

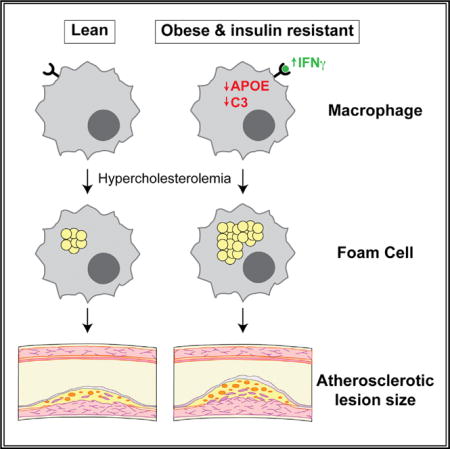
SUMMARY
Type 2 diabetes (T2D) is associated with increased risk for atherosclerosis; however, the mechanisms underlying this relationship are poorly understood. Macrophages, which are activated in T2D and causatively linked to atherogenesis, are an attractive mechanistic link. Here, we use proteomics to show that diet-induced obesity and insulin resistance (obesity/IR) modulate a pro-atherogenic “macrophage-sterol-responsive-network” (MSRN), which, in turn, predisposes macrophages to cholesterol accumulation. We identify IFNγ as the mediator of obesity/IR-induced MSRN dysregulation and increased macrophage cholesterol accumulation and show that obesity/IR primes T cells to increase IFNγ production. Accordingly, myeloid cell-specific deletion of the IFNγ receptor (Ifngr1−/−) restores MSRN proteins, attenuates macrophage cholesterol accumulation and atherogenesis, and uncouples the strong relationship between hyperinsulinemia and aortic root lesion size in hypercholesterolemic Ldlr−/− mice with obesity/IR, but does not affect these parameters in Ldlr−/− mice without obesity/IR. Collectively, our findings identify an IFNγ-macrophage pathway as a mechanistic link between obesity/IR and accelerated atherogenesis.
Graphical Abstract
In Brief: Obesity and insulin resistance are major risk factors for cardiovascular disease, but the underlying mechanisms are poorly understood. Reardon et al. show that obesity and insulin resistance induce an IFNγ-macrophage pathway that exacerbates foam cell formation and aortic lesion size in atherosclerotic mice.
INTRODUCTION
Compared to non-diabetics, patients with type 2 diabetes (T2D) have a 4-fold increased risk for cardiovascular disease (CVD) during their lifetime, and have a greater overall plaque burden and higher rate of multi-vessel disease (Beckman et al., 2002; Gore et al., 2015; Haffner et al., 1998; Hayward et al., 2015). Indeed, the 7-year incidence of first myocardial infarction (MI) or death in type 2 diabetics is ~6-fold higher than in type 2 diabetics, and the 5-year mortality rate following an MI is nearly double for patients with T2D compared to non-diabetics.
Although patients with T2D face a significant risk for developing CVD, the mechanisms underlying this risk are poorly understood. In epidemiological studies, traditional risk factors, such as smoking, hypertension, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, total cholesterol, and triglyceride levels, do not explain the risk associated with CVD in type 2 diabetic patients (Beckman et al., 2002; Gore et al., 2015; Haffner et al., 1998; Hayward et al., 2015). Moreover, intervention studies showed that increased mortality is observed even when plasma cholesterol levels are aggressively lowered with statin treatment, hypertension is controlled, or with aggressive glycemic control (Banach et al., 2016; Gore et al., 2015).
Macrophages may represent an important cellular link between T2D and atherosclerosis. Macrophages are inappropriately activated during diet-induced obesity and insulin resistance (obesity/IR) (Chawla et al., 2011; Kratz et al., 2014; McNelis and Olefsky, 2014), and macrophages that accumulate excess cholesterol (foam cells) are causatively linked to initiation, progression, and rupture of atherosclerotic plaques (Li and Glass, 2002; Moore et al., 2013; Tabas and Bornfeldt, 2016). For example, their inability to clear cholesterol leads to the formation of foam cells, their defective clearance of apoptotic cells in the artery wall promotes necrotic core formation and increases plaque complexity, and their increased secretion of proteases destabilizes atherosclerotic plaques and promotes plaque vulnerability. Thus, the idea that obesity/IR alters macrophages in a way that promotes atherogenesis is an attractive hypothesis.
Although attractive, this hypothesis has been difficult to test experimentally because diets that promote obesity/IR in atherosclerotic mice also elevate plasma cholesterol levels. Thus, it has been challenging to distinguish macrophage pathways driven by hypercholesterolemia, from those controlled by the concomitant obesity/IR that develops when Ldlr−/− or Apoe−/− mice are fed the Western-type diet.
To overcome this problem, we combined genetic and dietary interventions to study macrophages in the presence of hypercholesterolemia and obesity/IR, alone or in combination. Using an unbiased proteomics approach, we identified an obesity/IR-driven interferon-gamma (IFNγ) pathway that targets the macrophage-sterol-responsive-network (MSRN) (Becker et al., 2010) to promote foam cell formation. We show that blocking this pathway in macrophages in vivo (by deleting Ifngr1 in myeloid cells) normalized MSRN proteins and attenuated foam cell formation and atherosclerosis in Ldlr−/− mice with obesity/IR, but had no effect on these parameters in the absence of obesity/IR. Collectively, our studies identify an IFNγ -macrophage pathway as a mechanistic link between obesity/IR and accelerated atherogenesis.
RESULTS
Obesity/IR Targets the MSRN and Promotes Foam Cell Formation
To determine if obesity/insulin resistance (IR) alters macrophages to promote atherosclerosis, we used genetic models and dietary interventions to study macrophages in mice with (1) “obesity/IR only,” (2) “hypercholesterolemia (HC) and obesity/IR,” and (3) “HC only” (Hartvigsen et al., 2007) (Figure 1A). This approach allowed us to test whether obesity/IR was sufficient to alter macrophages in normocholesterolemic mice and necessary to produce similar changes in hypercholesterolemic mice.
Figure 1. Obesity/IR Targets the MSRN, Promotes Macrophage Cholesterol Loading, and Exacerbates Atherosclerosis.
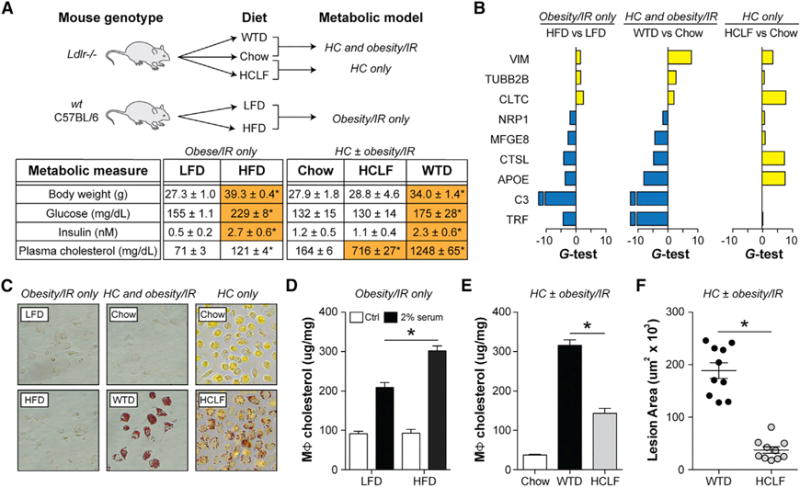
(A–F) “Obesity/IR only” model: WT C57BL/6 mice were fed a low-fat diet (LFD) or high-fat diet (HFD) for 9 weeks. “HC ± obesity/IR” models: Ldlr−/− mice were fed a chow diet, a Western-type diet (WTD), or a high-cholesterol-low-fat (HCLF) diet for 12 weeks.
