an irreversible inhibitor of BTK to a reversible inhibitor of BTK. Is this change, from an irreversible to a reversible (yet still functional) inhibitor, all that is needed to drive clinical resistance? Prior experiences with reversible inhibitors such as imatinib and erlotinib/gefitinib cannot help with this question. Prior experience with the irreversible inhibitor afatinib suggests that point mutations at a gatekeeper threonine residue can be associated with resistance [22], and acquired resistance to osimertinib is also associated with point mutations in EGFR and is discussed further in section 4.3.
A fifth complicating factor is that ibrutinib’s therapeutic effect may be both direct, due to effects on the neoplastic cells themselves, but also indirect, through effects on the microenvironment [23]. Ibrutinib cause downregulation of the PD-1 exhaustion marker on CD4+ and CD8+ T cells as well as downregulation of the concomitant ligand PD-L1 on neoplastic B cells [24]. Through nonspecific inhibition of ITK, ibrutinib alters differentiation of T cell subsets, skews T cells from a Th2-dominant to a Th1 and CD8+ cytotoxic population [25], and blocks the secretion of cytokines from activated T cells [17]. Ibrutinib also leads to expansion of effector and effector memory T cell subsets [26]. Since this broad activity against both the neoplastic cells and the supportive microenvironment may contribute to the clinical efficacy of ibrutinib, alterations within either of these cellular compartments might also contribute to resistance. [23, 27]
Resistance to targeted therapies is a new avenue of study within the CLL field, so there is not much prior experience to provide guidance. Venetoclax resistance in CLL, for example, is even less well understood than ibrutinib resistance. Preclinical evidence suggests that both upregulation of alternative anti-apoptotic BCL-2 family members [27, 28] and development of point mutations in BCL-2 [28, 29] could be associated with venetoclax resistance. However, whether any of these mechanisms are seen in vivo, and whether these findings are correlative or causative, has not been established. Thus, while experience with imatinib resistance in CML could guide investigation of resistance to bosutinib and dasatinib, no such prior experience is available with ibrutinib in CLL.
4. BTK and PLCG2 mutations are not necessary for acquired resistance
BTK and PLCG2 mutations are not strictly necessary for resistance to ibrutinib, as not all patients who relapse, either with RT or with CLL simple progression, have detectable BTK or PLCG2 mutations (Table 1) [5, 11, 18]. Other genetic alterations have been proposed to cause ibrutinib resistance in the approximately 20% of CLL patients with acquired resistance in the absence of BTK or PLCG2 mutations. For example, a del(8p) deletion reported in three cases of ibrutinib-resistant CLL leads to haploinsufficiency of the TRAIL receptor, possibly rendering the neoplastic cells resistant to TRAIL-induced apoptosis [18]. Supporting the conclusion that BTK and PLCG2 mutations are not necessary for ibrutinib resistance, studies in other B cell lymphomas have also identified resistance mechanisms that do not involve mutations in BTK or PLCG2. In Waldenstrom’s macroglobulinemia and mantle cell lymphoma (MCL), mutations in CARD11 have been reported in ibrutinib-resistant disease and some of these mutations can block sensitivity of MCL cell lines to ibrutinib in vitro [30, 31]. Activating mutations in CARD11 also predict primary resistance to ibrutinib in follicular lymphoma [32] and diffuse large B cell lymphoma [33]. In sum, BTK and PLCG2 mutations are not necessary for resistance to ibrutinib.
5. Characteristics of mutations that are sufficient for acquired resistance
If one considers only patients with CLL simple progression who possess detectable BTK and/or PLCG2 mutations at the time of resistance, however, the question remains: are these mutations associated with resistance or its primary cause? We now review common characteristics of genetic mutations that are sufficient for acquired resistance to a drug and investigate whether the BTK and PLCG2 mutations that have been identified share these characteristics.
5.1. Identification of similar mutations in multiple patients with acquired resistance
The identification of multiple mutations within the same gene, preferably occurring at similar locations within the gene (i.e. a “hotspot”), provides circumstantial evidence that the mutation seen could be sufficient for the development of resistance. This is because the repeated identification of similar mutations in independent patients implies convergent evolution and selection for functional drug-resistant mutants rather than the random accumulation of scattered passenger mutations during the progression of disease. This finding is strengthened when such mutations are identified by multiple independent investigators. In the case of BTK and PLCG2 mutations in ibrutinib-resistant disease, this characteristic is clearly demonstrated. Nearly all reported BTK mutations in the dozens of cases reported in the literature have occurred at the cysteine 481 hotspot [5, 8, 10], except for a few additional cases reported with mutations at T316, T474, and L528 [6, 11, 34]. While the reported PLCG2 mutations are more varied, and involve amino acid residues P664, R665, S707, L845, D993, D1140, and M1141 [5, 7, 10], they seem to cluster within particular domains [35], rather than being randomly distributed throughout the protein. These BTK/PLCG2 mutations are not the only recurrent genetic events seen in ibrutinib-resistant patients. A deletion in the short arm of chromosome 8, del(8p), was identified in three out of five patients with acquired resistance to ibrutinib in the absence of any BTK mutations [18]. Additionally, two separate investigators have reported a total of three ibrutinib-resistant patients (including one without any BTK mutation) with non-overlapping mutations in the gene PCLO [11, 18]. Nevertheless, the demonstration of genetic alterations or mutations within the same gene in more than one patient is not proof that such mutations are sufficient for resistance. For example, it is unclear how PCLO, a cytoskeletal matrix protein involved in neuronal synaptic signaling, may cause ibrutinib resistance; this lack of obvious mechanism may just reflect our limited understanding, however. Consequently, identification of similar mutations in multiple patients is consistent with the hypothesis that such mutations drive resistance, but could also be seen by random chance or if there is selection for such mutations without requiring that they drive the resistant phenotype.
