

SUMMARY
PARP inhibitors (PARPis) have been used to induce synthetic lethality in BRCA-deficient tumors in clinical trials with limited success. We hypothesized that RAD52-mediated DNA repair remains active in PARPi-treated BRCA-deficient tumor cells and that targeting RAD52 should enhance the synthetic lethal effect of PARPi. We show that RAD52 inhibitors (RAD52is) attenuated single-strand annealing (SSA) and residual homologous recombination (HR) in BRCA-deficient cells. Simultaneous targeting of PARP1 and RAD52 with inhibitors or dominant-negative mutants caused synergistic accumulation of DSBs and eradication of BRCA-deficient but not BRCA-proficient tumor cells. Remarkably, Parp1−/−; Rad52−/− mice are normal and display prolonged latency of BRCA1-deficient leukemia compared with Parp1−/− and Rad52−/− counterparts. Finally, PARPi+RAD52i exerted synergistic activity against BRCA1-deficient tumors in immunodeficient mice with minimal toxicity to normal cells and tissues. In conclusion, our data indicate that addition of RAD52i will improve therapeutic outcome of BRCA-deficient malignancies treated with PARPi.
In Brief
Sullivan-Reed et al. show that simultaneous treatment with PARP and RAD52 inhibitors exerts dual synthetic lethality in BRCA-deficient tumors. Addition of RAD52 inhibitor should improve therapeutic outcome of BRCA-deficient malignancies treated with PARP inhibitor.
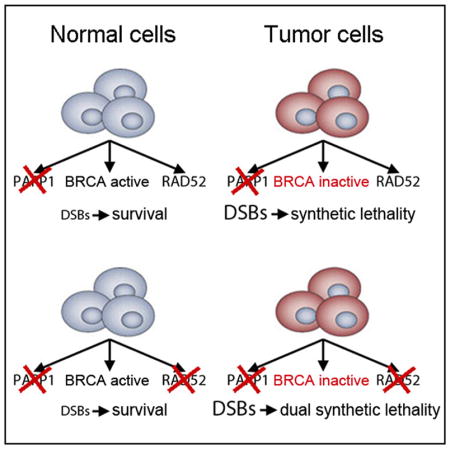
INTRODUCTION
Numerous reports indicate that tumor cells accumulate high levels of spontaneous and drug-induced DNA damage, but they survive because of enhanced or altered DNA repair activities (Bartkova et al., 2005). PARP1 may prevent accumulation of potentially lethal DNA double-strand breaks (DSBs) by playing a key role in base excision repair (BER), single-strand break (SSB) repair, and alternative non-homologous end-joining (Alt-NHEJ) and/or by facilitating MRE11-mediated recruitment of RAD51 to promote stalled replication fork restart (Metzger et al., 2013; Ying et al., 2012). Homologous recombination (HR), which depends mostly on BRCA1-PALB2-BRCA2-RAD51 paralogs-RAD51-RAD54 (BRCA-HR), and RAD52-dependent single-strand annealing (RAD52-SSA) play an important role in DSB repair in proliferating cells (Kass and Jasin, 2010).
The hypothesis that cancer cells are addicted to particular DNA repair pathways is supported by selective targeting of tumor cells by recently developed novel drugs and compounds against specific DNA repair mechanisms (Nickoloff et al., 2017). The success of the PARP inhibitor (PARPi) olaparib in BRCA1- and BRCA2-deficient breast tumors has established a proof of concept of personalized cancer therapy using synthetic lethality (Lord et al., 2015). Unfortunately, therapeutic effect is usually short-lived, and tumor cells become unresponsive to PARPi because of compensatory mechanisms such as restoration of HR via secondary mutations in BRCA2, PALB2, RAD51 paralogs (RAD51C, RAD51D), or loss of 53BP1, impaired drug uptake, and/or enhanced drug efflux (Lord and Ashworth, 2013). In concordance, we showed that BRCA-deficient breast carcinoma cells and leukemia cells could not be completely eradicated by PARPi (Nieborowska-Skorska et al., 2017). Therefore, more robust and rapid elimination of BRCA-deficient tumor cells is required to prevent time-dependent emergence of PARPi-resistant or refractory clones.
It has been suggested that RAD52-dependent HR pathways involving RAD51 (RAD52-HR) and/or RAD52-SSA can act as backups to the main BRCA-mediated HR pathway (BRCA-HR) (Stark et al., 2004; Wray et al., 2008). We hypothesized that RAD52-HR and/or RAD52-SSA represent potential escape route(s) from PARPi-mediated synthetic lethality in BRCA-deficient cells and that simultaneous inhibition of PARP and RAD52-dependent DNA repair pathways would trigger more effective “dual” synthetic lethality.
RESULTS
Inhibition of RAD52 Attenuated Residual HR Activity in PARPi-Treated BRCA-Deficient Tumor Cell Lines
BRCA1/2-deficient and BRCA1/2-proficient cells carrying DR-GFP recombination reporter cassette were co-transfected with pCBASceI (encoding I-Sce1 endonuclease generating a DSB in the reporter cassette) and pDsRed (transfection efficiency control) expression plasmids. As expected, BRCA1 and BRCA2 deficiencies were associated with reduced HR measured by the percentage of GFP+ cells in DsRed+ population, but residual HR activity was consistently detectable in BRCA-deficient cells (Figures 1A and 1B). PARPis olaparib and talazoparib did not affect HR activities in BRCA-deficient and proficient cells. However, a previously described RAD52i, 6-hydroxy-DL-dopa (Dopa) (Chandramouly et al., 2015), abrogated residual HR activity in naive and PARPi-treated BRCA-deficient cells without affecting BRCA-proficient counterparts.
Figure 1. RAD52 Inhibitor 6-OH-Dopa Attenuated HR and SSA in BRCA1/2-Deficient Cells Treated with PARP Inhibitor Olaparib.

