

Abstract
Heart failure (HF) is a complication of multiple cardiac diseases and is characterized by impaired contractile and electric function. Patients with HF are not only limited by reduced contractile function but are also prone to life-threatening ventricular arrhythmias. HF itself leads to remodeling of ion channels, gap junctions, and intracellular calcium handling abnormalities that in combination with structural remodeling, e.g., fibrosis, produce a substrate for an arrhythmogenic disorders. Not only ventricular life-threatening arrhythmias contribute to increased morbidity and mortality but also atrial arrhythmias, especially atrial fibrillation (AF), are common in HF patients and contribute to morbidity and mortality. The distinct ion channel remodeling processes in HF and in channelopathies associated with HF will be discussed. Further basic research and clinical studies are needed to identify underlying molecular pathways of HF pathophysiology to provide the basis for improved patient care and individualized therapy based on individualized ion channel composition and remodeling.
Keywords: Arrhythmia, Channelopathy, Heart failure, Ion channel, Pathophysiology
Introduction
Heart failure (HF) is a complication of multiple cardiac diseases and is characterized by impaired contractile and electric function. Patients with HF are not only limited by circulatory failure but are prone to life-threatening ventricular arrhythmias as well in up to 50% of cases (Kjekshus 1990).
HF itself is accompanied by remodeling of ion channels, gap junctions, and intracellular calcium handling abnormalities that in combination with structural remodeling, e.g., fibrosis, produce a substrate for arrhythmogenesis. Arrhythmogenesis on the basis of remodeling processes in HF is not limited to the ventricles. Rather, atrial arrhythmias, especially atrial fibrillation (AF), are common in HF patients and contribute to morbidity and mortality (Ehrlich et al. 2002, Mene-Afejuku et al. 2018). Nevertheless, this review focuses on ventricular changes remodeling of ion channels, and channelopathies, and their impact on structural remodeling. By contrast, ion channel remodeling in AF and its potential implications for arrhythmogenesis and treatment have been assessed in depth elsewhere (Dobrev and Ravens 2003; Nattel et al. 2008; Workman et al. 2008; Schotten et al. 2011; Grunnet et al. 2012; Heijman et al. 2014; Hancox et al. 2016).
The ion channel composition of each cardiac cell contributes to the formation of the cardiac action potential that is important for propagation of excitation. In addition, ion channel complexes form the basis for excitation contraction coupling including calcium-induced calcium release and mechanical contraction. Changes in action potential duration and QT interval in LQTS patients not only affect electrical but also mechanical function of the heart, highlighting the role of mechano-electrical dysfunction in channelopathies.
Moreover, ion channel remodeling processes may lead to HF development. Ion channels have recently been implicated in structural heart development, e.g., in patients carrying HCN4 mutations associated with left ventricular non-compaction, as outlined in this article. Ion channel remodeling processes in HF and in channelopathies associated with HF will be discussed below.
Ion channels and cardiac action potential
The cardiac action potential (AP) provides the basis for conduction of electricity throughout the heart and provides the basis for electro mechanic coupling which is essential for normal heart function. The AP is shaped by its underlying ionic currents, shown in Fig. 1, and transporters [for detailed reviews see Nerbonne and Kass 2005] and varies depending on the specific electric localization including the sinoatrial node, atria, atrioventricular node, His-Purkinje system, and ventricles shown in Fig. 2 (Schram 2002, Nerbonne and Kass 2005). Phase 0 is the initial depolarization that is carried by the fast sodium current INa, followed by a brief rapid repolarization (Phase 1), carried by Itof/s and the plateau phase (Phase 2) which is balanced by calcium inflow through L-type calcium currents (ICa,L) and repolarizing potassium currents; the latter also terminate the action potential in Phase 3, the final repolarization. Phase 4 constituted the resting state with a potential around − 80 to − 90 mV and is mainly held by inwardly rectifying potassium currents (IK1).
Fig. 1.