5.2. In vitro demonstration that enzymes carrying the mutations are drug-resistant
Mutations that are sufficient to drive progression must be resistant to the drug of interest in vitro. This has been demonstrated for both the BTK C481S and PLCG2 R665W and L845F mutations. In the case of BTK C481, a mutation to serine is the most commonly reported amino acid substitution; mutations to arginine, alanine, phenylalanine and tyrosine have also been reported [5, 10, 11]. All abolish the thiol group to which ibrutinib binds, and this leads to loss of covalent binding of a fluorescently tagged ibrutinib to BTK C481S and C481A [8]. BTK C481S is still functional and is still inhibited by ibrutinib, but the drug acts as a reversible inhibitor rather than an irreversible inhibitor [7, 8]. Cellular assays suggest that the IC50 for ibrutinib is 1006nM for the C481S mutant compared to 2.2nM for nonmutant BTK (the latter value must be interpreted with caution in light of the IC50 discussion above) [8]. These findings have been recapitulated in primary patient samples. For example, peripheral blood mononuclear cells taken from a CLL patient at baseline were treated with ibrutinib and showed loss of autophosphorylation of BTK upon either continuous exposure to the drug or brief exposure followed by washout. A second sample taken when the same patient had developed a BTK C481S mutation, however, showed loss of BTK auto-phosphorylation only upon continuous exposure to ibrutinib, but not with washout of the drug, consistent with conversion to a reversible inhibitor [7], Not all reported BTK mutations have similar functional consequences. When phenylalanine, arginine or tyrosine are substituted for C481, the enzyme has greatly reduced (and in some cases undetectable) autophosphorylation and activation of downstream PLCγ2 in cellular assays [36], raising questions as to how such mutations could drive clinical resistance. Additionally, the reported BTK T316A mutation seen in two ibrutinib resistant patients [11, 34] has also been found in the germline of a patient with a clinical phenotype consistent with a mild form of X linked agammaglobulinemia [37], suggesting that the mutation is a hypomorph. In sum, while C481S has been definitively established as a drug-resistant mutant in vitro, the evidence is not as conclusive for other less frequent BTK mutations.
Phospholipase Cγ2 is critical for BCR signaling [38]. PLCγl is widely expressed in all tissues but the expression of PLCγ2 is primarily restricted to hematopoietic cells. BCR crosslinking results in tyrosine phosphorylation and activation of PLCγ2 specifically [39]. While BTK directly phosphorylates and activates PLCγ2, the activity of other kinases such as SYK and LYN are necessary for signal transduction from the BCR to PLCγ2 [40]. A SH2 domain extending from amino acids 646 to 735 in PLCγ2 plays an autoinhibitory role. When there is active BCR signaling, the SH2 domain of PLCy2 binds to phosphorylated tyrosines on adaptor proteins of the signaling complex, thus relieving this domain’s repression of catalytic activity. Mutations within the SH2 domain inactivate its inhibitory function, leading to hyperfunctional PLCγ2 [41, 42]. Prior work had demonstrated the hypermorphic activity of germline S707Y mutations [41], and work with the DT40 cell line and primary CLL cells has shown that PLCG2 R665W and L845F mutations are also hypermorphs [7, 43]. With these mutations present, calcium influx upon BCR stimulation is sustained in the presence of ibrutinib [7]. In primary patient samples with PLCG2 mutations, PLCγ2 remains in an active phosphorylated state even upon treatment of the samples with ibrutinib [7]. These mutants are also hyper-responsive to upstream agonist signals, such as those provide by the GTPase Rac2 [43]. However, over a dozen different mutations in PLCG2 have been described, many lying outside of the SH2 domain, and the effect of many of these mutations on protein function are unclear (Table 2) [35]. For example, the C2 domain of PLCG2 mediates interactions with membrane phospholipids in a calcium-dependent fashion. Mutations in a conserved hotspot (amino acids 1140–1144) within this domain may alter its electrostatic properties and facilitate membrane interactions that otherwise would not occur. However, these mutations almost exclusively occur in the setting of other BTK or PLCG2 mutations, possibly within the same cell [35], raising the question of whether they can independently activate the pathway or whether their role is more complex [35]. In sum, PLCG2 mutations, at least those within the inhibitory SH2 domain, represent a method of sustaining signaling through the BCR pathway even in the presence of ibrutinib, and in vitro are drug-resistant mutants.
