

Abstract
Autophagy is a cellular process that mediates selective degradation of cellular components in lysosomes. Autophagy may protect against neuronal apoptosis, which is induced in a number of neurodegenerative diseases. Thus, compounds that modulate autophagy could be beneficial to treat neurological disorders characterized by apoptosis such as Parkinson’s and Alzheimer’s diseases, as well as human-immunodeficiency virus-dementia complex. In this paper, we review new and old evidence on the role of autophagy in neuronal cell survival and we present evidence that human-immunodeficiency virus may have adapted strategies to alter autophagic pathways in neurons. Moreover, we discuss the usefulness of drugs that facilitate autophagic clearance of proteins that are associated with neurodegeneration.
Keywords: Autophagy, apoptosis, beclin-1, gp120, lysosomes, Parkin
INTRODUCTION
Autophagy (or macroautophagy) is a normal physiological mechanism that plays a crucial role in the degradation of cellular components and damaged organelles. In addition, autophagy has been involved in mammalian cell-autonomous defense mechanisms against pathogens, including bacteria and viruses [1–3]. Autophagy comprises formation of autophagic vacuoles, called autophagosomes, which contain cellular debris and delivery them to lysosomes for proteolytic degradation. Autophagy can also regulate lipid metabolism (lypophagy) including cholesterol, or remove damaged mitochondria (mitophagy). Thus, autophagy is implicated in a number of human physiological and pathological conditions.
There are two other types of autophagy, chaperone-mediated autophagy and microautophagy. The former utilizes the chaperone heat-shock cognate protein 70 for specific substrate recognition. Proteins are then delivered to the lysosomal membrane where they are unfolded and translocated into the lysosomal lumen. Microautophagy, on the other hand, promotes direct engulfment of cytoplasmic substrates by the lysosomes through invagination of the lysosomal membrane [4].
Baseline autophagy is a dynamic multi-step process, comprising several compartments, including the pre-autophagosomal sequestering phagophore [5] and the autophagosome [6]. The autophagosome can fuse with endosomes to form amphisomes [7, 8], which may be involved in the degradation of internalized species via endocytosis (Fig. 1). Autophagosomes and amphisomes may then fuse with lysosomes to generate autolysosomes or autophagolysosomes, respectively. Therefore, autolysosomes can potentially clear accumulating intracellular debris, like misfolded proteins and defective organelles, while autophagolysosomes may degrade internalized species via endocytosis and the endosomal pathways (i.e. extracellular amyloid, pathogens). Although, autophagy can be a cellular survival mechanism to eliminate unwanted materials, it can also be detrimental depending on the cellular environment. For example, cell death may occur when the autophagic flux is blocked, when autophagosome fusion with lysosomes is inhibited, or as a result of loss of the proteolytic lysosomal functions [9, 10].
Fig. 1. Schematic pathways of autophagy.

Baseline autophagy is a dynamic multi-step process, comprising several compartments, including the pre-autophagosomal sequestering phagophore and the autophagosome, which is enriched in LC3. The autophagosome can fuse with endosomes to form the amphisome, which may be involved in the degradation of internalized species via endocytosis. Either autophagosomes or amphisomes may then fuse with the lysosome to generate autolysosomes or autophagolysosomes, respectively. Autolysosomes clear accumulating intracellular debris, like misfolded proteins and defective organelles, while autophagolysosomes may degrade internalized species via endocytosis and the endosomal pathways (i.e. extracellular amyloid, pathogens).
AUTOPHAGY: A PROCESS INVOLVING MULTIPLE PROTEINS
Autophagosome formation is initiated via phosphorylation of several proteins, including autophagy-related (Atg)–Unc51-like kinase complex [11, 12], which is phosphorylated by the serine/threonine protein kinase mammalian target of Rapamycin (mTOR) complex 1 (TORC1) and activated by AMP-activated protein kinase [13]. Unc51-like kinase phosphorylation may trigger molecular cascades to enwrap cytoplasmic materials, resulting in isolation of a sequestering membrane [14], which is enriched with complexes containing ubiquitin-like proteins [15]. Ubiquitin is a small (8.5 KDa) protein that tags and targets other proteins to proteosomes for their disposal [16]. A second process involves the conjugation of microtubule associated protein 1 light chain 3 (LC3) to phosphatidylethanolamine. LC3 undergoes post-translational C-terminal cleavage resulting in the cytosolic LC3-I form, which is then conjugated to phosphatidylethanolamine to generate the autophagosome associated LC3-II form. LC3-II mediates membrane tethering and hemifusion essential for the expansion and closure of phagophores to form autophagosomes. Thus, the levels of LC3-II are often used as a tool to determine the amount of autophagosomes and therefore induction of autophagy [17, 18]. Autophagosome clearance, on the other hand, can be initiated via translocation of a multiprotein complex containing Beclin-1, a Bcl-2-homology domain only protein, and class III phosphoinositide 3-kinase (also known as lipid kinase Vps34) from the cytoskeleton [19, 20] to pre-autophagosomal structures. The activity of hVps34 is enhanced by its interaction with Beclin-1 which, in turn, forms a macromolecule complex consisting of Beclin-1/Atg6, Atg14L and hVps15. Nevertheless, Beclin-1 may bind to anti-apoptotic proteins such as Bcl-2 and Bcl-XL and negatively regulates autophagy [18, 21].