(A) Metabolic parameters.
(B) Proteomics analysis of peritoneal macrophages. Differentially abundant proteins (yellow, up; blue, down) were identified based on the G-test (G > 1.5) and t test (p < 0.05).
(C) Oil-red-O staining of peritoneal macrophages.
(D) Cholesterol levels in peritoneal macrophages treated with/without 2% serum from WTD-fed Ldlr−/− mice.
(E) Cholesterol levels in peritoneal macrophages.
(F) Aortic root lesion area.
Results are mean ± SEM. *p < 0.05 (t test). n = 5–10 mice/group. See also Figures S1 and S2 and Table S1.
We used an unbiased proteomics approach to study how the various conditions affected protein abundance patterns in elicited peritoneal macrophages, whose responses to atherosclerosis modifying perturbations in vivo are reasonable surrogates for artery wall macrophages (Becker et al., 2010; Li et al., 2004).
We started with the “obesity/IR only” model, where C57BL/6 mice were fed a low-fat diet (LFD) or high-fat diet (HFD) for 9 weeks. Proteomic analyses of purified peritoneal macrophages (Figure S1) identified 27 secreted proteins that were altered by obesity/IR (false discovery rate [FDR] < 5%) (Figure S2). Nine of those proteins resided in a proatherogenic, MSRN (Figure 1B; Table S1) we previously identified in foam cells from hypercholesterolemic Ldlr−/− mice (Becker et al., 2010).
Although we previously identified the MSRN as a “sterol-responsive” protein network, MSRN protein changes in the “obesity/IR model only” occurred independent of changes in macrophage cholesterol or triglyceride levels (Figures 1C and S2), raising the possibility that a significant portion of this proatherogenic network was actually regulated by obesity/IR.
To test this, we asked whether eliminating obesity/IR could correct the 9 MSRN proteins in hypercholesterolemic Ldlr−/− mice. All 9 MSRN proteins were similarly dysregulated in the “HC and obesity/IR” model, but most were normalized or counter-regulated in the “HC only” model (Figure 1B; Table S1), even though macrophages were loaded with cholesterol (Figure 1C). For example, APOE levels were lowered in Western-type diet (WTD) fed Ldlr−/− cholesterol-low-fat mice but raised in high-(HCLF) diet-fed mice, findings that agree with the induction of APOE in macrophages loaded with cholesterol in vitro (Figure S2) (Basu et al., 1981).
APOE promotes reverse cholesterol efflux in macrophages, and its expression by macrophages restrains atherosclerosis (Fazio et al., 2002). The suppression of APOE by obesity/IR prompted us to determine whether obesity/IR promotes foam cell formation.
We first tested this in the “obesity/IR” only model. Since wild-type (WT) C57BL/6 mice fed the HFD have low plasma cholesterol levels we could not test this in vivo. Instead, we isolated peritoneal macrophages from these mice, exposed them to high levels of atherogenic lipoproteins (2% serum from WTD-fed Ldlr−/−), and found that obesity/IR induced by the HFD enhanced cholesterol accumulation (Figure 1D).
Next, we investigated whether eliminating obesity/IR from hypercholesterolemic mice (“HC ± obesity/IR” models) attenuated foam cell formation in vivo. Peritoneal macrophages from HCLF diet-fed Ldlr−/− mice had lower cholesterol levels than WTD-fed mice (Figure 1E). As previously described (Hartvigsen et al., 2007), aortic root lesion area was also lessened in HCLF diet-fed Ldlr−/− mice (Figure 1F). However, plasma cholesterol levels were lowered in HCLF diet-fed mice (see Figure 1A), high lighting the problem of dissociating effects of obesity/IR and hypercholesterolemia. We overcame this by identifying the regulator of the MSRN, deleting it, and showing attenuation of macrophage cholesterol loading and atherosclerosis only in the presence of obesity/IR (see below).
MSRN Protein Dysregulation Promotes Macrophage Foam Cell Formation
Why did macrophages from obese/IR mice accumulate more cholesterol? One possibility is that this was due to defects in non-MSRN proteins involved in cholesterol metabolism. Arguing against this, mRNA levels of Abca1, Abcg1, Lxra, Cd36, Sra1, or Srebp2 were unaffected or regulated to oppose cholesterol accumulation in all of the models tested (Figures 2A and 2B).
Figure 2. MSRN Protein Dysregulation Promotes Macrophage Foam Cell Formation.
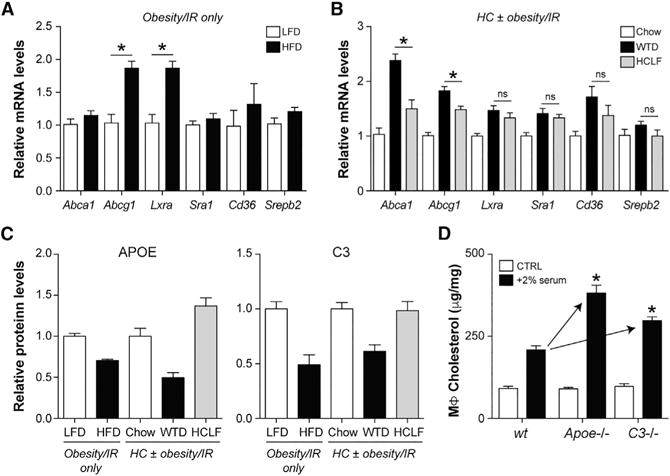
(A and B) Peritoneal macrophage mRNA levels for several genes involved in cholesterol metabolism in WT C57BL/6 mice fed a LFD or a HFD (“obesity/IR only” model, A) or in Ldlr−/− mice fed chow, a WTD, or a HCLF diet (“HC ± obesity/IR” models, B).
(C) Peritoneal macrophage media protein levels for APOE and C3.
(D) Peritoneal macrophages were isolated from 8-week-old WT, Apoe−/−, or C3−/− C57BL/6 mice fed a LFD. Macrophages were treated were with and without 2% serum from WTD-fed Ldlr−/− mice, and cholesterol levels were quantified.
Results are mean ± SEM.
*p < 0.05 (t test). n = 5 mice/group.
Increased cholesterol accumulation could also be due to altered MSRN proteins such as APOE and C3, which were lowered by obesity/IR in WT and Ldlr−/− mice (Figure 2C), and previously implicated in lipid metabolism (Barbu et al., 2015; Fazio et al., 2002). Peritoneal macrophages from lean 8-week-old Apoe−/− or C3−/− C57BL/6 mice accumulated more cholesterol (relative to WT) following exposure to 2% serum from WTD-fed Ldlr−/− mice (Figure 2D). Thus, obesity/IR may promote macrophage cholesterol accumulation by suppressing MSRN proteins such as APOE and C3.