(A and B) BRCA2-mutated VC8 cells (BRCA2−) and BRCA2 wild-type V79 cells (BRCA2+) (A) and BRCA1−/− murine ES clone 17 cells (BRCA1−) and BRCA1 wild-type clone 92B cells (BRCA1+) (B) carrying DR-GFP cassette were co-transfected with I-SceI and DsRed cDNAs, followed by treatment with 5 μM olaparib (Ola), 50 nM talazoparib (Tala), and/or 10 μM 6-OH-dopa (Dopa), or were left untreated (Control). Results represent mean percentage of GFP+DsRed+ cells in DsRed+ population ± SD from three independent experiments; *p < 0.05 in comparison with untreated control.
(C) BRCA1-mutated HCC1937 cells (BRCA1−) and HCC1937 expressing wild-type BRCA1 (BRCA1+) were treated with 3 μg/mL cisplatin (Control) and cisplatin combined with 5 μM olaparib (Ola) and/or 10 μM 6-OH-dopa (Dopa). Results represent percentage of cells with more than ten RAD51 foci from three independent experiments (100 cells/experiment were evaluated); *p < 0.05 in comparison with untreated control.
(D) BRCA2−/− murine ES clone 42E cells (BRCA2−) and BRCA2 wild-type clone 40b cells (BRCA2+) carrying SA-GFP cassette were co-transfected with I-SceI and DsRed cDNAs, followed by treatment with 1.25 μM olaparib (Ola) and/or 20 μM 6-OH-dopa (Dopa), or were left untreated (Control). Results represent mean percentage of GFP+DsRed+ cells in DsRed+ population ± SD from three independent experiments; *p < 0.05 in comparison with untreated control.
See also Figure S1.
In addition, RAD51 foci could be applied as a surrogate marker for HR activity (Oplustilova et al., 2012). We employed BRCA1-deficient HCC1937 cells, in which RAD51 foci formation depends on RAD52 (Lok et al., 2013). We detected that RAD52i Dopa, but not PARPi olaparib, inhibited cisplatin-induced RAD51 foci formation in BRCA1-deficient HCC1937 cells but not in BRCA1-proficient counterparts (Figure 1C). Moreover, Dopa reduced RAD51 foci formation in olaparib-treated BRCA1-deficient HCC1937 cells. As expected, RAD52i Dopa inhibited SSA activity in BRCA2-deficient and BRCA2-proficient cells and also in olaparib-treated cells (Figure 1D).
The importance of RAD52 was further supported by significantly higher relapse-free survival probability of the patients with leukemias displaying low expression levels of one of the BRCA-HR gene (BRCA1, BRCA2, PALB2, RAD51) and RAD52 gene compared with those with leukemias expressing high levels of these genes (Figure S1). In concordance, RAD52i reduced HR and SSA activities in BRCA-deficient tumor cells when applied alone or in combination with PARPi, suggesting that RAD52 promotes survival of PARPi-treated BRCA-deficient cells. This observation provided justification for the investigation of the potency of dual synthetic lethal effect exerted by simultaneous targeting of PARP and RAD52 in BRCA-deficient tumor cells.
RAD52i Enhanced the Synthetic Lethal Effect of PARPi, Causing Complete Eradication of BRCA-Deficient Tumor Cells In Vitro
Simultaneous treatment of BRCA1-deficient MDA-MB-436 tumor cells with PARPi olaparib and RAD52i Dopa (Chandramouly et al., 2015) resulted in enhanced accumulation of DSBs detected by neutral comet assay and γ-H2AX immunofluorescence and synergistic inhibition of cell growth in comparison with treatment with individual inhibitors, while BRCA-proficient counterparts were not affected by these inhibitors (Figure 2A). Similar growth inhibitory effect in BRCA1-deficient MDA-MB-436 cells was exerted by olaparib combined with RAD52i D-I03 (Huang et al., 2016) (Figure 2B). Moreover, combination of PARPi olaparib and RAD52i F79 aptamer (Cramer-Morales et al., 2013) synergistically inhibited the growth of BRCA1-deficient UWB1.289, MDA-MB-436, and HCC1937 tumor cell lines and BRCA2-deficient Capan-1 tumor cells compared with individual treatments (Figure 2C). BRCA-proficient counterparts and non-transformed NIH 3T3 cells were not affected by the inhibitors. PARPi and RAD52i did not cause downregulation of PARP1 and RAD52 proteins implicating functional inhibition of the targeted proteins (Figure S2A).
Figure 2. Simultaneous Targeting RAD52 and PARP1 Enhanced Synthetic Lethality in BRCA-Deficient Carcinoma Cell Lines and Abrogated the Emergence of Resistant Clones.

(A) BRCA1-deficient MDA-MB-436 cells and BRCA1-reconstituted counterparts were treated with 1 μM Ola and/or 5 μM Dopa. Results represent (left) mean percentage of comet tail DNA ± SD (100–150 cells) and (middle) mean percentage of γ-H2AX-positive cells ± SD (triplicate experiment) detected after 24 hr, and (right) mean percentage of viable cells ± SD relative to untreated counterparts (three experiments) detected at day 5. *p < 0.05 and **p = 0.06 in comparison with cells treated with individual drugs (olaparib, Dopa) using the response additivity approach.
(B) MDA-MB-436 BRCA1-deficient and BRCA1-proficient cells were treated on day 0 and day 2 with increasing concentrations of olaparib in absence or presence of D-I03 (1 μM). Results represent mean percentage of clonogenic cells ± SD relative to untreated counterparts (three experiments).
(C) Indicated BRCA1/2-deficient cells and BRCA1/2-reconstituted counterparts were treated with 1μM olaparib (Ola) and/or 1μM F79 aptamer (F79) added at 0 and 2 days, followed by trypan blue counting at day 5. Mean percentage of viable cells ± SD relative to untreated counterparts (three experiments). *p < 0.04 in comparison with cells treated with individual drugs using the response additivity approach.