Several cellular stress signals may induce autophagy, including alterations of amino acid concentration, growth factors such as insulin, ATP, hypoxia, and accumulation of molecular and cellular debris [22]. However, it appears that different cell types differentially respond to stress signals. For example, inhibition of TORC1, which induces autophagy in non-neuronal cells, mediates translational control necessary for memory reconsolidation, long-term potentiation [23], and dendritic arborization [24] in the central nervous system (CNS). Therefore, TORC1 inhibition may serve different functions in peripheral dividing cells (muscle, blood, liver) versus post-mitotic neurons. This property of TORC1 is important because pharmacological inhibitors of TORC1 kinase activity such as rapamycin and analogs can be used as possible anti-cancer drugs as well as to ameliorate diseases of the CNS.
AUTOPHAGY AND CNS DISEASES
Neurons are unique among other cell types in that they do not re-enter the cell cycle and thus cannot use mitosis as a method for clearing abnormally aggregated proteins. When proteins aggregate and accumulate in the cytoplasm, cytotoxicity emerges. Inefficient recognition of aggregated proteins or defective organelles has been described in models of neurodegeneration [25], whereby molecular steps of autophagy are activated, but autophagosome clearance, due to inefficient fusion with the lysosome, is defective [9]. Thus, accumulation of abnormal protein aggregates has been suggested to be a central common mechanism underlying human neurodegenerative diseases. These include Alzheimer’s, Parkinson’s, and Huntington’s diseases, amyotrophic lateral sclerosis, and others. All these diseases have in common the pathological hallmark of the presence of intraneuronal ubiquitylated protein inclusions or aggregates. Consequently, clearance of protein aggregates could provide a novel therapeutic approach for these diseases. Can up-regulation of autophagy be used as a therapeutic strategy for neurodegenerative diseases? To address these questions, we present a brief summary of autophagy in CNS.
Alzheimer’s disease
Alzheimer’s disease (AD) is characterized by degeneration of neurons and synapses in the neocortex and some subcortical areas, such as the hippocampus and basal forebrain. Cholinergic neurons are particularly affected. Amyloid β-peptide (Aβ) plaques and intraneuronal aggregates of hyper-phosphorylated tau proteins are believed to cause loss of neurons in AD. Neurons in the AD brain exhibit autophagosomes containing hyper-phosphorylated Tau and Aβ [26] which may be due to deficient lysosomal degradation [27]. Thus, it has been suggested that impaired clearance of autophagosomes contributes to AD. Indeed, deletion of Beclin-1 in mice decreases neuronal autophagy and promotes neuronal loss whereas overexpression of Beclin-1 induces autophagy and inhibits amyloid deposition and associated pathology [28].
Several studies reported that inhibition of mTOR leads to induction of autophagy [29–31], and increases Aβ degradation by the lysosomal system [32]. Conversely, accumulation of intraneuronal Aβ also increases mTOR activity [33] and accumulation of autophagic vacuoles [34, 35], while inhibition of mTOR attenuates Aβ levels in a mouse model of AD [33], suggesting mTOR involvement in autophagic induction. In patients with AD, Beclin-1 levels are altered [36, 37], suggesting inefficient execution of Beclin-1-dependent autophagy and increased Aβ and p-Tau accumulation. Lentiviral Beclin-1 expression activates autophagy and improves neurodegenerative pathology in animal models of AD [36]. Intraneuronal Aβ accumulates in the brain of AD mice and may lead to lysosomal pathology, while enhancement of lysosomal proteolytic degradation may increase the rate of autophagic protein clearance [38]. Thus, up-regulation of autophagy is considered a therapeutic strategy for AD.
The final step of autophagy following induction and maturation of autophagosomes is lysosomal degradation, which is indispensable for clearance of cellular debris. Autophagosome accumulation can be obtained by elevation of lysosomal pH. With an altered pH, lysosomes cannot recycle/degrade efficiently, resulting in an excess storage of various molecules within the lysosomes [39]. In AD, there is evidence that aberrant lysosomal function leads to abnormal sphingolipid ceramide metabolism. This abnormal accumulation of undigested material within neurons can lead to cell death. Thus, autophagy impairment in AD could lead to protein accumulation as well as impaired lysosomal function.
Parkinson’s disease
In addition to AD, dysfunction in autophagy is common in several other neurodegenerative diseases [40–43], including Parkinson’s disease (PD) and Lewy body dementia which are characterized by intracellular accumulation of α–Synuclein (Syn). It has been shown that in degenerating dopamine neurons in PD brains, there is a breakdown of lysosomal membranes [39] and mislocalization of lysosomal receptors and autophagy components. Because Syn is primarily degraded in lysosomes by multiple pathways, including chaperone-mediated autophagy [44–46], it has been proposed that a lack of Syn degradation is a pathogenic event in PD.
