

Abstract
The human T-cell leukemia virus type-1 (HTLV-1) is an oncoretrovirus that infects and transforms CD4+ T-cells and causes adult T-cell leukemia/lymphoma (ATLL) –an aggressive lymphoproliferative disease that is highly refractive to most anticancer therapies. The HTLV-1 proviral genome encodes several regulatory products within a conserved 3′ nucleotide sequence, known as pX; however, it remains unclear how these factors might cooperate or dynamically interact in virus-infected cells. Here we demonstrate that the HTLV-1 latency-maintenance factor p30II induces the TP53-induced glycolysis and apoptosis regulator (TIGAR) and counters the oxidative stress, mitochondrial damage, and cytotoxicity caused by the viral oncoproteins Tax and HBZ. The p30II protein cooperates with Tax and HBZ and enhances their oncogenic potential in colony transformation/foci-formation assays. Further, we have shown that TIGAR is highly expressed in HTLV-1-induced tumors associated with oncogene dysregulation and increased angiogenesis in an in vivo xenograft model of HTLV-1-induced T-cell lymphoma. These findings provide the first evidence that p30II likely collaborates as an ancillary factor for the major oncoproteins Tax and HBZ during retroviral carcinogenesis.
Keywords: HTLV-1, Tax, HBZ, p30II, TIGAR, ROS, ATLL, Lymphoma, Angiogenesis, Xenograft
Graphical Abstract
Introduction
The human T-cell leukemia virus type-1 (HTLV-1) infects and oncogenically transforms CD4+ T-cells and is the etiological agent of adult T-cell leukemia/lymphoma (ATLL), a rare, yet often fatal hematological malignancy that occurs in 3–5% of infected individuals (Bangham and Ratner, 2015; Panfil et al., 2016; Johnson et al., 2001). There is currently no effective medical treatment or cure for ATLL; and the advanced acute and lymphoma-stages of disease are generally considered terminal, with average post-diagnosis life-expectancies of 6 months-2 years.
The HTLV-1 is endemic in tropical equatorial regions, particularly, Southeast Asia (e.g., Japan, Taiwan, Philippines, and Malaysia), the Middle East, Northern and Central Africa, Central and South America, Australo-Melanesia, and certain islands of the Caribbean; and it is estimated there are approximately 10–20 million HTLV-1-infected carriers worldwide (Bangham and Ratner, 2015; Panfil et al., 2016; Johnson et al., 2001). The HTLV-1 genome encodes several regulatory/nonstructural proteins (i.e., Tax, Rex, HBZ, p8I, p12I, p13II, and p30II) within a highly conserved 3′ nucleotide sequence, known as pX, which is retained in the majority of ATLL isolates. However, it remains unclear whether these factors functionally cooperate or act independently during viral pathogenesis.
The retroviral transactivator Tax regulates HTLV-1 gene expression and activates host cellular transcriptional pathways, including CREB/ATF, Serum-response factor/p67, and NF-κB (Zhao et al., 1992; Suzuki et al., 1993; Smith and Greene, 1990; Sun et al., 1994; Geleziunas et al., 1998). Tax also impairs the functions of several key cell-cycle modulators and interferes with DNA-damage repair pathways (Johnson et al., 2001). The Tax protein forms dimers and interacts with CREB/ATF and the transcriptional coactivators, p300/CREB-binding protein (CBP) and the p300/CBP-associated factor (P/CAF) on three 21-base-pair repeat Tax-responsive elements (TREs) within the 3′ U3 region of the HTLV-1 promoter to activate proviral gene expression (Zhao et al., 1992; Suzuki et al., 1993; Kwok et al., 1996; Harrod et al., 1998; Geiger et al., 2008; Harrod et al., 2000). Also, Tax induces NF-κB-signaling by stimulating K63-linked polyubiquitination and activates the IκB-kinase (IκK) complex through interactions with the IκK scaffolding subunit, NF-κB essential modulator (NEMO)/IκK-γ, and recruitment of the TGFβ-activated kinase 1 (TAK1; Sun et al., 1994; Geleziunas et al., 1998; Yamaoka et al., 1998; Harhaj et al., 2007; Wu and Sun, 2007; Ho et al., 2015). The activation of NF-κB-signaling is essential for the proliferation and survival of HTLV-1-infected cells.
Tax is generally considered to be the major oncoprotein of HTLV-1 (Bangham and Ratner 2015; Panfil et al., 2016; Johnson et al., 2001). Transgenic animals expressing tax develop T-cell lymphomas, large granular lymphocytic leukemia, and exhibit polyarthropathy and high osteolytic bone turnover associated with hypercalcemia (Hasegawa et al., 2006; Grossman et al., 1995; Grossman and Ratner, 1997; Gao et al., 2005; Saggioro et al., 1997; Ruddle et al., 1993). It is widely assumed that Tax promotes the early-stage immortalization of a subset of HTLV-1-infected T-cells which leads to the onset of ATL (Bangham and Ratner, 2015; Johnson et al., 2001). Tax expression is critical for the lymphoproliferative and immortalizing activity of HTLV-1 (Xie et al., 2006; Mahgoub et al., 2018). However, Tax alone is poorly oncogenic and does not efficiently immortalize primary huPBMCs in vitro (Bellon et al., 2010), suggesting it likely cooperates with other viral and/or cellular factors to promote T-cell leukemogenesis.
The antisense HTLV-1 basic domain/leucine zipper factor, HBZ, negatively regulates Tax-dependent proviral gene expression and induces T-cell lymphoproliferation through the modulation of cellular pathways (Arnold et al., 2006; Arnold et al., 2008; Panfil et al., 2016; Li et al., 2009; Hiven et al, 2005; Kawatsuki et al., 2016; Ma et al., 2013). HBZ interacts with CREB and the transcriptional coactivator p300 and inhibits the formation of Tax-CREB-p300 transcription complexes on the 21-base-pair repeat elements in the viral promoter (Lemasson et al., 2007; Clerc et al., 2008). Both the hbz mRNA and protein have been shown to promote increased T-cell lymphoproliferation (Satou et al., 2006; Arnold et al., 2008; Kawatsuki et al., 2016). Interestingly, Choudhary and Ratner, 2011 reported that hbz transcripts could indirectly induce Tax expression by suppressing the expression of the latency-maintenance factor p30II. The HBZ protein has been shown to bind to nuclear p65RelA and inhibit Tax-dependent NF-κB-signaling (Zhao et al., 2009). HBZ also interacts with the AP-1 family transcription factor, JunD, and augments expression of the human telomerase reverse transcriptase (hTERT) subunit which could contribute to the long-term proliferation and immortalization of HTLV-1-infected cells (Kuhlmann et al., 2007). Transgenic animals expressing hbz develop CD4+ T-cell lymphomas and inflammatory lesions on their skin and lung tissues, as a result of the increased production of inflammatory factors (e.g., IFN-γ, TGF-β, and Foxp3) and proliferative signals, such as Wnt5a (Mitagami et al., 2015; Satou et al., 2011; Zhao et al., 2011; Ma et al., 2013). Esser et al., 2017 have further shown that transgenic mice expressing hbz under the control of the granzyme B promoter develop T-cell lymphomas and exhibit leukemic lymphoproliferation, associated with osteolytic bone lesions and increased levels of inflammatory cytokines and bone-remodeling factors.
Both Tax and HBZ are known to induce aberrant lymphoproliferation as well as cellular apoptosis (Nicot and Harrod, 2000; Hall et al., 1998; Los et al., 1998; Chen et al., 1997; Yamada, T., 1996; Kawatsuki et al., 2016), suggesting that other pX-encoded factors may help alleviate the cytotoxic effects of these viral oncoproteins. Zhi et al., 2011 have demonstrated that the hyperactivation of NF-κB-signaling by Tax causes permanent G1-phase growth-arrest and cellular senescence associated with expression of the cyclin dependent kinase-inhibitors, p21WAF1 and p27Kip1. Moreover, the Tax-dependent activation of NF-κB results in oxidative stress and the production of nitric oxide through the inducible nitric oxide synthase (iNOS; Baydoun et al., 2015), which could induce a pro-oxidant state and lead to apoptosis in Tax-expressing cells (Takahashi et al., 2013; Los et al., 1998).
The Tax protein is undetectable in freshly isolated ATLL samples (Bangham and Ratner, 2015; Johnson et al., 2001); and Mahgoub et al., 2018 have shown that Tax expression alternates between on and off states in HTLV-1-transformed cells. Billman et al., 2017 have also demonstrated the tax and hbz genes are differentially transcribed in bursts in HTLV-1-infected T-cell clones derived from asymptomatic individuals or HAM patients. While the HTLV-1 latency protein p30II is undetectable in clinical isolates, primary ATLL lymphocytes express the alternatively-spliced p30II mRNA (Koralnik et al., 1992; Cereseto et al., 1997) and HTLV-1-infected individuals contain cytotoxic T-lymphocytes specific for pX ORF-II (i.e., p30II) peptides (Pique et al., 2000), which suggests low levels of p30II may suppress proviral gene expression (Zhang et al., 2001; Zhang et al., 2000; Michael et al., 2006; Nicot et al., 2004; Younis et al., 2004; Younis et al., 2006; Bai et al., 2012; Edwards et al., 2011) and promote the proliferation of latently-infected cells through cooperation with viral and/or cellular oncoproteins (Awasthi et al., 2005; Romeo et al., 2015; Romeo et al., 2018).
p30II (also known as Tax-ORF-II, or Tof-II) is an arginine and serine/threonine-rich nucleocytoplasmic protein, comprised of 241 amino acid residues, which is transcribed from a doubly-spliced pX mRNA (Koralnik et al., 1992; Koralnik et al., 1993; Awasthi et al., 2005; Romeo et al., 2015). The p30II protein negatively regulates Tax-dependent LTR transactivation and HTLV-1 proviral gene expression through transcriptional and posttranscriptional mechanisms (Zhang et al., 2001; Zhang et al., 2000; Michael et al., 2006; Nicot et al., 2004; Younis et al., 2004; Younis et al., 2006; Bai et al., 2012; Edwards et al., 2011). The p30II protein interacts with RNA Pol II elongation complexes and is cotranscriptionally recruited to its downstream nascent RNA-target (Younis et al., 2006), and inhibits the nuclear export of tax/rex mRNAs (Nicot et al., 2004; Younis et al., 2004; Younis et al., 2006; Bai et al., 2012; Edwards et al., 2011). p30II also contains a functional transcriptional activation domain (TAD), spanning aa residues 100–179, which binds to p300/CBP and differentially regulates CREB-dependent transcription (Zhang et al., 2001; Zhang et al., 2000; Michael et al., 2006). Zhang et al., 2001 have demonstrated that p30II interacts with the kinase-inducible exchange (KIX) region of p300/CBP and interferes with the formation of Tax-CREB-p300 transcription complexes on the 21-base-pair repeat TREs within the HTLV-1 promoter. It is believed that p30II helps latently HTLV-1-infected cells evade detection by host immune-surveillance pathways for the establishment of persistent infections. Indeed, several studies have demonstrated that p30II is required for the maintenance of high proviral titers and the survival of HTLV-1-infected cells in in vivo models of pathogenesis (Bartoe et al., 2000; Edwards et al., 2011; Valeri et al., 2010).
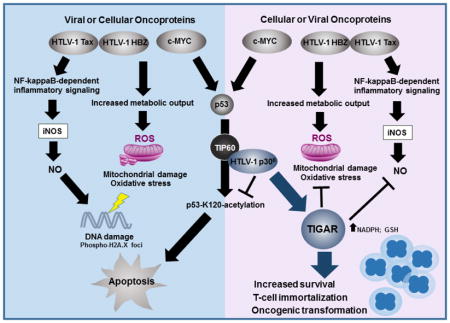
Our studies have shown that p30II cooperates with the c-Myc oncoprotein and induces aberrant S-phase cell-cycle entry and long-term proliferation beyond crisis (>4 months) in transduced huPBMCs (Awasthi et al., 2005; Romeo et al., 2015; Romeo et al., 2018). We have also previously demonstrated that aa residues 99–154 of p30II biochemically interact with the MYST-family acetyltransferase TIP60; and p30II is present in c-Myc/TIP60-containing NuA4 transcription complexes recruited to Myc-responsive E-box enhancer elements within the cyclin D2 promoter (Awasthi et al., 2005; Romeo et al., 2015). Moreover, we have shown that p30II cooperates with c-Myc and induces oncogenic cell transformation/foci formation in vitro, dependent upon p30II-interactions with the TIP60 acetyltransferase (Awasthi et al., 2005; Romeo et al., 2015; Romeo et al., 2018). TIP60 is a transcriptional cofactor for both c-Myc and the p53 tumor suppressor (Frank et al., 2003; Patel et al., 2004; Gorrini et al., 2007; Awasthi et al., 2005; Zhao et al., 2016; Tang et al., 2006; Sykes et al., 2006; Kurash et al., 2008); and the TIP60-mediated acetylation of p53 on lysine residue K120 differentially modulates the induction of p53-dependent pro-apoptotic genes (Tang et al., 2006; Sykes et al., 2006; Kurash et al., 2008). We recently demonstrated that p30II activates p53 and inhibits p53-K120-acetylation, and induces the expression and mitochondrial localization of the TP53-induced glycolysis and apoptosis regulator (TIGAR; Romeo et al., 2018). The cooperation between p30II and c-Myc was dependent upon p53-regulated transcriptional activity and TIGAR functions (Romeo et al., 2018). The TIGAR protein is highly expressed in HTLV-1-transformed T-cell-lines as well as primary ATLL clinical isolates associated with c-Myc oncogene dysregulation (Romeo et al., 2018). The TIGAR is a p53-regulated glycolytic enzyme (i.e., a fructose-2,6-bisphosphatase) and antioxidant effector that prevents the accumulation of damaging ROS by increasing the intracellular levels of the anabolic cofactor NADPH and reduced glutathione (GSH; Bensaad et al., 2006; Bensaad et al., 2009; Cheung et al., 2012; Cheung et al., 2016; Li and Jogl, 2009). Many human malignancies contain elevated TIGAR expression which often correlates with therapy-resistance and poor prognoses (Peña-Rico et al., 2011; Qian et al., 2016; Hong et al., 2016; Cheung et al., 2013; Yin et al., 2014). Cheung et al., 2016 have shown that c-Myc upregulates TIGAR associated with Wnt-signaling in a APC−/− transgenic model for intestinal cell proliferation. In Romeo et al., 2018, we demonstrated that TIGAR is highly expressed in hematological malignancies (i.e., ATLL, acute lymphoblastic leukemia, and multiple myeloma) concomitant with c-Myc oncogene dysregulation.
Here we demonstrate that the HTLV-1 p30II protein cooperates with the major viral oncoproteins Tax and HBZ and suppresses Tax/HBZ-induced oxidative stress, mitochondrial damage/mitophagy, and cytotoxicity dependent upon TIGAR functions. Using the infectious HTLV-1 ACH.wildtype and ACH.p30II mutant proviral clones (Kimata et al., 1994; Bartoe et al., 2000), we demonstrate that p30II is required to prevent the oxidative DNA-damage caused by other viral products. Importantly, our results further show that p30II enhances the oncogenic potential of Tax and HBZ in colony transformation/foci-formation assays, suggesting these pX-encoded factors may functionally collaborate during HTLV-1-induced carcinogenesis.
Results
HTLV-1 p30II suppresses Tax and HBZ-induced oxidative stress and mitochondrial toxicity
The increased metabolic activity and energy requirements of proliferating cells expressing cellular or viral oncoproteins can lead to the accumulation of cytotoxic byproducts (e.g., ROS) which cause oxidative damage to genomic DNA, proteins and lipid constituents. Indeed, the HTLV-1 transactivator protein Tax stimulates the production of ROS and induces a pro-oxidant state in T-lymphocytes associated with the induction of cellular apoptosis (Takahashi et al., 2013; Los et al., 1998). Kinjo et al., 2010 have shown that Tax induces ROS production which results in genomic DNA-damage and cell senescence, as determined by staining for senescence associated Beta-galactosidase (SA-β-gal) expression. Also, the hyperactivation of NF-κB-signaling by Tax causes permanent G1-phase growth-arrest and cellular senescence which can be countered through co-expression of the viral antisense protein, HBZ (Zhi et al., 2011; Ho et al., 2012). Both Tax and HBZ induce aberrant cell proliferation as well as apoptosis (Nicot and Harrod, 2000; Hall et al., 1998; Los et al., 1998; Chen et al., 1997; Yamada, T., 1996; Kawatsuki et al., 2016). Thus, to determine whether the HTLV-1 Tax and HBZ oncoproteins cause oxidative stress and damage to cellular mitochondria, Jurkat lymphocytes were transfected with expression constructs for Tax or HBZ (Myc-tagged), or an empty CβS vector, and then stained with the membrane-permeant dye, JC-1 (Molecular Probes), which forms aggregates and emits red fluorescence on healthy mitochondria, but emits cytoplasmic green fluorescence under conditions of mitochondrial membrane depolarization due to oxidative damage. As a positive control, the cells were treated with a chemical uncoupler of the electron transport chain, CCCP (Sigma-Aldrich). Flow cytometry was performed using a BD FACSCaliber instrument to quantify the relative ratios of JC-1 green fluorescence (X-axis) to red fluorescence (Y-axis) in logarithmic mode. The data in Figs. 1A and 1B demonstrate that both Tax and HBZ resulted in notably higher green-to-red fluorescence ratios than did cells containing an empty CβS vector control. The Tax-expressing cells also exhibited a higher mixed JC-1 red/green fluorescent signal (Figs. 1A and 1B). To further validate these findings, HT-1080 human fibrosarcoma cells were transfected with HTLV-1 Tax or HBZ (Myc-tagged) expression constructs, or an empty CβS vector, and live-imaging confocal-microscopy was used to visualize the JC-1 red and green fluorescent signals. Certain samples were treated with CCCP as a positive control. Representative micrographs shown in Fig. 1C demonstrate that cells transfected with the empty CβS vector exhibited a higher red-to-green fluorescence ratio which correlated with less mitochondrial membrane depolarization, as compared to either the Tax or HBZ-expressing cells, or cells treated with the chemical uncoupler CCCP. The expression of the HTLV-1 Tax and HBZ (Myc-tagged) proteins in the transfected Jurkat and HT-1080 cells was detected by SDS-PAGE and immunoblotting (Figs. 1D and 1E). The relative input levels of Actin are shown for comparison. These data demonstrate that the major viral oncoproteins, Tax and HBZ, induce mitochondrial oxidative damage, and suggest that other pX-encoded products may help counter Tax/HBZ-induced oxidative stress and cytotoxicity during retroviral carcinogenesis.