IFNγ Targets MSRN Proteins In Vitro and In Vivo
Next, we sought to determine how obesity/IR alters MSRN proteins. We considered the possibility that hyperglycemia and/or hyperinsulinemia was responsible, but found that exposing peritoneal macrophages to high levels of glucose and insulin, individually or in combination, had no effect on APOE or MFGE8 (Figure 3A).
Figure 3. IFNγ Targets MSRN Proteins and Promotes Macrophage Cholesterol Accumulation.
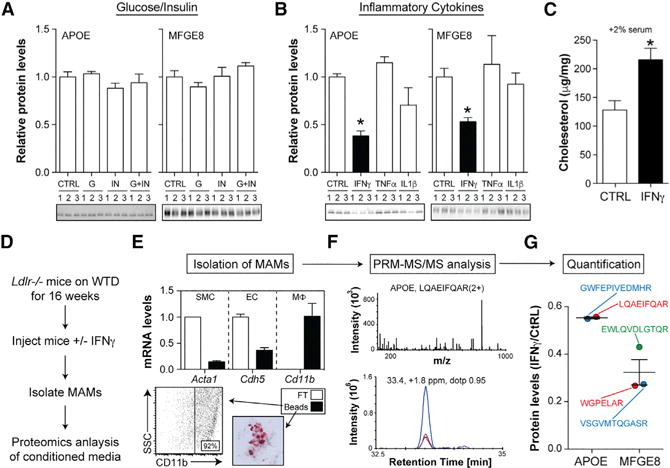
(A and B) APOE and MFGE8 levels in peritoneal macrophages treated with (A) vehicle (ctrl), 300 mg/dL glucose (G) or 10 nM insulin (I), alone or in combination (G+I), or (B) 12 ng/mL IFNγ, tumor necrosis factor alpha (TNF-α), or interleukin-1β (IL-1β) for 24 hr.
(C) Cholesterol levels in control and IFNγ -treated macrophages exposed to 2% serum from WTD-fed Ldlr−/− mice.
(D) Ldlr−/− mice were fed a WTD for 16 weeks and injected with vehicle or IFNγ, and murine aortic macrophages (MAMs) were isolated.
(E) MAM purity was assessed by qRT-PCR for smooth muscle cells (SMC), endothelial cells (EC), and macrophage (Mϕ) markers in anti-CD11b bound (beads) and unbound (FT) cells, flow cytometry, and Oil-red-O staining.
(F) Representative LC-MS/MS spectrum and ion chromatograms from parallel reaction monitoring.
(G) Relative quantification of APOE and MFGE8 for all peptides in IFNγ -injected versus control mice.
Results are mean ± SEM.
*p < 0.05 (t test); n = 3–5 mice/group. See also Figures S3 and S4.
In addition to elevated glucose/insulin, patients with T2D also exhibit low-grade inflammation (Donath and Shoelson, 2011). We tested various cytokines for their ability to target MSRN proteins in peritoneal macrophages, and found that only IFNγ lowered APOE and MFGE8 (Figure 3B). IFNγ also lowered both MSRN proteins in bone-marrow-derived macrophages and human monocyte-derived macrophages in vitro (Figure S3). Moreover, treatment with IFNγ, increased macrophage cholesterol levels following exposure to 2% serum from WTD-fed Ldlr−/− mice (Figure 3C).
To determine if IFNγ could alter MSRN proteins in vivo, we developed a mass spectrometry approach to interrogate murine aortic macrophages (MAMs) (Figure 3D). Ldlr−/− mice were fed a WTD for 16 weeks to allow MAMs to accumulate in the artery wall. At this time, mice were injected with IFNγ (IP, 25 mg/kg) or vehicle control; MAMs were purified (Figure 3E); and MSRN protein levels in the conditioned media were quantified by proteomics.
Because of the limited number of MAMs in mice, we first developed a selected reaction monitoring method (Hoofnagle et al., 2012) to quantify several MSRN proteins in small numbers of macrophages. After validating that this approach could reliably quantify APOE in 50,000 macrophages in vitro (Figure S4), we analyzed the abundance of APOE and MFGE8 in MAM-conditioned media in vivo. Results showed that IFNγ lowered both APOE and MFGE8 levels in MAMs in vivo (Figures 3F and 3G). Thus, IFNγ is capable of targeting MSRN proteins in murine artery wall macrophages.
IFNγ Induction during Obesity/IR Targets the MSRN without Inducing Host Defense Genes
Since IFNγ targeted MSRN proteins and enhanced cholesterol loading in vitro, we reasoned that it was required for obesity/IR to elicit similar effects in vivo. We first determined if IFNγ levels were raised in the “obesity/IR only” model. Because plasma IFNγ levels were undetectable, we quantified IFNγ production by splenic T cells and found that obesity/IR induced IFNγ (Figure 4A).
Figure 4. Deleting Ifngr1 Corrects MSRN Proteins and Macrophage Cholesterol Levels in the “Obesity/IR Only” Model.
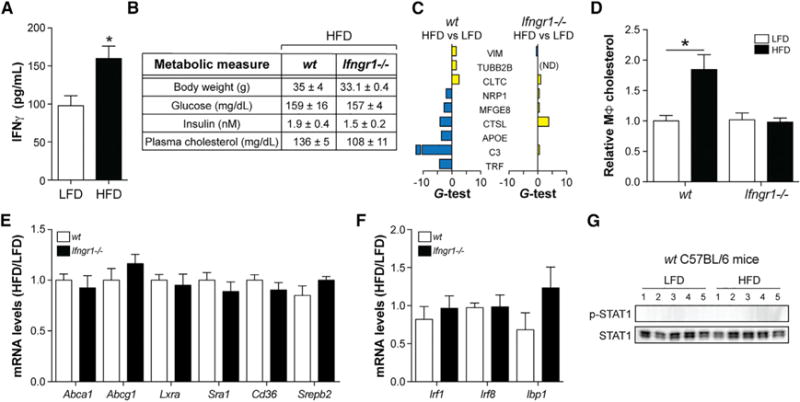
(A–G) WT and Ifngr1−/− mice were fed a LFD or HFD for 9 weeks.
(A) IFNγ production by splenic T cells in WT mice.
(B) Metabolic parameters. (C–F) Peritoneal macrophage MSRN protein levels (C), cholesterol levels following treatment with 2% serum from WTD-fed Ldlr−/− mice (D), cholesterol metabolism gene levels (E), and IFNγ -target gene levels (F).
(G) p-STAT1/STAT1 levels.
Results are mean ± SEM. *p < 0.05 (t test); n = 4–10 mice/group. See also Table S1.
Next, we investigated whether blocking IFNγ signaling (by knocking out its receptor, Ifngr1−/−) normalized MSRN proteins and macrophage cholesterol accumulation in the “obesity/IR only” model. Deleting Ifngr1 did not correct metabolic parameters in obese/IR mice (Figure 4B), a finding that, when combined with previous work (Rocha et al., 2008), suggests that IFNγ does not appreciably contribute to insulin resistance.