PD is also characterized by mitochondrial dysfunction which is believed to be the major pathological feature in this disease [47]. Thus, one would expect to see mitophagy in PD brains. Nevertheless, a functional correlation between recessive early-onset PD caused by mutation in the Parkin gene [48, 49] and the PINK1 gene and a defect in mitophagy has been established. Parkin is an E3 ubiquitin ligase that facilitates proteasomal degradation of misfolded proteins [50] and its expression contributes to autophagic clearance in several animal models [51, 52]. PINK1 encodes for PTEN-induced putative kinase 1, a mitochondrial serine/threonine-protein kinase. PINK1 mutations in the kinase domain fail to translocate Parkin to mitochondria and to induce mitochondrial aggregation [53–56] and enhance autophagy in vitro [57]. Parkin is crucial for neuronal survival because experimental findings have shown that Parkin over-expression attenuates Aβ1-42 and Syn toxicity [37, 58]. Moreover, activation of autophagy improves dopaminergic cell survival in Parkin deficient and Tau over-expressing mice [59]. Parkin ubiquitinligase activity modulates Beclin-1 LC-3 mediated autophagy [60] and lentiviral delivery of Parkin activates Beclin-1-mediated autophagy in mice over-expressing β-amyloid precursor protein [34]. More recently it was demonstrated that autophagosome accumulation is cleared via treatment with tyrosine kinase inhibitors (TKIs) that increase the Parkin-Beclin-1 interaction, leading to amyloid clearance in neurodegeneration models [37, 61, 62]. Taken together, these findings suggest that autophagy-associated cell death should be carefully interpreted with distinction between induction and/or arrest of autophagy.
AUTOPHAGY AND THE HUMAN IMMUNODEFICIENCY VIRUS TYPE 1
Several publications demonstrated that some intracellular pathogens (i.e., bacteria, viruses, protozoa) are targeted for autophagosomes, which acidify and destroy invading pathogens in peripheral macrophages [63]. However, microbes and other pathogens often have specific adaptations to overcome autophagic clearance. This appears to be the case for the Human Immunodeficiency Virus type-1 (HIV). Reduced HIV levels are associated with a blockade in the processing of a key precursor protein for virus assembly, Gag, which localized to LC3 [64], suggesting that Gag may alter autophagosome maturation in macrophages. A functional genomics screen also linked autophagy proteins with increased HIV replication [65], further suggesting that HIV exploits multiple host proteins during infection. Thus, HIV not only thwarts the host defense mechanisms, but also uses immune defense for its own protection. Specific pathogen effectors targeting autophagy are well described outside the CNS [63], but the interplay between autophagy and HIV in the CNS is not very well understood. For example, many of the viral factors characterized so far, including HIV, mainly target Beclin-1 or other parts of the molecular cascade of autophagy to inhibit autophagosome maturation [66]. Paradoxically, induction of early stages of autophagy in HIV-infected macrophages promotes Gag processing and promotes infectious HIV proliferation [66], suggesting that inhibition of autophagosome maturation is beneficial in early stages but may prevent autophagic degradation of the virus. Furthermore, unlike macrophages that are normally spared, T cells infected with HIV die due to impaired autophagy [67]. Therefore, a better understanding of the role of autophagy in HIV infection in the CNS is imperative to assist in the design of therapies that alleviate HIV-associated neurodegeneration.
HIV associated dementia complex
Acquired Immunodeficiency Syndrome is common in HIV positive subjects. Less common but equally devastating is the development of HIV associated dementia complex (HAD), an array of neurological alterations characterized by motor and cognitive abnormalities [68, 69]. Neuroimaging studies of HAD brains show atrophy of the cortex and subcortical regions such as the caudate nucleus and hippocampus [70]. It is now apparent that these patients may be suffering from protracted forms of HIV encephalitis (HIVE), a neuroinflammatory condition characterized by the presence of HIV infected microglial cells, formation of microglial nodules, multinucleated giant cells, astrogliosis and myelin loss [71]. In addition, these subjects exhibit profound neuronal degeneration and loss of synaptodendritic connections, which persist despite the highly active combination antiretroviral therapy (HAART) [72]. Paradoxically, as HIV-infected individuals live longer because of HAART, the prevalence of HAD is increasing. In addition, viral persistence in these subjects has been demonstrated in cerebrospinal fluid even when treated with HAART that lowers HIV RNA to undetectable levels in plasma [72]. Nevertheless, HAD, like other neurodegenerative diseases, develops over time. Therefore, one therapeutic strategy to prevent HAD is to modify biological events downstream of the etiopathological processes at a point when neurons are already infected but before they are committed to die. Key to develop such therapeutic compounds is the understanding of the molecular and cellular mechanisms of how HIV leads to HAD.
HIV is a retrovirus that, after initial infection, produces profound T cell depletion [73]. The end result is severe cellular immunodeficiency, with the development of opportunistic infections and cancers. Macrophages may also serve as an important reservoir for HIV, especially because HIV infection is latent and not cytopathic in these cells, in contrast to T lymphocytes. HIV utilizes CD4 as its primary receptor followed by engagement with chemokine receptors CXCR4 and CCR5 [74–76]. CCR5 is found in macrophages and mediates macrophage (M-tropic) HIV infection, whereas T-tropic HIV binds to CXCR4, which is abundant in T cells [77]. There are also HIV isolates that use both CCR5 and CXCR4 [78]. The importance of chemokine receptors in HIV pathogenesis is underscored by the observation that individuals deficient in CCR5 are resistant to infection by HIV [79]. HIV penetrates the CNS a few weeks after infection. Microglia, unlike neurons, express CD4, CXCR4, and CCR5 [80] and thus, are the primary target cells for HIV in the brain [81]. However, astrocytes can also be infected [82]. Yet, numerous in vitro and in vivo studies have revealed that HIV can cause neuronal cell death [83–86]. Thus, it has been suggested that infected microglia and infiltrated macrophages could produce several factors that are directly or indirectly toxic to neurons [84]. Moreover, astrocyte function could become disturbed in HIV positive subjects and influence neuronal cell viability. For instance, the uptake of neurotoxic glutamate could be impaired [87]. Furthermore, several HIV proteins have been shown to be released from HIV-infected microglia and/or present extracellularly in the HIV-infected brain. These proteins include at least i) the envelope glycoprotein gp120, which is crucial for the initial binding of the virions to CD4 and chemokine receptors, ii) the activator of HIV transcription Tat, and iii) Nef, the regulatory protein that promotes viral replication [88, 89]. Among these proteins, gp120 appears to be the most potent neurotoxin with lethal activities in the picomolar range [90, 91]. Most importantly, either transgenic mice expressing gp120 [92] or neurons exposed to recombinant gp120 [93] exhibit neuronal cell loss and dendritic simplification, which are pathological features of HAD [94].