(D) Indicated BRCA1/2-deficient cells were left untreated (black) or continuously treated with 2.5 μM olaparib (blue), 20 μM Dopa (green), and olaparib+Dopa (red) for 28 days. Results represent mean cumulative number of viable cells ± SD (triplicate experiments).
(E) BRCA1-deficient and BRCA1 reconstituted MBA-MB-436 cells and BRCA2-deficient and BRCA2 reconstituted EUFA423 cells were transfected with PARP1 and/or RAD52 wild-type or PARP1(E988K) and/or RAD52(F79A) dominant-negative mutants. Results represent relative growth of the cells expressing dominant-negative mutant(s) relative to these expressing wild-type proteins from at least three experiments. *p < 0.02 compared with BRCA1/2-proficient counterparts using Student’s t test; **p ≤ 0.01 compared with BRCA1/2-deficient cells transfected with individual mutants using the response additivity approach.
See also Figures S2 and S3.
Hence, these data show consistent and selective killing of BRCA-deficient cells by dual treatment with PARPi (i.e., olaparib, talazoparib) and one of three different RAD52i (i.e., F79 aptamer, Dopa, D-I03), which validates RAD52 as an attractive target for inhibition along with PARPi.
The effect of dual synthetic lethality may be triggered selectively in BRCA-deficient cells. MLL-AF9-positive leukemia cells are BRCA proficient, but they are sensitive to PARPi, especially when combined with standard cytotoxic drug (Maifrede et al., 2017b). However, these cells did not respond favorably to the combination of PARPi + RAD52i (Figure S2B), most likely because PARP1-dependent but not RAD52-mediated mechanisms play a major role in their DNA repair.
Importantly, long-term continuous treatment (28 days) with PARPi (olaparib) + RAD52i (Dopa) led to eradication of BRCA1-and BRCA2-deficient cells, while individual agents generated partial inhibition of the growth rate (Figure 2D). At the end of 28 days of continuous treatment, double-treated cells were washed out of the drugs and incubated in drug-free medium. Remarkably, no living cells were retrieved even after 14 days of culture.
We also examined the effect of combination treatment on leukemia cells deficient in another BRCA-HR gene, RAD54 (Mazin et al., 2010). Simultaneous short-term treatment of RAD54−/− Nalm-6 human leukemia cell line but not RAD54+/+ parental cells with PARPi olaparib and RAD52i Dopa enhanced accumulation of DSBs assessed by neutral comet assay and γ-H2AX immunofluorescence and eliminated significantly more cells, as determined by trypan blue exclusion test, compared with individual drugs (Figures S2C–S2E). Moreover, long-term (35 days) combinatorial treatment resulted in complete elimination of RAD54−/− Nalm-6 cells, whereas individual inhibitors exerted only partial effect (Figure S2F). At the end of the experiment, living RAD54−/− Nalm-6 cells were not detected after a 14 day incubation of double-treated cells in drug-free medium.
In addition, a combination of olaparib and Dopa exerted a much stronger inhibitory effect against BRCA2-deficient Burkitt lymphoma (BL)-derived Epstein-Barr virus (EBV)-positive B cell lines Mutu and Raji (Maifrede et al., 2017a) compared with individual inhibitor treatment (Figure S2G). At the same time, BRCA2-proficient EBV-immortalized lymphoblastoid cell lines (LCL1 and LCL2) established from healthy donor cells were not sensitive to these inhibitors.
To rule out possible off-target effects of the small-molecule inhibitors, dominant-negative mutants of PARP1 and RAD52 were used. Ectopic expression of catalytically inactive PARP1(E988K) mutant downregulated protein PARylation and expression of DNA binding-defective RAD52(F79A) mutant reduced RAD52 foci formation (Figure S3). PARP1(E988K) and RAD52(F79A) when expressed individually were able to selectively inhibit the growth of BRCA1-deficient MDA-MB-436 cells and BRCA2-deficient EUFA423 cells (Figure 2E). Importantly, co-expression of PARP1(E988K) and RAD52(F79A) mutants exerted synergistic growth inhibitory effect in MDA-MB-436 cells and EUFA423 cells, whereas BRCA1/2-reconstituted counterparts were not affected.
In conclusion, our data show that simultaneous targeting of PARP1 and RAD52 by small-molecule inhibitors or dominant-negative mutants facilitates accumulation of toxic DSBs, resulting in selective elimination of tumor cells displaying deficiencies in BRCA-mediated HR. Moreover, prolonged combinatorial treatment completely eradicated BRCA-deficient cells, demonstrating a highly robust method for selective killing of these cells.
Combination of PARPi and RAD52i Eliminated BRCA-Deficient Primary Leukemia Cells More Efficiently Than Individual Inhibitors
We and others reported previously that several oncogenes, such as BCR-ABL1, AML1-ETO, and IGH-MYC, but not FLT3(ITD) and MLL-AF9, inhibit BRCA1/2 protein expression, resulting in synthetic lethality triggered by PARP and/or RAD52 inhibitors used individually (Cramer-Morales et al., 2013; Fan et al., 2010; Maifrede et al., 2017a; Nieborowska-Skorska et al., 2017; Podszywalow-Bartnicka et al., 2014). Here we tested the effect of combination PARPi+RAD52i treatment on BRCA1/2-deficient and BRCA1/2-proficient patient leukemia cells. Three different RAD52is and two PARPis were applied to exclude the possibility that the effect depends on non-specific properties of an individual inhibitor.