On the other hand, deleting Ifngr1 blocked the ability of obesity/IR to alter MSRN proteins (Figure 4C; Table S1) and increased macrophage cholesterol accumulation following treatment with 2% serum from WTD-fed Ldlr−/− mice (Figure 4D). This lower cholesterol accumulation could not be explained by changes in Abca1, Abcg1, Lxra, Cd36, Sra1, or Srebp2, all of which were unaffected by Ifngr1 deletion (Figure 4E).
Although these data support a model wherein IFNγ alters MSRN proteins which, in turn, predisposes macrophages to excessive cholesterol accumulation, IFNγ also alters hundreds of host defense genes (Shtrichman and Samuel, 2001) that could potentially explain this effect. Surprisingly, obesity/IR did not induce IFNγ -target genes (Irf1, Irf8, and Ibp1) or phosphorylation of STAT1 (required for target gene induction) in macrophages from WT C57BL/6 mice, nor were IFNγ -target genes lowered when Ifngr1 was deleted (Figures 4F and 4G). Thus, the effects of IFNγ on the MSRN during obesity/IR are specific.
IFNGR1 is essential for IFNγ signaling, and we confirmed that IFNγ could not target MSRN proteins or host defense genes in Ifngr1−/− macrophages in vitro (Figure S5). How could blocking IFNγ signaling in vivo normalize MSRN proteins without altering known IFNγ -target genes or signaling? Much of our understanding of how IFNγ affects macrophages is based on doses (~10 ng/mL) that are much higher than those in patients with T2D (~50 pg/mL) (Mirhafez et al., 2015; Nosratabadi et al., 2009) or those produced by T cells in obese/IR mice (see Figure 4A).
We reasoned that “metabolic disease appropriate” doses of IFNγ specifically target the MSRN. Consistent with this interpretation, lower IFNγ doses (30–120 pg/mL) suppressed APOE (Figure 5A), but did not induce STAT1 phosphorylation, Irf8 expression, or macrophage bacterial killing, all of which required higher doses (1.2–12 ng/mL) (Figures 5B–5D).
Figure 5. “Metabolic Disease Appropriate” Doses of IFN γ Specifically Target MSRN Proteins.
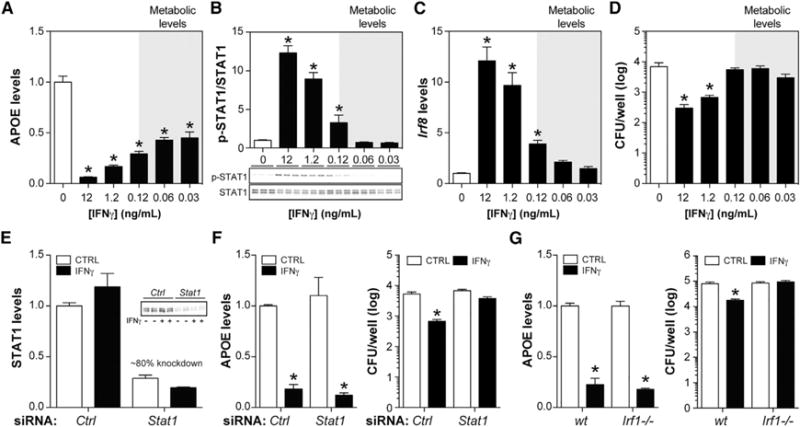
(A–D) Macrophages were treated with varying levels of IFNγ.
(A) APOE levels.
(B) p-STAT1/STAT1 levels.
(C) Irf8 levels.
(D) Number of P. aeruginosa remaining after incubation with macrophages.
(E) Efficiency of STAT1 knockdown in macrophages treated with control or Stat1 siRNA. STAT1 levels were quantified 30 min after IFNγ exposure.
(F and G) Effects of IFNγ on APOE levels and number of P. aeruginosa remaining after incubation with siRNA control or siRNA Stat1 macrophages (F) and WT or Irf1−/− macrophages (G).
Results are mean ± SEM. *p < 0.05 (t test); n = 3–10 biological replicates/group. See also Figure S5.
Following ligand binding, IFNGR1 heterodimerizes with IFNGR2, resulting in phosphorylation and nuclear translocation of STAT1 to induce interferon regulatory factors (IRFs) that upregulate additional genes essential to host defense (Ikushima et al., 2013). Our demonstration that MSRN dysregulation and host defense pathways can be dissociated by altering IFNγ dose implies that distinct pathways (downstream of IFNGR1) control these diverse functional properties.
To begin to test this hypothesis, we investigated whether knocking down Stat1 with small interfering RNA (siRNA), or knocking out Irf1 (Irf1−/−), two key components of the host defense pathway, could block IFNγ’s effect on the MSRN. We treated siRNA-control and siRNA-Stat1 (~80% knockdown) (Figure 5E) or WT and Irf1−/− macrophages with 12 ng/mL IFNγ. Lowering STAT1 levels or knocking out IRF1 blocked the ability of IFNγ to induce bacterial killing by macrophages, but had no effect on its ability to suppress APOE (Figures 5F and 5G). These findings suggest that IFNγ might alter MSRN proteins through a mechanism that is independent of the canonical pathway required for host defense. Although this interpretation is consistent with our in vivo findings (see Figure 4), further experimentation will be required to delineate this putative mechanism.
Myeloid Cell Deletion of Ifngr1 Attenuates Atherosclerosis in Ldlr−/− Mice Only when Obesity/IR Is Present
Our findings suggested that obesity/IR was required to induce IFNγ and its proatherogenic actions on macrophages. Furthermore, atherogenic effects of IFNγ are well supported in mouse studies (Ramji and Davies, 2015). Based on these findings, we hypothesized that obesity/IR induces IFNγ production, which, in turn, alters MSRN proteins to promote macrophage cholesterol accumulation and atherogenesis in the presence of hypercholesterolemia. This hypothesis makes several predictions.
First, IFNγ should be elevated in the “HC and obesity/IR” model, but not the “HC only” model. Indeed, we found that IFNγ production by splenic T cells was increased in Ldlr−/− mice fed the WTD, but not the HCLF diet (Figure 6A), and these IFNγ levels were similar to those observed in the “obesity/IR only” model (see Figure 4A).
Figure 6. Macrophage IFNGR1 Is Required for Obesity/IR to Target MSRN Proteins, Increase Macrophage Cholesterol Accumulation, and Promote Atherosclerosis.