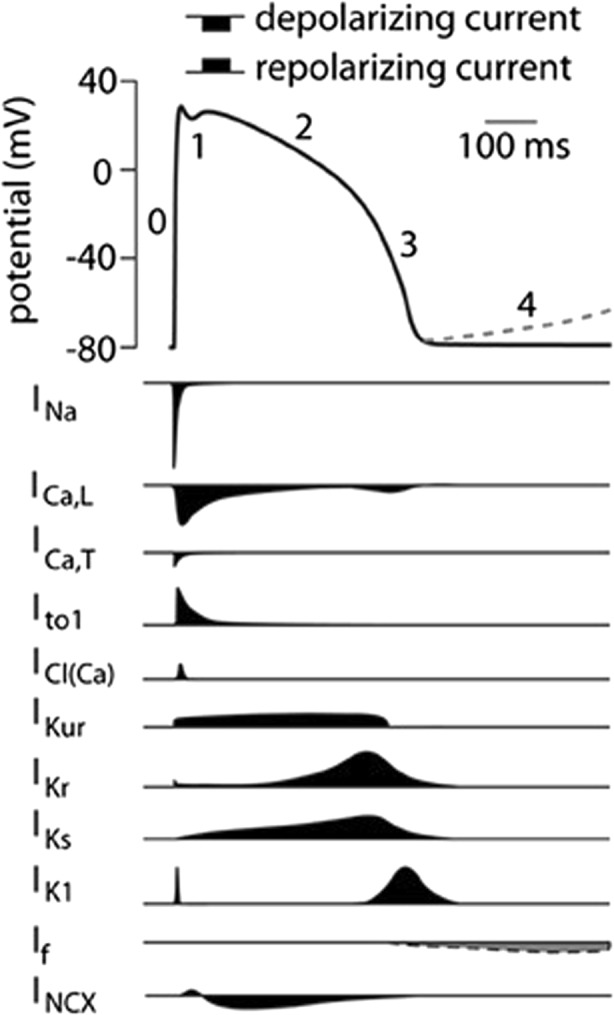
Cardiac action potential and its underlying ionic currents. Schematic representation of a human ventricular action potential. Numbers denote the different phases of the ventricular action potential outlined above. The dashed line represents phase 4 depolarization normally present in cells from the conduction system and not in ventricular cardiomyocytes. Underlying ionic membrane currents are depicted below. INa, Na+ current; ICa,L, L-type Ca2+ current; ICa,T, T-type Ca2+ current; Ito1, transient outward current type 1; ICl(Ca), Ca2+ activated Cl− current, also called Ito2; IKur, ultra rapid component of the delayed rectifier K+ current, IKr, rapid component of the delayed rectifier K+ current; IKs, slow component of the delayed rectifier K+ current; IK1, inward rectifier K+ current; If, funny current; INCX, Na+/Ca2+ exchange current (adapted from Hoekstra et al. 2012)
Fig. 2.

Morphologies of cardiac action potential throughout the heart. Schematic drawing of cardiac action potential morphologies in the SA and AV node and atrial and ventricular action potentials. RA right atrium, LA left atrium, RV right ventricle, LV left ventricle [adapted from Chahal and Somers, 2016]
Various forms of cardiac diseases and rhythm disturbances may lead to changes in ion channels and transporters. These alterations can be part of homeostatic response mechanism or may worsen cardiac dysfunction by maladaptive processes through further rhythm disturbances (Nattel et al. 2007).
Mechanism of arrhythmogenesis in HF
Remodeling processes during the development of heart failure increase the susceptibility to arrhythmias. These remodeling processes include changes in ion channel composition, function, and localization as well as histological changes including fibrosis.
Ventricular tachyarrhythmias are responsible for sudden cardiac death in up to 50% of heart failure patients (Kjekshus 1990). Therefore, implantation of implantable cardioverter defibrillators was shown to improve survival in heart failure collectives (Al-Khatib et al. 2017).
Not only ventricular but also atrial arrhythmias in HF patients contribute to increased morbidity and mortality. In particular, the occurrence of atrial fibrillation increases with worsening ejection fraction (Ehrlich et al. 2002; Lugenbiel et al. 2015). Normal sinus rhythm may also be slowed or impaired in HF due to abnormal sinoatrial node function (Opthof et al. 2000; Sanders et al. 2004, and Zicha et al. 2005). In a dog model, tachypacing-induced heart failure led to downregulation of HCN4 and HCN2 expression and contributed to HF-induced sinus node dysfunction (Zicha et al. 2005), as shown in Fig. 3a.
Fig. 3.

Mechanism of arrhythmogenesis in HF. Schematic drawing of mechanism underlying arrhythmogenesis in heart failure. a Highlights the changes in the SA node. b, c The mechanism of the initiation of EADs and DADs. d Reentry supporting remodeling processes [reproduced with permission from Nattel et al. 2007]
Early afterdepolarizations (EADs; Fig. 3b) represent a different trigger of arrhythmias. Remodeling of ion channels leads to a prolongation of the AP by reduction of repolarizing currents (IKs, IK1, and Ito) and increasing of depolarizing plateau currents (INaL). In contrast, delayed afterdepolarizations (Fig. 3c) are caused by calcium handling abnormalities in diastole. In HF, DADs are promoted by increased leak from the sarcoplasmatic reticulum (SR) and increased function levels of the sodium-calcium antiporter that depolarizes the cardiomyocyte by transporting on Ca2+ ion out for three Na+ ions transported intracellularly. Delayed afterdepolarizations are also promoted by the decrease of IK1 current in HF and subsequent increase in diastolic membrane resistance. If these afterdepolarizations suffice to activate, a new action potential arrhythmias can be started.
Reentry mechanisms are prone by remodeling processes that lead to slowed conduction and spatial inhomogeneities in conduction velocities. Reduction of connexin 43 and alteration in connexin 43 localization and spatially increased variability of action potential duration (Akar and Rosenbaum 2003) can increase susceptibility to reentry.
Specific ion channel changes and ion channel mutations leading to channelopathies and impaired cardiac function are described in the following sections.
Ion channel remodeling in heart failure
Multiple ion channels are remodeled during the development of heart failure. Several animal models of heart failure have been used to assess specific ion channel alternations in past studies. These models include mostly rapid ventricular pacing. Also, human samples obtained from HF patients were assessed. For a more detailed review, see, e.g., Nattel et al. 2007.