Fig. 1.

The HTLV-1 p30II protein suppresses Tax/HBZ-induced ROS production through the activation of TIGAR. (A) Jurkat T-cells were transfected with either an empty CβS vector, or expression constructs for HTLV-1 Tax or HBZ, and then stained with the JC-1 dye (Molecular Probes) to detect changes in mitochondrial membrane potential. The JC-1 dye forms aggregates on regions of high mitochondrial polarization (red signal), whereas depolarized regions associated with oxidative damage are indicated by green-fluorescence. The chemical uncoupler, CCCP (Sigma-Aldrich), was included as a positive control to induce mitochondrial membrane depolarization. The JC-1-associated red and green fluorescent signals were quantified by flow cytometry. (B) Graph depicting the relative JC-1 red, green, and red/green (yellow) fluorescent signals in transfected Jurkat cells as shown in A. The data represent the mean +/- standard deviation (error bars) from three independent experiments. (C) HT-1080 fibrosarcoma cells were transfected with an empty CβS vector, or expression constructs for HTLV-1 Tax or HBZ (Myc-tagged), and confocal microscopy was performed to visualize JC-1 red and green fluorescence associated with alterations in mitochondrial membrane polarization. CCCP was included as a positive control. (D and E) The expression of the HTLV-1 Tax and HBZ proteins in transfected Jurkat T-cells (D) and HT-1080 cells (E) was detected by immunoblotting. Relative Actin levels are shown for comparison. (F) The accumulation of intracellular ROS in HT-1080 cells cotransfected with either an empty CβS vector, or expression constructs for HTLV-1 Tax, HBZ and/or p30II (HA-tagged), was detected by staining the cells with the fluorescent chemical ROS probe, CM-H2DCFDA (Molecular Probes), and the percentages of CM-H2DCFDA-positive cells were quantified using fluorescence-microscopy, as compared to the total numbers of cells visualized with a DIC phase-contrast filter under a 20x objective lens (see micrographs in Supplemental Fig. S1B). (G) To determine if the activation of TIGAR contributes to the suppression of Tax/HBZ-induced ROS production by p30II, the cells were cotransfected as in F and then repeatedly transfected with a siRNA-tigar oligonucleotide or scrambled RNA (scrRNA) negative control. A pcDNA3.1-FLAG-tagged-TIGAR expression construct was included in some samples to determine the effects of overexpressing the TIGAR protein. The samples were then stained with CM-H2DCFDA and the relative percentages of CM-H2DCFDA-positive cells were determined using fluorescence-microscopy as in F. (H) The specificity of the siRNA-tigar oligonucleotide to inhibit TIGAR expression was determined by transfecting HT-1080 cells with siRNA-tigar, a scrRNA control, or empty CβS vector and then immunoblotting to detect the endogenous TIGAR protein. Alternatively, the cells were cotransfected with pcDNA3.1-FLAG-tagged-TIGAR and either an empty CβS vector, siRNA-tigar, or a scrRNA control and the FLAG-tagged TIGAR protein was detected by immunoblotting using a monoclonal Anti-FLAG M2 primary antibody. Relative Actin levels are shown for comparison. All the data is representative of at least three independent experiments. The data in B, F, and G represent the mean of the experiments ± standard deviation (error bars).
We recently demonstrated in Romeo et al., 2018 that the HTLV-1 p30II protein activates p53, inhibits TIP60-dependent p53-K120-acetylation, and induces the expression and mitochondrial localization of TIGAR, associated with c-Myc oncogenic dysregulation in HTLV-1-transformed T-cells and ATLL clinical isolates. Representative micrographs showing the induction and mitochondrial expression of the TIGAR protein by lentiviral-HTLV-1 p30II-GFP in transduced human HFL1 fibroblasts, compared to a lentiviral-GFP negative control, are provided in supplementary Fig. S1A. We therefore investigated if p30II could prevent the intracellular accumulation of damaging ROS and counter the oxidative stress and cytotoxicity caused by the viral oncoproteins, Tax and HBZ. For these experiments, HT-1080 fibrosarcoma cells (which contain wildtype p53 and express inducible TIGAR; Romeo et al., 2018) were cotransfected with various combinations of expression constructs for Tax, HBZ, p30II (HA-tagged), or an empty CβS vector control, and the production of ROS was detected by staining the cultures with the fluorescent chemical ROS-probe, CM-H2DCFDA (Molecular Probes). The relative percentages of ROS-positive cells per well were quantified using fluorescence-microscopy and compared to the total numbers of cells visible in the DIC phase-contrast images (Fig. 1F and Supplemental Fig. S1B). The results in Figs. 1F and S1B demonstrate that both Tax and HBZ induced high intracellular ROS levels as detected by CM-H2DCFDA-staining. The co-expression of HBZ, together with the transactivator protein Tax, also resulted in significant ROS production (Figs. 1F and S1B), in agreement with the mitochondrial membrane depolarization induced by these factors (Figs. 1A and 1B). However, co-expression of the p30II protein prevented the intracellular accumulation of damaging ROS by either Tax or HBZ, as compared to the empty CβS vector control (Figs. 1F and S1B). Consistent with the role of TIGAR in suppressing mitochondrial ROS, the overexpression of FLAG-tagged TIGAR inhibited ROS production by the viral oncoproteins Tax and HBZ (Fig. 1G). The ability of p30II to cooperate with Tax and HBZ was dependent upon TIGAR functions (Romeo et al., 2018), as the targeted siRNA-tigar-knockdown of TIGAR expression significantly countered the suppression of ROS levels by p30II (Fig. 1G). The cotransfection of a scrambled RNA (scrRNA) negative control oligonucleotide did not alter p30II’s ability to counter the ROS production and oxidative stress induced by Tax and HBZ (Fig. 1G). The siRNA-tigar-knockdown of endogenous TIGAR protein expression in the transfected HT-1080 cells was demonstrated by immunoblotting (Fig. 1H, left panels); and the specificity of the siRNA-tigar oligonucleotide, compared to the scrRNA control, was further confirmed by inhibiting the expression of FLAG-tagged TIGAR in cotransfected cells (Fig. 1H, right panels; Bensaad et al., 2006).
The TIGAR protein inhibits cellular autophagy under conditions of nutrient starvation or metabolic stress (Bensaad et al., 2009); and Hoshino et al., 2012 have shown that TIGAR inhibits mitophagy in cardiomyocytes following ischemic injury. The HTLV-1 transactivator protein Tax induces autophagy which is required for viral replication, dependent upon the activation of IκK-signaling and recruitment of Beclin-1 into lipid raft microdomains (Tang et al., 2013; Wang et al., 2014; Ren et al., 2015). The p30II protein is important for the maintenance of latency and a high proviral titer in vivo; and p30II negatively regulates Tax-dependent viral gene expression (Bartoe et al., 2000; Valeri et al., 2010; Nicot et al., 2004; Edwards et al., 2011; Sinha-Datta et al., 2007; Younis et al., 2006; Zhang et al., 2001; Michael et al., 2006; Lairmore et al., 2011; Silverman et al., 2004). We therefore tested whether the activation of TIGAR by p30II (Romeo et al., 2018) might inhibit cellular autophagy and mitophagy resulting from oxidative damage caused by Tax and/or HBZ. HT-1080 cells expressing Tax or HBZ were transduced with lentiviral-HTLV-1 p30II (HA) or an empty pLenti vector, and then immunostained with primary antibodies against TIM23 and the autophagy marker, Beclin-1. The Beclin-1 protein contains a leucine-rich nuclear export signal and its cytoplasmic translocation and localization in autophagic vesicles is required for the induction of cellular autophagy (Liang et al., 2001). The relative percentages of cells with nuclear Beclin were quantified using confocal microscopy (Figs. 2A and 2B). As shown in the representative micrographs in Fig. 2A and the data in Fig. 2B, transduction with a lentiviral-HTLV-1 p30II (HA) expression vector resulted in the nuclear accumulation of Beclin-1 and significantly prevented Tax and HBZ-induced autophagy. To determine if p30II could also inhibit Tax and/or HBZ-induced mitochondrial damage or mitophagy, HT-1080 cells expressing Tax or HBZ were transduced with lentiviral-HTLV-1 p30II (HA) or an empty pLenti vector, and then immunostained using primary antibodies that recognize the mitochondrial protein TOM20 and the marker of mitophagy, SQSTM1. The relative percentages of cells containing mitochondrial SQSTM1/TOM20-positive foci (arrows in the inset images in Fig. 2C) were quantified using confocal microscopy. The transduction with lentiviral-HTLV-1 p30II (HA) effectively blocked the induction of mitochondrial damage/mitophagy by Tax and HBZ, as compared to the empty lentiviral vector control (Figs. 2C and 2D), suggesting that p30II cooperates with these major viral oncoproteins.
Fig. 2.

The HTLV-1 p30II protein prevents Tax/HBZ-induced oxidative damage and mitophagy, dependent upon TIGAR functions. (A and B) HT-1080 cells expressing the HTLV-1 Tax or HBZ oncoproteins were transduced with an empty pLenti vector or lentiviral-HTLV-1 p30II (HA-tagged) expression vector. The samples were then fixed and immunostained to visualize the TIM23 (red signal) and Beclin-1 (blue signal) proteins by confocal microscopy. Beclin-1 was present within cytoplasmic autophagosomal vesicles that colocalized with mitochondria (TIM23-labeled structures) in the cytoplasm of Tax or HBZ-expressing cells transduced with the empty vector. However, lentiviral HTLV-1 p30II inhibited Tax- and HBZ-induced autophagy/mitophagy and correlated with the predominantly nuclear distribution of Beclin-1. The graphs in A (right panels) give a three-dimensional representation of the subcellular distributions of TIM23 and Beclin-1 in the micrographs shown. The data in B show the percentages of cells with nuclear Beclin-1 and represent the mean ± standard deviation (error bars) from three independent experiments. (C and D) HT-1080 cells expressing HTLV-1 Tax or HBZ were transduced with an empty pLenti vector or lentiviral-HTLV-1 p30II (HA) particles, and then fixed and immunostained to detect TOM20 (red signal) and the mitophagy marker SQSTM1 (blue signal) by confocal microscopy. The arrows in the enlarged inset images at right indicate SQSTM1-positive mitophagic structures in the Tax or HBZ-expressing cells transduced with the empty vector. The data in D show the relative percentages of SQSTM1/TOM23-positive cells and represent the mean ± standard deviation (error bars) from three independent experiments. (E) The transiently-amplified, infectious HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were serially passaged over 4-weeks and the release of extracellular HTLV-1 particles into the culture supernatants was measured, relative to a p19Gag protein standard, by performing Anti-HTLV-1 p19Gag ELISAs (Zeptometrix). (F) The relative expression of TIGAR protein in the HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones was detected by SDS-PAGE and immunoblotting. Actin levels are shown for comparison. (G) The production of intracellular ROS in the HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant clones transfected with siRNA-tigar, a scrRNA negative control, or empty CβS vector was detected using the fluorescent chemical ROS-probe, CM-H2DCFDA, and then quantified by fluorescence-microscopy. CCCP was included as a positive control. The phenotypic impairment of cells containing the ACH.p30II mutant provirus was rescued either through transfection with pcDNA3.1-FLAG-TIGAR, or transducing them with lentiviral-HTLV-1 p30II (HA), as compared to an empty lentiviral vector. (H) The levels of oxidative damage and mitophagy in the HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were determined using immunofluorescence-confocal microscopy to quantify the relative percentages of SQSTM1/TOM20-positive cells. The effects of inhibiting TIGAR expression were determined by transfecting these cells with siRNA-tigar or a scrRNA negative control. The overexpression of TIGAR (through transfection with pcDNA3.1-FLAG-TIGAR) significantly countered the mitochondrial oxidative damage in the ACH.p30II mutant clone. All the data is representative of at least three independent experiments. The data in E, G, and H represent the mean of the experiments ± standard deviation (error bars).
HT-1080 clones containing the infectious HTLV-1 ACH.p30II mutant provirus, defective for p30II production, exhibit increased oxidative mitochondrial damage and cytotoxicity
We next sought to determine if p30II protects against the oxidative and metabolic toxicity induced by pX-encoded viral oncoproteins in the context of the full-length HTLV-1 provirus. For these experiments, we used the transiently-amplified HT-1080 clones which contain the infectious HTLV-1 ACH.wt or ACH.p30II mutant (defective for p30II production) proviruses that were described in Romeo et al., 2018. There was no detectable difference in the production of extracellular p19Gag-containing virus particles by the HT-1080/ACH.wt and HT-1080/ACH.p30II mutant clones, as determined by performing Anti-HTLV-1 p19Gag ELISAs on culture supernatants following serial-passage over a period of 4-weeks (Fig. 2E). The results in Fig. 2F demonstrate that the HT-1080/ACH.wt proviral clone exhibited increased TIGAR protein expression, compared to the HT-1080/ACH.p30II mutant or untreated HT-1080 cells, in agreement with the p30II-dependent induction of TIGAR as shown in supplementary Fig. S1A (Romeo et al., 2018). We then compared the relative levels of intracellular ROS between the HT-1080/ACH.wt and HT-1080/ACH.p30II mutant proviral clones by staining the cultures with CM-H2DCFDA. Certain samples were treated with CCCP as a positive control for oxidative stress (Fig. 2G). The results in Fig. 2G show that the HT-1080/ACH.p30II mutant transfected with an empty CβS vector contained significantly higher ROS levels than the HT-1080/ACH.wt cells with the CβS vector. Also, the siRNA-tigar-knockdown of TIGAR expression increased the intracellular accumulation of ROS in the HT-1080/ACH.wt clone, as compared to cells transfected with a scrRNA negative control (Fig. 2G). The transfection with a siRNA-tigar oligonucleotide, however, did not lead to greater ROS production in HT-1080/ACH.p30II mutant cells (Fig. 2G). We further observed that either transduction with lentiviral-HTLV-1 p30II (HA) or the overexpression of FLAG-tagged TIGAR countered the production of ROS by the HT-1080/ACH.p30II mutant clone, as compared to the ACH.p30II mutant transduced with an empty pLenti vector (Fig. 2G). Consistent with the increased oxidative stress observed in the HT-1080.ACH.p30II mutant clone, defective for p30II production (Fig. 2G), the results in supplementary Fig. S2 demonstrate that the HT-1080/ACH.p30II mutant exhibited a greater degree of mitochondrial membrane depolarization (i.e., higher green/red-fluorescence ratio in merged images), compared to the HT-1080/ACH.wt clone. CCCP was included as a positive control.
We then examined whether the oxidative stress in the HT-1080/ACH.p30II mutant proviral clone was correlated with increased mitochondrial damage or mitophagy in these cells. As shown in Fig. 2H, the HT-1080/ACH.p30II mutant proviral clone transfected with an empty CβS vector exhibited a higher percentage of SQSTM1/TOM23-positive cells, relative to the HT-1080/ACH.wt clone transfected with the CβS vector. The mitochondrial damage induced by the ACH.p30II mutant was countered through overexpression of the FLAG-tagged TIGAR protein (Fig. 2H). Further, the siRNA-tigar-knockdown of TIGAR expression resulted in increased mitophagy in the HT-1080/ACH.wt proviral clone, as compared to ACH.wt cells transfected with a scrRNA negative control (Fig. 2H).