Gp120 neurotoxicity
The ability of gp120 to promote pruning of the synaptic network has been widely accepted [92]. However, debate exists on whether gp120 induces neuronal cell damage directly or indirectly. The characterization of the neurotoxic mechanisms of gp120 is paramount for a drug discovery program against HAD. Gp120 has been shown to interact with neuronal chemokine receptors CXCR4 and CCR5 to activate neurotoxic signals/pathways or inflammatory responses [91, 95, 96]. These pathways converge on caspase-3, a member of the cysteine-aspartic acid protease family that plays a crucial role in the execution of neuronal cell apoptosis, which is well recognized in HIV [94]. Apoptosis has numerous physiological functions. In mature organisms, these functions include, among others, negative selection in the immune system, removal of damaged or compromised cells during tissue turnover, protection from viral infection, and protection from cancer. Apoptosis serves its most important physiological role in the nervous system during neurogenesis, when apoptosis reduces by ~50% the number of excess neurons that do not innervate a given target and thus sculpts the developing nervous system [97]. However, when apoptosis occurs inappropriately in the adult CNS where neurons cannot proliferate or regenerate it creates loss of neurons and other neurodegenerative pathologies, including AD, PD, and HAD. Pro-apoptotic molecules or signaling pathways induced by gp120 through activation of chemokine receptors include ceramide [98], p38/mitogen-activated protein kinase, c-Jun terminal kinase and extracellular signal-regulated kinase [95]. Conversely, pharmacological inhibition of the c-Jun terminal kinase pathway prevents gp120-mediated apoptosis [99]. These findings are important because they point to specific signal transduction pathways as potential therapeutic targets. However, drugs that block signaling in all cells may be detrimental as these pathways, especially extracellular signal-regulated kinase, are necessary also for neuronal cell differentiation and survival [100, 101]. In addition, CXCR4 or CCR5 receptor inhibitors, either synthetic or naturally occurring, do not cross the blood-brain barrier. Therefore, additional studies must be carried out to reveal the numerous biological and molecular events that occur after HIV/gp120 binds to chemokine receptors.
Gp120 endocytosis and autophagy
Endocytosis is a fundamental cellular function involved in nutrient uptake, pathogen removal, and transporting signaling molecules from extracellular environment to intracellular cytoplasm. Endocytosis includes phagocytosis, which internalizes large particles, and pinocytosis, which internalizes membrane proteins, lipids, and fluid with soluble molecules [102]. Neurons, through the endocytosis process control intracellular trafficking for internalizing neurotrophic factors, for recycling or down-regulating receptors, for neurotransmitter uptake or for directing information to intracellular biosynthetic pathways. Recent data show a novel mechanism of T-tropic gp120 toxicity that followed gp120 endocytosis by CNS [103] and peripheral [104] neurons. The endocytotic process is not unique for gp120, as other neurotoxic proteins promote neuronal cell degeneration, including prions, Aβ, and Syn. What is unique for T-tropic gp120 is the fact that this protein relies on the chemokine receptor CXCR4 to be internalized. Gp120 is then axonally transported both anterogradely or retrogradely to accumulate inside neurons, either around microtubules or perinuclearly inside lysosomes [103]. Neurons internalizing gp120 undergo axonal retraction before showing signs of apoptosis. Indeed, quantitative image analysis revealed that neurons internalizing gp120 exhibit shorter processes compared to controls within 6 hours [93]. Thus, gp120 appears to initiate intracellular processes that are crucial for neuronal cell survival before affecting the apoptotic pathway. For instance, accumulation of gp120 may impair neuronal cell homeostasis which involves anterograde axonal transport of proteins. Moreover, anterograde axonal transport must be balanced by a similar rate of retrograde transport and autophagic clearance. Because a connection exists between pathways that regulate autophagy and apoptosis, a simple question is whether gp120 is neurotoxic because it impairs autophagy in neurons.