Combination of sub-optimal concentrations of olaparib and Dopa exerted a strong effect against primary BRCA1/2-deficient leukemia and lymphoma cells expressing BCR-ABL1, AML1-ETO, or IGH-MYC but not against BRCA1/2-proficient normal cells and leukemia cells expressing FLT3(ITD) (Figure 3A). Moreover, long-term continuous treatment (28 days) of the cells from very aggressive BCR-ABL1-positive CML-blast phase (CML-BP) with PARPi (olaparib) + RAD52i (Dopa) led to eradication of leukemia cells, while individual agents generated only partial inhibition (Figure 3B). At the end of 28 days of continuous treatment, double-treated cells were washed out of the drugs and incubated in drug-free medium. Remarkably, no living cells were retrieved even after 14 days of culture. In addition, CML-BP cells were sensitive to the combination of sub-optimal concentrations PARPi talazoparib + RAD52i D-I03, and addition of imatinib (BCR-ABL1 kinase inhibitor) to talazoparib + D-I03 resulted in complete eradication of clonogenic cells (Figure 3C).
Figure 3. A Combination of PARP1 and RAD52 Inhibitors Exerted Stronger Synthetic Lethality Than Individual Inhibitors against Primary Cells from BRCA1/2-deficient Hematopoietic Malignancies.

(A) Lin−CD34+ cells from healthy donors (n = 4–8), BRCA-proficient FLT3(ITD)-positive AML (n = 3 or 4), and from BRCA1-deficient BCR-ABL1-positive CML-CP (n = 3–5), BRCA2-deficient AML1-ETO-positive AML (n = 3–6) and BRCA2-deficient IGH-MYC-positive BL cells were treated with 2.5 μM olaparib (Ola) and/or 2.5 μM Dopa.
(B) BRCA1-deficient BCR-ABL1-positive CML-BP cells were left untreated (black) or treated with 2.5 μM olaparib (blue), 20 μM Dopa (green), and olaparib + Dopa (red) for 28 days. Mean cumulative number ± SD of viable cells.
(C) CML-BP cells were treated with 2.5 nM talazoparib (Tala), 2.5 μM D-I03, and/or 1 μM imatinib (Im).
(D) BRCA pathway-deficient and BRCA pathway-proficient AML (n = 3 of each) were treated with 0.125 μg/mL daunorubicin (DNR), 1.25 μM olaparib, and/or 1.25 μM F79.
(E) BRCA pathway-deficient and BRCA pathway-proficient t-MDS/AML (n = 3 of each) were treated with 1.25 μM olaparib and/or 1.25 μM F79.
Cells were treated at 0 and 48 hr, followed by counting in trypan blue (B) or plating in methylcellulose at 96 hr (D); colonies were counted after 7–10 days (A, C, and E). Results represent mean percentage ± SD of surviving colonies/cells. *p < 0.05 in comparison with single and dual treatments, respectively, using Student’s t test; **p = 0.02 compared with DNR+Ola, DNR+F79, Ola, and F79 using the response additivity approach.
Using DR-GFP recombination reporter cassette and/or BRCA1/2 expression and foci formation, we were able to identify several BRCA-deficient and BRCA-proficient samples from individual acute myeloid leukemia (AML) and therapy-related myelodysplastic syndrome (t-MDS) patients (Cramer-Morales et al., 2013; Nieborowska-Skorska et al., 2017). These cells were treated with sub-optimal concentrations of the cytotoxic drug daunorubicin (DNR), PARPi olaparib, and/or RAD52i F79 aptamer. The combination of olaparib + F79 aptamer exerted much stronger inhibitory effect than individual compounds against AML cells from BRCA-deficient but not BRCA-proficient patients (Figures 3D and 3E). Triple combination of DNR + olaparib + F79 aptamer exerted synergistic effect compared with dual combinations of these compounds (Figure 3D).
In conclusion, combination of PARPi+RAD52i exerted stronger inhibitory effect than individual inhibitors specifically in BRCA-deficient leukemia and lymphoma cells. Addition of standard therapeutic drug (e.g., imatinib for CML-BP and DNR for AML) enhanced the effect of PARPi+RAD52i, suggesting a potentially beneficial therapeutic application of this approach.
Simultaneous Targeting of PARP1 and RAD52 Exerted a Synergistic Effect against BRCA-Deficient Tumors in Mice
BCR-ABL1 oncogenic tyrosine kinase induces translational repression and degradation of BRCA1 protein, which makes leukemia cells sensitive to PARPi and RAD52i (Cramer-Morales et al., 2013; Dkhissi et al., 2015; Nieborowska-Skorska et al., 2017; Podszywalow-Bartnicka et al., 2014). To study the effect of PARPi+RAD52i against BRCA1-deficient BCR-ABL1-positive leukemias, we employed a tet-off SCLtTA/p210BCR-ABL1 transgenic mouse model, which upon withdrawal of tetracycline develops CML-CP-like disease (Koschmieder et al., 2005). Bone marrow cells from SCLtTA/p210BCR-ABL1 transgenic mice cultured in the absence of tetracycline were sensitive to PARPi olaparib and RAD52i Dopa, but the combination of these two inhibitors exerted a synergistic anti-leukemia effect (Figure 4A). As expected, BCR-ABL1 protein was expressed and BRCA1 protein was downregulated in the absence of tetracycline (Figure 4B).
Figure 4. The Effect of Simultaneous Inactivation of RAD52 and PARP1 on Leukemo-genesis in BRCA1-Deficient BCR-ABL1 Transgenic Mice.

(A) Sensitivity of clonogenic bone marrow cells from SCLtTA;p210BCR-ABL1 mice (n = 3) to 5 μM olaparib (Ola) and/or 20 μM 6-OH-dopa (Dopa) in the absence of tetracycline. Results represent mean percentage of clonogenic cells ± SD from three mice in triplicates; *p < 0.001 in comparison with untreated cells using Student’s t test; **p = 0.016 compared with individual drugs using the response additivity approach.