To determine if p30II helps to prevent oxidative genomic DNA-damage induced by other viral oncoproteins (Tax and HBZ) in the context of the full HTLV-1 provirus, the HT-1080/ACH.wt and HT-1080/ACH/p30II mutant proviral clones were labeled using a fluorescent Click-iT Alex Fluor 594-TUNEL kit (Molecular Probes) and subsequently immunostained with a primary antibody that recognizes the Ser139-phosphorylated histone variant, H2A.X, which localizes to sites of damaged chromatin. The average numbers of nuclear phospho-H2A.X foci per cell (arrows in the inset images in Fig. 3A) were quantified using confocal microscopy. Hoechst 33342 nuclear-staining was used to visualize cell nuclei (Fig. 3A). As positive controls, HT-1080 cells were treated with either CCCP or DNAse I (Figs. 3A and 3B). The HT-1080/ACH.p30II mutant proviral clone exhibited significantly more genomic DNA-damage and phospho-H2A.X foci than the HT-1080/ACH.wt clone (Figs. 3A and 3B). We next labeled the HT-1080/HTLV-1 ACH proviral clones using a fluorescent TUNEL kit and immunostained them with a primary antibody against TOM20 to detect oncogene-induced mitochondrial DNA-damage. As shown in Figs. 3C and 3D, the HT-1080/ACH.p30II mutant exhibited increased numbers of TUNEL/TOM20-positive foci (arrows in the inset images in Fig. 3D), compared to the HT-1080/ACH.wt clone or untreated HT-1080 cells. Samples treated with CCCP or DNAse I were included as positive controls (Figs. 3C and 3D). Based upon these data, we conclude that the HTLV-1 latency-maintenance factor p30II inhibits Tax/HBZ-induced ROS-production, oxidative DNA-damage and mitophagy through the activation of TIGAR.
Fig. 3.

HT-1080 cells containing the infectious HTLV-1 ACH.p30II mutant provirus, defective for p30II production, exhibit increased genomic and mitochondrial DNA-damage as compared to the wildtype ACH provirus. (A and B) The parental HT-1080 cell-line and the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were stained using a Click-iT Alexa Fluor 594-TUNEL kit (Molecular Probes; red signal) and a rabbit polyclonal primary antibody that recognizes the Ser139-phosphorylated histone variant H2A.X (Anti-Phospho-Ser139-H2A.X; green signal) which localizes at sites of genomic DNA-damage. The chemical uncoupler CCCP and DNAse I were included as positive controls. The cell nuclei were stained with the fluorescent dye, Hoechst 33342 (Molecular Probes; blue signal). The arrows in the enlarged inset panels at right indicate Phospho-Ser139-H2A.X foci in the merged images. The data in B represent the mean ± standard deviation (error bars) from three independent experiments. (C and D) HT-1080 cells and the HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were stained using a fluorescent TUNEL kit (red signal) and a rabbit polyclonal Anti-TOM20 primary antibody (green signal) to detect mitochondrial DNA-damage resulting from oxidative stress. The cell nuclei were stained with Hoechst 33342. Positive control samples were treated with either CCCP or DNAse I. The arrows in the enlarged inset panels at right indicate sites of mitochondrial DNA-damage. The graph in C shows the relative numbers of TUNEL/TOM20-positive foci per cell; and the data represent the mean ± standard deviation (error bars) from three independent experiments. (E and F) The ability of the HTLV-1 p30II protein to protect against Tax/HBZ-induced cytotoxicity was determined by either (E) transfecting HT-1080 cells with Tax or HBZ expression constructs and then transducing them with lentiviral-HTLV-1 p30II (HA) particles or an empty lentiviral vector, or (F) cotransfecting the cells with expression constructs for Tax, HBZ, and/or p30II (HA). Certain samples were repeatedly cotransfected with a siRNA-tigar oligonucleotide or scrRNA control using HiPerFect transfection reagent. The cells in E were also treated with staurosporine (100 nM) as a positive control to induce apoptosis. Apoptotic cells were detected by staining them with Annexin V-FITC (green signal) and propidium iodide (PI; red signal); and the relative percentages of Annexin V-FITC +/− PI-positive cells were quantified by fluorescence-microscopy, as compared to the total numbers of cells visualized using a DIC phase-contrast filter. All the data is representative of at least three independent experiments; and the data in F represent the mean of the experiments ± standard deviation (error bars).
The viral oncoproteins Tax and HBZ induce aberrant cellular proliferation as well as apoptosis (Nicot and Harrod, 2000; Hall et al., 1998; Los et al., 1998; Chen et al., 1997; Yamada, T., 1996; Kawatsuki et al., 2016). As our results have shown that p30II suppresses Tax/HBZ-induced oxidative stress (Figs. 1F, 1G, 2A–2D, Supplemental Fig. S1B), we next investigated whether p30II could prevent Tax and/or HBZ-induced programmed cell death/apoptosis. HT-1080 cells expressing Tax or HBZ were transduced with lentiviral-HTLV-1 p30II (HA) or an empty pLenti vector, and then stained with Annexin V-FITC and propidium iodide (PI) to visualize apoptotic cells (Annexin V-FITC and/or PI-positive) using fluorescence-microscopy. Staurosporine-treated cells were included as a positive control for apoptosis. The representative micrographs in Fig. 3E demonstrate that less apoptosis was observed in the Tax or HBZ-expressing cells transduced with lentiviral-HTLV-1 p30II (HA), versus the pLenti vector control. To better quantify the effects of p30II upon Tax/HBZ-induced apoptosis, HT-1080 cells were cotransfected with various expression constructs for HTLV-1 Tax, HBZ, or p30II, or an empty CβS vector and the percentages of Annexin V-FITC and/or PI-positive cells were quantified using fluorescence-microscopy, relative to the total numbers of cells visible with a DIC phase-contrast filter. The viral transactivator Tax induced more apoptosis than HBZ (Figs. 3E and 3F); however, the co-expression of p30II (HA) significantly countered Tax/HBZ-induced cellular apoptosis (Figs. 3E and 3F). The siRNA-knockdown of TIGAR expression, by repeatedly transfecting the cells with a siRNA-tigar oligonucleotide, inhibited p30II’s ability to prevent Tax/HBZ-induced apoptosis as compared to a scrRNA negative control (Fig. 3F). These results demonstrate that p30II suppresses Tax/HBZ-induced oxidative damage and cytotoxicity through the activation of TIGAR, and suggests these pX-encoded factors may functionally collaborate during retroviral carcinogenesis.
HTLV-1 p30II protects against Tax/HBZ-induced cellular senescence and cooperates with these viral oncoproteins to induce oncogenic foci-formation in vitro
The aberrant activation of cellular oncogenes can lead to increased metabolic toxicity and cumulative physiological and oxidative stress resulting in cellular senescence or apoptosis (Vafa et al., 2002; Maya-Mendoza et al., 2015; Olenchock and Vander Heiden, 2013; Gorrini et al., 2007; Hermeking and Eick, 1994; Zindy et al., 1998). The HTLV-1 transactivator Tax has been shown to induce apoptotic cell death (Nicot and Harrod, 2000; Hall et al., 1998; Los et al., 1998; Chen et al., 1997; Yamada, T., 1996); and Giam and colleagues have demonstrated that Tax induces cellular senescence in HeLa cells associated with NF-κB hyperactivation which can be countered by the HBZ protein (Ho et al., 2012; Zhi et al., 2011; Kuo and Giam, 2006). Importantly, Kawatsuki et al., 2016 have shown that HBZ interacts with the Rb/E2F-1 complex and induces both cell proliferation and apoptosis in transduced primary T-cells, as well as HBZ-expressing transgenic mice. We therefore investigated whether p30II might counter Tax and/or HBZ-induced cellular senescence as a result of oncogene-associated oxidative and physiological stress. As shown in Figs. 4A and 4B, both Tax and HBZ induced significant cellular senescence, compared to the empty CβS vector in transfected cells, as determined by staining with an X-Gal solution to detect SA-β-gal expression (blue-stained cells in the micrographs). In agreement with Zhi et al., 2011, the co-expression of HBZ markedly inhibited Tax-induced cell senescence (Figs. 4A and 4B). However, co-expression of the HTLV-p30II-GFP was more effective at preventing Tax-induced cellular senescence; and p30II-GFP also countered HBZ-induced senescence (Figs. 4A and 4B). This difference could be attributable to p30II’s better ability to suppress Tax-induced ROS production relative to HBZ (see Figs. 1F, 1G, and Supplement Fig. S1B), and may allude to distinct roles for NF-κB hyperactivation and oxidative stress in mediating Tax-induced cellular senescence (Ho et al., 2012; Zhi et al., 2011). The expression of the HTLV-1 Tax, HBZ (Myc-tagged), and p30II-GFP in the transfected cells was detected by SDS-PAGE and immunoblotting (Fig. 4C).
Fig. 4.

The HTLV-1 p30II protein inhibits Tax/HBZ-induced cellular senescence and cooperates with these major viral oncoproteins to induce oncogenic foci-formation in vitro. (A and B) To determine if p30II prevents Tax/HBZ-induced cellular senescence in HeLa cells as reported in Ho et al., 2012 and Kuo and Giam, 2006, the cells were cotransfected with an empty CβS vector or expression constructs for HTLV-1 Tax, HBZ, and/or p30II-GFP in various combinations. The cells were then cultured for five days and subsequently stained using an X-Gal solution to detect senescence-associated Beta-galactosidase (SA-β-gal) expression (blue signal in micrographs). DIC phase-contrast is included in the merged images for reference. The data in A represent the mean ± standard deviation (error bars) from three independent experiments. (C) The expression of the HTLV-1 Tax, p30II-GFP, and HBZ (Myc-tagged) proteins was detected by SDS-PAGE and immunoblotting. The relative levels of Actin are shown for comparison. (D) The parental HT-1080 cells and transiently-amplified HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were cotransfected with an empty CβS vector, or expression constructs for HTLV-1 p30II-GFP or TIGAR (FLAG-tagged). Alternatively, the HT-1080/ACH.p30II mutant cells were transduced with lentiviral-HTLV-1 p30II (HA) particles or an empty pLenti vector. Certain samples were also repeatedly transfected with a siRNA-tigar oligonucleotide or scrRNA negative control. The cells were then stained with an X-Gal solution to detect SA-β-gal expression and analyzed by microscopy using a 20x objective lens to quantify the numbers of senescent cells per well. The data in D represent the mean ± standard deviation (error bars) from three independent experiments. (E-H) To determine whether the HTLV-1 p30II protein cooperates with the viral oncoproteins Tax and HBZ, the HFL1 human fibroblast cell-line (a pseudodiploid cell-line with a stable karyotype which grows as a uniform monolayer) was cotransfected with an empty CβS vector, or expression constructs for HTLV-1 Tax, HBZ, or p30II-GFP in various combinations. The stably transfected cells were monitored over a three-week period for the formation of oncogenically-transformed colonies (i.e., loss of contact-inhibition; see DIC phase-contrast images in H). The transformed colonies were fixed and stained with a methylene blue solution; and the numbers of transformed colonies per well were quantified by microscopy using a 10x objective. The expression of HTLV-1 p30II-GFP within transformed colonies was visualized by direct fluorescence-microscopy (top panels in H). The data in E, F, and G represent the mean ± standard deviation (error bars) from three independent experiments.
We next compared the relative levels of cellular senescence induced by the infectious HT-1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones which express Tax, HBZ, and/or p30II in the context of the full-length provirus. The chemical uncoupler CCCP was included as a positive control and the parental HT-1080 cells (UT) are also shown (Fig. 4D). The HT-1080/ACH.wt provirus cotransfected with an empty CβS vector control did not exhibit significant senescence, as compared to the HT-1080/ACH.p30II mutant provirus with the empty vector (Fig. 4D), which suggests that p30II expression is necessary to protect against the oxidative and physiological stress induced by viral oncoproteins (Tax and HBZ) in the context of the full-length provirus. The cellular senescence induced by the HT-1080/ACH.p30II mutant was phenotypically countered by either transducing these cells with lentiviral-HTLV-1 p30II (HA) particles or cotransfecting them with a HTLV-1 p30II-GFP expression construct (Fig. 4D). We further demonstrated that inhibiting TIGAR expression, using a siRNA-tigar oligonucleotide, induced cell senescence in the HT-1080/ACH.wt clone and blocked the ability of lentiviral-HTLV-1 p30II to rescue the phenotypic impairment of the HT-1080/ACH.p30II mutant, compared to cells transfected with a scrRNA negative control (Fig. 4D). To confirm these findings, we also demonstrated that overexpression of the TIGAR protein through transfection with the pcDNA3.1-TIGAR (FLAG-tagged) construct significantly countered the induction of cellular senescence by the HT-1080/ACH.p30II mutant proviral clone (Fig. 4D).
The roles of the various HTLV-1 pX-encoded factors during retroviral carcinogenesis are yet to be fully elucidated, and it remains unclear whether these proteins cooperatively interact to deregulate cellular pathways associated with oncogenic transformation and the development of neoplastic disease. We have previously demonstrated that HTLV-1 p30II cooperates with the c-Myc oncoprotein through the activation of p53-dependent pro-survival signals (e.g., TIGAR) and induces oncogenic foci formation in vitro (Romeo et al., 2018; Romeo et al., 2015; Awasthi et al., 2005). We therefore tested whether p30II, Tax, and/or HBZ might functionally cooperate to induce oncogenic cellular transformation and foci-formation in vitro. For these experiments, the HFL1 human fibroblast cell-line (which has a stable pseudodiploid karyotype, contains wildtype p53, and grows as a uniform monolayer) was cotransfected with various combinations of expression constructs for HTLV-1 Tax, HBZ, and p30II-GFP, or an empty CβS vector, and oncogenic foci-formation resulting from the loss of contact-inhibition was monitored over a period of three weeks. As shown in Figs. 4E and 4F, the HTLV-1 p30II-GFP construct alone induced approximately 25–30 transformed colonies. The p30II-GFP fusion significantly cooperated with Tax (Tax + p30II-GFP produced >90 colonies) and HBZ (HBZ + p30II-GFP produced approximately 45 colonies), as compared to either Tax or HBZ in combination with an empty CβS vector control (Figs. 4E and 4F). Surprisingly, the HBZ protein only moderately cooperated with Tax (Tax + HBZ produced approximately 15 colonies), compared to Tax + empty CβS vector (produced approximately 10 colonies) or HBZ + CβS vector (produced 9 colonies; Fig. 4G). These results agree with the observation by Esser et al., 2017 that the Tax protein was more oncogenic than HBZ in a granzyme B promoter-driven transgenic mouse model of tumorigenesis. The expression of the HTLV-1 p30II-GFP fusion within transformed colonies containing Tax or HBZ was visualized by direct fluorescence-microscopy (Fig. 4H).
These findings demonstrate that p30II likely functions as an ancillary factor for the viral oncoproteins Tax and HBZ and suppresses their metabolic oxidative toxicity through induction of the antioxidant effector TIGAR.
TIGAR is highly expressed in HTLV-1-induced xenograft tumor tissues associated with oncogene dysregulation and angiogenesis
By intraperitoneally engrafting immune-compromised NOD/scid mice with the tumorigenic HTLV-1-transformed SLB1 and MET-1 lymphoma T-cell-lines (Liu et al., 2002; Zimmerman et al., 2011; Harrod, R., 2011; Chen et al., 2009; Ju et al., 2014), we established a highly penetrant in vivo model of HTLV-1-induced T-cell lymphoma. As shown in Figs. 5A–5D, several experimental animals exhibited abdominal distension (arrow in the left panel of Fig. 5A) associated with the development of HTLV-1-induced primary effusion lymphomas (or PELs), also known as body-cavity-based lymphomas –a type of Non-Hodgkin’s lymphoma often associated with poor clinical prognoses in humans. These tumors arise due to the attachment of tumor cells to the mesothelium which then overgrows, as a result of heterotypic signaling, and forms a fluid-filled membranous mass which contains tumor ascites (Figs. 5A–5D). Primary effusion lymphomas are frequently associated with oncogenic viruses, in particular, Kaposi’s sarcoma-associated herpesvirus (KSHV)/human herpesvirus-8 (HHV-8) and Epstein-Barr virus (EBV; Gloghini et al., 2017; Sanchez-Martin et al., 2017; Fassone et al., 2000; Nador et al., 1996) and have been reported in HTLV-1+ ATLL patients with advanced lymphoma-stage disease (Shimamura et al., 1991; Nomura et al., 2002; Dabaja et al., 2002; Okada et al., 2004). The PEL tumors were highly vascularized (Fig. 5B); and we also observed aberrant outgrowths on the livers of some animals (arrows in Fig. 5C). Many experimental animals developed solid T-cell lymphomas as shown in Figs. 5E–5G, occasionally accompanied by enlargement of the liver and/or spleen (Fig. 5F, right panels). The plasma load of infectious HTLV-1 particles in the peripheral bloodstream of the engrafted animals was monitored by performing Anti-HTLV-1 p19Gag ELISAs on blood samples collected at 2-week intervals from the facial veins (Fig. 5H). The animals engrafted with either the HTLV-1+ SLB1 or MET-1 lymphoma T-cell-lines exhibited increased mortality rates, as compared to the Vehicle control groups (Figs. 5I and 5J); however, there was no discernable difference in the mortality rates between SLB1 and MET-1-engrafted mice.
Fig. 5.