Autophagy plays a crucial role in gp120-induced T cell death, using CXCR4 [105]. Interestingly, autophagy in T cells does not require CD4. In addition, a CXCR4 antagonist inhibits the formation of autophagic vacuoles supporting the concept that autophagy in these cells requires the presence of CXCR4. Beclin-1 and LC3 are higher in the cortex of young HIVE subjects when compared to aged HIVE [106]. Thus, progressive neurodegeneration, and loss of synaptic processes seen in these subjects may be associated with decreased autophagic clearance. This notion is supported by data showing that Beclin-1 induction by gene transfer in gp120 transgenic mice restores autophagy and reduces neuroinflammation and neurodegeneration. In addition, p53, which is also activated by gp120, is another activator of autophagy [107]. Finally, the ubiquitin proteasome system, which is altered in HAD subjects [108] and is crucial for protein degradation is also implicated in autophagy [109]. Thus, gp120 may play a role in altering autophagy pathways causing neuronal cell death. This would be in line with the notion that impaired autophagy leads to neurodegenerative diseases.
Gp120 may impair neuronal survival per se by inducing proteinopathies, as we have observed accumulation of gp120 inside neurons [103]. To answer this question we exposed primary rat cortical neurons to gp120 and determined Beclin-1 levels in a time-dependent manner. Neurons treated with gp120 (5nM) exhibited a time-dependent increase in Beclin-1, which was induced by 3 hours relative to actin or GAPDH (Fig. 2). This effect was similar to that observed with Rapamycin. However, by 6 hours, Beclin-1 levels returned to control levels, suggesting that gp120 may alter autophagy only for a limited period of time. We also measured the level of LC3 in primary neurons treated with gp120. LC3 is initially synthesized in an unprocessed form, proLC3, but sequential conjugation of Atgs leads to its modification into LC3-II, which is a marker of autophagosomes [110, 111]. Western blot analysis was used to determine the conversion of LC3-I to LC3-II (Fig. 3). Gp120 induced a time-dependent increase in LC3-I conversion to LC3-II but the effect lasted only for 6 hours, suggesting that gp120 induces autophagosome formation early after infection, which may then be fused into the lysosomes, where gp120 was previously detected [103]. The molecular steps of autophagy can be activated, but autophagosome clearance remains insufficient, due to inefficient fusion with the lysosome [9]. In fact, while gp41, a subunit of the envelop protein has been shown to undergo ubiquitination in non-neuronal cells [112] ubiquitin-dependent gp120 degradation has not been yet established in neurons. Lack of gp120 degradation in the CNS, may reflect absence of autophagic degradation in glial and neuronal cells or defects in lysosomal proteolysis, leading to intracellular accumulation of gp120. Such gain of function of gp120 could be deleterious for neurons because it can lead to alterations of many intracellular processes including endoplasmic reticulum stress, mitochondria failure, and axonal retraction. Therefore, it is imperative to better understand the effects of HIV infection and/or gp120 on neurons, which die after exposure to gp120, compared to astrocytes that accumulate gp120 and survive. Taken together this leads to speculation whether gp120 may be able to alter autophagy in brain astrocytes as it does in macrophages; and whether neurons fail to sequester gp120 in a similar fashion to T cells. These speculations lead us to think that neuronal autophagy may be different than the poorly understood astrocytic autophagy. Furthermore, the observed accumulation of gp120 in lysosomes may reflect inefficiency in proteolytic enzymes or alteration of lysosomal paucity, leading to lysosomes to be overwhelmed by gp120 proteins (Fig. 4). However, other HIV proteins, i.e. Tat, Gag, Nef may affect the autophagic clearance machinery in a different way compared to gp120. In addition, LC3-II and Beclin-1 alone may not be sufficient to estimate autophagy. Thus, more markers (e.g. ATG) and experimental approach (e.g. subcellular fractionation, mitophagy) are needed to characterize a working “mechanism” for gp120-mediated neuronal cell death.
Fig. 2. HIV/gp120 evokes a short-time activation of Beclin-1.
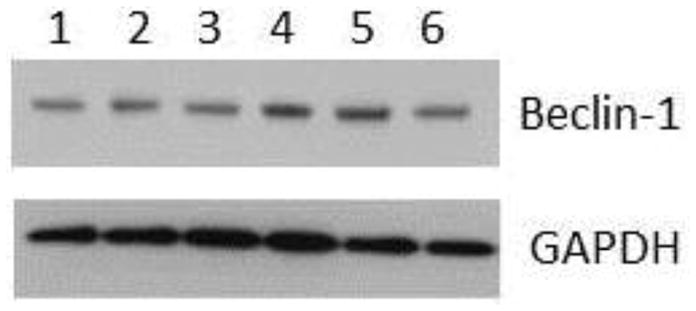
Cortical neurons were exposed to gp120 (5 nM) for various times. Lysates were prepared in RIPA buffer and Beclin-1 was analyzed by Western blot on 4–12% NuPAGE SDS gel. 1=DMSO; 2=boiled gp120; 3=gp120 1 hour; 4=gp120 3 hours; 5=rapamycin 3 hours; 6=gp120 6 hours. Results were replicated in three independent experiments.
Fig. 3. HIV/gp120 promotes LC3-I conversion to LC3-II.

Neurons were treated with 5nM gp120 for the indicated times. Total cell lysates in RIPA buffer were analyzed by Western blot on 4–12% NuPAGE SDS gel using monoclonal anti-LC3 antibody. The level of LC3-II was increased relative to LC3-I after 3 hours of treatment (n=3) but LC3-II levels returned to control at 18 hours, suggesting autophagic induction up to 3 hours and an autophagosome clearance at 18 hours.
Fig. 4. Possible homology of HIV manipulation of autophagic mechanisms between the immune system and CNS.