Table 2:
Mutational Frequencies of Clones in Patients with Reported Summed VAFs of Greater Than 30%
| Patient | Number of Clones | Clonal Frequency (VAFs) | Reference |
|---|---|---|---|
| 1 | 5 |
PLCG2 L845fs (14.9%), PLCG2 R742P (6.1%), PLCG2 R665W (5.5%), PLCG2 S707P (5.3%), PLCG2 S707F (3.3%) |
[6] |
| 2 | 1 | PLCG2 R665W (45%) | |
| 3 | 1 | BTK C481F (84%) | |
| 4 | 4 |
PLCG2 L845F (23.8%), BTK C481S (6.8%), PLCG2 S707Y (6.6%), PLCG2 R665W (2%) |
|
| 5 | 1 | BTK C481S (37.8%) | |
| 6 | 1 | BTK C481S (47.5%) | |
| 7 | 1 | BTK C481S (74.7%) | |
| 8 | 1 | BTK C481S (56.5%) ( | [5] |
| 9 | 2 | BTK C481S (71%), BTK C481R (18.1%) | |
| 10 | 1 | BTK C481S (53%) | |
| 11 | 2 | BTK C481A (45.3%), BTK C481S (42.5%) | |
| 12 | 1 | BTK C481S (94.8%) | |
| 13 | 1 | BTK C481S (34.9%) | |
| 14 | 2 | BTK C481S (26.9%), PLCG2 (13.2%) | |
| 15 | 1 | BTK C481S (45.8%)1 | |
| 16 | 1 | BTK C481S (52%)1 | |
| 17 | 1 | BTK C481S (83.4%) | |
| 18 | 1 | BTK C481S (52.4%) | |
| 19 | 1 | PLCG2 R665W (35.1%) | |
| 20 | 2 | PLCG2 (D993Y), BTK C481F (8.5%) | |
| 21 | 1 | BTK C481F (100%) | |
| 22 | 1 | BTK C481S (80%) | |
| 23 | 1 | BTK C481S (38.8%) | |
| 24 | 1 | BTK C481S (85.6%) | |
| 25 | 4 |
BTK C481S (78.2%), PLCG2 L845F (4.7%), PLCG2 R665W (0.26%), PLCG2 S707Y (0.17%) |
[10] |
| 26 | 5 |
PLCG2 L845F (18.3%), BTK C481R (15.8%), BTK C481S (8.8%),2 PLCG2 R665W (7.3%), BTK C481S (7.2%)2 |
|
| 27 | 1 | BTK C481S (57.3%) | |
| 28 | 2 |
BTK C481S (32.6%) PLCG2 6nt deletion (below limit of detection) |
|
| 29 | 3 |
BTK C481S (90.7%),2
BTK C481S (8.5%),2
BTK C481R (2.5%) |
[11] |
Denotes samples that were CD19+ selected prior to sequencing.
Sequencing indicates that different nucleotides were used to encode for the same mutated amino acid; thus, more than one clone with the reported amino acid change was present.
5.3. Analogy to drug-resistance mutations in other proteins
BTK shares homology with other tyrosine kinases, and prior experience with irreversible inhibitors of these other kinases may inform our understanding of BTK inhibitors. Osimertinib is an irreversible EGFR inhibitor that covalently binds to cysteine 797 of EGFR and has been studied for the treatment of EGFR-mutant lung cancer [44, 45]. C797S mutations have been described in patients with acquired resistance to osimertinib. In vitro, C797S mutations prevent covalent binding of osimertinib and shift the IC50 from the nanomolar to micromolar range [46, 47]. While there is not yet clinical data to prove that targeting mutant EGFR carrying a C797S mutation will lead to disease regression in osimertinib-resistant patients, this mutation is presumably causing clinical resistance to the drug since it occurs in the oncogene that is known to drive this cancer. Thus, a C797S mutation is hypothesized to be both necessary and sufficient for osimertinib-resistant, EGFR-mutant lung cancer. This hypothesis could be extended to BTK and ibrutinib-resistant CLL. Cysteine 481 of BTK is homologous to cysteine 797 in EGFR, and correspondingly the BTK C481S mutation is analogous to the EGFR C797S mutation. This is supportive of the hypothesis that the BTK C481S mutation could potentially drive resistance to ibrutinib in CLL in the same manner that the EGFR C797S mutation drives resistance to osimertinib in EGFR-mutant lung cancer.