Tumor development and secondary organ-involvement in an in vivo xenograft model of HTLV-1-induced T-cell lymphoma. (A–G) Immunocompromised NOD/scid mice (12 animals per sample group, n=12) were anesthetized by gas inhalation with 3% isoflurane/O2 and then intraperitoneally engrafted with the tumorigenic HTLV-1-transformed T-cell lymphoma cell-lines, SLB1 or MET-1, in 500 μl of the vehicle (i.e., calcium/magnesium-free phosphate buffered saline, pH 7.4) using a 27-gauge tuberculin syringe. As a negative control, one group of animals (n=12) was injected with the vehicle alone. The animals were monitored for 8–12 weeks for signs of tumor growth (e.g., abdominal distension or the visible formation of masses) or changes to their overall health status. Following 2-weeks and every 2-weeks thereafter, the experimental animals were anesthetized and 75 μl of blood was collected from the facial vein using a sterile No. 2 lancet. The animals were eventually sacrificed and necropsies were performed to harvest and analyze tumor tissues and any affected secondary organs (e.g., spleen, liver, and pancreas). Several experimental animals exhibited abdominal distension (left panel in A) associated with the development of HTLV-1-induced primary effusion lymphomas (PEL), or body-cavity-based lymphomas (arrows in A and D). The PEL tumors were highly vascularized (B); and we also observed abnormal growths on the livers of some animals with HTLV-1-induced PEL (arrows in C). (E–G) Many animals engrafted with either the HTLV-1-transformed SLB1 or MET-1 cells developed solid T-cell lymphomas (arrows in E, F, and G), which were occasionally accompanied by enlargement of the spleen (lower right panel in F) and/or liver. (H) Representative results are shown for Anti-HTLV-1 p19Gag ELISAs (Zeptometrix) performed on three different SLB1-engrafted animals to measure their relative plasma loads of infectious HTLV-1 particles present in the circulating blood at the indicated time-points, as compared to a p19Gag protein standard. The red asterisk indicates the same animal as shown in A. The data in H represent the mean ± standard deviation (error bars) from three independent Anti-HTLV-1 p19Gag ELISA experiments. (I and J) Kaplan-Meier survival plots are shown for experimental NOD/scid animals that were intraperitoneally engrafted with 5×105 of the HTLV-1-transformed SLB1 (I) or MET-1 (J) lymphoma cells, as compared to the Vehicle control group (n=12 for each sample group).
Our in vivo xenograft model of HTLV-1-induced lymphoma reproduces many of the hallmark clinical characteristics of ATLL, including multi-organ involvement, hepato- and splenomegaly, lymphadenopathy, and the development of lymphoid tumors. Indeed, the infiltration of HTLV-1+ tumor cells with abnormal/polylobate or pleomorphic nuclei into secondary tissues (i.e., spleen, pancreas, and liver) was observed in 100% of the engrafted animals, including those without detectable tumors, as determined by H&E staining and/or immunostaining using a primary antibody that recognizes the human proliferating cell antigen, Ki67 (huKi67; Figs. 6A, 6E–6G). In most cases, the spleen was the predominant organ that exhibited the highest degree of tumor cell-infiltration or metastasis, compared to the pancreas or liver (Fig. 6E).
Fig. 6.

The TIGAR protein is highly expressed in tumor tissues with oncogene dysregulation in animals engrafted with HTLV-1-transformed SLB1 or MET-1 lymphoma T-cells. (A) Tissue sections were prepared from HTLV-1-induced tumors (PEL and solid lymphomas) and affected secondary tissues (spleen, pancreas, and liver) which were then stained with hematoxylin and eosin (H&E) to detect tumor cells with abnormal polylobate or pleomorphic nuclei and a large nuclear-to-cytoplasmic ratio (arrows in micrographs). (B) The HTLV-1+ lymphoma tumor tissues were immunostained with primary antibodies that recognize the human proliferating antigen Ki67 (huKi67; red signal), c-Myc oncoprotein (green signal), and human TIGAR protein (green signal) and the expression of these factors was analyzed by confocal microscopy. DAPI nuclear-staining (blue signal) was included to visualize the surrounding murine cells. (C and D) Graphs depicting the relative percentages of experimental animals, engrafted with 5×105 or 1×106 of the HTLV-1-transformed SLB1 or MET-1 cells, that developed tumors after 12-weeks compared to the Vehicle control group (n=12 animals per sample group). (E) Representative images showing the infiltration of secondary tissues (i.e., spleen, pancreas, and liver) by huKi67-positive (red signal) HTLV-1-transformed SLB1 tumor cells. DAPI nuclear-staining (blue signal) is provided for reference and the visualization of murine stromal cells. (F and G) Graphs depicting the percentages of HTLV-1+ engrafted animals which exhibited the infiltration of secondary tissues by tumor cells, based upon H&E-staining and/or Anti-huKi67 immunostaining results as shown in E (n=10 animals). All of the data in A, B, E, F, and G are representative of at least three sections for each tumor or secondary tissue analyzed.
Approximately 84–100% of the HTLV-1+ SLB1 or MET-1-engrafted experimental animals developed T-cell lymphomas, in contrast to the Vehicle control groups in which no tumors were observed (Figs. 6C and 6D). As we have previously demonstrated that the HTLV-1 p30II protein induces TIGAR and cooperates with the c-Myc oncoprotein (Romeo et al., 2018; Romeo et al., 2015; Awasthi et al., 2005), we next investigated whether TIGAR and/or c-Myc are dysregulated in the lymphoma tumor tissues of SLB1 and MET-1-engrafted animals. Tissue sections were prepared from the processed lymphoma samples and the engrafted HTLV-1+ cells were detected by immunofluorescence-confocal microscopy using a primary antibody against the huKi67 antigen (Fig. 6B). DAPI nuclear-staining was included to visualize the surrounding murine cells. Interestingly, the majority of the tumor mass in all of the HTLV-1-induced lymphomas was predominantly comprised of murine mesenchymal cells, including endothelial progenitors (see Fig. 7D), whereas the HTLV-1+ tumor cells (i.e., huKi67-positive) made up approximately 10–25% of this heterogeneous tissue (Fig. 6B). This underscores the aggressive chemoattractant nature of the engrafted HTLV-1+ lymphoid cells. Consistent with our findings in Romeo et al., 2018, all of the huKi67-positive tumor tissues contained elevated c-Myc expression and high levels of the TIGAR protein (Fig. 6B). We were surprised to discover that two experimental animals with HTLV-1+ lymphomas also developed rare splenic hemangioma tumors (arrow in Fig. 7A), associated with the secondary infiltration of tumor cells and splenomegaly. Splenic hemangiomas are derived from the overgrowth of endothelial progenitor cells (Kutok and Fletcher, 2003; Willcox et al., 2000), which prompted us to speculate that the engrafted HTLV-1+ tumor lymphocytes might promote increased angiogenesis and the vascularization of lymphoma tissues. Cheung et al., 2012 have demonstrated that TIGAR activates HIF-1α expression associated with mitochondrial targeting of the TIGAR protein under hypoxic conditions. Conversely, Rajendran et al., 2013 have shown that HIF-1α and the transcriptional cofactor P/CAF upregulate tigar gene expression, dependent on wildtype p53, through hypoxia-responsive elements present within the tigar promoter. As shown in Figs. 7B and 7D, both the splenic-infiltrating HTLV-1+ tumor cells as well as those resident in lymphoma tissues (i.e., huKi67-positive), expressed high levels of TIGAR associated with induction of the angiogenic markers, HIF-1α and vascular endothelial growth factor (VEGF). This correlated with increased angiogenesis and the recruitment of murine endothelial progenitors into the splenic tissues (Fig. 7B, right panels) and HTLV-1+ T-cell lymphomas (Fig. 7D, right panels). The endothelial origin of the hemangioma tumors was confirmed by immunostaining the processed tissue sections with antibodies that specifically recognize the surface markers of murine endothelial progenitors: CD31/platelet and endothelial cell adhesion molecule 1 (PECAM1) and Flk1/vascular endothelial growth factor receptor-2 (VEGFR2; Fig. 7C).
Fig. 7.

The HTLV-1+ lymphoma tumor cells contain high levels of TIGAR and express the angiogenic markers, HIF-1α and VEGF. (A) A rare splenic hemangioma tumor (arrow) in an HTLV-1+ SLB1-engrafted animal associated with a solid T-cell lymphoma and splenomegaly. (B) Representative data demonstrating that infiltrating HTLV-1-transformed SLB1 tumor cells (huKi67-positive; red signal) in the spleens of engrafted animals strongly expressed the TIGAR protein (green signal) as well as the angiogenic factors, HIF-1α and VEGF (green signals). This correlated with the increased recruitment of murine endothelial progenitors (CD31/PECAM1-positive and Flk1/VEGFR2-positive; green signals) into the splenic tissues of HTLV-1+ engrafted animals, as compared to the normal spleens of Vehicle control animals (right panels in B). DAPI nuclear-staining (blue signal) is shown for reference. (C) The splenic hemangioma in A was processed, sectioned and immunostained using antibodies that specifically recognize the surface markers of murine endothelial progenitors: CD31/PECAM1 and Flk1/VEGFR2 (green signals), to confirm the endothelial origin of this membranous tumor. (D) Representative images of HTLV-1+ lymphoma tumor tissues (huKi67-positive; red signals) which express TIGAR, HIF-1α, and VEGF (green signals), associated with the increased recruitment of murine endothelial precursor cells (CD31/PECAM1 and Flk1/VEGFR2-positive; green signals in the micrographs at right) into the tumor stroma. DAPI nuclear-staining (blue signal) is shown for reference. All of the data in B and D are representative of at least three sections from tissues obtained from three different animals (n=3). The splenic hemangiomas in A and C are rare tumors and were only observed in two HTLV-1+ experimental animals associated with lymphomas (solid and PEL) and splenomegaly. The data in C is representative of at least three sections for the tumor tissues shown.
These results demonstrate that TIGAR is highly expressed in infiltrating and lymphoma-resident huKi67-positive tumor cells in our in vivo xenograft model of HTLV-1-induced T-cell lymphoma, and correlates with oncogene dysregulation and the increased recruitment of endothelial cell mediators of angiogenesis.
Discussion
The HTLV-1 encodes several regulatory and accessory factors within a highly conserved pX region, yet it remains unclear how these products coordinately interact or contribute to the dysregulation of cellular proliferative pathways during viral carcinogenesis. This study reports on the cooperative dynamics between Tax, p30II, and HBZ and mechanistically demonstrates that p30II suppresses the oxidative and metabolic toxicity induced by the viral oncoproteins Tax/HBZ in a manner dependent upon TIGAR functions (Figs. 1G, 2A–2D, 2G, 2H, 3A–3F, 4A–4D, S1A; Romeo et al., 2018). The HTLV-1 transactivator Tax activates cellular transcriptional pathways, including CREB/ATF, Serum-response factor/p67, and NF-κB (Zhao and Giam, 1992; Harrod et al., 1998; Harrod et al., 2000; Smith and Greene, 1990; Sun et al., 1994), and subverts host negative growth-regulatory signals and DNA-damage repair pathways associated with aberrant lymphoproliferation and the development of neoplastic disease. It is generally believed that Tax contributes to the early-stage immortalization of a subset of HTLV-1-infected cells, as the viral transactivator is undetectable in fresh ATLL clinical isolates –whereas HBZ is persistently expressed throughout the course of disease. Interestingly, it was recently demonstrated that Tax expression is heterogeneous within HTLV-1-transformed cells and sporadically alternates between on and off states (Mahgoub et al., 2018). Using RNA-fluorescence in situ hybridization, Billman et al., 2017 showed that the expression of tax and hbz occurs in alternating transcriptional bursts in patient-derived HTLV-1-infected T-cell clones.
The hyperactivation of NF-κB by Tax increases the expression of the cyclin dependent kinase-inhibitors p21WAF1 and p27Kip1 and induces permanent G1-arrest and cellular senescence (Ho et al., 2012; Zhi et al., 2011). Further, Baydoun et al., 2015 have reported that Tax induces DNA double-strand breaks, dependent upon NF-κB-activation, as a result of the production of nitric oxide by iNOS. Tax recruits the ring finger protein 8 (RNF8) and E3 ubiquitin ligase, Ubc13, and stimulates K63-linked polyubiquitination and activates IκK signaling through interactions with the IκK-γ/NEMO subunit and TAK1 in HTLV-1-infected cells (Ho et al., 2015; Wu and Sun, 2007; Harhaj et al., 2007; Geleziunas et al., 1998; Yamaoka et al., 1998). Choi and Harhaj, 2014 have demonstrated that Tax mediates the mitochondrial interaction between the E3 ubiquitin ligase TRAF6 and the anti-apoptotic myeloid cell leukemia-1 (MCL-1) protein, dependent upon IκK complex activation, which resulted in K63-polyubiquitination and the increased stability of MCL-1. Tax/TRAF6/MCL-1 molecular interactions were required for the in vitro immortalization and survival of Tax-expressing primary T-cells and HTLV-1-transformed ATLL cell-lines (Choi and Harhaj, 2014). Moreover, Swains et al., 2010 have shown that immune-activated HTLV-1 LTR-tax transgenic CD4+ T-cells, engrafted into immune-compromised NSG mice, contain high levels of MCL-1 and resulted in a leukemia-like proliferative phenotype with diffuse lymphoid infiltration. It is intriguing to speculate that the p30II-induced mitochondrial targeting of TIGAR might prevent oxidative damage to mitochondrial membrane proteins and potentially could influence Tax/TRAF6/MCL-1 interactions and contribute to the oncogenic cooperation between p30II and Tax (see Figs. 4E and 4H).
The HTLV-1 antisense factor, HBZ, inhibits Tax-dependent LTR transactivation (Lemasson et al., 2007) and was shown to be required for the persistence of HTLV-1-infected CD4+ T-cell clones in an in vivo rabbit model of pathogenesis (Arnold et al., 2006; Kannian et al., 2012; Li et al., 2009). HBZ also activates cellular proliferative pathways, including E2F, JunD, TGF-β, and Wnt5a (Kawatsuki et al., 2016; Zhao et al., 2011; Ma et al., 2013), increases expression of the hTERT subunit, (Kuhlmann et al., 2007), and induces T-cell lymproliferation (Satou et al., 2006; Arnold et al., 2008). Panfil et al., 2018 recently demonstrated that the E3 ubiquitin ligase, UBR5, interacts with the central basic region of HBZ and ubiquitinates and modulates the stability and turnover of this viral oncoprotein. Transgenic mice expressing HBZ from a granzyme B promoter developed T-cell lymphomas in association with splenomegaly and osteolytic bone lesions, which correlated with diffuse infiltration by HBZ-expressing tumor cells and the upregulation of inflammatory cytokines and bone-remodeling factors (Esser et al., 2017). Moreover, others have shown that HBZ-expressing transgenic mice exhibit increased Foxp3 and IFN-γ-mediated inflammatory signaling which promoted the development of lymphoid tumors in these animals (Mitagami et al., 2015; Satou et al., 2011). Arnold et al., 2008 have demonstrated that HTLV-1-transformed SLB1 cells, transduced with a lentiviral short hairpin-RNA construct to inhibit HBZ expression, exhibited reduced proliferative potential, tumor growth, and organ infiltration in engrafted immune-deficient mice. However, HBZ, as well as the related HTLV-2 antisense synologue APH-2, suppress Tax-dependent NF-κB activation (Zhao et al., 2009; Panfil et al., 2016); and Zhi et al., 2011 have reported that HBZ counters the cellular senescence resulting from Tax-mediated hyperactivation of NF-κB. Our results demonstrate that p30II co-expression significantly blocks the oxidative stress, cell senescence and apoptosis induced by Tax and HBZ (Figs. 1F, 1G, 3E, 3F, 4A–4D, and S1B); see also Kawatsuki et al., 2016; Kuo and Giam, 2006; Nicot and Harrod, 2000; Hall et al., 1998; Los et al., 1998), alluding to potential coordinate interplay between these pX-encoded factors. While the ability of the viral transactivator Tax to induce both cellular proliferation and apoptosis may seem paradoxical in nature, it is possible that Tax-mediated programmed cell-death could represent a self-selective mechanism whereby HTLV-1-infected T-cells expressing high levels of Tax would be eliminated by the host immune response and only those cells with low levels of Tax expression might persist by evading immune recognition. In this regard, the p30II protein likely serves dual functions by promoting viral latency through the suppression of Tax-dependent transactivation, and supporting the survival of HTLV-1-infected cells through the activation of p53-dependent pro-survival/antioxidant signals.