Potassium channels
Potassium channels are the most important ion channels contributing to repolarization in cardiomyocytes. A consistent feature of cardiomyocytes from patients or animals with HF is prolonged action potential duration (Nuss et al. 1999; Akar and Rosenbaum 2003; Janse 2004), and it becomes obvious that K+ channel remodeling is of great importance for repolarization abnormalities. Some of these changes in HF mimic inherited channelopathies that cause long QT syndrome. The interplay between the various K+ channel subtypes during the progression to HF is dynamic. The underlying transcriptional, posttranscriptional, and posttranslational remodeling of the individual K+ channel types changes their activity and their significance respectively, and they must be viewed together to understand their role in maintaining a stable heart rhythm.
Studies that examined changes in K+ channels in ventricular cardiomyocytes are listed in Table 1. Differences in remodeling are seen which may be attributed to different animal models or stages of HF. The so far most consistent observation is a downregulation of Ito in HF. This occurs by a downregulation on mRNA and protein level (Kaab et al. 1998; Zicha et al. 2004). Results for the principal IKr subunit ERK show unaltered expression levels. Most studies also confirmed a downregulation in the inward-rectifier current IK1.
Table 1.
Altered K+ currents in heart failure in ventricular cardiomyocytes
| Current | Changes in HF | References |
|---|---|---|
| Ito | ↓ | Beuckelmann et al. (1993), Kääb et al. (1996), Rozanski et al. (1997), Li et al. (2000), Tsuji et al. (2006), Pogwizd et al. (2001), Li et al. (2002), Zicha et al. (2004), Rose et al. (2005) |
| IKs | ↓ | Tsuji (2000), Tsuji et al. (2006) , Li et al. (2000), Li et al. (2002) |
| IKr | ↓ | Tsuji (2000), Tsuji et al. (2006) |
| → | Li et al. (2000), Li et al. (2002) | |
| ISS | → | |
| IK1 | ↓ | Beuckelmann et al.( 1993), Kääb et al. (1996), Li et al. (2000), Pogwizd et al. (2001), Li et al. (2002), Rose et al. (2005) |
| → | Rozanski et al. (1997), Tsuji (2000) | |
| IK2P | ↓ | Schmidt et al. (2017) |
Two-pore-domain potassium (K2P) channels modulate cellular excitability. The significance of stretch-activated cardiac K2P channels (K2P2.1, TREK-1, KCNK2; K2P4.1, TRAAK, KCNK4; K2P10.1, TREK-2, KCNK10) has only been studied in recent years. HF- and AF-associated downregulation of KCNK2 (K2P2.1) mRNA and protein levels suggest a mechanistic contribution to cardiac arrhythmogenesis (Schmidt et al. 2017).
Sodium channels
INa is important for the fast upstroke of the cardiac action potential (phase 0) and defines propagation velocity in normal cardiac tissue (Schram 2002). In HF peak, INa has been shown to be reduced by posttranslational reduction of the alpha subunit Nav1.5 and other posttranslational modifications (Ufret-Vincenty et al. 2001). Slowed cardiac conduction favors reentry mechanism and contributes to inefficiently synchronized cardiac conduction.
Abnormalities in inactivation of INa lead to a late persistent sodium current that antagonizes repolarization and leads to APD prolongation and EADs that cause arrhythmia (Ufret-Vincenty et al. 2001; Yu et al. 2018).
Chloride channels
Chloride channels in the heart may influence both repolarization and depolarization (Bers and Lederer, 2008). They have been implicated in cardiac arrhythmogenesis, myocardial hypertrophy, and heart failure, as well as cardioprotection against ischaemia–reperfusion (Duan 2009). The swelling sensitive chloride channel appears to be of particular interest. ICl,swell is distributed throughout the heart and is activated by osmotic and hydrostatic increase in cell volume, by changes in membrane tension, and by direct mechanical stretch (Baumgarten and Clemo 2003). ICl,swell leads to APD shortening, resting membrane potential depolarization, and reduces cell volume. In HF cardiomyocytes, constitutively active swelling-sensitive chloride channel currents ICl, swell have been observed (Clemo et al. 1999).
Altered calcium handling and calcium currents
Significant changes in Ca2+ handling occur during the development of HF (Hasenfuss et al. 1997, Lugenbiel et al. 2015). Detailed reviews have been provided by Bers and Guo (2005) and Beuckelmann and Erdmann (1992)).
The L-type-current itself has been studied in HF samples with different results, some showing a decrease of ICaL (Ouadid et al. 1995; Mukherjee et al. 1998; Li et al. 2000) and others not reporting significant changes. The membrane density of the L-type Ca2+ currents is decreased (Mukherjee et al. 1998, Chen 2002, He 2001); however, phosphorylation increases and leads to increases in single-channel open probability.
Proteins involved in Ca2+-induced Ca2+ release, cycling, and storage of Ca2+ in the SR are changed in activity (Fig. 4). Ryanodyne receptors (RyRs) are hyperphosphorylated by protein kinase A (PKA) and/or calmodulin-dependent kinase II (CamKII), causing abnormal Ca2+ leaks to the SR. NCX activity is enhanced promotion of the occurrence for triggered activity by DADs (Vermeulen et al. 1994, Pogwizd et al. 2001, Pogwizd and Bers 2002). Finally, SERCA function is reduced, both by reduction in expression level as well as the concomitant dephosphorylation of phospholamban that detaches from SERCA and reduces transporting capacities of Ca2+ to the SR. These changes in Ca2+ handling lead to higher diastolic Ca2+ and increased diastolic Ca2+ loss from the SR which finally impairs Ca2 +-induced Ca2+ release mechanism and excitation–contraction coupling with reduced contractile force.