(B–D) SCLtTA;p210BCR-ABL1;Parp1−/−; Rad52−/− (BA;Parp1−/−;Rad52−/−), SCLtTA;p210 BCR-ABL1;Parp1−/− (BA;Parp1−/−), SCLtTA; p210BCR-ABL1;Rad52−/− (BA;Rad52−/−), and SCLtTA;p210BCR-ABL1 (BA) mice were assayed for (B) expression of BRCA1, BCR-ABL1, and actin proteins in the absence (−) or presence (+) of tetracycline; (C) clonogenic activity of bone marrow cells from BA;Parp1−/−;Rad52−/−, BA; Parp1−/−, BA;Rad52−/−, and BA mice (at least three mice per group) (results show mean ± SD number of BCR-ABL1-dependent colonies [tet− – tet+]; *p < 0.02 compared with BA and **p < 0.05 compared with BA;Parp1−/− and BA;Rad52−/−); and (D) Kaplan-Meier survival curves of BA;Parp1−/−;Rad52−/− (n = 19), BA;Parp1−/− (n = 27), BA;Rad52−/− (n = 16), and BA (n = 63) mice.
See also Figure S4.
To test the effect of PARP1 and RAD52 inhibition on BRCA1-deficient BCR-ABL1 leukemia in genetic settings, we generated Parp1−/−;Rad52−/− mice, which did not display any detectable defects in various inspected organs, including bone marrow (Figures S4A–S4D). Next, we obtained SCLtTA/p210BCR-ABL1/Parp1−/−Rad52−/−, SCLtTA/p210BCR-ABL1/Parp1−/−, SCLtTA/p210BCR-ABL1/Rad52−/−, and SCLtTA/p210BCR-ABL1 mice. Bone marrow cells from these mice cultured in the absence of tetracycline expressed BCR-ABL1 kinase and displayed downregulation of BRCA1 protein (Figure 4B).
BCR-ABL1-mediated clonogenic potential was calculated as the difference between the numbers of colonies that grew in the absence (BCR-ABL1 kinase expressed) and presence (BCR-ABL1 kinase not expressed) of tetracycline. Bone marrow cells from SCLtTA/p210BCR-ABL1/Parp1−/− Rad52−/− mice displayed the lowest BCR-ABL1-dependemt clonogenic potential compared with that from SCLtTA/p210 BCR-ABL1/Parp1−/−, SCLtTA/p210BCR-ABL1/Rad52−/−, and SCLtTA/p210BCR-ABL1 animals (Figure 4C).
As expected, in the absence of tetracycline, SCLtTA/p210BCR-ABL1 animals succumbed to CML-CP-like disease in 73.7 ± 5.6 days (Figures 4D and S4E–S4I). Development of lethal disease in SCLtTA/p210BCR-ABL1/Parp1−/− mice (120.0 ± 8.4 days) and SCLtTA/p210BCR-ABL1/Rad52−/− mice (99.9 ± 17.6 days) was significantly prolonged in comparison with SCLtTA/p210BCR-ABL1 animals (p < 001 and p = 0.03, respectively). Importantly, mean survival time of SCLtTA/p210BCR-ABL1/Parp1−/−Rad52−/− mice (148.4 ± 11.7 days) was significantly prolonged in comparison with SCLtTA/p210BCR-ABL1/Parp1−/− (p < 0.04) and SCLtTA/p210BCR-ABL1/Rad52−/− (p < 0.05) animals. Remarkably, 33% of SCLtTA/p210BCR-ABL1/Parp1−/−Rad52−/− mice did not develop detectable leukemia during 200 days of observation, which demonstrates the ability of dual PARP1 and RAD52 inhibition to actually prevent onset of the disease.
To test the anti-tumor effect of PARPi+RAD52i against disseminated tumor, previously selected BRCA-deficient AML patient cells were injected into immunodeficient NGS mice (Cramer-Morales et al., 2013; Nieborowska-Skorska et al., 2017). Upon engraftment, the mice were treated with PARPi talazoparib, RAD52i F79 aptamer, or talazoparib + F79 aptamer for 7 consecutive days. Talazoparib was applied in vivo because it demonstrated better pharmacokinetic properties than olaparib in rodents (Shen et al., 2013). F79 aptamer was used because Dopa may cause severe neurobehavioral and other side effects (Eskow Jaunarajs et al., 2010).
Although mice treated with talazoparib and F79 aptamer individually had diminished percentages of hCD45+ leukemia cells in peripheral blood, the combination of talazoparib and F79 aptamer caused an additional 2.6- to 3.8-fold reduction in the number of leukemia cells (Figure 5A). Untreated mice succumbed to leukemia after 37.3 ± 2.9 days, whereas those treated with F79 aptamer or talazoparib survived for 57.2 ± 3.5 days (p < 0.002) and 70.3 ± 2.5 days (p < 0.001), respectively (Figure 5B). Combinatorial treatment of talazoparib + F79 aptamer prolonged survival of leukemic mice to 116.7 ± 9.1 days (p < 0.001 compared with individual treatment).
Figure 5. The Effect of a Combination of PARP1 and RAD52 Inhibitors against BRCA1-Deficient Primary AML Xenograft in NSG Mice and against BRCA1-Deficient Solid Tumor Growth in Nude Mice.

(A and B) NSG mice were inoculated i.v. with 106 BRCA1-deficient primary AML xenograft cells. One week later, the animals were treated with vehicle (Control), F79 aptamer (F79), talazoparib (Tala), or F79 + Tala (six mice per group) for 7 consecutive days.
(A) Representative plots of peripheral blood leukocyte (PBL) from treated mice; mean percentage ± SD of human CD45+ AML cells in peripheral blood leukocytes 3 weeks after leukemia injection; *p ≤ 0.005 and **p < 0.001 in comparison with Control and single-compound treatment, respectively, using Student’s t test.
(B) Kaplan-Meier survival curves.
(C and D) Nu/nu mice were inoculated s.c. with 106 BRCA1-deficient MDA-MB-436 cells (C) and BRCA1-proficient MDA-MB-436 +BRCA1 cells (D). Tumor-bearing animals were treated with D-I03, talazoparib (Tala), or D-I03+Tala for 7 consecutive days (three to five mice per group). Results represent mean ± SD fold increase of tumor volume; *p < 0.05 compared with individual agents.
See also Figures S5–S7 and Table S1.