5.4. Mutual exclusivity of drug-resistant mutations
If multiple different mutations are sufficient for the clinical phenotype of drug resistance, then they are typically mutually exclusive. While multiple drug resistant mutations can occur within the same patient, they would not appear within the same cell. This is because the selective pressure driving the appearance of the first drug-resistant mutation is lost once the mutation has occurred, thus abrogating the need for the development of any additional mutations. If, however, two or more mutations co-occur within the same cell, this suggests that one is indispensable for drug resistance or that the individual mutations alone are each too weak to cause resistance. In CLL patients with simple progression, putative resistance-causing mutations can appear together in up to half of all patients with such mutations [10]; combinations include multiple BTK mutations, multiple PLCG2 mutations, or BTK mutations in conjunction with PLCG2 mutations (Table 1) [5, 6, 10, 11]. Since most of the deep sequencing studies of ibrutinib-resistant patients have been done on genomic DNA isolated from pooled whole blood (or CD19+ cells) rather than single cells, these studies cannot distinguish between single mutations present in different subclones or multiple mutations present in single cells. Burger et al. performed single cell sequencing and did find that, in a patient with four distinct PLCG2 mutations, each mutation occurred in distinct subclones [18]. Ahn et al. detected three distinct BTK variants in a male patient, and since BTK is present on the X chromosome, these mutations must have occurred in different clones (assuming no pathologic genomic duplications) [10]. Additionally, different co-occurring mutations often follow different kinetics, suggesting - although not proving - that they are present in different cell populations. The data available now support the conclusion that BTK and PLCG2 mutations are mutually exclusive within the same cell.
5.5. Increase in the allelic frequency of drug-resistant mutations with continued exposure to the drug
The presence of a mutation prior to the development of clinical resistance does not necessarily imply that the mutation is the cause of clinical resistance. Stronger evidence that a mutation is sufficient for resistance would include demonstration that the allelic fraction of the mutation increases over time with continued exposure to the drug. At the very least, this implies that there is a growth advantage for cells with the mutation; at the most, this is consistent with the hypothesis that those cells with the growth advantage are causing clinical relapse. In CLL, available evidence suggests that the fraction of cancer cells with either BTK or PLCG2 mutations does increase over time with exposure to ibrutinib. Ahn et al. identified BTK and PLCG2 mutations up to 15.4 months (median 8 months, range: 2.9–15.4 months) prior to the development of simple clinical progression, and in general the variant allele frequencies of the BTK and PLCG2 mutations increased over time [10]. Woyach et al. identified BTK or PLCG2 mutations prior to simple clinical progression in 18 of 20 tested patients who were eventually found to have such mutations at relapse [5]. The median time from mutation detection to clinical relapse was 9.3 months (range: 3–18 months), consistent with the data from Ahn. Furthermore, Woyach et al. prospectively followed a cohort of 112 ibrutinib-treated CLL patients by sequencing the entire BTK and PLCG2 coding regions every three months. In this cohort, eight patients experienced clinical relapse, and all eight had expansion of a BTK mutated clone prior to relapse. Furthermore, eight additional patients who had not yet relapsed also had detectable BTK mutant clones that were expanding at the time of publication [5].
The above data showing expansion of BTK and PLCG2 mutant clones over time is strengthened by the finding that ibrutinib does cause some CLL subclones to involute. For example, Kadri et al. found that pretreatment clones carrying del(17p) and TP53 mutations had disappeared at relapse in two patients [11]. Both patients had instead developed BTK mutant clones without TP53 abnormalities that had expanded over time. Involution is clearly possible on ibrutinib, but it has not been reported in clones carrying BTK or PLCG2 mutations. There appears to be positive selection for these clones in the presence of ibrutinib, and they may even be driving resistance to the drug.
5.6. High allelic frequencies of the drug-resistant mutation at the time and site of clinical relapse
Acquired resistance to targeted therapies is heterogenous. Multiple subpopulations of cells can exist within the same patient. These subpopulations likely have a range of sensitivities to the drug, and the mechanisms by which they acquire resistance may be different. Despite this heterogeneity, the clone causing clinical progression typically achieves dominance at the time of relapse. For example, in patients with EGFR-mutant lung cancer that has developed acquired resistance to osimertinib, the clone containing the C797S mutation is responsible for a significant fraction of cell free tumor DNA at the time of relapse, and is thus presumably the dominant clone [46]. In CML, clones carrying point mutations that render BCR-ABL insensitive to imatinib can make up nearly 100% of the cancer cell fraction at the time of relapse [19, 48]. However, there is a wide variability in the fraction of cancer cells bearing BTK or PLCG2 mutations at the time of CLL simple progression on ibrutinib.
Woyach et al. reported variant allele frequencies (VAFs) in 40 patients with CLL simple progression [5]. Intriguingly, seventeen patients had summed VAFs (of all BTK/PLCG2 mutations present) of less than 30% at the time of progression, including seven patients with VAFs less than 10% at the time of progression (Table 1). The majority of patients in Ahn et al. had both BTK and PLCG2 mutations at the time of progression; even if the VAFs of all the mutations are summed, four out of eight patients with BTK/PLCG2 mutations had total VAFs less than 10% at relapse (Table 1) [10].