The HTLV-1 latency-maintenance factor p30II inhibits Tax-dependent proviral gene expression through transcriptional and posttranscriptional mechanisms (Zhang et al., 2001; Zhang et al., 2000; Michael et al., 2006; Nicot et al., 2004; Younis et al., 2004; Younis et al., 2006; Bai et al., 2012; Edwards et al., 2011). The p30II protein binds to the KIX domain of the transcriptional coactivators/acetyltransferases, p300/CBP, and interferes with the formation of Tax-CREB-p300 transactivation complexes on the 21-base-pair repeat TREs within the HTLV-1 promoter (Zhang et al., 2001). Michael et al., 2006 have demonstrated that lysine residue K106 within the p300/CBP-binding domain of p30II (aa residues 100–179) mediates the transcriptional repression of the HTLV-1 LTR. Moreover, p30II posttranscriptionally inhibits Tax-dependent HTLV-1 gene expression by inhibiting the nuclear export of the pX-encoded tax/rex mRNA (Nicot et al., 2004; Younis et al., 2004; Younis et al., 2006). As Lin et al., 2017 have shown that the stability of tax/rex transcripts modulates HTLV-1 gene expression, it remains unclear whether the p30II-tax/rex mRNA axis might affect the stability of these transcripts. Lentiviral p30II interacts with the 20S proteasome activator, REGγ, and reduces the intracellular levels of the pro-apoptotic ataxia telangiectasia mutated (ATM) kinase and increases the survival of transduced Jurkat lymphocytes (Anupam et al., 2011; Doueiri et al., 2012). The p30II protein has also been shown to interact with the ETS-family transcription factor PU.1 and inhibit the expression of Toll-like receptors 3 and 4 (TLR3/TLR4) which could suppress interferon-mediated cytokine signaling and contribute to the persistence of HTLV-1-infected monocytes and dendritic cells in vivo (Fenizia et al., 2014; Datta et al., 2006). Although the p30II protein is not detectable in primary ATLL samples, several studies have demonstrated p30II is required for the maintenance of latently-HTLV-1-infected cells in in vivo models of pathogenesis (Bartoe et al., 2000; Edwards et al., 2011; Valeri et al., 2010); and fresh ATLL isolates express the alternatively-spliced p30II mRNA (Koralnik et al., 1992; Cereseto et al., 1997) and HTLV-1-infected individuals contain cytotoxic T-lymphocytes specific for pX ORF-II (i.e., p30II) peptides (Pique et al., 2000). It is possible that low-level persistent expression of p30II is necessary for the maintenance of proviral latency and to counter the oxidative toxicity induced by the viral oncoproteins Tax and HBZ (Figs. 1F, 1G, 2A–2D, 3E, 3F, 4A–4C, and S1B). One might speculate that p30II could also contribute to the higher levels of viral antigens expressed during the earliest stages of infection, associated with increased Tax-dependent transactivation and Tax-specific seroreactivity in HTLV-1-infected asymptomatic individuals and/or HAM patients. Intriguingly, Choudary and Ratner 2011 have demonstrated that hbz antisense gene expression promotes increased Tax protein production by suppressing p30II mRNA levels, which suggests RNA-regulation could influence the cooperative dynamics between these pX-encoded factors. However, Silverman et al., 2004 observed that rabbits inoculated with T-cell-clones harboring the infectious HTLV-1 ACH.p30II mutant provirus, defective for p30II production, which contains a premature stop codon that only minimally disrupts the p30II mRNA sequence, exhibited in vivo genetic reversion back to the wildtype ACH sequence as a result of immune selection in this animal model of pathogenesis. These findings underscore the importance of the p30II protein for the persistence and survival of HTLV-1-infected cells in vivo.
We have previously demonstrated that amino acid residues 99–154 of p30II interact with the MYST-family acetyltransferase TIP60; and p30II is present in c-Myc/TIP60 NuA4 transcription complexes –together with TRRAP, TIP48/49, and hGCN5, recruited to Myc-responsive E-box enhancer elements within the cyclin D2 gene promoter (Awasthi et al., 2005). The p30II protein cooperates with c-Myc and increases S-phase cell-cycle entry and oncogenic foci-formation in vitro (Awasthi et al., 2005; Romeo et al., 2015). We have also shown that lentiviral-HTLV-1 p30II induces aberrant T-cell lymphoproliferation and the long-term proliferation of transduced primary huPBMCs (Romeo et al., 2018). TIP60 is a transcriptional cofactor for both c-Myc and the p53 tumor suppressor (Frank et al., 2003; Patel et al., 2004; Gorrini et al., 2007; Awasthi et al., 2005; Zhao et al., 2016; Tang et al., 2006; Sykes et al., 2006; Kurash et al., 2008), and the TIP60-dependent acetylation of p53 on lysine residue K120 differentially modulates the induction of p53-regulated pro-apoptotic genes (Sykes et al., 2006; Kurash et al., 2008). In Romeo et al., 2018, we recently demonstrated that p30II activates p53 and inhibits p53-K120-acetylation, and induces the mitochondrial expression of TIGAR which is required for the oncogenic cooperation between p30II/c-Myc. Here we have demonstrated that p30II suppresses the oxidative stress, mitochondrial damage, and cytotoxicity induced by the viral oncoproteins Tax and HBZ in a TIGAR-dependent manner (Figs. 1F, 1G, 2A–2H, 3A–3F, 4A–4D, and S1B). Both Tax and HBZ induced mitochondrial membrane depolarization (Figs. 1A–1E) and ROS production which was significantly countered by p30II co-expression (Figs. 1F, 1G, and S1B). Moreover, lentiviral p30II prevented Tax/HBZ-induced cellular autophagy and mitophagy (Figs. 2A–2D), consistent with TIGAR’s role in suppressing ROS-induced autophagy (Bensaad et al., 2009). In the context of the infectious full-length HTLV-1 ACH provirus, we have demonstrated that p30II is required for the suppression of oxidative genomic and mitochondrial DNA-damage, as well as cell senescence induced by other viral oncoproteins (Figs. 2E–2H, 3A–3D, 4D). The p30II protein also cooperated with Tax and HBZ in oncogenic cell transformation/foci-formation experiments (Figs. 4E–4H). Surprisingly, the combinations of Tax+p30II or HBZ+p30II resulted in higher oncogenic potential and more transformed colonies in in vitro foci-formation assays, as compared to Tax+HBZ (Figs. 4E–4H). Whereas both HBZ and p30II effectively suppressed Tax-induced cellular senescence (Fig. 4A), only p30II inhibited Tax-induced ROS production (Fig. 1F, 1G, and S1B), alluding to potential pivotal roles for p30II and TIGAR during viral carcinogenesis. The expression of most viral gene products in ATLL clinical isolates is nearly undetectable; whereas acute and lymphoma-stage ATLL patient lymphocytes have been shown to contain overexpression of the c-Myc oncoprotein and wildtype p53. Thus, it is conceivable p30II might alternately cooperate with either viral and/or cellular oncoproteins through the activation of p53 and TIGAR during specific stages of HTLV-1 pathogenesis or ATLL malignant disease progression.
The TIGAR protein was highly expressed associated with c-Myc oncogene dysregulation within the huKi67-positive lymphoma cells and infiltrating tumor cells in the secondary tissues of immune-deficient NOD/scid animals engrafted with the HTLV-1+ SLB1 or MET-1 cell-lines (Figs. 6B, 7B, and 7D; Liu et al., 2002; Chen et al., 2009; Ju et al., 2014; Zimmerman et al., 2011). These animals developed aggressive lymphoid tumors (i.e., PELs and solid T-cell lymphomas) and exhibited diffuse infiltration of secondary tissues by lymphoid tumor cells which was occasionally accompanied by organomegaly and enlargement of the liver and/or spleen (Figs. 5A–5G, 6A, 6C–6G, 7B and 7D). Two experimental animals were found to have splenic hemangiomas in association with splenomegaly and the development of HTLV-1-induced T-cell lymphomas (Fig. 7A). The elevated expression of TIGAR in the lymphoma-resident and infiltrating huKi67-positive tumor cells correlated with induction of the pro-angiogenic markers, HIF-1α and VEGF (Figs. 7B and 7D), and the increased recruitment of murine CD31(PECAM1)/Flk1(VEGFR2)-positive endothelial progenitors into these tissues (Figs. 7B and 7D, right panels). As Cheung et al., 2012 have shown that TIGAR activates HIF-1α and localizes to mitochondria under hypoxic conditions, it is possible the upregulation of TIGAR expression could promote angiogenesis and stromal chemoattractant signaling by HTLV-1+ tumor cells in our in vivo xenograft model of HTLV-1-induced T-cell lymphoma. Few studies to date have investigated angiogenic signaling or vasculogenesis in the context of HTLV-1-infected cells and ATLL (Kchour et al., 2008; Mitra-Kaushik et al., 2004; El-Sabban et al., 2002; Bazarbachi et al., 2004). Indeed, the recruitment of endothelial progenitors by HTLV-1+ lymphoma cells expressing TIGAR, HIF-1α, and VEGF could contribute to aggressive hematogenous metastasis and the infiltration of secondary tissues by tumor lymphocytes.
These findings allude to a likely key ancillary role for p30II in retroviral carcinogenesis by promoting the survival of Tax and/or HBZ-expressing cells, and further suggest that therapeutically targeting TIGAR functions could be a plausible strategy to sensitize ATLL tumor cells to oncogene-induced oxidative damage.
Materials and methods
Cell-lines
The Jurkat E6.1 T-lymphoblast cell-line (ATCC No. TIB-152) was cultured in RPMI-1640 medium (ATCC No. 30-2001), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Biowest), penicillin (100 U/ml), streptomycin sulfate (100 μg/ml), and gentamycin sulfate (20 μg/ml; Invitrogen), and incubated at 37 C under 5% CO2. The HT-1080 human fibrosarcoma cell-line (ATCC No. CCL-121) was cultured in collagen-treated tissue-culture flasks (Greiner) in Eagle’s Minimum Essential Medium (EMEM; ATCC No. 30-2003), supplemented with 10% heat-inactivated FBS, penicillin, streptomycin sulfate, and gentamycin sulfate at 37 C and 5% CO2. The transiently-amplified HT-1080/HTLV-1 ACH.wildtype (wt) and HT-1080/HTLV-1 ACH.p30II mutant-expressing proviral clones (Romeo et al., 2018; Kimata et al., 1994; Bartoe et al., 2000) were cultured under the same conditions as the parental HT-1080 cell-line; and the continuous production of extracellular virus particles was confirmed after serially passaging the cultures for 4-weeks by performing Anti-HTLV-1 p19Gag enzyme-linked immunosorbent assays (ELISAs; Zeptometrix) on culture supernatants which were filtered through 0.22 μm cellulose acetate syringe filters. The levels of infectious p19Gag-containing particles released into the HT-1080/ACH.wt and HT-1080/ACH.p30II culture supernatants were compared to the virus-producing HTLV-1-transformed SLB1 lymphoma cell-line as a positive control. HeLa adenocarcinoma cells (ATCC No. CCL-2) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; ATCC No. 30-2002), supplemented with 10% heat-inactivated FBS and antibiotics, at 37 C in a humidified incubator under 5% CO2. The immortalized HFL1 human fetal lung fibroblast cell-line (ATCC No. CCL-153) was grown in collagen-treated tissue-culture flasks in F-12K medium (ATCC No. 30-2004), supplemented with 10% heat-inactivated FBS, penicillin, streptomycin sulfate, and gentamycin sulfate, and incubated at 37 C and 5% CO2. The HTLV-1-transformed SLB1 T-cell lymphoma cell-line (Liu et al., 2002) was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; ATCC No. 30-2005), supplemented with 10% heat-inactivated FBS and antibiotics, at 37 C and 10% CO2. The tumorigenic HTLV-1-transformed MET-1 T-cell lymphoma cell-line was serially propagated in NOD/scid mice (Charles River Laboratories), and was kindly provided by S. Niewiesk (The Ohio State University) and T. Waldmann (NCI/NIH) and has been described in Chen et al., 2009 and Ju et al., 2014. All cell-lines tested negative for mycoplasma contamination.
Plasmids and antibodies
The RcCMV-HTLV-1 Tax plasmid has been described in Harrod et al., 1998 and Harrod et al., 2000 and was kindly provided by C.Z. Giam (The Uniformed Services University of the Health Sciences). The pcDNA-HTLV-1 HBZ-MycHis-tagged expression construct (Hiven et al., 2005; Clerc et al., 2008) was provided by I. Lemasson (East Carolina University). The pMH-HTLV-1 p30II (HA-tagged) and pEGFP-N3-HTLV-1 p30II-Green Fluorescent Protein (p30II-GFP) expression constructs (Nicot et al., 2004) were kindly provided by G. Franchini (NCI/NIH). The plasmids encoding the full-length HTLV-1 ACH.wt and ACH.p30II mutant (defective for p30II production) proviral clones have been previously described in Kimata et al., 1994 and Bartoe et al., 2000. The pcDNA3.1-FLAG-tagged-TIGAR expression construct (Bensaad et al., 2006) was generously provided by K. Vousden (The Francis Crick Institute). The following primary antibodies were used throughout: goat polyclonal Anti-HTLV-1 Tax (vC-12; Santa Cruz Biotechnology), goat polyclonal Anti-Actin (I-19; Santa Cruz Biotechnology), mouse monoclonal Anti-Beclin-1 (E-8; Santa Cruz Biotechnology), goat polyclonal Anti-TIM23 (C-19; Santa Cruz Biotechnology), rabbit polyclonal Anti-TOM20 (FL-145; Santa Cruz Biotechnology), mouse monoclonal Anti-SQSTM1 (D-3; Santa Cruz Biotechnology), rabbit polyclonal Anti-Phospho-Ser139-H2A.X (Santa Cruz Biotechnology), rabbit polyclonal Anti-GFP (FL; Santa Cruz Biotechnology), Anti-HTLV-1 p19Gag (Zeptometrix), mouse monoclonal Anti-TIGAR (G-2; Santa Cruz Biotechnology), rabbit polyclonal Anti-Human TIGAR (described in Bensaad et al., 2006), rabbit polyclonal Anti-Human Ki67 (huKi67, H-300; Santa Cruz Biotechnology), mouse monoclonal Anti-FLAG M2 (Sigma-Aldrich), mouse monoclonal Anti-c-Myc (9E10; Santa Cruz Biotechnology), mouse monoclonal Anti-CD31/PECAM1 (H-3; Santa Cruz Biotechnology), mouse monoclonal Anti-Flk1/VEGFR2 (A-3; Santa Cruz Biotechnology), mouse monoclonal Anti-HIF-1α (28b; Santa Cruz Biotechnology), and mouse monoclonal Anti-VEGF (C-1; Santa Cruz Biotechnology). The fluorescence-conjugated secondary antibodies (Anti-Mouse IgG [H+L]-Dylight 488; Anti-Rabbit IgG [H+L]-Dylight 488; Anti-Rabbit IgG [H+L]-Rhodamine Red; Anti-Goat IgG [H+L]-Rhodamine Red) used for immunofluorescence-microscopy experiments and horseradish peroxidase (HRP)-conjugated secondary antibodies (Anti-Mouse IgG [H+L]-HRP; Anti-Rabbit IgG [H+L]-HRP; Anti-Goat IgG [H+L]-HRP) used for chemiluminescence-based immunoblotting were purchased from Jackson ImmunoResearch Laboratories, Inc. The Anti-Mouse IgG-Pacific Blue secondary antibody was purchased from Molecular Probes/Invitrogen, Inc.
Lentiviral transduction and siRNA-knockdown of TIGAR expression
The pLenti-HTLV-1 p30II (HA-tagged) lentiviral vector was generated using the pLenti-6.2/V5-DEST cloning vector (Invitrogen) and expresses a translationally-optimized version of the HTLV-1 p30II protein and has been previously described in Romeo et al., 2015 and Romeo et al., 2018. The VSV-Env-pseudotyped virus particles were packaged in 293FT HEK cells (Invitrogen) and the supernatants were clarified by centrifugation at 2,500 rpm at 4 C for 10 minutes in a swinging bucket rotor and passed through a 0.22 μm polyethersulfone filter. The infectious particles were then concentrated by ultracentrifugation on a 20% (w/v) sucrose-TSE (10 mM Tris [pH 7.5], 100 mM NaCl, 10 mM EDTA) cushion at 44,000 rpm and 4 C for 4 hours using a Beckman 70.1 Ti rotor. The pellets were resuspended in TNE (10 mM Tris [pH 7.4], 100 mM NaCl, 1 mM EDTA) buffer and stored in aliquots at −80 C until ready for experimental use.
To inhibit TIGAR expression in the HT-1080/HTLV-1 ACH.wt and HT-1080/ACH.p30II mutant-expressing proviral clones (Romeo et al., 2018), the cells were repeatedly transfected with a 2′-O,4′aminoethylene bridged nucleic acid (BNA)-modified small-interfering RNA oligonucleotide targeted against tigar transcripts (siRNA-tigar) with the sequence: 5′-AUAUACCAGCAGCUGCUGC-3′, or with a 2′-O-methyl-uridine-modified scrambled RNA (scrRNA) negative control with the sequence: 5′-UUACCGAGACCGUACGUAU-3′ (Biosynthesis). All RNA transfections were carried out using HiPerFect transfection reagent (Qiagen) as per the manufacturer’s recommended protocol. The siRNA-knockdown of TIGAR protein expression was confirmed by SDS-PAGE and immunoblotting using a rabbit polyclonal Anti-TIGAR antibody (Bensaad et al., 2006). The specificity of the siRNA-tigar oligonucleotide was further verified by transfecting HT-1080 cells with a pcDNA3.1-TIGAR (FLAG-tagged) expression construct and then transfecting them with siRNA-tigar, followed by SDS-PAGE and immunoblotting using a monoclonal Anti-FLAG M2 primary antibody and HRP-conjugated Anti-Mouse IgG secondary antibody and chemiluminescence detection using a Pierce Supersignal West Pico kit.