HIV proteins, including Gag, inhibit autophagosome maturation, turning the cell defense system to their advantage in macrophages. However, T cells die leading to AIDS. HIV proteins may imitate this process in astrocytes, leading to storage of the virus in astrocytes, thus providing a reservoir for future supply. However, neurons cannot clear viral proteins via autophagy. This mechanism may lead to neurodegeneration and HAD.
POTENTIAL INTERVENTION
The possibility of autophagic clearance of molecular and cellular debris from neurons was demonstrated in models of neurodegenerative diseases, including PD and AD Parkin-Beclin-1 interaction, leading to increased proteasome function and autophagosomal maturation, which is impaired in HIV. Therefore, Nilotinib and Bosutinib might be used as brain-penetrant drugs to ameliorate autophagic gp120, and perhaps as HIV proteins, clearance.
CONCLUSION
HIV proteins form proteinacious species and impair autophagic clearance in a similar fashion to accumulating or misfolded proteins in neurodegenerative diseases. Although the effects of HIV on autophagy are very well understood in the peripheral organs, there is very little information how viral proteins may behave in the CNS, leading to neuroinflammation and degenerative death. Most importantly, it is necessary to understand how gp120 and other HIV proteins may differentially affect autophagy in neurons versus astrocytes. The short term increase in autophagic markers, including Beclin-1 and LC3-II, in neurons may suggest a [34, 35, 51, 113, 114]. More recently we demonstrated that autophagosome accumulation can also be cleared via treatment with TKIs, which penetrate the brain and facilitate autophagic clearance in neurodegeneration models [37, 61, 62]. TKIs such as Imatinib are effective and well-tolerated treatments in patients with Philadelphia chromosome-positive chronic myeloid leukemia [115, 116]. Nilotinib is a second generation Abelson TKI inhibitor, which was approved by the US Food and Drug Administration in 2007 [117–119] following Imatinib resistance and intolerance [115]. Another dual Src/Abelson TKI inhibitor TKI, Bosutinib, (SKI-606) was also approved by the US Food and Drug Administration in 2012 [120–122]. A much lower dose of these drugs can promote autophagic clearance of accumulating proteins, leading to translocation of amyloid from autophagosomes and clearance within lysosomes [37, 61, 62]. Short term administration of these drugs improved amyloid pathology, protected against cell death and enhanced cognitive and motor performance in AD and PD models, respectively. TKIs increased functional parallel process with HIV-infected macrophages (Fig. 4), which are unable to clear the virus due to its highly adaptive nature to overcome lysosomal degradation. It is possible that HIV proteins either impair autophagosome maturation or alter lysosomal degradation in the CNS, ultimately leading to gp120 accumulation and neuronal cell death. Finally, autophagy can be exploited therapeutically via treatment of brain-penetrant and clinically tolerated TKIs that promote autophagic clearance of molecular and cellular debris.
Acknowledgments
This work was supported by HHS AG30378 grant and Georgetown University funding to Charbel Moussa and HHS NS079172 and NS074916 grants to Italo Mocchetti.
LIST OF ABBREVIATIONS
- mTOR
Mammalian target of Rapamycin
- CNS
Central nervous system
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- Syn
α synuclein
- HIV
Human immunodeficiency virus type-1
- HAD
HIV associated dementia
- LC3
Light chain protein-3
- HIVE
HIV encephalitis
- HAART
Highly active combination antiretroviral therapy
- Aβ
Beta-amyloid
- TKIs
Tyrosine kinase inhibitors
Footnotes
Send Orders for Reprints to reprints@benthamscience.net
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
DISCLAIMER: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
References
- 1.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–37. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 4.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seglen PO. Regulation of autophagic protein degradation in isolated liver cells. In: Glaumann H, Ballard FJ, editors. Lysosomes: Their Role in Protein Breakdown. London: Academic Press; 1987. pp. 369–414. [Google Scholar]
- 6.de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 7.Dunn WA., Jr Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–43. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 8.Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151:40–7. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- 9.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–87. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy. 2011;7:924–6. doi: 10.4161/auto.7.8.15860. [DOI] [PubMed] [Google Scholar]
- 14.Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 15.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–6. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–9. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Bartolomeo S, Corazzari M, Nazio F, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 21.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoica L, Zhu PJ, Huang W, et al. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci USA. 2011;108:3791–6. doi: 10.1073/pnas.1014715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jossin Y, Goffinet AM. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 2007;27:7113–24. doi: 10.1128/MCB.00928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong ES, Tan JM, Soong WE, et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–82. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulamek-Koziol M, Furmaga-Jablonska W, Januszewski S, et al. Neuronal autophagy: self-eating or self-cannibalism in Alzheimer’s disease. Neurochem Res. 2013;38:1769–73. doi: 10.1007/s11064-013-1082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keilani S, Lun Y, Stevens AC, et al. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32:5223–36. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaeger PA, Pickford F, Sun CH, et al. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5:e11102. doi: 10.1371/journal.pone.0011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Illi B, Dello Russo C, Colussi C, et al. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ Res. 2008;102:51–8. doi: 10.1161/CIRCRESAHA.107.157305. [DOI] [PubMed] [Google Scholar]