In addition, we tested the effect of PARPi talazoparib combined with RAD52i D-I03 against BRCA1-deficient MDA-MB-436 and BRCA1-proficient MDA-MB-436+BRCA1 carcinoma in nu/nu mice. Pharmacokinetic and toxicity studies indicated that maximal tolerated dose of D-I03 is ≥50 mg/kg, and t1/2 was 23.4 ± 17.4 hr, resulting in >1 μM maximal concentration in peripheral blood. Mice bearing subcutaneous tumors were treated with talazoparib, D-I03, or talazoparib + D-I03 for 7 consecutive days. PARPi and RAD52i when used individually reduced BRCA1-deficient MDA-MB-436 tumor growth in comparison with control vehicle-treated animals, whereas the combination of these compounds exerted stronger effect (Figure 5C). Talazoparib + D-I03 did not affect the growth of BRCA1-proficient tumors (Figure 5D) and did not exert any significant toxicity against normal tissues and organs (Figure S5; Table S1).
In conclusion, simultaneous genetic and pharmacological inhibition of PARP1 and RAD52 exerted stronger effect against BRCA-deficient tumors in vivo compared with individual inhibitors targeting PARP1 or RAD52. Single-cell RNA-sequencing (RNA-seq) analyses of untreated tumors revealed clonal heterogeneity in expression of DSB repair genes in tumor initiating cells and progenitor cells populations (Figure S6), suggesting that PARPi-resistant clones may be already present at diagnosis. However, at least some of these clones should be sensitive to the inhibitors such as RAD52i, which targets unrelated DSB repair pathway. In concordance, olaparib-resistant BRCA1-mutated/53BP1-deficient SUM149PT breast carcinoma cells, olaparib-resistant BRCA1-mutated MDA-MB-436 breast carcinoma cells, and talazoparib-resistant RAD54−/− Nalm6 leukemia cells were sensitive to RAD52i Dopa (Figure S7).
DISCUSSION
Patients with BRCA-deficient tumors who initially respond to PARPi often develop resistance, leading to cancer relapse (Lord and Ashworth, 2013). In addition, PARPi increase the probability of accumulation of additional chromosomal translocations in BRCA-deficient cells (Bunting et al., 2010), which may facilitate disease progression. Altogether, because PARPis have become widely used to treat BRCA pathway-deficient tumors, there is an urgent need to develop novel strategies to prevent development of drug resistance, for example by killing tumor cells more rapidly and robustly before resistance mechanisms are selected for.
PARPi-mediated synthetic lethality in BRCA pathway-deficient cells is associated with increased number of lethal DSBs (Bryant et al., 2005; Farmer et al., 2005). In BRCA-deficient cells, some of these DSBs can still be repaired by the alternative RAD52-HR and/or by RAD52-SSA (Liu and Heyer, 2011; Stark et al., 2004) potentially weakening the synthetic lethal effect of PARPi. In concordance, we detected RAD52-HR and SSA activities in PARPi-treated BRCA-deficient cells. The contribution of each mechanism may depend on the magnitude of DNA damage and capability of specific mutations in BRCA1/2 and/or in other genes (e.g., 53BP1) to engage RAD52 and/or RAD51. Moreover, the role of RAD52 in protecting cells from the toxicity of excessive DSBs is supported by recent reports demonstrating that RAD52 plays a vital role in break-induced replication and in transcript-RNA-templated DNA recombination and repair and that RAD52 acetylation is required for sustained RAD51 colocalization at DSBs (Keskin et al., 2014; Sotiriou et al., 2016; Yasuda et al., 2018).
We and others have shown that inhibition of RAD52 DNA binding activity exerted synthetic lethality in BRCA-deficient carcinomas and leukemia cells without affecting normal cells and tissues (Cramer-Morales et al., 2013; Feng et al., 2011). Therefore, RAD52 has proved to be an important target for therapeutic intervention in BRCA-deficient tumors.
In concordance, using three different approaches (inhibitors, dominant-negative mutants, and gene knockouts) we demonstrated that simultaneous targeting of PARP1 and RAD52 exerted synergistic effect (dual synthetic lethality) against a variety of BRCA-deficient solid tumor cells and leukemia cells. At the same time, BRCA-proficient tumor cells and normal cells and tissues were not significantly affected by the combinatorial treatment.
PARPi resistance is a common phenomenon (Sonnenblick et al., 2015), and PARPi+RAD52i-mediated dual synthetic lethality is an aggressive approach, which may lead to more effective elimination of BRCA-deficient malignant cells, thus limiting or preventing time-dependent emergence of preexisting and/or drug-induced resistant clones. In concordance, our data indicate that inhibition of RAD52 will improve therapeutic outcome of BRCA-deficient malignancies treated with PARPi while causing minimal toxicity to normal cells and tissues.
EXPERIMENTAL PROCEDURES
Further details about experimental procedures used in this work can be found in Supplemental Experimental Procedures.
Primary Cells
FLT3(ITD)-positive AML samples (bone marrow or blood) were obtained during routine investigation at the Department of Internal Medicine I, Division of Hematology and Hemostaseology, Medical University of Vienna. Isolated mononuclear cells were stored as intact cells in a biobank in liquid nitrogen until used for in vitro experiments and/or in xenotransplantation experiments. BCR-ABL1-positive CML, AML1-ETO-positive AML, IGH/MYC-positive BL primary samples, and BRCA1/2-deficient and BRCA1/2-proficient AML and t-MDS samples have been previously characterized (Maifrede et al., 2017a; Nieborowska-Skorska et al., 2017). Samples of normal hematopoietic cells were purchased from Cambrex Bio Science. Lin−CD34+ cells were obtained from mononuclear fractions by magnetic sorting using the EasySep negative selection human progenitor cell enrichment cocktail followed by human CD34-positive selection cocktail (StemCell Technologies) as described previously (Nieborowska-Skorska et al., 2017).