Fig. 4.

Cycling of Ca2+ in a ventricular myocyte and changes in heart failure. RyR ryanodine receptor, PLB phopholamban, NaCaX 3 Na+-1 Ca2+-antiporter, SR sarcoplasmatic reticulum [reproduced with permission from Pogwizd und Bers 2002]
Altered connexin expression and distribution
Connexins form pores and enable cardiomyocytes to form electrical cell-to-cell coupling. The expression of the main ventricular connexin isoform, connexin 43, is downregulated in heart failure which was experimentally shown to reduce cell-to-cell coupling by luciferase yellow transfer (Dupont et al. 2001, Ai and Pogwizd 2005, Akar et al. 2004). Reduction of connexin amount of 90% has shown to decrease conduction velocity to 50% (van Rijen et al. 2004). But not only changes in protein amount increase susceptibility to arrhythmias. Additionally, HF causes changes in phosphorylation state of connexins that impair function (Akar et al. 2004) and localization disorders from the intercalated discs to lateral membrane occur (Severs et al. 2008, Kostin 2007).
Functionally altered connexin expression and function leads to conduction slowing in the failing heart (Akar et al. 2004; Ai and Pogwizd 2005) which contributed to mechanical dysfunction and adverse ventricular remodeling. In addition, APD heterogeneity is promoted by altered connexin expression pattern that favor reentry (Poelzing and Rosenbaum 2004).
T-tubular remodeling and ion channel sub-localization
Cardiomyocytes are functionally well-adapted cells that propagate electrical activity across their long axis. In support of structured and fast electrical transmission of cardiac electricity, T-tubules promote ion fluxes, e.g., calcium handling and excitation–contraction coupling and influence conduction velocity (Bers and Lederer 2008, Fu and Hong 2016, Manfra et al. 2017). In heart failure and cardiomyopathies, a decrease in T-tubules has been observed (Caldwell et al. 2014). The protein bridging integrator 1 (BIN1) appears to be essential for normal T-tubule formation and is downregulated in failing hearts. BIN 1 affects calcium channel trafficking. Downregulation of BIN1 in a mouse model decreased T-tubule formation accompanied by prolonged APD and increased prevalence of ventricular arrhythmias (Hong et al. 2014).
In addition to changes in T-tubular count and morphology, HF is characterized by redistribution of L-type Ca2+ channels from the T-tubules to the surface membrane in animal models.
Channelopathies and heart failure
Channelopathies are inherited genetic disorders with mutations in mainly specific ion channels that predispose patients to arrhythmias which among others include LQTS, short-QT syndrome (SQTS), Brugada syndrome (BrS), and catecholaminergic polymorphic ventricular tachycardia (CPVT) (Bezzina et al. 2015) (Table 2). Although most mutations accounting for channelopathies are located in specific ion channel genes, other mutations occur in genes encoding proteins involved in ion channel synthesis, membrane trafficking, and/or posttranslational modifications and regulation (Curran and Mohler 2015). The protein MOG1 for example interacts with Nav1.5 by regulating Nav1.5 membrane expression, and silencing of MOG1 was associated with BrS (Kattygnarath et al. 2011).
Table 2.
Inherited channelopathies and underlying gene mutations
| Syndrome | Gene | Functional alteration | References |
|---|---|---|---|
| LQTS | KCNQ1 | IKr ↓ | Ackerman et al. (2011), Schwartz et al. (2012) |
| KCNH2 | IKr ↓ | ||
| SCN5A | INa↑ | ||
| ANK2 | INa,K ↑ | ||
| Brugada syndrome | SCN5A | INa↓ | Brugada and Brugada (1992), Antzelevitch et al. (2007) |
| GPD1L | INa↓ | ||
| SCN1B | INa↓ | ||
| CACNA1C | ICa↓ | ||
| CACNB2B | ICa↓ | ||
| SQTS | KCNQ1 | IKr ↑ | Brugada et al. (2004), Bellocq et al. (2004), Priori et al. (2005), Antzelevitch et al. (2007), Harrel et al. (2015) |
| KCNH2 | IKr ↑ | ||
| KCNJ2 | IK1↑ | ||
| CACNA1C | ICa ↓ | ||
| CACNB2B | ICa ↓ | ||
| CPVT | RYR2 CASQ2 |
Abnormal Ca2+ release from SR | Bhuiyan et al. (2007), Napolitano and Priori (2007), Mohamed et al. (2007) |
Channelopathy syndromes are often characterized by specific ECG abnormalities either at baseline or during specific stressing conditions such as exercise (in CPVT and LQTS), fever (BrS), or pharmacological challenge (BrS). Depending on the respective ion channel mutation, function and/or expression level of the respective ionic current will change with effect on the cardiac action potential (Garcia-Elias and Benito 2018). The list of familial arrhythmia syndromes has been expanded by two other syndromes, early repolarization syndrome (Gourraud et al. 2013) and idiopathic ventricular fibrillation (Alders et al. 2009), which will not be discussed in this review.