- 30.Martin M, Potente M, Janssens V, et al. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc Natl Acad Sci USA. 2008;105:4727–32. doi: 10.1073/pnas.0708455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simboeck E, Sawicka A, Zupkovitz G, et al. A phosphorylation switch regulates the transcriptional activation of cell cycle regulator p21 by histone deacetylase inhibitors. J Biol Chem. 2010;285(52):41062–73. doi: 10.1074/jbc.M110.184481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vingtdeux V, Chandakkar P, Zhao H, et al. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-{beta} peptide degradation. FASEB J. 2011;25(1):219–31. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011;20:2091–102. doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman AM, Moussa CE. The ubiquitin ligase parkin modulates the execution of autophagy. Autophagy. 2011;7:919–21. doi: 10.4161/auto.7.8.15814. [DOI] [PubMed] [Google Scholar]
- 36.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med. 2013;5:1247–62. doi: 10.1002/emmm.201302771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dehay B, Bove J, Rodriguez-Muela N, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 41.Pan T, Kondo S, Le W, Jankovic J. The role of autophagylysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–78. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 42.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 43.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Vicente M, Talloczy Z, Kaushik S, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–88. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–66. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 49.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–7. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 50.Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–5. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 51.Lonskaya I, Shekoyan AR, Hebron ML, et al. Diminished parkin solubility and co-localization with intraneuronal amyloid-beta are associated with autophagic defects in Alzheimer’s disease. J Alzheimers Dis. 2013;33:231–47. doi: 10.3233/JAD-2012-121141. [DOI] [PubMed] [Google Scholar]
- 52.Lonskaya I, Hebron ML, Algarzae NK, Desforges N, Moussa CE. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience. 2012;232C:90. doi: 10.1016/j.neuroscience.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 54.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park J, Kim Y, Chung J. Mitochondrial dysfunction and Parkinson’s disease genes: insights from Drosophila. Dis Model Mech. 2009;2:336–40. doi: 10.1242/dmm.003178. [DOI] [PubMed] [Google Scholar]
- 56.Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas KJ, McCoy MK, Blackinton J, et al. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20(1):40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moussa CE. Parkin attenuates wild-type tau modification in the presence of beta-amyloid and alpha-synuclein. J Mol Neurosci. 2009;37:25–36. doi: 10.1007/s12031-008-9099-x. [DOI] [PubMed] [Google Scholar]
- 59.Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–38. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 60.Chen D, Gao F, Li B, et al. Parkin Mono-ubiquitinates Bcl-2 and Regulates Autophagy. J Biol Chem. 2010;285:38214–23. doi: 10.1074/jbc.M110.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hebron ML, Lonskaya I, Moussa CE. Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy. 2013;9:1249–50. doi: 10.4161/auto.25368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hebron ML, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum Mol Genet. 2013;22:3315–28. doi: 10.1093/hmg/ddt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–49. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tucker KA, Reggiori F, Dunn WA, Jr, Klionsky DJ. Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J Biol Chem. 2003;278:48445–52. doi: 10.1074/jbc.M309238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brass AL, Dykxhoorn DM, Benita Y, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 66.Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–68. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gannage M, Dormann D, Albrecht R, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–80. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perumal MB, Dhanasekaran S. HIV associated dementia: role for neurosteroids. Med Hypotheses. 2012;78:672–4. doi: 10.1016/j.mehy.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Dahiya S, Irish BP, Nonnemacher MR, Wigdahl B. Genetic variation and HIV-associated neurologic disease. Adv Virus Res. 2013;87:183–240. doi: 10.1016/B978-0-12-407698-3.00006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dal Pan GJ, McArthur JH, Aylward E, et al. Patterns of cerebral atrophy in HIV-1-infected individuals: results of a quantitative MRI analysis. Neurology. 1992;42:2125–30. doi: 10.1212/wnl.42.11.2125. [DOI] [PubMed] [Google Scholar]
- 71.Spudich S, Gonzalez-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med. 2012;2:a007120. doi: 10.1101/cshperspect.a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Everall I, Vaida F, Khanlou N, et al. Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol. 2009;15:360–70. doi: 10.3109/13550280903131915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soudeyns H, Rebai N, Pantaleo GP, et al. The T cell receptor V beta repertoire in HIV-1 infection and disease. Semin Immunol. 1993;5:175–85. doi: 10.1006/smim.1993.1021. [DOI] [PubMed] [Google Scholar]
- 74.He J, Chen Y, Farzan M, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–9. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 75.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science. 1990;250:1139–42. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 76.Dragic T, Litwin V, Allaway GP, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 77.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 78.Doranz BJ, Rucker J, Yi Y, et al. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–58. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 79.Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–5. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 80.Jordan CA, Watkins BA, Kufta C, Dubois-Dalcq M. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J Virol. 1991;65:736–42. doi: 10.1128/jvi.65.2.736-742.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Albright AV, Shieh JT, Itoh T, et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73:205–13. doi: 10.1128/jvi.73.1.205-213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31:9456–65. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bachis A, Biggio F, Major EO, Mocchetti I. M- and T-tropic HIVs promote apoptosis in rat neurons. J Neuroimmune Pharmacol. 