Transgenic and Knockout Mice
Rad52−/− mice were obtained from Dr. M. Jasin (Memorial Sloan Kettering Cancer Center), and Parp1−/− mice and SCLtTA;p210BCR-ABL1 mice were used previously (Bolton-Gillespie et al., 2013; Nieborowska-Skorska et al., 2017). Rad52−/− were cross-bred with Parp1−/− mice to generate Parp1−/−;Rad52−/−, Parp1−/−;wt, Rad52−/−;wt, and wt/wt mice. These animals were cross-bred with SCLtTA;p210BCR-ABL1 mice to generate SCLtTA;p210BCR-ABL1;Parp1−/−;Rad52−/−, SCLtTA;p210 BCR-ABL1;Parp1−/−, SCLtTA;p210BCR-ABL1;Rad52−/−, and SCLtTA; p210BCR-ABL1;wt;wt mice. Transgenic and knockout mice were identified by PCR of tail snip DNA. Mice were provided with drinking water supplemented with 0.5 g/L tetracycline hydrochloride (Sigma-Aldrich), and leukemia was induced in 10- to 12-week-old male and female mice by withdrawal of tetracycline. CML-CP-like leukemia was characterized by splenomegaly and leukocytosis associated with expansion of mature myeloid cells as described previously (Bolton-Gillespie et al., 2013; Koschmieder et al., 2005).
In Vitro Treatment
PARPi olaparib and talazoparib (Selleckchem), RAD52i F79 aptamer (Cramer-Morales et al., 2013), 6-OH-dopa (Chandramouly et al., 2015), and D-I03 (Huang et al., 2016), DNR (Selleckchem), and BCR-ABL1 tyrosine kinase inhibitor imatinib (Selleckchem) were added to indicated cells for 3–5 days. Cell count and viability were determined by trypan blue exclusion. Clonogenic activity was assessed 7 days after plating of treated cells. Cell death and γ-H2AX staining were examined by flow cytometry after staining with Fixable Viability Dye eFluor 780 (eBioscience) and Alexa Fluor 647 anti-γ-H2AX (BD Biosciences) as described previously (Maifrede et al., 2017b). For long-term experiments, fresh inhibitors were added every 3–4 days, and cells were expanded in fresh medium every 7 days.
Clonogenic Assay
Freshly harvested Lin− murine bone marrow cells were plated in serum-free MethoCult-SF H4236 (StemCell Technologies) supplemented with TET System Approved Fetal Bovine Serum (Takara Bio USA) with and without 10 μg/mL tetracycline hydrochloride in the presence of a threshold concentrations (0.1 U/mL) of recombinant murine IL-3, IL-6, and SCF. Colonies were scored after 5–7 days. BRCA1-deficient and BRCA1-proficient counterpart MDA-MB-436 cells, cultured in RPMI + 10% fetal bovine serum (FBS), were seeded on day 0 in triplicate at 5,000 cells/well. On day 1, the cells were treated with 0, 10, 20, and 50 nM olaparib in the absence or presence of 1 μM D-I03. On day 3, the treatment was repeated with media refreshment. Cells were counted on day 4 using trypan blue exclusion and were immediately plated at a density of 500 cells/well in a six-well plate, in RPMI + 10% FBS. After 2 weeks, cell survival was determined using the clonogenic assay. For this purpose, the colonies were fixed and stained with 0.05% of 10 mg/mL ethidium bromide in 50% ethanol and visualized using an Alphaimager 3400 gel documentation system.
In Vivo Treatment
NSG mice (10- to 12-week-old males and females) were total-body irradiated (250 cGy) and inoculated intravenously (i.v.) with 1 × 106 BRCA-deficient AML primary leukemia xenograft cells. Two weeks later, mice were treated with vehicle (control), talazoparib (0.33 mg/kg/day by oral gavage for 7 days; Nieborowska-Skorska et al., 2017), F79 aptamer (2.5 mg/kg i.v.; Cramer-Morales et al., 2013), and a combination of talazoparib + F79 aptamer. Leukemia burden was analyzed by flow cytometry 7 days after the end of treatment. Human leukemia cells were detected by anti-human CD45 antibody as described previously (Nieborowska-Skorska et al., 2017). Median survival time was determined. Nude mice (10- to 12-week-old females) were injected subcutaneously (s.c.) with 1 × 106 BRCA1-deficient MDA-MB-436 cells or BRCA1-reconstituted counterparts. Once tumors reached a volume of 100 mm3, mice were treated with vehicle (control), talazoparib (0.33 mg/kg/day by oral gavage; Nieborowska-Skorska et al., 2017), D-I03 (50 mg/kg/day intraperitoneally [i.p.]), and a combination of talazoparib and D-I03 for 7 days. Tumors were measured weekly, and tumor volumes were calculated using the ellipsoid volume formula ([π/6] × length × width × height).
Statistical Analysis
Data are expressed as mean ± SD and were compared using the unpaired Student’s t test; p values less than 0.05 were considered to indicate statistical significance. Mean survival time of the mice ± SE was calculated using Kaplan-Meier log-rank survival analysis. The response additivity approach was used to study the synergistic effects (Slinker, 1998). This approach shows a positive drug combination effect when the observed combination effect (EAB) is greater than the expected additive effect by the sum of the individual effects (EA + EB). The combination index (CI) was calculated as CI = (EA + EB)/EAB. The p value for the possible synergistic effect is given by the significance of the interaction effect in a factorial ANOVA of the individual and combination effects.
Study Approval
Studies involving primary leukemias were approved by the ethics committee of the Medical University of Vienna and Temple University Lewis Katz School of Medicine and met all requirements of the Declaration of Helsinki. Animal studies were approved by the Temple University Institutional Animal Care and Use Committee.
Supplementary Material
Highlights.