Long-QT-syndrome
Clinically, long-QT-syndrome (LQTS) is characterized by prolongation of the QT-interval on the surface ECG that predispose patients to ventricular arrhythmias especially torsades-des-pointes-tachycardias. Patients present with syncope or during evaluation for risk of sudden cardiac death in families with affected members. The QT prolongation may be caused from either a decrease in repolarizing ionic currents or an increase in depolarizing currents late in the cardiac cycle (Moss and Kass 2005). Mutations in three ion channel genes KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) account for 75% of all LQTS cases (Ackerman et al. 2011, Schwartz et al. 2012). The genes associated with LQTS have been numerically ordered by the chronology of their discovery. Interestingly, triggers for arrhythmia differ among the LQTS subtypes. Genetic testing in affect patients and in first-degree relatives of patients who have a causative mutation for long-QT syndrome is recommended (Al-Khatib et al. 2017).
Channelopathies were initially thought to be mainly problems in cardiac electrophysiology. Nevertheless, recent studies show that patients with LQTS exhibit mechanical problems like diastolic dysfunction and prolonged contraction duration (Brado et al. 2017) and confirmed LQTS as an electromechanical disorder.
Short-QT-syndrome
Clinically, short-QT-syndrome (SQTS) is characterized by drastic shortening of the QT interval. As genetic causes gain-of-function mutations in potassium channel genes, KCNH2 (Brugada et al. 2004), KCNQ1 (Bellocq et al. 2004), KCNJ2 (Priori et al. 2005), or loss-of-function mutations in the L-type calcium channel (Antzelevitch et al. 2007) have been identified. These mutations shorten the effective refractory period and increase dispersion of repolarization and susceptibility to reentry in the ventricles and the atria.
Brugada syndrome
Clinically, Brugada syndrome is characterized by a typical ECG pattern (coved-type ST-segment elevation in right precordial leads and an increased risk to develop ventricular tachyarrhythmias and sudden cardiac death; Brugada and Brugada 1992). From experimental studies, it is known that the characteristic ECG pattern correlate with imbalances between depolarizing and repolarizing currents in the early phase of the cardiac action potential. Mutations in SCN5A account for the majority of Brugada syndrome cases with an identified gene mutation.
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a syndrome characterized by stress-induced polymorphic beats and ventricular tachycardias. CPVT-causing mutations encode the cardiac ryanodine receptor (RYR2) or for cardiac calsequestrin (CASQ2) in 60% of affected patients (Ackerman et al. 2011); in the remaining patients, the molecular basis still needs to be identified. Calcium leak from the sarcosplasmatic reticulum is assumed to be involved in the pathophysiology of AF in CPVT patients to trigger arrhythmia through calcium-handling abnormalities as outlined previously.
Ion channels cause electrical and structural overlap syndromes
Inherited arrhythmias were thought to be primarily electrical defects. Growing evidence shows that ion channel dysfunction can also contribute to myocardial disorders. As one example, the HCN4 overlap syndrome comprising sinus node dysfunction and non-compaction cardiomyopathy (Milano et al. 2014, Schweizer et al. 2014) is highlighted here to show the pathophysiological relevance of ion channels in both electrical and structural heart disease.
Sinus node dysfunction and biventricular non-compaction cardiomyopathy (NCCM) were diagnosed in a German family with an autosomal-dominant inheritance pattern. Sequencing revealed a novel hyperpolarization-activated cyclic nucleotide channel 4 (HCN4)-G482R mutation (Schweizer et al. 2014). HCN4, known to importantly contribute to cardiac pace-making, is expressed mainly in the sinoatrial node and the conduction system in the adult heart. Abundant expression of HCN4 throughout cardiac development suggests a putative contribution of this channel to signaling pathways involved in myocardial compaction (Fig. 5). Furthermore, “loss-of-function” mutations in HCN4 were also associated with atrial fibrillation and dilation of the aorta ascending (Duhme et al. 2013, Vermeer et al. 2016), supporting the view that ion channel mutations often do not solely show arrhythmic symptoms but contribute to complex electrical and structural phenotypes (Akhirome and Jay, 2015).
Fig. 5.

Spatial and temporal HCN4 activity. Schematic drawing of spatial and temporal expression pattern and activity of the cardiac pacemaker current HCN4 and possible explanation for the development of non-compaction cardiomyopathy. SAN sinoatrial node, C conus arteriosus, LA left atrium, LV left ventricle, RV right ventricle, RA right atrium, VCS ventricular conduction system [adapted with permission from Schweizer et al. 2014]
Impact of ion channels on HF therapy
Treatment of arrhythmias in HF
Arrhythmias contribute to significant morbidity and mortality in HF patients. Treatment of ventricular arrhythmias can be performed by diminishing potential causing factors as electrolyte imbalances and treatment of newly acquired ischemia by coronary interventions. Furthermore, acute ventricular arrhythmias can be terminated by electrical therapy, either cardioversion or defibrillation, and antiarrhythmic therapy or ablation regimen depending on the patient’s status.
Patients suffering from high risk for ventricular arrhythmias can be planned for primary prophylactic implantation of an implan.table cardioverter-defibrillator (ICD) according to the guidelines (Al-Khatib et al. 2017). Patients that already survived a sudden cardiac death are protected as well with an ICD for secondary prophylaxis.
Antiarrhythmic therapy concepts are limited and mostly include the class III antiarrhythmic drug amiodarone and have to take into considerations each patient’s individual risk profile.