It is difficult to see how a clone with a VAF of less than 10% can substantially contribute to resistance. One explanation could be that mutational assessment of peripheral blood does not adequately sample the site of resistance. As discussed above, the site of CLL simple progressions varies from patient to patient and is not always biopsied. Woyach et al. noted that of the seven patients with VAFs less than 10% at time of relapse, all but one had disease progression only in the lymph nodes [5]. In these patients, sampling of the peripheral blood presumably provides an inadequate representation of the genetic make-up of resistant cells within the lymph node. If this is the case, then without tissue from the node we can only speculate about the molecular mechanisms underpinning resistance. Alternatively, BTK/PLCG2 mutant clones at low VAFs may somehow exert a dominant effect on the more numerous wild-type CLL cells. A final explanation for the development of resistance with low BTK/PLCG2 mutant VAFs is that the mutant clones could co-occur with non-mutant clones that have developed alternative mechanisms for drug resistance. In this setting, the BTK/PLCG2 mutant clones may be bystanders or may only partially contribute to the phenotype of clinical progression. There is precedent for this in solid tumors, as patients with gefitinib/erlotinib-resistant EGFR-mutant lung cancer can have both EGFR T790M-mutant clones and non-mutant clones co-existing at the time of relapse [49, 50]. The existence of multiple BTK and/or PLCG2 clones in individual patients at the time of relapse supports the hypothesis that different resistant clones can co-exist at the same time. Perhaps BTK/PLCG2-mutant and non-mutant resistant clones can co-exist as well.
5.7. The Gold Standard: specific inhibition of the putative drug-resistant mutation with a kinase Inhibitor
The gold standard for demonstrating that a putative drug-resistance mutation is sufficient for clinical relapse is to specifically inhibit the mutation and demonstrate regression of disease. Two examples of mutations and corresponding drugs that meet this rigorous criteria include osimertinib, which has demonstrated efficacy in EGFR-mutant lung adenocarcinomas with T790M mutations [44], and ponatinib, which has demonstrated efficacy in CML with T315I mutations in BCR-ABL [51].
This question may be answered soon in the case of BTK C481 mutations. While the second generation BTK inhibitors, including acalabrutinib (ACP-196), tirabrutinib (GS-4059, ONO-4059), spebrutinib (CC-292), and BGB-3111, are all irreversible inhibitors that bind BTK C481 and are therefore not reported to have efficacy against the BTK C481S mutation [52], some drugs earlier in development do inhibit BTK C481S. The drug ARQ 531 is an ATP competitive inhibitor of BTK that blocks BCR signaling in vitro, even in primary patient samples possessing BTK C481S mutations [53]. A phase 1 dose escalation study of ARQ 531 is currently enrolling patients with lymphoid malignancies (NCT03162536). One caveat here is that ARQ 531, like ibrutinib [54], is a nonspecific inhibitor of other Src and Tec family kinases [53], and thus any effects in BTK C481S mutant disease could also be due to effects on those other kinases. Vecabrutinib (SNS-062) is a reversible BTK inhibitor that blocks autophosphorylation of BTK in cells expressing either BTK or BTK C481S [55]. It has modest selectivity for BTK (IC60 = 3nM); IC50 values for ITK, TEC, BLK, LCK, SRC, and NEK11 are all also under 100nM [55]. A phase 1a study has been performed in healthy subjects [56], and a phase 1b/2 dose escalation and cohort-expansion study in CLL patients is currently enrolling (NCT03037645). LOXO-305 (RXC005, REDX08608) reversibly inhibits both wild-type BTK and BTK C481S and additionally has > 100-fold selectivity against other Tec and Src kinase family members, but has not yet entered clinical trials [57]. Noncovalent BTK inhibitors, such as PRN1008 [58] and BMS986142 [59] have been tested in healthy volunteers in phase I trials. Noncovalent inhibitors may possess activity against BTK with C481 mutations [60], but PRN1008 and BMS986142 have not to our knowledge been tested in this context.
6. Conclusion
Ibrutinib is the prototypical member of a novel class of targeted therapies, BTK inhibitors, and has revolutionized the treatment of both front-line and relapsed/refractory CLL. While initial clinical experience with ibrutinib has been promising, with longer clinical follow-up it is clear that drug resistance in ibrutinib-treated patients will be a formidable problem. Relapsed CLL on ibrutinib manifests as RT, typically within the first two years of drug therapy, or as CLL simple progression, which occurs at increasing rates the longer a patient is on the drug.
Mutations in BTK and PLCG2 are seen in 80–90% of patients with CLL simple progression. Explanations for mutations that appear when patients are taking a drug could include the following, which are not necessarily mutually exclusive: (1) they appear by random chance, (2) they are associated with resistance (e.g. clones bearing the mutation are selected over time on the drug) but do not cause it, or (3) they are the direct cause of resistant disease. Significant circumstantial evidence implicates BTK and PLCG2 mutations as the cause of progressive disease on ibrutinib. Similar mutations occur in multiple patients, most commonly at the residue where ibrutinib covalently binds, and they confer in vitro resistance to the drug in those cases studied; whether less common BTK/PLCG2 mutations lead to in vitro drug resistance is not known. The allelic frequency of these mutations increases over time, implying that they have functional consequences that are selected for in the presence of ibrutinib. The BTK C481S mutation is analogous to the C797S mutation in EGFR that drives clinical resistance to the EGFR inhibitor osimertinib. However, BTK and/or PLCG2 mutations do not appear necessary for relapse, as about 10–20% of patients relapse without any detectable mutations in these genes. Furthermore, a substantial number of patients have clinical relapse with very low levels of BTK/PLCG2 mutations. Thus, for an individual patient with ibrutinib-resistant disease, particularly those with low levels of BTK/PLCG2 mutations in the peripheral blood, it remains difficult to determine whether such mutations are simply associated with resistance, the sole drivers of the resistant phenotype perhaps under-represented in peripheral blood sampling, or one of multiple concomitant mechanisms of resistance. Ultimately, to distinguish between these explanations, a drug that specifically targets the mutations of interest is needed. Drugs that inhibit BTK C481S are in development, suggesting that this question may be answered in the near future.