RAD52 inhibitors attenuate SSA and residual HR in BRCA-deficient cells
PARP + RAD52 inhibitors exert dual synthetic lethality in BRCA-deficient cells
RAD52 inhibitor improves the effect of PARP inhibitor in BRCA-deficient tumors
Acknowledgments
This work was funded by grants R01 CA186238 (T.S.), R01 CA188347, P30 CA056036 and the Drexel Coulter Program Award (A.V.M.), and R01 CA214799 and U.S. Department of Defense (DoD) grant OC130212 (N.J.). K.S.-R. was supported by grant F31 CA203161. M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar (HHMI-55108547).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.034.
AUTHORS CONTRIBUTIONS
Conceptualization, T.S.; Methodology, T.S., E.B.-G., and A.J.B.; Software, J.L. and K.M.-W.; Validation, E.B.-G. and K.S.-R., Formal Analysis, H.Z.; Investigation, K.S.-R., E.B.-G., Y.D., S.L., M.S., M.N.-S., K.H., E.A.B., and M. Moore; Resources, A.J.B., P.V., S.B., and R.B., Writing – Original Draft, T.S.; Writing –Review & Editing, T.S., A.V.M., and N.J.; Visualization, K.S.-R., E.B.-G., Y.D., K.H., E.A.B., J.L., and K.M.-W.; Supervision, M. Müschen, N.J., M.A.W., A.V.M., and T.S.; Project Administration, T.S.; Funding Acquisition, K.S., N.J., A.V.M., and T.S.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bolton-Gillespie E, Schemionek M, Klein HU, Flis S, Hoser G, Lange T, Nieborowska-Skorska M, Maier J, Kerstiens L, Koptyra M, et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013;121:4175–4183. doi: 10.1182/blood-2012-11-466938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, McDevitt S, Sullivan K, Kent T, Luz A, Glickman JF, Andrake M, Skorski T, Pomerantz RT. Small-molecule disruption of RAD52 rings as a mechanism for precision medicine in BRCA-deficient cancers. Chem Biol. 2015;22:1491–1504. doi: 10.1016/j.chembiol.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng H, Roy D, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122:1293–1304. doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dkhissi F, Aggoune D, Pontis J, Sorel N, Piccirilli N, LeCorf A, Guilhot F, Chomel JC, Ait-Si-Ali S, Turhan AG. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp Hematol. 2015;43:775–780. doi: 10.1016/j.exphem.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Eskow Jaunarajs KL, Dupre KB, Ostock CY, Button T, Deak T, Bishop C. Behavioral and neurochemical effects of chronic L-DOPA treatment on nonmotor sequelae in the hemiparkinsonian rat. Behav Pharmacol. 2010;21:627–637. doi: 10.1097/FBP.0b013e32833e7e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116:5298–5305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A. 2011;108:686–691. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, Wang CX, An WF, Spoonamore J, Metkar S, et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016;44:4189–4199. doi: 10.1093/nar/gkw087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. Transcript-RNA-templated DNA recombination and repair. Nature. 2014;515:436–439. doi: 10.1038/nature13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmieder S, Gottgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, Dayaram T, Geary K, Green AR, Tenen DG, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood. 2005;105:324–334. doi: 10.1182/blood-2003-12-4369. [DOI] [PubMed] [Google Scholar]
- Liu J, Heyer WD. Who’s who in human recombination: BRCA2 and RAD52. Proc Natl Acad Sci U S A. 2011;108:441–442. doi: 10.1073/pnas.1016614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- Maifrede S, Martin K, Podszywalow-Bartnicka P, Sullivan-Reed K, Langer SK, Nejati R, Dasgupta Y, Hulse M, Gritsyuk D, Nieborowska-Skorska M, et al. IGH/MYC translocation associates with BRCA2 deficiency and synthetic lethality to PARP1 inhibitors. Mol Cancer Res. 2017a;15:967–972. doi: 10.1158/1541-7786.MCR-16-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maifrede S, Martinez E, Nieborowska-Skorska M, Di Marcantonio D, Hulse M, Le BV, Zhao H, Piwocka K, Tempera I, Sykes SM, Skorski T. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017b;1:1467–1472. doi: 10.1182/bloodadvances.2017006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin AV, Mazina OM, Bugreev DV, Rossi MJ. Rad54, the motor of homologous recombination. DNA Repair (Amst) 2010;9:286–302. doi: 10.1016/j.dnarep.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MJ, Stoddard BL, Monnat RJ., Jr PARP-mediated repair, homologous recombination, and back-up non-homologous end joining-like repair of single-strand nicks. DNA Repair (Amst) 2013;12:529–534. doi: 10.1016/j.dnarep.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff JA, Jones D, Lee SH, Williamson EA, Hromas R. Drugging the cancers addicted to DNA repair. J Natl Cancer Inst. 2017;109:109. doi: 10.1093/jnci/djx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, Martinez E, Di Marcantonio D, Bolton-Gillespie E, Cramer-Morales K, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest. 2017;127:2392–2406. doi: 10.1172/JCI90825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, Lenobel R, Bartkova J, Lau A, O’Connor MJ, et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11:3837–3850. doi: 10.4161/cc.22026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, Wolanin K, Skowronek K, Nieborowska-Skorska M, Dasgupta Y, Skorski T, Piwocka K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13:3727–3741. doi: 10.4161/15384101.2014.965013. [DOI] [PubMed] [Google Scholar]
- Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19:5003–5015. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- Sonnenblick A, de Azambuja E, Azim HA, Jr, Piccart M. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Ré C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S, et al. Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol Cell. 2016;64:1127–1134. doi: 10.1016/j.molcel.2016.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J, Liu J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008;68:2699–2707. doi: 10.1158/0008-5472.CAN-07-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Kagawa W, Ogi T, Kato TA, Suzuki T, Dohmae N, Takizawa K, Nakazawa Y, Genet MD, Saotome M, et al. Novel function of HATs and HDACs in homologous recombination through acetylation of human RAD52 at double-strand break sites. PLoS Genet. 2018;14:e1007277. doi: 10.1371/journal.pgen.1007277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.