2009;4:150–60. doi: 10.1007/s11481-008-9141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14:1189–97. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- 85.Bertrand SJ, Aksenova MV, Mactutus CF, Booze RM. HIV-1 Tat protein variants: critical role for the cysteine region in synaptodendritic injury. Exp Neurol. 2013;248:228–35. doi: 10.1016/j.expneurol.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro. 2012;4:243–52. doi: 10.1042/AN20120017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bezzi P, Domercq M, Brambilla L, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–10. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 88.Bansal AK, Mactutus CF, Nath A, et al. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–9. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- 89.van Marle G, Henry S, Todoruk T, et al. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329:302–18. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 90.Meucci O, Miller RJ. gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J Neurosci. 1996;16:4080–8. doi: 10.1523/JNEUROSCI.16-13-04080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hesselgesser J, Taub D, Baskar P, et al. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8:595–8. doi: 10.1016/s0960-9822(98)70230-1. [DOI] [PubMed] [Google Scholar]
- 92.Toggas SM, Masliah E, Rockenstein EM, et al. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–93. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 93.Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci. 2012;32:9477–84. doi: 10.1523/JNEUROSCI.0865-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Famularo G, De Simone C, Marcellini S. Apoptosis: mechanisms and relation to AIDS. Med Hypotheses. 1997;48:423–9. doi: 10.1016/s0306-9877(97)90041-4. [DOI] [PubMed] [Google Scholar]
- 95.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:8212–6. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meucci O, Fatatis A, Simen AA, et al. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci USA. 1998;95:14500–5. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–7. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- 98.Haughey NJ, Cutler RG, Tamara A, et al. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–67. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- 99.Bodner A, Toth PT, Miller RJ. Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol. 2004;188:246–53. doi: 10.1016/j.expneurol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 100.Volosin M, Song W, Almeida RD, et al. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci. 2006;26:7756–66. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mocchetti I, Bachis A. Brain-derived neurotrophic factor activation of TrkB protects neurons from HIV-1/gp120-induced cell death. Crit Rev Neurobiol. 2004;16:51–7. doi: 10.1615/critrevneurobiol.v16.i12.50. [DOI] [PubMed] [Google Scholar]
- 102.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 103.Bachis A, Aden SA, Nosheny RL, Andrews PM, Mocchetti I. Axonal transport of human immunodeficiency virus type 1 envelope protein glycoprotein 120 is found in association with neuronal apoptosis. J Neurosci. 2006;26:6771–80. doi: 10.1523/JNEUROSCI.1054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Melli G, Keswani SC, Fischer A, Chen W, Hoke A. Spatially distinct and functionally independent mechanisms of axonal degeneration in a model of HIV-associated sensory neuropathy. Brain. 2006;129:1330–8. doi: 10.1093/brain/awl058. [DOI] [PubMed] [Google Scholar]
- 105.Espert L, Denizot M, Grimaldi M, et al. Autophagy and CD4+ T lymphocyte destruction by HIV-1. Autophagy. 2007;3:32–4. doi: 10.4161/auto.3275. [DOI] [PubMed] [Google Scholar]
- 106.Fields J, Dumaop W, Rockenstein E, et al. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol. 2013;19:89–101. doi: 10.1007/s13365-012-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, Dong XX, Cao Y, et al. p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur J Neurosci. 2009;30:2258–70. doi: 10.1111/j.1460-9568.2009.07025.x. [DOI] [PubMed] [Google Scholar]
- 108.Kragh CL, Ubhi K, Wyss-Coray T, Masliah E. Autophagy in dementias. Brain Pathol. 2012;22:99–109. doi: 10.1111/j.1750-3639.2011.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Nat Acad Sci USA. 2008;105:20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang W-P, Scott SV, Kim J, Klionsky DJ. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem. 2000;275:5845–51. doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- 112.Bultmann A, Eberle J, Haas J. Ubiquitination of the human immunodeficiency virus type 1 env glycoprotein. J Virol. 2000;74:5373–6. doi: 10.1128/jvi.74.11.5373-5376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khandelwal PJ, Dumanis SB, Feng LR, et al. Parkinson-related parkin reduces alpha-Synuclein phosphorylation in a gene transfer model. Mol Neurodegener. 2010;5:47. doi: 10.1186/1750-1326-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mahul-Mellier A-L, Fauvet B, Gysbers A, et al. c-Abl phosphorylates a-synuclein and regulates its degradation: implication for a-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum Mol Genet. 2014;23(11):2858–79. doi: 10.1093/hmg/ddt674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kantarjian HM, Giles F, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110:3540–6. doi: 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 116.de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol. 2008;26:3358–63. doi: 10.1200/JCO.2007.15.8154. [DOI] [PubMed] [Google Scholar]
- 117.Deremer DL, Ustun C, Natarajan K. Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin Ther. 2008;30:1956–75. doi: 10.1016/j.clinthera.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 118.Skorski T. BCR-ABL1 kinase: hunting an elusive target with new weapons. Chem Biol. 2011;18:1352–3. doi: 10.1016/j.chembiol.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahon FX, Hayette S, Lagarde V, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68:9809–16. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- 120.Keller VAG, Brummendorf TH. Novel aspects of therapy with the dual Src and Abl kinase inhibitor bosutinib in chronic myeloid leukemia. Expert Rev Anticancer Ther. 2012;12:1121–7. doi: 10.1586/era.12.84. [DOI] [PubMed] [Google Scholar]
- 121.Cortes JE, Kantarjian HM, Brummendorf TH, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118:4567–76. doi: 10.1182/blood-2011-05-355594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66:11314–22. doi: 10.1158/0008-5472.CAN-06-1199. [DOI] [PubMed] [Google Scholar]