Gene therapy for ion channel alterations in HF
Gene therapy has emerged as a powerful tool for targeting molecular mechanism implicated in heart failure. Refinements in vector technology, including the development of recombinant adeno-associated vectors, have allowed for safe, long-term, and efficient gene transfer to the myocardium. After success in animal models with mainly structural and calcium-handling-associated proteins as targets [junctophilin-2 (Reynolds et al. 2016) and S100A1 (Ritterhoff et al., 2015)] besides L-type Ca2+ current (Cingolani et al. 2007), first clinical trials in patients have already been started. However, after promising early-phase clinical trials, the more recent larger clinical trials targeting SERCA2 (CUPID gene therapy trial) (Greenberg et al. 2016) did not improve functional indices of heart failure. Unfortunately, transfection efficiency was not controlled in the study making it highly likely that transgene dosing and transfection rate were too low to provide beneficial effects and raise the need to re-evaluate vectors, delivery systems, targets, and endpoints (Hulot et al. 2016).
As new targets, ion channels also may play a major role despite major challenges that need to be faced. Individual cross-talk between ion channels, ion exchangers, and transporters has to be considered.
Conclusion
HF in general is a complex and heterogeneous disease in which various remodeling processes including changes in ion channel composition and function. Further basic research and clinical studies are needed to identify underlying molecular pathways of HF pathophysiology to provide the basis for improved patient care and individualized therapy based on individualized ion channel composition and remodeling.
Funding
This work was supported in part by grants from the University of Heidelberg, Faculty of Medicine (Physician Scientist Scholarship to A.K.R.), from the German Cardiac Society (DGK Scholarship to A.K.R) from the German Cardiac Society and the Hengstberger Foundation (Klaus-Georg and Sigrid Hengstberger Scholarship to D.T.), from the German Heart Foundation/German Foundation of Heart Research (F/08/14 to D.T.), from the Joachim Siebenreicher Foundation (to D.T.), and from the Ministry of Science, Research and the Arts Baden-Wuerttemberg (Sonderlinie Medizin to D.T.).
Compliance with ethical standards
D.T. reports receiving lecture fees/honoraria from Bayer Vital, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Medtronic, Pfizer Pharma, Sanofi-Aventis, St. Jude Medical, and ZOLL CMS, and research grant support from Daiichi Sankyo.
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
This article is part of a Special Issue on “Heart Failure Due to Non-Myofibrillar Defects” edited by Elisabeth Ehler and Katja Gehmlich.
Contributor Information
Ann-Kathrin Rahm, Email: ann-kathrin.rahm@med.uni-heidelberg.de.
Patrick Lugenbiel, Email: patrick.lugenbiel@med.uni-heidelberg.de.
Patrick A. Schweizer, Email: patrick.schweizer@med.uni-heidelberg.de
Hugo A. Katus, Email: sekretariat.katus@med.uni-heidelberg.de
Dierk Thomas, Phone: ++49 6221 568855, Email: dierk.thomas@med.uni-heidelberg.de.
References
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European heart rhythm association (EHRA) Europace. 2011;13(8):S. 1077–S. 1109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. 2005;96(1):S. 54–S. 63. doi: 10.1161/01.RES.0000152325.07495.5a. [DOI] [PubMed] [Google Scholar]
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93(7):S. 638–S. 645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95(7):S. 717–S. 725. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- Akhirome E, Jay PY. Rhythm genes sing more than one tune: noncanonical functions of cardiac ion channels. Circ Arrhythm Electrophysiol. 2015;8(2):261–262. doi: 10.1161/CIRCEP.115.002834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MW, Zeppenfeld K, Loh P, Koch KT, Demolombe S, Mannens MM, Bezzina CR, Wilde AA. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84(4):S. 468–S. 476. doi: 10.1016/j.ajhg.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, Gillis AM, Hlatky MA, Granger CB, Hammill SC, Joglar JA, Kay GN, Matlock DD, Myerburg RJ, Page RL (2017) 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the Heart Rhythm Society. Heart Rhythm 2017. 10.1010/j.jacc.2017.10.054
- Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):S. 442–S. 449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarten CM, Clemo HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol. 2003;82(1–3):S. 25–S. 42. doi: 10.1016/s0079-6107(03)00003-8. [DOI] [PubMed] [Google Scholar]
- Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, Baró I, Wilde AA (2004) Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109: 2394–2397 [DOI] [PubMed]
- Bers DM, Lederer WJ (2008) Excitation-contraction coupling and cardiac contractile force, 2nd edn. Springer, Dordrecht
- Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci. 2005;1047:S. 86–S. 98. doi: 10.1196/annals.1341.008. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Erdmann E. Ca2+-currents and intracellular Ca2+i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol. 1992;87(Suppl 1):S. 235–S. 243. doi: 10.1007/978-3-642-72474-9_19. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Näbauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73(2):S. 379–S. 385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116(12):S. 1919–S. 1936. doi: 10.1161/CIRCRESAHA.116.304030. [DOI] [PubMed] [Google Scholar]
- Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, Postma AV, van Langen I, Mannens MM, Wilde AA. Expanding spectrum of human RYR2-related disease new electrocardiographic, structural, and genetic features. Circulation. 2007;116(14):S. 1569–S. 1576. doi: 10.1161/CIRCULATIONAHA.107.711606. [DOI] [PubMed] [Google Scholar]
- Brado J, Dechant MJ, Menza M, Komancsek A, Lang CN, Bugger H, Foell D, Jung BA, Stiller B, Bode C, Odening KE. Phase-contrast magnet resonance imaging reveals regional, transmural, and base-to-apex dispersion of mechanical dysfunction in patients with long QT syndrome. Heart Rhythm. 2017;14(9):S. 1388–S. 1397. doi: 10.1016/j.hrthm.2017.04.045. [DOI] [PubMed] [Google Scholar]
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death a distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol. 1992;20(6):S. 1391–S. 1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):S. 30–S. 35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, Dibb KM, Trafford AW. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1) Circ Res. 2014;115(12):986–996. doi: 10.1161/CIRCRESAHA.116.303448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahal AA, Somers VK. Ion channel remodeling-a potential mechanism linking sleep apnea and sudden cardiac death. J Am Heart Assoc. 2016;5(8):e004195. doi: 10.1161/JAHA.116.004195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91(6):S. 517–S. 524. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- Cingolani E, Ramirez Correa GA, Kizana E, Murata M, Cho HC, Marbán E. Gene therapy to inhibit the calcium channel beta subunit physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ Res. 2007;101(2):S. 166–S. 175. doi: 10.1161/CIRCRESAHA.107.155721. [DOI] [PubMed] [Google Scholar]
- Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. 1999;84(2):S. 157–S. 165. doi: 10.1161/01.res.84.2.157. [DOI] [PubMed] [Google Scholar]
- Curran J, Mohler P. Alternative paradigms for ion channelopathies: disorders of ion channel membrane trafficking and posttranslational modification. Annu Rev Physiol. 2015;77:S. 505–S. 524. doi: 10.1146/annurev-physiol-021014-071838. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol. 2003;98(3):S. 137–S. 148. doi: 10.1007/s00395-003-0409-8. [DOI] [PubMed] [Google Scholar]
- Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J Physiol. 2009;587(Pt 10):S. 2163–S. 2177. doi: 10.1113/jphysiol.2008.165860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhme N, Schweizer PA, Thomas D, Becker R, Schröter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus HA, Koenen M. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J. 2013;34(35):2768–2775. doi: 10.1093/eurheartj/ehs391. [DOI] [PubMed] [Google Scholar]
- Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33(2):S. 359–S. 371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Nattel S, Hohnloser SH. Atrial fibrillation and congestive heart failure. Specific considerations at the intersection of two common and important cardiac disease sets. J Cardiovasc Electrophysiol. 2002;13(4):S. 399–S. 405. doi: 10.1046/j.1540-8167.2002.00399.x. [DOI] [PubMed] [Google Scholar]
- Fu Y, Hong T. BIN1 regulates dynamic t-tubule membrane. Biochim Biophys Acta. 2016;1863(7 Pt B):S. 1839–S. 1847. doi: 10.1016/j.bbamcr.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Elias A, Benito B (2018) Ion Channel disorders and sudden cardiac death. Int J Mol Sci 19(3) [DOI] [PMC free article] [PubMed]
- Gourraud J-B, Le Scouarnec S, Sacher F, Chatel S, Derval N, Portero V, Chavernac P, Sandoval JE, Mabo P, Redon R, Schott J-J, Le Marec H, Haïssaguerre M, Probst V. Identification of large families in early repolarization syndrome. J Am Coll Cardiol. 2013;61(2):S. 164–S. 172. doi: 10.1016/j.jacc.2012.09.040. [DOI] [PubMed] [Google Scholar]
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2)a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet (London, England) 2016;387(10024):S. 1178–S. 1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Bentzen BH, Sorensen US, Diness JG. Cardiac ion channels and mechanisms for protection against atrial fibrillation. Rev Physiol Biochem Pharmacol. 2012;162:S. 1–S.58. doi: 10.1007/112_2011_3. [DOI] [PubMed] [Google Scholar]
- Hancox JC, James AF, Marrion NV, Zhang H, Thomas D. Novel ion channel targets in atrial fibrillation. Expert Opin Ther Targets. 2016;20(8):S. 947–S. 958. doi: 10.1517/14728222.2016.1159300. [DOI] [PubMed] [Google Scholar]
- Harrell DT, Ashihara T, Ishikawa T, Tominaga I, Mazzanti A, Takahashi K, Oginosawa Y, Abe H, Maemura K, Sumitomo N, Uno K, Takano M, Priori SG, Makita N. Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. Int J Cardiol. 2015;190:S. 393–S. 402. doi: 10.1016/j.ijcard.2015.04.090. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Res Cardiol. 1997;92(S1):S. 87–S. 93. doi: 10.1007/BF00794072. [DOI] [PubMed] [Google Scholar]
- He J. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49(2):S. 298–S. 307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114(9):S. 1483–S. 1499. doi: 10.1161/CIRCRESAHA.114.302226. [DOI] [PubMed] [Google Scholar]
- Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol. 2012;3:346. doi: 10.3389/fphys.2012.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J, Gorelik J, Marbán E, Jan LY, Shaw RM (2014) Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. Jun;20(6):624-32 [DOI] [PMC free article] [PubMed]