Mitochondrial membrane depolarization and ROS analysis
The effects of the HTLV-1 Tax and HBZ oncoproteins upon mitochondrial membrane potential, as a result of viral oncogene-induced oxidative stress, were determined by staining transfected cells with the membrane-permeant dye, JC-1 (Molecular Probes) –which exists as cytoplasmic green-fluorescent monomers under conditions of mitochondrial membrane depolarization, but forms red-fluorescent aggregates in polarized mitochondria. HT-1080 cells were plated on 8-chamber CultureWell coverslips (Invitrogen) for live-imaging and then transfected with 250 ng of the RcCMV-HTLV-1 Tax or pcDNA-HTLV-1 HBZ-MycHis plasmids, or an empty CβS vector. Cells treated with 50 μM of the chemical uncoupler, carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich), were included as a positive control for mitochondrial depolarization. The transfected cells were stained with 2 μM of the JC-1 dye in 500 μl of complete medium for 12 minutes and incubated at 37 C under 5% CO2. The samples were washed with phosphate-buffered saline (PBS), pH 7.4, and then immediately analyzed by fluorescence-confocal microscopy to observe the red/green JC-1 fluorescent signals in Tax- or HBZ-expressing cells. Alternatively, Jurkat E6.1 T-lymphoblasts were transfected with 3 μg of the RcCMV-HTLV-1 Tax or pcDNA-HTLV-1 HBZ-MycHis expression constructs, or an empty CβS vector. Following 72 hours, the transfected cultures were stained with 2 μM of the JC-1 dye in 2 ml of complete medium for 12 minutes at 37 C and 10% CO2. The samples were pelleted by centrifugation at 2,500 rpm for 3 minutes, washed with PBS, and resuspended in 1 ml of PBS, pH 7.4 + D-Glucose (4.5 mg/ml) for subsequent analysis by flow-cytometry using a BD FACSCaliber instrument to quantify the relative ratios of red/green JC-1 fluorescence in the transfected cells. The samples were gated to exclude debris and apoptotic cells from the quadrant plots. The expression of HTLV-1 Tax and HBZ (Myc-tagged) proteins, as compared to an Actin loading control, in transfected Jurkat E6.1 lymphoblasts or HT-1080 cells was detected by SDS-PAGE and immunoblotting.
To determine if the HTLV-1 latency-maintenance factor p30II cooperates with the viral oncoproteins, Tax and/or HBZ, and prevents cytotoxicity and oxidative damage due to the intracellular accumulation of reactive oxygen species (ROS), we stained cotransfected cells with the fluorescent chemical ROS-probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Molecular Probes) and performed fluorescence-confocal microscopy. HT-1080 cells were plated on Corning 12-well tissue-culture plates and transfected with various combinations of RcCMV-HTLV-1 Tax, pMH-HTLV-1 p30II (HA-tagged), pcDNA-HTLV-1 HBZ-MycHis, or an empty CβS vector (500 ng each). The cultures were incubated for 72 hours and then stained with 2.5 μM of CM-H2DCFDA. The samples were rinsed 3X with PBS, pH 7.4, and fixed for 15 minutes at room temperature in a solution of 2% formaldehyde and 0.2% glutaraldehyde in PBS. Finally, the samples were washed 2X with PBS, mounting medium (KPL) and glass coverslips were added, and the relative percentages of CM-H2DCFDA-positive cells per well were quantified by counting in triplicate on a Nikon Eclipse TE2000-U confocal microscope using a 20x objective lens. The total numbers of cells per well were determined by microscopy and counting using a DIC phase-contrast filter.
Autophagy and mitophagy analysis
To determine whether the HTLV-1 p30II protein protects cells against Tax- and/or HBZ-induced autophagy and mitophagy (mitochondrial damage), HT-1080 fibrosarcoma cells were plated on collagen-treated 8-chamber tissue-culture slides (Nunc Permanox) and transfected with the RcCMV-HTLV-1 Tax or pcDNA-HTLV-1 HBZ-MycHis expression constructs, and then transduced with either the pLenti-HTLV-1 p30II (HA-tagged) virus or pLenti-6.2/V5-DEST empty vector. After 48 hours, the samples were immunostained using monoclonal antibodies that recognize the autophagy and mitophagy markers, Beclin-1 and SQSTM1, in combination with a fluorescent Pacific Blue-conjugated Anti-Mouse IgG secondary antibody. The mitochondria were visualized by co-immunostaining the samples with rabbit polyclonal Anti-TOM20 or goat polyclonal Anti-TIM23 primary antibodies and appropriate Rhodamine-Red-conjugated fluorescent secondary antibodies. The percentages of cells exhibiting nuclear localization of Beclin-1 (Liang et al., 2001), or percentages of SQSTM1/TOM20-positive cells were quantified using immunofluorescence-confocal microscopy and counting each well in triplicate.
Measuring cellular senescence and apoptosis
To determine how the HTLV-1 p30II or HBZ proteins influence Tax-induced rapid senescence in HeLa cells as described in Ho et al., 2012 and Kuo and Giam, 2006, 5×104 cells were plated on 8-chamber tissue-culture slides and then cotransfected with various combinations of pEGFP-N3-HTLV-1 p30II-GFP, pcDNA-HTLV-1 HBZ-MycHis-tagged, RcCMV-HTLV-1 Tax, or empty CβS vector as a negative control. The expression of HTLV-1 Tax, p30II-GFP, and HBZ-Myc-His-tagged proteins was confirmed by SDS-PAGE and immunoblotting. Alternatively, we measured the levels of Tax-induced cellular senescence in the HT-1080/HTLV-1 ACH.wt and HT-1080/ACH.p30II mutant-expressing proviral clones, as compared to the parental HT-1080 cell-line. The chemical uncoupler CCCP was included as a positive control. Certain samples were transfected with either CMV-TIGAR (FLAG-tagged; Bensaad et al., 2006) or an empty CβS vector; and transcomplementation of the HT-1080/ACH.p30II mutant proviral clone was accomplished by transfecting these cells with the pEGFP-N3-HTLV-1 p30II-GFP expression construct or transduction with the pLenti-HTLV-1 p30II (HA-tagged) lentivirus (Romeo et al., 2015; Romeo et al., 2018). The p30II-GFP was detected in transfected HT-1080/ACH.p30II cells by direct fluorescence-microscopy. To inhibit TIGAR expression, certain samples were repeatedly cotransfected with siRNA-tigar or a scrRNA negative control using HiPerFect transfection reagent. After five days, the cultures were stained using X-Gal to detect senescence-associated Beta-galactosidase (SA-β-gal) expression. The cells were washed with PBS, pH 7.4, and then fixed for 15 minutes at room temperature in a solution of 2% formaldehyde and 0.2% glutaraldehyde in PBS. The samples were washed 2X with PBS and subsequently stained with an X-gal-Staining Solution (37.2 mM citric acid/sodium phosphate, 140 mM NaCl, 2mM MgCl2, 0.5 mM potassium ferrocyanide, 0.5 mM potassium ferricyanide, 1 mg/ml X-gal, and 2% N-N-dimethylformamide, pH 6.0) at 37 C overnight in a dry incubator without CO2. The senescent cells (i.e., SA-β-gal-positive, blue cells) were visualized by microscopy and quantified by counting the wells in triplicate.
To determine if HTLV-1 p30II protects against Tax- or HBZ-induced cellular apoptosis, HT-1080 cells were plated on collagen-treated 8-chamber slides or 6-well tissue culture plates (Corning) and the cultures were transfected with either RcCMV-HTLV-1 Tax or pcDNA-HTLV-1 HBZ-MycHis-tagged constructs. After 48 hours, the cells were transduced with the pLenti-HTLV-1 p30II (HA-tagged) lentivirus or pLenti-6.2/V5-DEST empty lentiviral vector. Alternatively, the cells were cotransfected with various combinations of RcCMV-HTLV-1 Tax, pcDNA-HTLV-1 HBZ-MycHis-tagged, and pMH-HTLV-1 p30II (HA), and either siRNA-tigar or a scrRNA negative control. Staurosporine (100 nM)-treated cells were included as a positive apoptosis control. The cells were washed 2X with PBS, pH 7.4, and stained with fluorescein isothiocyanate (FITC)-conjugated Annexin-V and propidium iodide (PI) in Annexin V-Binding Buffer (BD-Pharmingen) for 15 minutes at room temperature in the dark. The samples were then gently washed 2X and resuspended in PBS, and the relative percentages of Annexin-V-FITC and/or PI-positive cells per well were determined using confocal fluorescence-microscopy and counting each well in triplicate. The total numbers of cells were quantified by microscopy using the DIC phase-contrast filter.
Fluorescence-based terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assays were performed to detect apoptosis and nuclear or mitochondrial DNA-damage, associated with oncogene-induced oxidative stress, in the HT-1080/HTLV-1 ACH.wt and HT-1080/ACH.p30II mutant-expressing proviral clones. In brief, 5×104 cells were plated on collagen-treated 8-chamber tissue-culture slides and then permeabilized and labeled using a Click-iT Alexa Fluor 594-TUNEL kit (Molecular Probes) as per the manufacturer’s recommended protocol. Deoxyribonuclease I (DNAse I)-treated cells were included as a positive control. The cell nuclei were stained using Hoechst 33342 (Molecular Probes). Genomic DNA-damage was detected by co-immunostaining the samples with a rabbit polyclonal Anti-Phospho-Ser139-H2A.X primary antibody and fluorescent Dylight 488-conjugated Anti-Rabbit IgG (H+L) secondary antibody; and mitochondrial DNA-damage was visualized by co-immunostaining the samples with a rabbit polyclonal Anti-TOM20 primary antibody and Dylight 488-conjugated fluorescent secondary antibody. The average numbers of TUNEL-positive or TUNEL/TOM20-positive foci per cell were determined by immunofluorescence-confocal microscopy and counting each visual field in triplicate using a 40x objective lens.
Microscopy
The color images for hematoxylin and eosin (H&E)-stained tissue sections, as well as for the detection of SA-β-galactosidase expression in cotransfected cells stained with X-gal, were generated using a Zeiss Axioimager Z2 microscope equipped with AxioCam HRm monochromatic and MRc color cameras and either Plan-Apochromat 20x/0.8 or EC Plan-NEOFLUAR 40x/1.3 oil-immersion objectives and Axiovision 4.8 software. Fluorescence-confocal microscopy was performed on a Zeiss LSM800 instrument equipped with an Airyscan super-resolution detector and stage CO2 incubator, using either Plan-Apochromat 40x/1.3 or Plan-Apochromat 63x/1.4 oil-immersion objectives and Zeiss ZEN system software. The visualization and quantification of ROS-positive cells and oncogenic foci-formation by HTLV-1 Tax, HBZ, and p30II-GFP in cotransfected cells were carried out using a Nikon Eclipse TE2000-U inverted microscope and D-Eclipse C1 confocal imaging system equipped with 633 nm and 543 nm He/Ne and 488 nm Ar lasers and a Plan-Apo 20x/0.75 objective lens.
Oncogenic transformation and foci-formation experiments
In vitro foci-formation experiments were performed as described in Awasthi et al., 2005, Romeo et al., 2015, and Romeo et al., 2018 using the HFL1 human fibroblast cell-line which exhibits contact-inhibition, has a stable pseudodiploid karyotype with wildtype p53 status, and typically grows as a monolayer. To assess the oncogenic potential and cooperation between the various pX-encoded proteins of HTLV-1, 6×104 cells were plated on Corning 12-well tissue-culture plates, pretreated with rat tail collagen (100 μg/ml in PBS, pH 7.4; Roche), and cultured in F-12K medium supplemented with 10% FBS, penicillin, streptomycin sulfate, gentamycin sulfate, and high-glucose (4.5 mg/ml) in a humidified incubator at 37 C and 5% CO2. The cells were cotransfected with various combinations of HTLV-1 Tax, p30II (GFP-tagged), or HBZ expression constructs (325 ng each), or an empty CβS vector using SuperFect (Qiagen) transfection reagent, and the cultures were monitored for the appearance of oncogenically-transformed colonies over a three-week period. The expression of p30II-GFP within transformed foci was visualized by direct fluorescence-microscopy. The foci were fixed with 70% ethanol/PBS, washed 2X with PBS, and stained with a 0.025% methylene blue solution for 15 minutes at room temperature for improved colony visualization. Then the plates were washed 3X with PBS and the numbers of transformed colonies were counted in triplicate on an inverted Nikon TE2000 microscope using a 10x objective.
In vivo tumorigenesis studies and Anti-HTLV-1 p19Gag ELISAs
For the in vivo tumorigenesis studies, immunocompromised NOD/scid mice (NOD.CB17-Prkdcscid/NcrCrl; from Charles River Laboratories: 12 animals per sample group) were weighed and then anesthetized by gas inhalation with 3% isoflurane/O2 administered using a SurgiVet model 100 key-lock vaporizer connected to an induction chamber, rodent nose cone, and two passive F/AIR halogenated anesthetic gas scavenging canisters. The animals were engrafted with either 5×105 or 1×106 of the HTLV-1+ SLB1 or MET-1 lymphoma T-cells in 500 μl of vehicle (Ca2+/Mg2+-free PBS, pH 7.4) by intraperitoneal injection using a sterile tuberculin syringe with a 27-gauge needle. A negative control group was injected with the vehicle alone. The animals were allowed to recover on a heated circulating thermal water-pad; and then they were housed and maintained under BSL-2 containment in an Allentown NexGen ventilated cage-racking system. The experimental animals were monitored daily over a period of 12-weeks for signs of tumor development (e.g., abdominal distension and/or lethargy) or changes in their overall health status (e.g., rough coat, severe weight loss, or difficulties eating, drinking or with ambulation). Blood collections (75 μl from the facial vein) were performed at 2-week intervals on anesthetized animals using a No. 2 lancet in order to monitor the levels of extracellular virus particles present in the circulating blood through Anti-HTLV-1 p19Gag ELISAs (Zeptometrix). The ELISAs were performed in Corning 96-well microtiter plates and read on a Berthold Tristar plate-reader at 450 nm in Absorbance mode; and the concentrations of blood-associated p19Gag-containing HTLV-1 particles were determined by comparing their absorbance values to a p19Gag protein standard. At the end of 12-weeks, the animals were humanely euthanized by the initial inhalation of 30% CO2 until loss-of-consciousness was achieved, followed by 70% CO2 to induce death. Cervical dislocation was used as a secondary physical procedure. The carcasses were weighed and necropsies were performed using a surgical microscope mounted under a Class II laminar-flow biosafety cabinet to harvest tumor tissues and any affected secondary organs (e.g., spleen, liver, and kidneys). Fine needle aspirations were performed on HTLV-1-induced primary effusion lymphomas/body cavity-based lymphomas using a 27-gauge syringe needle to isolate tumor ascites. The lymphoma tumors were weighed on a Mettler Toledo XP26 microanalytical balance and measured using a digital caliper. The tumor volumes were determined using the calculation: (D x d2)/2, where ‘D’ equals the longest diameter and ‘d’ equals the shortest diameter. The tissues were processed and fixed for 4 days in 15X volume of 10% neutral-buffered formalin (3.75% formaldehyde, 33.3 mM sodium phosphate monobasic, 45.8 mM sodium phosphate dibasic, in distilled deionized water, pH 6.8) at room temperature. The tissues were then dehydrated by incubating the samples with gentle agitation at room temperature in 50X volume of 70% ethanol for 1 hour, 95% ethanol for 1.5 hours, 100% ethanol for 1.5 hours, and xylene for 1 hour. The samples were then embedded in Histoplast IM paraffin (Thermo Scientific) using Fisher Tissue Path IV cassettes on a HistoStar tissue embedding system and stored at −80 C until ready for experimental use. The in vivo tumorigenesis studies and blood collections in immunocompromised mice as well as humane euthanasia and necropsy procedures were performed under a protocol approved by the SMU Institutional Animal Care and Use Committee.