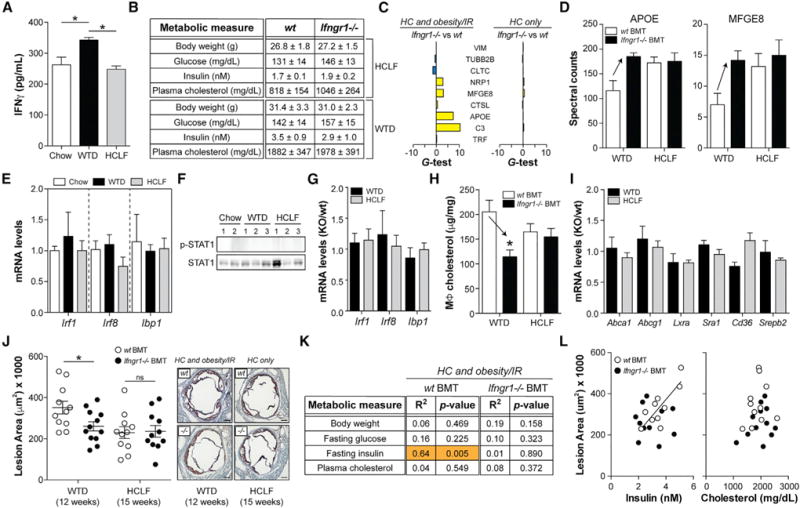
(A–L) Ldlr−/− mice transplanted with WT or Ifngr1−/− bone marrow cells were fed a chow diet, a WTD, or a HCLF diet for up to 15 weeks.
(A) IFNγ production by splenic T cells in WT mice.
(B) Metabolic parameters.
(C–I) Peritoneal macrophage MSRN protein levels (C and D), IFNγ -target gene levels in WT mice (E), p-STAT1/STAT1 levels in WT mice (F), IFNγ -target gene levels in WT and Ifngr1−/− mice (G), cholesterol levels (H), and cholesterol metabolism gene levels (I).
(J) Aortic root lesion area and representative images. Scale bar, 200 mm.
(K) Relationships between metabolic parameters and aortic root lesion area.
(L) Relationships between fasting insulin or plasma cholesterol levels and aortic root lesion area.
Results are mean ± SEM. *p < 0.05 (t test); n = 3–12 mice/group. See also Figure S6 and Table S1.
Second, myeloid cell deletion of Ifngr1 should normalize MSRN proteins in the “HC and obesity/IR” model, but not the “HC only” model. To test this, we created mice lacking Ifngr1 in myeloid cells by transplanting Ifngr1−/− (or WT) bone marrow cells into Ldlr−/− mice (Figure S6), and fed them a chow diet, a WTD, or a HCLF diet. Deleting Ifngr1 in myeloid cells did not correct metabolic parameters (Figure 6B), which mimicked our findings in the “obesity/IR only” model. In contrast, deleting Ifngr1 in myeloid cells restored 5/9 MSRN proteins in WTD-fed Ldlr−/− mice, but had no effect on these proteins in HCLF diet-fed mice (Figures 6C and 6D; Table S1).
Third, correction of MSRN proteins in myeloid cell Ifngr1−/− mice should be specific. Similar to the “obesity/IR only model,” IFNγ -target genes and STAT1 phosphorylation were not elevated in Ldlr−/− mice fed the WTD or HCLF diet (Figures 6E and 6F), and IFNγ -target genes were not lowered when Ifngr1 was deleted from myeloid cells (Figure 6G).
Fourth, myeloid cell deletion of Ifngr1 should attenuate macrophage cholesterol accumulation in the “HC and obesity/IR” model, but not the “HC only” model. Deleting Ifngr1 in myeloid cells lowered macrophage cholesterol levels in WTD-fed Ldlr−/− mice, but not in HCLF-diet-fed mice (Figure 6H). The reduced foam cell formation in WTD-fed mice could not be explained by changes in Abca1, Abcg1, Lxra, Cd36, Sra1, or Srebp2, all of which were unaffected in Ifngr1-deficient macrophages (Figure 6I).
Fifth, this hypothesis predicts that myeloid cell deletion of Ifngr1 should lessen atherosclerotic lesion size in the “HC and obesity/IR” model, but not the “HC only” model. Deleting Ifngr1 in myeloid cells reduced aortic root lesion area in Ldlr−/− mice fed the WTD for 12 weeks (Figures 6J and S6), and this reduction could not be explained by changes in metabolic parameters (see Figure 6B) or plasma lipoprotein distribution (Figure S6). In contrast, lesion area was not lowered in mice fed the HCLF diet for 15 weeks (to compensate for delayed lesion formation) (Figures 6J and S6).
Finally, this hypothesis predicts that blocking IFNγ action on macrophages should abolish the relationship between obesity/IR and atherosclerosis (Gruen et al., 2006). As shown in Figures 6K and 6L, plasma insulin levels, but not plasma cholesterol levels, were positively correlated to aortic root lesion size in WTD-fed Ldlr−/− mice. This relationship was abolished in mice transplanted with Ifngr1−/− cells (Figures 6K and 6L), even though metabolic parameters were not corrected (see Figure 6B).
DISCUSSION
Compared to non-diabetics, patients with T2D have a 4-fold increased risk for cardiovascular disease, which accounts for ~70% of the morbidity in these patients (Gore et al., 2015; Haffner et al., 1998). Despite these staggering statistics, mechanisms explaining this risk are poorly understood, partly because diets that induce obesity/IR and T2D in Ldlr−/− and Apoe−/− mice fed a WTD, the most common murine models of atherosclerosis, also elevate plasma cholesterol levels. Thus, it has been challenging to ascertain direct effects of T2D, which is important because the increased cardiovascular events in these patients cannot be fully explained by hypercholesterolemia (Costa et al., 2006).
To overcome this problem, we used genetic (WT, Ldlr−/− mice) and dietary (HFD, WTD, HCLF diets) interventions to study macrophages from mice with obesity/IR only, HC and obesity/IR, and HC only. Using this approach, we identified an IFNγ -macrophage-MSRN pathway that promotes foam cell formation and atherogenesis, only in the context of obesity/IR.
Three lines of evidence support the specificity of this pathway for obesity/IR. First, we demonstrated that obesity/IR is both necessary and sufficient to induce IFNγ production by T cells. Second, blocking IFNγ signaling in macrophages (by Ifngr1−/− bone marrow transplantation) attenuated atherosclerosis in Ldlr−/− mice with obesity/IR, but did not affect lesion size in Ldlr−/− mice without obesity/IR. Third, blocking IFNγ signaling in macrophages abolished the strong relationship between hyperinsulinemia and aortic root lesion size, even though the extent of obesity/IR was unaffected in the mice.
These findings suggest that some of the mechanisms regulating atherosclerosis in the presence and absence of obesity/ IR are distinct and identify IFNγ as a T2D-specific driver of atherogenesis. Therapies targeting pro-inflammatory cytokines in patients with increased risk of cardiovascular disease are underway (Ridker, 2016). In addition to these efforts, our work suggests that anti-IFNγ therapeutics might be particularly beneficial for lowering risk in patients with T2D.
On a molecular level, we showed that IFNγ suppressed specific anti-atherogenic MSRN proteins (i.e., APOE and C3), which predispose macrophages to increased cholesterol accumulation. Macrophages from obese/IR mice had reduced APOE and C3 levels, but did not exhibit deficits in other cholesterol metabolism genes, including Abca1, Abcg1, Cd36, Lxra, Sra1, and Srepb2. Similarly, ablating Ifngr1 attenuated foam cell formation and restored APOE and C3 levels, but had no effect on the cholesterol metabolism genes. Thus, the suppression of APOE and C3, and perhaps other MSRN proteins, represents a key mechanism by which IFNγ and obesity/IR promote foam cell formation.