7. Expert Commentary
Ibrutinib is a nonspecific kinase inhibitor, inhibiting 19 kinases with an IC50 <100nM [54]. While it is certainly effective for the treatment of CLL, this lack of specificity raises questions concerning its ultimate mechanism of action. Activating mutations in BTK are not found in CLL, and thus the absolute necessity for signaling through BTK for CLL cell proliferation and survival is unclear. The finding that ibrutinib-resistant BTK mutations appear to be selected over time in ibrutinib-treated patients confirms that the drug is blocking its target in vivo. However, this does not necessarily prove that blockade of BTK is responsible for the drug’s efficacy. Determining whether acquired BTK and PLCG2 mutations are the drivers of ibrutinib resistance is thus not merely an academic exercise. Rather, this distinction is very important for the development of future therapeutics in the field. If it can be definitively confirmed that such mutations are the drivers of ibrutinib resistance, then this validates ongoing efforts to design inhibitors of such mutations. More importantly, it validates the principle that blockade of the BCR signaling pathway is a viable therapeutic strategy and opens up the opportunity for the development of other agents targeting this pathway.
There is strong evidence that BTK and PLCG2 mutations can be drivers of ibrutinib resistance. The recurrence of similar mutations in different patients is suggestive of convergent evolution. The most commonly identified mutations are resistant to ibrutinib in vitro. These mutations appear mutually exclusive with one another at a cellular level and increase in frequency over time in patients treated with ibrutinib, suggesting functional consequences for such mutations. The identification of patients who start without any detectable BTK mutations and then have a high frequency of BTK mutations at relapse implies that such mutations are responsible for relapse. However, patients who do not have a high frequency of such mutations in the peripheral blood also experience relapse. Ultimately, three conclusions could explain these disparate findings. One explanation is that in some patients, BTK/PLCG2 mutations may be exclusively responsible for progression, but in other patients they may co-exist with alternative mechanisms of acquired resistance that dominantly drive the phenotype of clinical progression. If this is the case, the two groups of patients may have differing sensitivity to next-generation BTK inhibitors and therefore identifying markers to distinguish between the two groups will be necessary. A second explanation is that even small levels of BTK/PLCG2 mutant clones may have a dominant effect, promoting drug resistance in other cells that do not contain the mutations either through direct effects in the microenvironment or soluble factors. A third explanation is that resistance in CLL can be a localized phenomenon. In other words, BTK/PLCG2 mutations are not identified as present in high frequencies in some patients perhaps because the correct location was not sampled. If resistance is localized, this raises interesting possibilities. Could localized therapy for resistant disease (e.g. radiation to a growing node) extend the time that a patient receives clinical benefit from ibrutinib? Hopefully, future research may provide evidence to support or negate these hypotheses.
8. Five-year view
Multiple unanswered questions remain regarding resistance to ibrutinib in CLL and will hopefully be answered in the next few years. The efficacy of drugs that can inhibit the BTK C481S mutation will be tested in patients with ibrutinib-resistant disease. If these prove successful, then questions about when to initiate such therapy could be explored. The use of inhibitors that target both wild-type BTK and BTK C481S in the front-line setting may delay the development of resistance and prolong progression-free survival (PFS), in the same way that front-line osimertinib prolongs PFS compared to gefitinib/erlotinib in EGFR-mutant lung cancer [45][57]. Additional BTK inhibitors, including reversible noncovalent inhibitors, are entering clinical trials, and it remains to be seen whether the resistance that develops with these agents bears any similarity to the resistance that develops on ibrutinib. The effects of time-limited therapy with BTK inhibitors on the kinetics of relapse are currently unknown; combinatorial treatment with regimens including BTK inhibitors may be one fruitful way of delaying or eliminating relapse. None of the current next generation BTK inhibitors target PLCG2 mutations; this remains an unmet need. Efforts focused on BTK and PLCG2 mutations should not preclude the search for other mechanisms of ibrutinib resistance, including the possibilities of alternative secondary resistance mutations, gene amplifications, or deletions (such as the previously mentioned del(8p)). The cohort of patients who relapse without BTK/PLCG2 mutations (including those with RT) deserve further study, perhaps with more effort to biopsy areas of resistant disease. Given the unquestionable efficacy of ibrutinib, a full understanding of why and how resistance develops is critical to best avoiding it.
9. Key Issues:
Reasons for ibrutinib discontinuation include drug intolerance, Richter’s transformation, and simple progression of CLL.
CLL simple progression appears to occur at exponentially increasing rates over time and has been associated with the detection of BTK and PLCG2 hotspot mutations in the peripheral blood.