Tissue processing and sectioning
Thin-sections (0.5 mm) were prepared for immunofluorescence and histological analyses by cutting the paraffin-embedded tumor tissues (or secondary organs) using an automated Microm HM360 rotary microtome. The tissue sections were mounted on glass microscope slides and deparaffinized by treating the samples 2X with xylene for 3 minutes at room temperature, and then with a 1:1 solution of xylene/ethanol for 3 minutes, followed by 2X treatment with 100% ethanol for 3 minutes, 95% ethanol for 3 minutes, 70% ethanol for 3 minutes, 50% ethanol for 3 minutes, and rinsed 2X with distilled deionized water. The samples were then incubated in fixative solution (2% formaldehyde and 0.2% glutaraldehyde in PBS, pH 7.4) for 15 minutes at room temperature, washed 2X with PBS, and stained with hematoxylin and eosin (H&E) for subsequent microscopy analysis. For H&E staining: the samples were incubated in Hematoxylin+ reagent (Fisher Healthcare) for 4 minutes with gentle agitation, rinsed with deionized water, and incubated for 5 minutes in Bluing Reagent (Fisher Healthcare). The samples were rinsed 7X with Clarifier 1 solution (Fisher Healthcare) and then incubated 2X for 1 minute in Clarifier 2 solution (Fisher Healthcare). They were rinsed again by incubating the samples in deionized water for 2 minutes with gentle shaking. Then the samples were incubated for 20 seconds in Eosin-Y reagent (Fisher Healthcare), rinsed 4X with 95% ethanol, incubated 3X in 95% ethanol for 5 minutes, 3X in 100% ethanol for 5 minutes, and then 3X in xylene for 15 minutes before the addition of mounting medium (KPL) and glass coverslips. In order to detect the engrafted HTLV-1+ SLB1 or MET-1 tumor lymphocytes, the sections were co-stained using a rabbit polyclonal antibody against the human proliferating cell antigen, Ki67 (huKi67, H-300; Santa Cruz Biotechnology), together with 4′6-diamidino-2-phenyl-indole, dihydrochloride (DAPI; Molecular Probes) nuclear-staining to visualize the murine stromal cells by immunofluorescence-confocal microscopy.
Statistical analysis
The statistical significance of experimental datasets was determined based on their Student’s t-distribution values in two-tailed unpaired tests (alpha =0.05) and calculated P-values using the Shapiro-Wilk normality test and Graphpad Prism 7.03 software. Error bars represent the mean standard deviation between independent experiments. The significance values were defined as: ****P<0.0001, ***P<0.0002, **P<0.0021, *P<0.0332. For in vivo tumorigenesis studies, the numbers of animals needed for each sample group to produce robust, statistically significant results were determined using PASS 14 Sample size statistical software. Two-sided t-tests were performed assuming equal variance (alternative hypothesis: Ha: δ ≠ 0) and with standard deviations of σ = 1, 5, or 10 millimeters, and difference in means of δ = 2 to 10 by 1 (millimeters). Alpha =0.05. Based on these calculations, a sample size of 12 animals (N) per experimental test group (N1 + N2) will detect a difference of 2 millimeters in tumor volume with a standard deviation of 1.0 and an actual power of 0.87642.
Supplementary Material
Highlights.
The HTLV-1 oncoproteins Tax and HBZ induce oxidative damage and autophagy/mitophagy
HTLV-1 p30II activates TIGAR and suppresses Tax/HBZ-induced ROS and cytotoxicity
HTLV-1 p30II cooperates with Tax and HBZ and induces oncogenic foci-formation
TIGAR is highly expressed in HTLV-1+ xenograft tumors with oncogene dysregulation
The expression of TIGAR in HTLV-1+ lymphoma tissues is associated with angiogenesis
Acknowledgments
The authors thank K. Vousden for kindly providing the pcDNA3.1-TIGAR (FLAG-tagged) expression construct and rabbit polyclonal Anti-TIGAR antibody. We thank I. Lemasson for the pcDNA-HBZ-MycHis plasmid, P. Green for the HTLV-1-transformed SLB1 T-cell lymphoma cell-line, and S. Niewiesk and T. Waldmann for the HTLV-1-transformed MET-1 T-cell lymphoma cell-line. We also thank M.D. Lairmore for helpful suggestions and generous advice for the establishment of our in vivo model of HTLV-1-induced lymphomagenesis. This work was supported by grants from the National Cancer Institute/National Institutes of Health 1R15CA202265-01A1 and University Research Council to RH. TH is a recipient of a 2015 American Society for Microbiology-Eugene & Millicent Goldschmidt Graduate Student Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anupam R, Datta A, Kesic M, Green-Church K, Shkriabai N, Kvaratskhelia M, Lairmore MD. Human T-lymphotropic virus type 1 p30 interacts with REGgamma and modulates ATM (ataxia telangiectasia mutated) to promote cell survival. J Biol Chem. 2011;286:7661–7668. doi: 10.1074/jbc.M110.176354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood. 2006;107:3976–3982. doi: 10.1182/blood-2005-11-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood. 2008;112:3788–3797. doi: 10.1182/blood-2008-04-154286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, Motloch P, Takemoto S, Taguchi H, Cole MD, Lüscher B, Dittrich O, Tagami H, Nakatani Y, McGee M, Girard AM, Gaughan L, Robson CN, Monnat RJ, Jr, Harrod R. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol. 2005;25:6178–6198. doi: 10.1128/MCB.25.14.6178-6198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XT, Sinha-Datta U, Ko NL, Bellon M, Nicot C. Nuclear export and expression of human T-cell leukemia virus type 1 tax/rex mRNA are RxRE/Rex dependent. J Virol. 2012;86:4559–4565. doi: 10.1128/JVI.06361-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangham CR, Ratner L. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr Opin Virol. 2015;14:93–100. doi: 10.1016/j.coviro.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoe JT, Albrecht B, Collins ND, Robek MD, Ratner L, Green PL, Lairmore MD. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J Virol. 2000;74:1094–1100. doi: 10.1128/jvi.74.3.1094-1100.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydoun HH, Cherian MA, Green P, Ratner L. Inducible nitric oxide synthase mediates DNA double strand breaks in human T-cell leukemia virus type 1-induced leukemia/lymphoma. Retrovirology. 2015;12:71. doi: 10.1186/s12977-015-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarbachi A, Abou Merhi R, Gessain A, Talhouk R, El-Khoury H, Nasr R, Gout O, Sulahian R, Homaidan F, de Thé H, Hermine O, El-Sabban ME. Human T-cell lymphotropic virus type 1-infected cells extravasate through the endothelial barrier by a local angiogenesis-like mechanism. Cancer Res. 2004;64:2039–2046. doi: 10.1158/0008-5472.can-03-2390. [DOI] [PubMed] [Google Scholar]
- Bellon M, Baydoun HH, Yao Y, Nicot C. HTLV-I Tax-dependent and –independent events associated with immortalization of human primary T lymphocytes. Blood. 2010;115:2441–2448. doi: 10.1182/blood-2009-08-241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–3026. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Billman MR, Rueda D, Bangham CRM. Single-cell heterogeneity and cell-cycle-related viral bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. 2017;2:87. doi: 10.12688/wellcomeopenres.12469.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereseto A, Berneman Z, Koralnik I, Vaughn J, Franchini G, Klotman ME. Differential expression of alternatively spliced pX mRNAs in HTLV-I-infected cell lines. Leukemia. 1997;11:866–870. doi: 10.1038/sj.leu.2400665. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang M, Ju W, Waldmann TA. Effective treatment of a murine model of adult T-cell leukemia using depsipeptide and its combination with unmodified daclizumab directed toward CD25. Blood. 2009;113:1287–1293. doi: 10.1182/blood-2008-04-149658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zachar V, Zdravkovic M, Guo M, Ebbesen P, Liu X. Role of the Fas/Fas ligand pathway in apoptotic cell death induced by the human T cell lymphotropic virus type I Tax transactivator. J Gen Virol. 1997;78:3277–3285. doi: 10.1099/0022-1317-78-12-3277. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. 2013;25:463–477. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Lee P, Ceteci F, Nixon C, Blyth K, Sansom OJ, Vousden KH. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 2016;30:52–63. doi: 10.1101/gad.271130.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci, USA. 2012;109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Harhaj EW. HTLV-1 Tax stabilizes MCL-1 via TRAF6-dependent K63-linked polyubiquitination to promote cell survival and transformation. PLoS Pathog. 2014;10:e1004458. doi: 10.1371/journal.ppat.1004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary G, Ratner L. The HTLV-1 hbz antisense gene indirectly promotes tax expression via down-regulation of p30(II) mRNA. Virology. 2011;410:307–315. doi: 10.1016/j.virol.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc I, Polakowski N, André-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem. 2008;283:23903–23913. doi: 10.1074/jbc.M803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabaja BS, Ha CS, Thomas DA, Wilder RB, Gopal R, Cortes J, Bueso-Ramos C, Hess MA, Cox JD, Kantarjian HM. The role of local radiation therapy for mediastinal disease in adults with T-cell lymphoblastic lymphoma. Cancer. 2002;94:2738–2744. doi: 10.1002/cncr.10552. [DOI] [PubMed] [Google Scholar]
- Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J Biol Chem. 2006;281:23414–23424. doi: 10.1074/jbc.M600684200. [DOI] [PubMed] [Google Scholar]
- Doueiri R, Anupam R, Kvaratskhelia M, Green KB, Lairmore MD, Green PL. Comparative host protein interactions with HTLV-1 p30 and HTLV-2 p28: insights into difference in pathobiology of human retroviruses. Retrovirology. 2012;9:64. doi: 10.1186/1742-4690-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, Franchini G. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses. 2011;3:861–885. doi: 10.3390/v3060861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sabban ME, Merhi RA, Haidar HA, Arnulf B, Khoury H, Basbous J, Nijmeh J, de Thé H, Hermine O, Bazarbachi A. Human T-cell lymphotropic virus type 1-transformed cells induce angiogenesis and establish gap junctions with endothelial cells. Blood. 2002;99:3383–3389. doi: 10.1182/blood.v99.9.3383. [DOI] [PubMed] [Google Scholar]
- Esser AK, Rauch DA, Xiang J, Harding JC, Kohart NA, Ross MH, Su X, Wu K, Huey D, Xu Y, Vij K, Green PL, Rosol TJ, Niewiesk S, Ratner L, Weilbaecher KN. HTLV-1 viral oncogene HBZ induces osteolytic bone disease in transgenic mice. Oncotarget. 2017;8:69250–69263. doi: 10.18632/oncotarget.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassone L, Bhatia K, Gutierrez M, Capello D, Gloghini A, Dolcetti R, Vivenza D, Ascoli V, Lo Coco F, Pagani L, Dotti G, Rambaldi A, Raphael M, Tirelli U, Saglio G, Magrath IT, Carbone A, Gaidano G. Molecular profile of Epstein-Barr virus infection in HHV-8-positive primary effusion lymphoma. Leukemia. 2000;14:271–277. doi: 10.1038/sj.leu.2401651. [DOI] [PubMed] [Google Scholar]
- Fenizia C, Fiocchi M, Jones K, Parks RW, Ceribelli M, Chevalier SA, Edwards D, Ruscetti F, Pise-Masison CA, Franchini G. Human T-cell leukemia/lymphoma virus type 1 p30, but not p12/p8, counteracts toll-like receptor 3 (TLR3) and TLR4 signaling in human monocytes and dendritic cells. J Virol. 2014;88:393–402. doi: 10.1128/JVI.01788-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Deng H, Zhao H, Hirbe A, Harding J, Ratner L, Weilbaecher K. HTLV-1 Tax transgenic mice develop spontaneous osteolytic bone metastases prevented by osteoclast inhibition. Blood. 2005;106:4294–4302. doi: 10.1182/blood-2005-04-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger TR, Sharma N, Kim YM, Nyborg JK. The human T-cell leukemia virus type 1 tax protein confers CBP/p300 recruitment and transcriptional activation properties to phosphorylated CREB. Mol Cell Biol. 2008;28:1383–1392. doi: 10.1128/MCB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geleziunas R, Ferrell S, Lin X, Mu Y, Cunningham ET, Jr, Grant M, Connelly MA, Hambor JE, Marcu KB, Greene WC. Human T-cell leukemia virus type 1 Tax induction of NF-kappaB involves activation of the IkappaB kinase alpha (IKKalpha) and IKKbeta cellular kinases. Mol Cell Biol. 1998;18:5157–5165. doi: 10.1128/mcb.18.9.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloghini A, Volpi CC, Gualeni AV, Dolcetti R, Bongarzone I, De Paoli P, Carbone A. Multiple viral infections in primary effusion lymphoma: a model of viral cooperation in lymphomagenesis. Expert Rev Hematol. 2017;10:505–514. doi: 10.1080/17474086.2017.1326815. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, Gasco M, Scanziani E, Capra M, Mai S, Nuciforo P, Crook T, Lough J, Amati B. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–1067. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- Grossman WJ, Kimata JT, Wong FH, Zutter M, Ley TJ, Ratner L. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc Natl Acad Sci, USA. 1995;92:1057–1061. doi: 10.1073/pnas.92.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman WJ, Ratner L. Cytokine expression and tumorigenicity of large granular lymphocytic leukemia cells from mice transgenic for the tax gene of human T-cell leukemia virus type I. Blood. 1997;90:783–794. [PubMed] [Google Scholar]
- Hall AP, Irvine J, Blyth K, Cameron ER, Onions DE, Campbell ME. Tumours derived from HTLV-I tax transgenic mice are characterized by enhanced levels of apoptosis and oncogene expression. J Pathol. 1998;186:209–214. doi: 10.1002/(SICI)1096-9896(1998100)186:2<209::AID-PATH162>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Harhaj NS, Sun SC, Harhaj EW. Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa B kinase. J Biol Chem. 2007;282:4185–4192. doi: 10.1074/jbc.M611031200. [DOI] [PubMed] [Google Scholar]
- Harrod R. Inhibiting HDACs in a preclinical model of HTLV-1-induced adult T-cell lymphoma. Leuk Res. 2011;35:1436–1437. doi: 10.1016/j.leukres.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod R, Kuo YL, Tang Y, Yao Y, Vassilev A, Nakatani Y, Giam CZ. P300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J Biol Chem. 2000;275:11852–11857. doi: 10.1074/jbc.275.16.11852. [DOI] [PubMed] [Google Scholar]
- Harrod R, Tang Y, Nicot C, Lu HS, Vassilev A, Nakatani Y, Giam CZ. An exposed KID-like domain in human T-cell lymphotropic virus type 1 Tax is responsible for the recruitment of coactivators CBP/p300. Mol Cell Biol. 1998;18:5052–5061. doi: 10.1128/mcb.18.9.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Sawa H, Lewis MJ, Orba Y, Sheehy N, Yamamoto Y, Ichinohe T, Tsunetsugu-Yokota Y, Katano H, Takahashi H, Matsuda J, Sata T, Kurata T, Nagashima K, Hall WW. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med. 2006;12:466–472. doi: 10.1038/nm1389. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hiven P, Frédéric M, Arpin-André C, Basbous J, Gay B, Thébault S, Mesnard JM. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J Cell Sci. 2005;118:1355–1362. doi: 10.1242/jcs.01727. [DOI] [PubMed] [Google Scholar]
- Ho YK, Zhi H, Bowlin T, Dorjbal B, Philip S, Zahoor MA, Shih HM, Semmes OJ, Schaefer B, Glover JN, Giam CZ. HTLV-1 Tax stimulates ubiquitin E3 ligase, ring finger protein 8, to assemble lysine 63-linked polyubiquitin chains for TAK1 and IKK activation. PLoS Pathog. 2015;11:e1005102. doi: 10.1371/journal.ppat.1005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YK, Zhi H, DeBiaso D, Philip S, Shih HM, Giam CZ. HTLV-1 tax-induced rapid senescence is driven by the transcriptional activity of NF-κB and depends on chronically activated IKKα and p65/RelA. J Virol. 2012;86:9474–9483. doi: 10.1128/JVI.00158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Xia Y, Zhu Y, Zhao HH, Zhu H, Xie Y, Fan L, Wang L, Miao KR, Yu H, Miao YQ, Wu W, Zhu HY, Chen YY, Xu W, Qian SX, Li JY. TP53-induced glycolysis and apoptosis regulator protects from spontaneous apoptosis and predicts poor prognosis in chronic lymphocytic leukemia. Leuk Res. 2016;50:72–77. doi: 10.1016/j.leukres.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012;52:175–184. doi: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Harrod R, Franchini G. Molecular biology and pathogenesis of the human T-cell leukaemia/lymphotropic virus type-1 (HTLV-1) Int J Exp Pathol. 2001;82:135–147. doi: 10.1046/j.1365-2613.2001.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Zhang M, Petrus M, Maeda M, Pise-Masison CA, Waldmann TA. Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukemia. Retrovirology. 2014;11:43. doi: 10.1186/1742-4690-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannian P, Yin H, Doueiri R, Lairmore MD, Fernandez S, Green PL. Distinct transformation tropism exhibited by human T lymphotropic virus type 1 (HTLV-1) and HTLV-2 is the result of postinfection T cell clonal expansion. J Virol. 2012;86:3757–3766. doi: 10.1128/JVI.06900-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawatsuki A, Yasunaga JI, Mitobe Y, Green PL, Matsuoka M. HTLV-1 bZIP factor protein targets the Rb/E2F-1 pathway to promote proliferation and apoptosis of primary CD4(+) T cells. Oncogene. 2016;35:45-9-4517. doi: 10.1038/onc.2015.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kchour G, Tarhini M, Sharifi N, Farid R, Khooei AR, Shirdel A, Afshari JT, Sadeghian A, Otrock Z, Hermine O, El-Sabban M, Bazarbachi A. Increased microvessel density in involved organs from patients with HTLV-I associated adult T cell leukemia lymphoma. Leuk Lymphoma. 2008;49:265–270. doi: 10.1080/10428190701760060. [DOI] [PubMed] [Google Scholar]