IFNγ -induced changes to MSRN proteins are insufficient to cause foam cell formation (or atherogenesis) in the absence of hypercholesterolemia. Instead, they create a molecular susceptibility that, in the presence of hypercholesterolemia, increases macrophage cholesterol accumulation, a central event in atherogenesis. From this perspective, obesity/IR can be conceptualized as a perturbation that “sensitizes macrophages to atherogenic lipoproteins,” which may help to explain why type 2 diabetics generally require more aggressive cholesterol lowering to achieve therapeutic benefit (Banach et al., 2016; Hoe and Hegele, 2015).
Importantly, deleting Ifngr1 did not affect metabolic parameters in normocholesterolemic (WT) or hypercholesterolemic (Ldlr−/−) mice, which agrees with previous studies using Ifng−/− mice (Rocha et al., 2008). The uncoupling of the metabolic and pro-atherogenic functions of IFNγ might help to explain the curious relationship between metabolic dysfunction and atherosclerotic risk in patients with T2D. While elevated HBA1c levels are an excellent predictor of atherosclerotic risk, therapeutically lowering HBA1c does not necessarily alleviate this risk (Hayward et al., 2015). These data suggest that T2D induces factor(s) that promote atherogenesis, but that these factor(s) are not necessarily corrected by insulin sensitizing interventions. Recent studies showed that systemic and adipose tissue inflammation persists following bariatric surgery despite correction of hyperglycemia and insulin sensitivity (Kratz et al., 2016). Based on these observations and our work, we speculate that IFNγ might be one of these factors; however, additional studies will be needed to confirm this hypothesis.
IFNγ -producing immune cells are prominent in human and murine atheromas (Libby et al., 2013), and atherogenic effects of IFNγ are well supported in mouse studies (Ramji and Davies, 2015). However, the dietary conditions supporting its induction, the molecular changes it induces in vivo, and the cellular target(s) that mediates its pro-atherogenic effects are incompletely understood.
We showed that the induction of IFNγ requires the presence of obesity/IR. Surprisingly, the IFNγ levels produced during obesity/IR specifically targeted macrophage MSRN proteins, without inducing canonical signaling through STAT1 or its target genes involved in host defense. The latter required IFNγ doses several orders of magnitude higher than those observed in obese/IR mice or patients with T2D. Similarly, ablating Ifngr1 (the ligand binding receptor) corrected MSRN proteins and attenuated foam cell formation and atherosclerosis, but did not affect established IFNγ -target genes.
These findings suggest that the proatherogenic actions of IFNγ on macrophages may be independent of its canonical signaling pathway, which is supported by previous studies showing that myeloid cell deletion of Stat1 or Ifngr2, two key components of this signaling pathway (Ikushima et al., 2013), failed to attenuate atherogenesis (Boshuizen et al., 2016; Lim et al., 2008). Consistent with this hypothesis, previous studies showed that IFNγ alters expression of many genes in Stat1−/− macrophages (Gil et al., 2001), suggesting that STAT1-independent mechanisms significantly contribute to the effects of this cytokine on macrophage gene expression and function.
Future studies aimed at delineating this putative host-defense-independent mechanism and its contribution to atherosclerosis and infection models may inform therapeutic approaches to target IFNγ biology to treat cardiovascular disease in patients with T2D without predisposing them to opportunistic infections.
EXPERIMENTAL PROCEDURES
Mice
All animal studies were approved by the University of Chicago IACUC (ACUP#72209) and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. WT (CD45.1 or CD45.2), Ldlr−/−, Irf1−/−, and Ifngr1−/− mice on the C57BL/6 background came from Jackson Labs. For diet-induced obesity (DIO) studies, WT and Ifgnr1−/− male mice (8 weeks of age) were placed on an LFD (20:50503, PicoLab) or 60% HFD (D12492, Research Diets) for 9 weeks. For atherosclerosis studies, Ldlr−/− mice (8 weeks of age) or chimeric mice (6 weeks post-transplantation) were placed on an LFD (20:50503, PicoLab), low-fat high cholesterol (Envigo TD02026), or Western-type diet (TD96121, Harlan Teklad) for up to 16 weeks.
Blood Measurements
Serum insulin levels were measured by ELISA (Millipore), and blood glucose levels were measured with a One Touch Ultra 2 glucometer (Lifescan) following a 3-hr fast. Total plasma cholesterol levels and cholesterol levels within the very low density lipoprotein (VLDL), LDL, and HDL fractions were obtained by an Amplex Red Cholesterol Assay Kit (Invitrogen). Lipoproteins were separated by fast protein liquid chromatography on two Superose 6 size-exclusion columns in tandem (GE Lifescience).
Bone Marrow Transplantation
Bone marrow cells (5 × 106) collected by PBS perfusion of the femurs and tibia of 8-week-old Ifngr1−/− or WT (CD45.1) male mice were injected into the retro-orbital sinus of lethally irradiated (9.5 Gy ionizing radiation) 8-week-old male Ldlr−/− recipient mice. Mice were maintained on a Uniprim diet for 1 week before and 2 weeks after transplant and allowed to recover for 6 weeks before initiation of diets. For WT transplants, engraftment was determined by flow cytometry using the ratio of CD45.1-positive (donor) to CD45.2-positive (recipient) bone marrow cells. For Ifngr1−/− transplants, engraftment was quantified by PCR as the ratio of Ifngr1−/− (donor) to WT (recipient) signal. See also Figure S6 and Table S2.
Quantification of Atherosclerosis
Anesthetized mice were perfused with PBS followed by 4% paraformaldehyde with 5% sucrose in PBS. The heart and upper vasculature were excised, cleaned of adventitia, and imbedded in optimal cutting temperature (OCT), and serial 10-mm sections in the aortic root were collected. Sections, beginning at the appearance of the coronary artery and aortic valve leaflets, were stained with Oil Red O/Fast green, and digital images were captured using a Nikon Eclipse Ti2 microscope. Aortic root lesion area was quantified by cross-sectional analysis of four sections/mouse (spaced 100 mm apart) using NIS Elements AR software.
Isolation and Analysis of BMDMs and Elicited Peritoneal Macrophages
Bone-marrow-derived macrophages (BMDMs) were prepared by culturing bone marrow cells in 30% L cell-conditioned media for 7 days (Kratz et al., 2014). Elicited peritoneal macrophages were isolated by lavaging the peritoneal cavity with PBS containing 2% BSA (endotoxin-free) 5 days after 4% thioglycolate injection (3 mL/mouse) (Becker et al., 2010). Cells were plated in serum-free DMEM for 2 hr, washed with PBS, and cultured in DMEM with 10% fetal calf serum (FCS) for 24 hr prior to use. Macrophage-conditioned media were prepared by treating cells with glucose and/or insulin or cytokines for 24 hr, washing cells with PBS, and incubating them in serum-free media for an additional 24 hr. MSRN proteins in the macrophage-conditioned media were quantified by immunoblotting.