While mutations that drive acquired resistance have been clearly identified for drugs targeting disease defining mutations , such as erlotinib in EGFR-mutant lung cancer and imatinib in chronic myelogeneous leukemia, ibrutinib and CLL have unique features that complicate identification of resistance mutations. For example, CLL cells are dependent on the B cell receptor signaling pathway, rather than on a single driving oncogene. Furthermore, nodal sites of simple disease progression that develop on ibrutinib are not often biopsied. Ibrutinib is also an irreversible, rather than reversible, kinase inhibitor, and it is unclear how this may affect the development of resistance.
BTK and PLCG2 mutations have many characteristics of mutations that could drive acquired resistance to ibrutinib, including their ability to confer in vitro resistance to the drug, their occurrence in multiple patients often in multiple clones, and their increasing allelic frequency over time with continued exposure to the drug. However, not all patients with acquired clinical resistance have high allelic frequencies of these mutations, and confirmation that these mutations drive resistance will require direct targeting of the mutations with a selective inhibitor.
Funding
BL Lampson is supported by the NIH Training Grant 2 T32 CA 9172–42 A1. Jennifer Brown is supported by National Institutes of Health (NCI 1R01CA213442–01A1, NIH 1U10CA180861–03, and P01CA206978–01), and the Susan and Gary Rosenbach Fund for Lymphoma Research, the Melton Family Fund for CLL Research and the Okonow/Lipton Family Lymphoma Research Fund.
Footnotes
Declaration of interest
JR Brown has served as a consultant for AstraZeneca, Acerta, Gilead, Infinity, Janssen, Pharmacyclics, Roche/Genentech, and TG Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Brown JR, Hillmen P, O’Brien S, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2017. June 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014. July 17;371(3):213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med. 2015. December 17;373(25):2425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mato AR, Hill BT, Lamanna N, et al. Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol. 2017. May 01;28(5):1050–1056. [DOI] [PubMed] [Google Scholar]
- 5. **.Woyach JA, Ruppert AS, Guinn D, et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J Clin Oncol. 2017. May 01;35(13):1437–1443. This comprehensive study is the largest report to date of BTK and PLCG2 mutations in pateints resistant to ibrutinib, describing the mutational profile and variant allele frequencies both prior to and at the time of clinical resistance to the drug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. *.Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol. 2015. April;1(1):80–7. This study reports long term clinical follow-up of patients treated on ibrutinib, including rates of ibrutinib discontinuation, Richter’s transformation, and simple progression on the drug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. *.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014. June 12;370(24):2286–94. This was one of the first reports of BTK and PLCG2 mutations in patients with acquired resistance to ibrutinib and details the in vitro phenotypes of BTK C481S and PLCG2 R665W and L845F mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014. March 20;123(12):1810–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015. March 26;125(13):2062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. *.Ahn IE, Underbayev C, Albitar A, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017. March 16;129(11):1469–1479. This study provides BTK and PLCG2 mutational profile data on 15 ibrutinib-resistant patients independent of the larger OSU cohort. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. *.Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Advances. 2017;1(12):715–727. This study examines mutations in nodal tissue obtained from patients with Richter’s transformation on ibrutinib and compares the findings to BTK and PLCG2 mutations over time in the peripheral blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008. June 15;111(12):5446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosti G, Castagnetti F, Gugliotta G, et al. Tyrosine kinase inhibitors in chronic myeloid leukaemia: which, when, for whom? Nat Rev Clin Oncol. 2017. March;14(3):141–154. [DOI] [PubMed] [Google Scholar]
- 14.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006. November 01;12(21):6494–501. [DOI] [PubMed] [Google Scholar]
- 15.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016. May 19;127(20):2391–405. [DOI] [PubMed] [Google Scholar]
- 16.Davids MS, Brown JR. Targeting the B cell receptor pathway in chronic lymphocytic leukemia. Leuk Lymphoma. 2012. December;53(12):2362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011. June 9;117(23):6287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016. May 20;7:11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michor F, Hughes TP, Iwasa Y, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005. June 30;435(7046):1267–70. [DOI] [PubMed] [Google Scholar]
- 20.Sanderson K Irreversible kinase inhibitors gain traction. Nat Rev Drug Discov. 2013. September;12(9):649–51. [DOI] [PubMed] [Google Scholar]
- 21.Singh J, Petter RC, Baillie TA, et al. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011. April;10(4):307–17. [DOI] [PubMed] [Google Scholar]
- 22.Campo M, Gerber D, Gainor JF, et al. Acquired Resistance to First-Line Afatinib and the Challenges of Prearranged Progression Biopsies. J Thorac Oncol. 2016. November;11(11):2022–2026. [DOI] [PubMed] [Google Scholar]