- Kimata JT, Wong FH, Wang JJ, Ratner L. Construction and characterization of infectious human T-cell leukemia virus type 1 molecular clones. Virology. 1994;204:656–664. doi: 10.1006/viro.1994.1581. [DOI] [PubMed] [Google Scholar]
- Kinjo T, Ham-Terhune J, Peloponese JM, Jr, Jeang KT. Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J Virol. 2010;84:5431–5437. doi: 10.1128/JVI.02460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralnik IJ, Fullen J, Franchini G. The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J Virol. 1993;67:2360–2366. doi: 10.1128/jvi.67.4.2360-2366.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralnik IJ, Gessain A, Klotman ME, Lo Monico A, Berneman ZN, Franchini G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc Natl Acad Sci, USA. 1992;89:8813–8817. doi: 10.1073/pnas.89.18.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann AS, Villaudy J, Gazzolo L, Castellazzi M, Mesnard JM, Duc Dodon M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT) Retrovirology. 2007;4:92. doi: 10.1186/1742-4690-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YL, Giam CZ. Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. EMBO J. 2006;25:1741–1752. doi: 10.1038/sj.emboj.7601054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Kutok JL, Fletcher CD. Splenic vascular tumors. Semin Diagn Pathol. 2003;20:128–139. doi: 10.1016/s0740-2570(03)00011-x. [DOI] [PubMed] [Google Scholar]
- Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih H, Connor LM, Marriott SJ, Goodman RH. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature. 1996;380:642–646. doi: 10.1038/380642a0. [DOI] [PubMed] [Google Scholar]
- Lairmore MD, Anupam R, Bowden N, Haines R, Haynes RA, 2nd, Ratner L, Green PL. Molecular determinants of human T-lymphotropic virus type 1 transmission and spread. Viruses. 2011;3:1131–1165. doi: 10.3390/v3071131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh MH, Thébault S, Barbeau B, Nyborg JK, Mesnard JM. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol. 2007;81:1543–1553. doi: 10.1128/JVI.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator) J Biol Chem. 2009;284:1748–1754. doi: 10.1074/jbc.M807821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kesic M, Yin H, Yu L, Green PL. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol. 2009;83:3788–3797. doi: 10.1128/JVI.02315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Yu J, Brown K, Levine B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001;61:3443–3449. [PubMed] [Google Scholar]
- Lin HC, Simon PJ, Ysla RM, Zeichner SL, Brewer G, Rabson AB. RNA stability regulates human T cell leukemia virus type 1 gene expression in chronically-infected CD4 T cells. Virology. 2017;508:7–17. doi: 10.1016/j.virol.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Dole K, Stanley JR, Richard V, Rosol TJ, Ratner L, Lairmore M, Feuer G. Engraftment and tumorigenesis of HTLV-1 transformed T cell lines in SCID/bg and NOD/scid mice. Leuk Res. 2002;26:561–567. doi: 10.1016/s0145-2126(01)00169-2. [DOI] [PubMed] [Google Scholar]
- Los M, Khazaie K, Schulze-Osthoff K, Baeuerle PA, Schirrmacher V, Chlichlia K. Human T cell leukemia virus-I (HTLV-I) Tax-mediated apoptosis in activated T cells requires an enhanced intracellular prooxidant state. J Immunol. 1998;161:3050–3055. [PubMed] [Google Scholar]
- Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene. 2013;32:4222–4230. doi: 10.1038/onc.2012.450. [DOI] [PubMed] [Google Scholar]
- Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, Matsuoka M. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci, USA. 2018;115:e1269–1278. doi: 10.1073/pnas.1715724115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, Merchut-Maya JM, Hodny Z, Bartkova J, Christensen C, Bartek J. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol. 2015;9:601–616. doi: 10.1016/j.molonc.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael B, Nair AM, Datta A, Hiraragi H, Ratner L, Lairmore MD. Histone acetyltransferase (HAT) activity of p300 modulates human T lymphotropic virus type 1 p30II-mediated repression of LTR transcriptional activity. Virology. 2006;354:225–239. doi: 10.1016/j.virol.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitagami Y, Yasunaga J, Kinosada H, Ohshima K, Matsuoka M. Interferon-γ promotes inflammation and development of T-cell lymphoma in HTLV-1 bZIP factor transgenic mice. PLoS Pathog. 2015;11:e1005120. doi: 10.1371/journal.ppat.1005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra-Kaushik S, Harding J, Hess J, Schreiber R, Ratner L. Enhanced tumorigenesis in HTLV-1 tax-transgenic mice deficient in interferon-gamma. Blood. 2004;104:3305–3311. doi: 10.1182/blood-2004-01-0266. [DOI] [PubMed] [Google Scholar]
- Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM. Primary effusion lymphoma: a distinct clinicopathologic entity associated with Kaposi’s sarcoma-associated herpes virus. Blood. 1996;88:645–656. [PubMed] [Google Scholar]
- Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R, Princler GL, Derse D, Misteli T, Franchini G. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med. 2004;10:197–201. doi: 10.1038/nm984. [DOI] [PubMed] [Google Scholar]
- Nicot C, Harrod R. Distinct p300-responsive mechanisms promote caspase-dependent apoptosis by human T-cell lymphotropic virus type 1 Tax protein. Mol Cell Biol. 2000;20:8580–8589. doi: 10.1128/mcb.20.22.8580-8589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Sakai I, Narumi H, Yasukawa M, Fujita S, Hato T, Watanabe S. Adult T-cell leukemia with pleural effusion and infiltration in bilateral anterior chambers. Nihon Naika Gakkai Zasshi. 2002;91:2476–2478. doi: 10.2169/naika.91.2476. [DOI] [PubMed] [Google Scholar]
- Okada F, Ando Y, Kondo Y, Matsumoto S, Maeda T, Mori H. Thoracic CT findings of adult T-cell leukemia or lymphoma. Am J Roentgenol. 2004;182:761–767. doi: 10.2214/ajr.182.3.1820761. [DOI] [PubMed] [Google Scholar]
- Olenchock BA, Vander Heiden MG. Pyruvate as a pivot point for oncogene-induced senescence. Cell. 2013;153:1429–1430. doi: 10.1016/j.cell.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panfil AR, Al-Saleem J, Howard CM, Shkriabai N, Kvaratskhelia M, Green PL. Stability of the HTLV-1 antisense-derived protein, HBZ, is regulated by the E3 ubiquitin-protein ligase, UBR5. Front Microbiol. 2018;9:80. doi: 10.3389/fmicb.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panfil AR, Dissinger NJ, Howard CM, Murphy BM, Landes K, Fernandez SA, Green PL. Functional comparison of HBZ and the related APH-2 protein provides insight into human T-cell leukemia virus type 1 pathogenesis. J Virol. 2016;90:3760–3772. doi: 10.1128/JVI.03113-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panfil AR, Martinez MP, Ratner L, Green PL. Human T-cell leukemia virus-associated malignancy. Curr Opin Virol. 2016;20:40–46. doi: 10.1016/j.coviro.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, Blobel GA, McMahon SB. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol. 2004;24:10826–10834. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Rico MA, Calvo-Vidal MN, Villalonga-Planells R, Martínez-Soler F, Giménez-Bonafé P, Navarro-Sabaté A, Tortosa A, Bartrons R, Manzano A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol. 2011;101:132–139. doi: 10.1016/j.radonc.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Pique C, Ureta-Vidal A, Gessain A, Chancerel B, Gout O, Tamouza R, Agis F, Dokhélar MC. Evidence for the chronic in vivo production of human T cell leukemia virus type I Rof and Tof proteins from cytotoxic T lymphocytes directed against viral peptides. J Exp Med. 2000;191:567–572. doi: 10.1084/jem.191.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S, Li J, Hong M, Zhu Y, Zhao H, Xie Y, Huang J, Lian Y, Li Y, Wang S, Mao J, Chen Y. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J Hematol Oncol. 2016;9:128. doi: 10.1186/s13045-016-0360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran R, Garva R, Ashour H, Leung T, Stratford I, Krstic-Demonacos M, Demonacos C. Acetylation mediated by the p300/CBP-associated factor determines cellular energy metabolic pathways in cancer. Int J Oncol. 2013;42:1961–1972. doi: 10.3892/ijo.2013.1907. [DOI] [PubMed] [Google Scholar]
- Ren T, Takahashi Y, Liu X, Loughran TP, Sun SC, Wang HG, Cheng H. HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene. 2015;34:334–345. doi: 10.1038/onc.2013.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo M, Hutchison T, Malu A, White A, Kim J, Gardner R, Smith K, Nelson K, Bergeson R, McKee R, Harrod C, Ratner L, Lüscher B, Martinez E, Harrod R. The human T-cell leukemia virus type-1 p30II protein activates p53 and induces the TIGAR and suppresses oncogene-induced oxidative stress during viral carcinogenesis. Virology. 2018;518:103–115. doi: 10.1016/j.virol.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, Harrod CK, Barnett B, Ratner L, Lairmore MD, Martinez E, Lüscher B, Robson CN, Henriksson M, Harrod R. Acetylation of the c-Myc oncoprotein is required for cooperation with the HTLV-1 p30II accessory protein and the induction of oncogenic cellular transformation by p30II/c-Myc. Virology. 2015;476:271–288. doi: 10.1016/j.virol.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH, Li CB, Horne WC, Santiago P, Trojano N, Jay G, Horowitz M, Baron R. Mice transgenic for HTLV-I LTR-tax exhibit tax expression in bone, skeletal alterations, and high bone turnover. Virology. 1993;197:196–204. doi: 10.1006/viro.1993.1580. [DOI] [PubMed] [Google Scholar]
- Saggioro D, Rosato A, Esposito G, Rosenberg MP, Harrison J, Felber BK, Pavlakis GN, Chieco-Bianchi L. Inflammatory polyarthropathy and bone remodeling in HTLV-I Tax-transgenic mice. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14:272–280. doi: 10.1097/00042560-199703010-00012. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin D, Uldrick TS, Kwak H, Ohnuki H, Polizzotto MN, Annunziata CM, Raffeld M, Wyvill KM, Aleman K, Wang V, Marshall VA, Whitby D, Yarchoan R, Tosato G. Evidence for a mesothelial origin of body cavity effusion lymphomas. J Natl Cancer Inst. 2017;109:djx016, 1–11. doi: 10.1093/jnci/djx016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci, USA. 2006;103:720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou Y, Yasunaga J, Zhao T, Yoshida M, Miyazato P, Takai K, Shimizu K, Ohshima K, Green PL, Ohkura N, Yamaguchi T, Ono M, Sakaguchi S, Matsuoka M. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7:e1001274. doi: 10.1371/journal.ppat.1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura R, Ishibashi H, Morioka E, Teshima T, Kudo J, Hirata Y, Shibuya T, Niho Y. Assessment of abdominal involvement of adult T-cell leukemia/lymphoma by ultrasonography: comparison among four clinical types. J Clin Ultrasound. 1991;19:485–492. doi: 10.1002/jcu.1870190806. [DOI] [PubMed] [Google Scholar]
- Silverman LR, Phipps AJ, Montgomery A, Ratner L, Lairmore MD. Human T-cell lymphotropic virus type 1 open reading frame II-encoded p30II is required for in vivo replication: evidence of in vivo reversion. J Virol. 2004;78:3837–3845. doi: 10.1128/JVI.78.8.3837-3845.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha-Datta U, Datta A, Ghorbel S, Dodon MD, Nicot C. Human T-cell lymphotropic virus type I rex and p30 interactions govern the switch between virus latency and replication. J Biol Chem. 2007;282:14608–14615. doi: 10.1074/jbc.M611219200. [DOI] [PubMed] [Google Scholar]
- Smith MR, Greene WC. Identification of HTLV-I tax trans-activator mutants exhibiting novel transcriptional phenotypes. Genes Dev. 1990;4:1875–1885. doi: 10.1101/gad.4.11.1875. [DOI] [PubMed] [Google Scholar]
- Sun EC, Elwood J, Beraud C, Greene WC. Human T-cell leukemia virus type I Tax activation of NF-kappa B/Rel involves phosphorylation and degradation of I kappa B alpha and RelA (p65)-mediated induction of the c-rel gene. Mol Cell Biol. 1994;14:7377–7384. doi: 10.1128/mcb.14.11.7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Fujisawa JI, Toita M, Yoshida M. The trans-activator tax of human T-cell leukemia virus type 1 (HTLV-1) interacts with cAMP-responsive element (CRE) binding and CRE modulator proteins that bind to the 21-base-pair enhancer of HTLV-1. Proc Natl Acad Sci, USA. 1993;90:610–614. doi: 10.1073/pnas.90.2.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaims AY, Khani F, Zhang Y, Roberts AI, Devadas S, Shi Y, Rabson AB. Immune activation induces immortalization of HTLV-1 LTR-Tax transgenic CD4+ T cells. Blood. 2010;116:2994–3003. doi: 10.1182/blood-2009-07-231050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Higuchi M, Makokha GN, Matsuki H, Yoshita M, Tanaka Y, Fujii M. HTLV-1 Tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood. 2013;122:715–725. doi: 10.1182/blood-2013-03-493718. [DOI] [PubMed] [Google Scholar]
- Tang SW, Chen CY, Klase Z, Zane L, Jeang KT. The cellular autophagy pathway modulates HTLV-1 replication. J Virol. 2013;87:1699–1707. doi: 10.1128/JVI.02147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, Nicot C, Cecchinato V, Ruscetti F, Franchini G. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood. 2010;116:3809–3817. doi: 10.1182/blood-2010-05-284141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhou J, Shi J, Zhang Y, Liu S, Liu Y, Zheng D. Human T-cell leukemia virus type 1 Tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J Virol. 2014;88:2786–2798. doi: 10.1128/JVI.03025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox TM, Speer RW, Schlinkert RT, Sarr MG. Hemangioma of the spleen: presentation, diagnosis, and management. J Gastrointest Surg. 2000;4:611–613. doi: 10.1016/s1091-255x(00)80110-9. [DOI] [PubMed] [Google Scholar]
- Wu X, Sun SC. Retroviral oncoprotein Tax deregulates NF-kappaB by activating Tak1 and mediating the physical association of Tak1-IKK. EMBO Rep. 2007;8:510–515. doi: 10.1038/sj.embor.7400931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Yamamoto B, Haoudi A, Semmes OJ, Green PL. PDZ binding motif of HTLV-1 Tax promotes virus-mediated T-cell proliferation in vitro and persistence in vivo. Blood. 2006;107:1980–1988. doi: 10.1182/blood-2005-03-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T. The human T-cell leukemia virus type I (HTLV-I) tax protein induces apoptosis. Nihon Rinsho. 1996;54:1855–1859. [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- Yin L, Kufe T, Avigan D, Kufe D. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood. 2014;123:2997–3006. doi: 10.1182/blood-2013-11-539395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis I, Boris-Lawrie K, Green PL. Human T-cell leukemia virus open reading frame II encodes a posttranscriptional repressor that is recruited at the level of transcription. J Virol. 2006;80:181–191. doi: 10.1128/JVI.80.1.181-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis I, Khair L, Dundr M, Lairmore MD, Franchini G, Green PL. Repression of human T-cell leukemia virus type 1 and type 2 replication by a viral mRNA-encoded posttranscriptional regulator. J Virol. 2004;78:11077–11083. doi: 10.1128/JVI.78.20.11077-11083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Nisbet JW, Albrecht B, Ding W, Kashanchi F, Bartoe JT, Lairmore MD. Human T-lymphotropic virus type 1 p30(II) regulates gene transcription by binding CREB binding protein/p300. J Virol. 2001;75:9885–9895. doi: 10.1128/JVI.75.20.9885-9895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Nisbet JW, Bartoe JT, Ding W, Lairmore MD. Human T-lymphotropic virus type 1 p30(II) functions as a transcription factor and differentially modulates CREB-responsive promoters. J Virol. 2000;74:11270–11277. doi: 10.1128/jvi.74.23.11270-11277.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LJ, Giam CZ. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc Natl Acad Sci, USA. 1992;89:7070–7074. doi: 10.1073/pnas.89.15.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LJ, Loewenstein PM, Green M. Ad E1A 243R oncoprotein promotes association of proto-oncogene product MYC with the NuA4/Tip60 complex via the E1A N-terminal repression domain. Virology. 2016;499:178–184. doi: 10.1016/j.virol.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, Matsuoka M. HTLV-1 bZIP factor enhances TGF-β signaling through p300 coactivator. Blood. 2011;118:1865–1876. doi: 10.1182/blood-2010-12-326199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Yasunaga J, Satou Y, Nakao M, Takahashi M, Fujii M, Matsuoka M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood. 2009;113:2755–2764. doi: 10.1182/blood-2008-06-161729. [DOI] [PubMed] [Google Scholar]
- Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ. NF-κB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011;7:e1002025. doi: 10.1371/journal.ppat.1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B, Sargeant A, Landes K, Fernandez SA, Chen CS, Lairmore MD. Efficacy of novel histone deacetylase inhibitor, AR42, in a mouse model of, human T-lymphotropic virus type 1 adult T-cell lymphoma. Leuk Res. 2011;35:1491–1497. doi: 10.1016/j.leukres.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.