Isolation and Analysis of Human Monocyte-Derived Macrophages
Human peripheral blood was obtained from healthy volunteers as approved by the University of Chicago Institutional Review committee (IRB16-0321) and following obtaining written consent. Monocytes were isolated using anti-CD14 coupled magnetic beads and differentiated into human monocyte-derived macrophages by treating with M-CSF for 7 days as previously described (Kratz et al., 2014). Cells were treated for 24 h with vehicle or IFNγ, and effects on media APOE levels were determined by immunoblotting.
Isolation and Analysis of Murine Aortic Macrophages
Aortic macrophages were isolated from Ldlr−/− mice fed a Western diet for 16 weeks. Mice were injected intraperitoneally (i.p.) with PBS or IFNγ (25 mg/kg = 100 U/kg) (Whitman et al., 2000) 48 hr and 24 hr prior to euthanasia. Anesthetized mice were perfused with cold PBS with 20 U/mL heparin, and the upper vasculature was isolated and dissected of adipose tissue and paraaortic lymph nodes. The intima in the arch and innominate artery were excised and incubated in digestion media containing collagenase, hyaluronidase, and DNase1 in Hank’s balanced salt solution (HBSS) for 1 hr (Butcher et al., 2011). Macrophages were isolated using anti-CD11b microbeads (Miltenyl Biotek) and plated in a 96-well dish in serum-free DMEM for 24 hr. Macrophage purity was assessed by flow cytometry on a Cantos II (BD Biosciences) and analyzed by FlowJo software (v.10.4.1).
Macrophage Cholesterol Loading
Macrophages were incubated for 24 hr in serum-free medium supplemented with or without 2% serum from Ldlr−/− mice fed a WTD for 12 weeks. Cellular lipids were extracted using isopropanol, dried, and solubilized in methanol: chloroform (2:1), and proteins were solubilized with 0.1% NaOH. Cholesterol levels were quantified using an Amplex Red Cholesterol Assay Kit (Invitrogen) and normalized to total cellular protein, which was measured using a Pierce BCA.
Macrophage siRNA Treatment
Macrophages were transfected with control siRNA or Stat1 siRNA (Silencer Select, Ambion) using Lipofectamine RNAiMax (Invitrogen) and analyzed 48 hr post-transfection as previously described (Becker et al., 2010). STAT1 knockdown was confirmed by immunoblotting.
Macrophage Bacterial Killing
Macrophages were incubated with P. aeruginosa for 2 hr to allow for phagocytosis, treated with gentamicin to kill non-internalized bacteria, incubated for various times, and lysed, and live bacteria were plated on agar to quantify the number of colony-forming units (Hilbi et al., 2001).
Shotgun Proteomics
Conditioned media from 100,000 macrophages were diluted 1:1 with 1% sodium deoxycholate in 100 mM ammonium bicarbonate. Samples were denatured, reduced with 5 mM DTT by heating at 65 C for 1 hr, and alkylated with 15 mM iodoacetamide; excess iodoacetamide was quenched with additional 5 mM DTT; and samples were digested with trypsin (Promega, Madison, WI, USA) at a 1:20 w/w ratio overnight at 37 C. Samples were precipitated with 1% trifluoroacetic acid (TFA), desalted by solid phase extraction, dried down, and reconstituted with 0.1% formic acid in 5% acetonitrile. Digested peptides were injected on a trap column (40 × 0.1 mm, Reprosil C18, 5 mm, Dr. Maisch, Germany), desalted, and separated on a pulled-tip analytical column (400 × 0.075 mm, Reprosil C18, 5 mm, Dr. Maisch, Germany) heated to 50 C with a 3-segment linear gradient of acetonitrile, 0.1% FA in acetonitrile (Solution B), and 0.1% FA in water (Solution A) as follows: 0–2 min 1%–5% Solution B, 2–150 min 5%–25% Solution B, and 150–180 min 25%–35% Solution B (Waters NanoACQUITY).
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was acquired on Orbitrap Fusion Lumos (Thermo Scientific) operated in a data-dependent mode on charge states 2–4 with a 2-s cycle time, dynamic exclusion for 30 s, higher-energy collisional dissociation (HCD) fragmentation (normalized collision energy [NCE] 30%) and LC-MS/MS acquisition in the Or-bitrap. MS spectra were acquired at resolution 120,000, and LC-MS/MS spectra (precursor selection window 1.6 Da) were acquired at a resolution of 30,000. Peptides and proteins were identified using the Comet search engine (Eng et al., 2015), with PeptideProphet and ProteinProphet validation (search criteria included a 20-ppm tolerance window for precursors and products and Cys alkylation and methionine oxidation as fixed and variable modifications, respectively). Proteins considered for analysis had to be identified in at least 4 of 5 biological replicates for one dietary condition.
Targeted Proteomics
Proteins of interest were quantified using liquid chromatography Parallel Reaction Monitoring (PRM) MS on a Thermo Orbitrap Fusion Lumos tribrid mass spectrometer (Themo Scientific) connected to a NanoACQUITY HPLC (Waters). Several peptides from each proteins of interest were monitored by selecting their precursor ions in the quadrupole analyzer (selection window 1.6 Da) and full scan LC-MS/MS after HCD fragmentation (NCE 29%) in the Or-bitrap analyzer with high resolution (15,000). Data were processed using Skyline software (MacLean et al., 2010). Identity of the chromatographic peaks was ascertained by matching the PRM LC-MS/MS spectrum to the spectra from the shotgun experiment (dot product > 0.9 and mass precision < 5 ppm). See also Figure S4.
IFNγ Production by T Cells
Isolated splenocytes (0.5 × 106 cells) were treated with anti-CD3 and anti-CD28 antibodies (eBioscience/Invitrogen) to activate T cells. After 48 hr, IFNγ levels in the media were measured using a Mouse IFNγ Ready-Set-Go ELISA Kit (eBioscience/Invitrogen).
Antibodies for Flow and Immunoblotting
The following were used for flow cytometry: anti-CD45.1, anti-CD45.2, anti-CD11b, anti-F4/80, and anti-CD11c (eBioscience/Invitrogen). The following were used for immunoblotting: rabbit anti-APOE (Abcam); goat anti-MFGE8 (R&D Systems); and rabbit anti-STAT1, rabbit anti-P-STAT1 (701), and rabbit anti-GAPDH (Cell Signaling).
qRT-PCR
RNA was isolated using QIAGEN Midi-Prep Kits and RT with Quantiscript (QIAGEN) using random hexamers (Invitrogen). mRNA levels were measured with specific primers (Table S2) using SYBR green on a One Step Plus system (Applied Biosystems). Relative levels of each target gene were calculated using the DDCt formula and 18S RNA as a control.
Statistics
For proteomics studies, differentially expressed proteins were identified using a combination of the unpaired two-tailed Student’s t test (p < 0.05) and G-test (G > 1.5) with correction for false discovery (FDR < 5%) as previously described (Heinecke et al., 2010). For all other studies, statistical significance (p < 0.05) was determined by an unpaired two-tailed Student’s t test using Prism GraphPad software (v.6.0h). Data are presented as means ± SEM.
Supplementary Material
Highlights.