- 23.Maffei R, Fiorcari S, Martinelli S, et al. Targeting neoplastic B cells and harnessing microenvironment: the “double face” of ibrutinib and idelalisib. J Hematol Oncol. 2015. May 29;8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondo K, Shaim H, Thompson PA, et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia. 2017. October 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013. October 10;122(15):2539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long M, Beckwith K, Do P, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017. August 1;127(8):3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogler M, Butterworth M, Majid A, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009. April 30;113(18):4403–13. [DOI] [PubMed] [Google Scholar]
- 28.Tahir SK, Smith ML, Hessler P, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it [journal article]. BMC Cancer. 2017. June 02;17(1):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fresquet V, Rieger M, Carolis C, et al. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014. June 26;123(26):4111–9. [DOI] [PubMed] [Google Scholar]
- 30.Wu C, de Miranda NF, Chen L, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget. 2016. June 21;7(25):38180– 38190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood. 2017. May 4;129(18):2519–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartlett NL, Costello BA, LaPlant BR, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood. 2018. January 11;131(2):182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015. August;21(8):922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma S, Galanina N, Guo A, et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget. 2016. October 18;7(42):68833–68841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones D, Woyach JA, Zhao W, et al. PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia. 2017. July;31(7):1645–1647. [DOI] [PubMed] [Google Scholar]
- 36.Hamasy A, Wang Q, Blomberg KE, et al. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia. 2017. January;31(1):177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graziani S, Di Matteo G, Benini L, et al. Identification of a Btk mutation in a dysgammaglobulinemic patient with reduced B cells: XLA diagnosis or not? Clin Immunol. 2008. September;128(3):322–8. [DOI] [PubMed] [Google Scholar]
- 38.Wilde JI, Watson SP. Regulation of phospholipase C gamma isoforms in haematopoietic cells: why one, not the other? Cell Signal. 2001. October;13(10):691–701. [DOI] [PubMed] [Google Scholar]
- 39.Coggeshall KM, McHugh JC, Altman A. Predominant expression and activation-induced tyrosine phosphorylation of phospholipase C-gamma 2 in B lymphocytes. Proc Natl Acad Sci U S A. 1992. June 15;89(12):5660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurosaki T, Maeda A, Ishiai M, et al. Regulation of the phospholipase C-gamma2 pathway in B cells. Immunol Rev. 2000. August; 176:19–29. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012. October 05;91(4):713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ombrello MJ, Remmers EF, Sun G, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012. January 26;366(4):330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walliser C, Hermkes E, Schade A, et al. The Phospholipase Cgamma2 Mutants R665W and L845F Identified in Ibrutinib-resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J Biol Chem. 2016. October 14;291(42):22136–22148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015. April 30;372(18):1689–99. [DOI] [PubMed] [Google Scholar]
- 45.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 2018. January 11;378(2):113–125. [DOI] [PubMed] [Google Scholar]
- 46.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015. June;21(6):560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ercan D, Choi HG, Yun CH, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015. September 01;21(17):3913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Branford S, Rudzki Z, Parkinson I, et al. Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood. 2004. November 01;104(9):2926–32. [DOI] [PubMed] [Google Scholar]
- 49.Suda K, Murakami I, Katayama T, et al. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res. 2010. November 15;16(22):5489–98. [DOI] [PubMed] [Google Scholar]
- 50.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013. April 15;19(8):2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosomepositive leukemias. N Engl J Med. 2013. November 07;369(19):1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu J, Liu C, Tsui ST, et al. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016. September 02;9(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reiff SD, Mantel R, Smith LL, et al. The Bruton’s Tyrosine Kinase (BTK) Inhibitor ARQ 531 Effectively Inhibits Wild Type and C481S Mutant BTK and Is Superior to Ibrutinib in a Mouse Model of Chronic Lymphocytic Leukemia. Blood. 2016;128(22):3232–3232. [Google Scholar]
- 54.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010. July 20;107(29):13075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fabian CA, Reiff SD, Guinn D, et al. Abstract 1207: SNS-062 demonstrates efficacy in chronic lymphocytic leukemia in vitro and inhibits C481S mutated Bruton tyrosine kinase. Cancer Research. 2017;77(13 Supplement):1207–1207. [Google Scholar]
- 56.Neuman LL, Ward R, Arnold D, et al. First-in-Human Phase 1a Study of the Safety, Pharmacokinetics, and Pharmacodynamics of the Noncovalent Bruton Tyrosine Kinase (BTK) Inhibitor SNS-062 in Healthy Subjects. Blood. 2016;128(22):2032–2032. [Google Scholar]
- 57.Guisot NES, Best SA, Wright V, et al. REDX08608, a Novel, Potent and Selective, Reversible BTK Inhibitor with Efficacy and Equivalent Potency Against Wild -Type and Mutant C481S BTK. Blood. 2016;128(22):4399–4399. [Google Scholar]
- 58.Smith PF, Krishnarajah J, Nunn PA, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. 2017. November;83(11):2367–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee SK, Xing J, Catlett IM, et al. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur J Clin Pharmacol. 2017. June;73(6):689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson AR, Kohli PB, Katewa A, et al. Battling Btk Mutants With Noncovalent Inhibitors That Overcome Cys481 and Thr474 Mutations. ACS Chem Biol. 2016. October 21;11(10):2897–2907. [DOI] [PubMed] [Google Scholar]