Obesity and insulin resistance induces IFNγ production by T cells
IFNγ targets macrophage MSRN proteins and promotes foam cell formation
Deleting myeloid cell Ifngr1 attenuates atherosclerosis in obese/IR Ldlr−/− mice
Deleting myeloid cell Ifngr1 does not affect atherosclerosis in lean Ldlr−/− mice
Acknowledgments
This research was supported by the NIH (R01DK102960, R01HL131028, P30DK020595, and P30DK017047) and the American Heart Association (10SDG3600027).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.010.
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: all authors. Performed the experiments: C.A.R., A.L., K.Q.S., G.Z., C.C., H.J.-E., D.C., I.B., T.V., and A.H. Critically reviewed the manuscript: all authors. Wrote the manuscript: L.B. and C.A.R.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Banach M, Nikolic D, Rizzo M, Toth PP. IMPROVE-IT: what have we learned? Curr Opin Cardiol. 2016;31:426–433. doi: 10.1097/HCO.0000000000000305. [DOI] [PubMed] [Google Scholar]
- Barbu A, Hamad OA, Lind L, Ekdahl KN, Nilsson B. The role of complement factor C3 in lipid metabolism. Mol Immunol. 2015;67:101–107. doi: 10.1016/j.molimm.2015.02.027. [DOI] [PubMed] [Google Scholar]
- Basu SK, Brown MS, Ho YK, Havel RJ, Goldstein JL. Mouse macrophages synthesize and secrete a protein resembling apolipoprotein E. Proc Natl Acad Sci USA. 1981;78:7545–7549. doi: 10.1073/pnas.78.12.7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker L, Gharib SA, Irwin AD, Wijsman E, Vaisar T, Oram JF, Heinecke JW. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010;11:125–135. doi: 10.1016/j.cmet.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis:epidemiology, pathophysiology,and management. JAMA. 2002;287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- Boshuizen MC, Neele AE, Gijbels MJ, van der Velden S, Hoeksema MA, Forman RA, Muller W, Van den Bossche J, de Winther MP. Myeloid interferon-γ receptor deficiency does not affect atherosclerosis in LDLR(−/−) mice. Atherosclerosis. 2016;246:325–333. doi: 10.1016/j.atherosclerosis.2016.01.026. [DOI] [PubMed] [Google Scholar]
- Butcher MJ, Herre M, Ley K, Galkina E. Flow cytometry analysis of immune cells within murine aortas. J Vis Exp. 2011;53:2848. doi: 10.3791/2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa J, Borges M, David C, Vaz Carneiro A. Efficacy of lipid lowering drug treatment for diabetic and non-diabetic patients: meta-analysis of randomised controlled trials. BMJ. 2006;332:1115–1124. doi: 10.1136/bmj.38793.468449.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Eng JK, Hoopmann MR, Jahan TA, Egertson JD, Noble WS, MacCoss MJ. A deeper look into Comet–implementation and features. J Am Soc Mass Spectrom. 2015;26:1865–1874. doi: 10.1007/s13361-015-1179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio S, Babaev VR, Burleigh ME, Major AS, Hasty AH, Linton MF. Physiological expression of macrophage apoE in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J Lipid Res. 2002;43:1602–1609. doi: 10.1194/jlr.m200108-jlr200. [DOI] [PubMed] [Google Scholar]
- Gil MP, Bohn E, O’Guin AK, Ramana CV, Levine B, Stark GR, Virgin HW, Schreiber RD. Biologic consequences of Stat1-independent IFN signaling. Proc Natl Acad Sci USA. 2001;98:6680–6685. doi: 10.1073/pnas.111163898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore MO, McGuire DK, Lingvay I, Rosenstock J. Predicting cardiovascular risk in type 2 diabetes: the heterogeneity challenges. Curr Cardiol Rep. 2015;17:607. doi: 10.1007/s11886-015-0607-7. [DOI] [PubMed] [Google Scholar]
- Gruen ML, Saraswathi V, Nuotio-Antar AM, Plummer MR, Coenen KR, Hasty AH. Plasma insulin levels predict atherosclerotic lesion burden in obese hyperlipidemic mice. Atherosclerosis. 2006;186:54–64. doi: 10.1016/j.atherosclerosis.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- Hartvigsen K, Binder CJ, Hansen LF, Rafia A, Juliano J, Hörkkö S, Steinberg D, Palinski W, Witztum JL, Li AC. A diet-induced hypercholesterolemic murine model to study atherogenesis without obesity and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2007;27:878–885. doi: 10.1161/01.ATV.0000258790.35810.02. [DOI] [PubMed] [Google Scholar]
- Hayward RA, Reaven PD, Emanuele NV, VADT Investigators Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:978. doi: 10.1056/NEJMc1508386. [DOI] [PubMed] [Google Scholar]
- Heinecke NL, Pratt BS, Vaisar T, Becker L. PepC: proteomics software for identifying differentially expressed proteins based on spectral counting. Bioinformatics. 2010;26:1574–1575. doi: 10.1093/bioinformatics/btq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbi H, Segal G, Shuman HA. Icm/dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol. 2001;42:603–617. doi: 10.1046/j.1365-2958.2001.02645.x. [DOI] [PubMed] [Google Scholar]
- Hoe E, Hegele RA. Lipid management in diabetes with a focus on emerging therapies. Can J Diabetes. 2015;39(Suppl 5):S183–S190. doi: 10.1016/j.jcjd.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Hoofnagle AN, Becker JO, Oda MN, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58:777–781. doi: 10.1373/clinchem.2011.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78:105–116. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–625. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz M, Hagman DK, Kuzma JN, Foster-Schubert KE, Chan CP, Stewart S, van Yserloo B, Westbrook EO, Arterburn DE, Flum DR, Cummings DE. Improvements in glycemic control after gastric bypass occur despite persistent adipose tissue inflammation. Obesity (Silver Spring) 2016;24:1438–1445. doi: 10.1002/oby.21524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WS, Timmins JM, Seimon TA, Sadler A, Kolodgie FD, Virmani R, Tabas I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–951. doi: 10.1161/CIRCULATIONAHA.107.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Mirhafez SR, Pasdar A, Avan A, Esmaily H, Moezzi A, Mohebati M, Meshkat Z, Mehrad-Majd H, Eslami S, Rahimi HR, et al. Cytokine and growth factor profiling in patients with the metabolic syndrome. Br J Nutr. 2015;113:1911–1919. doi: 10.1017/S0007114515001038. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosratabadi R, Arababadi MK, Hassanshahi G, Yaghini N, Pooladvand V, Shamsizadeh A, Zarandi ER, Hakimi H. Evaluation of IFN-gamma serum level in nephropatic type 2 diabetic patients. Pak J Biol Sci. 2009;12:746–749. doi: 10.3923/pjbs.2009.746.749. [DOI] [PubMed] [Google Scholar]
- Ramji DP, Davies TS. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26:673–685. doi: 10.1016/j.cytogfr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118:145–156. doi: 10.1161/CIRCRESAHA.115.306656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103:467–476. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R, Samuel CE. The role of gamma interferon in antimicrobial immunity. Curr Opin Microbiol. 2001;4:251–259. doi: 10.1016/s1369-5274(00)00199-5. [DOI] [PubMed] [Google Scholar]
- Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.