- Hulot JS, Ishikawa K, Hajjar RJ. Gene therapy for the treatment of heart failure promise postponed. Eur Heart J. 2016;37(21):S. 1651–S. 1658. doi: 10.1093/eurheartj/ehw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse M. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61(2):S. 208–S. 217. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Kääb S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78(2):S. 262–S. 273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98(14):S. 1383–S. 1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- Kattygnarath D, Maugenre S, Neyroud N, Balse E, Ichai C, Denjoy I, Dilanian G, Martins RP, Fressart V, Berthet M, Schott JJ, Leenhardt A, Probst V, Le Marec H, Hainque B, Coulombe A, Hatem SN, Guicheney P. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011;4(3):S. 261–S. 268. doi: 10.1161/CIRCGENETICS.110.959130. [DOI] [PubMed] [Google Scholar]
- Kjekshus J. Arrhythmias and mortality in congestive heart failure. Am J Cardiol. 1990;65(19):42I–48I. doi: 10.1016/0002-9149(90)90125-k. [DOI] [PubMed] [Google Scholar]
- Kostin S. Zonula occludens-1 and connexin 43 expression in the failing human heart. J Cell Mol Med. 2007;11(4):S. 892–S. 895. doi: 10.1111/j.1582-4934.2007.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101(22):S. 2631–S. 2638. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283(3):H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA, Thomas D. Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS One. 2015;10(3):e0116395. doi: 10.1371/journal.pone.0116395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfra O, Frisk M, Louch WE. Regulation of cardiomyocyte T-tubular structure: opportunities for therapy. Curr Heart Fail Rep. 2017;14(3):S. 167–S. 178. doi: 10.1007/s11897-017-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mene-Afejuku TO, López PD, Akinlonu A, Dumancas C, Visco F, Mushiyev S, Pekler G (2018) Atrial Fibrillation in Patients with Heart Failure: Current State and Future Directions. Am J Cardiovasc Drugs. 10.1007/s40256-018-0276-1 [DOI] [PubMed]
- Milano A, Vermeer AM, Lodder EM, Barc J, Verkerk AO, Postma AV, van der Bilt IA, Baars MJ, van Haelst PL, Caliskan K, Hoedemaekers YM, Le Scouarnec S, Redon R, Pinto YM, Christiaans I, Wilde AA, Bezzina CR. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol. 2014;64(8):S. 745–S. 756. doi: 10.1016/j.jacc.2014.05.045. [DOI] [PubMed] [Google Scholar]
- Mohamed U, Napolitano C, Priori SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007;18(7):S. 791–S. 797. doi: 10.1111/j.1540-8167.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Kass RS. Long QT syndrome from channels to cardiac arrhythmias. J Clin Invest. 2005;115(8):S. 2018–S. 2024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Hewett KW, Walker JD, Basler CG, Spinale FG. Changes in L-type calcium channel abundance and function during the transition to pacing-induced congestive heart failure. Cardiovasc Res. 1998;37(2):S. 432–S. 444. doi: 10.1016/s0008-6363(97)00128-4. [DOI] [PubMed] [Google Scholar]
- Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4(5):S. 675–S. 678. doi: 10.1016/j.hrthm.2006.12.048. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87(2):S. 425–S. 456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1(1):S. 62–S. 73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):S. 1205–S. 1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Kääb S, Kass DA, Tomaselli GF, Marbán E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Phys. 1999;277(1 Pt 2):H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- Opthof T, Coronel R, Rademaker HM, Vermeulen JT, Wilms-Schopman FJ, Janse MJ. Changes in sinus node function in a rabbit model of heart failure with ventricular arrhythmias and sudden death. Circulation. 2000;101(25):S. 2975–S. 2980. doi: 10.1161/01.cir.101.25.2975. [DOI] [PubMed] [Google Scholar]
- Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol. 1995;25(2):S. 282–S. 291. doi: 10.1097/00005344-199502000-00014. [DOI] [PubMed] [Google Scholar]
- Poelzing S, Rosenbaum DS (2004) Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 287(4):H1762–70 [DOI] [PubMed]
- Pogwizd SM, Bers DM. Calcium cycling in heart failure. The arrhythmia connection. J Cardiovasc Electrophysiol. 2002;13(1):S. 88–S. 91. doi: 10.1046/j.1540-8167.2002.00088.x. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure roles of sodium-calcium exchange, inward rectifier potassium current, and residual -adrenergic responsiveness. Circ Res. 2001;88(11):S. 1159–S. 1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96: 800–807 [DOI] [PubMed]
- Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, Barreto-Torres G, Thuerauf DJ, Doroudgar S, Glembotski CC, Wehrens XH. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int J Cardiol. 2016;225:S. 371–S. 380. doi: 10.1016/j.ijcard.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritterhoff J, Völkers M, Seitz A, Spaich K, Gao E, Peppel K, Pleger ST, Zimmermann WH, Friedrich O, Fink RH, Koch WJ, Katus HA, Most P. S100A1 DNA-based inotropic therapy protects against proarrhythmogenic ryanodine receptor 2 dysfunction. Mol Ther. 2015;23(8):S. 1320–S. 1330. doi: 10.1038/mt.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O'Rourke B, Kass DA, Marbán E, Tomaselli GF. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005;288(5):H2077–H2087. doi: 10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanski GJ, Xu Z, Whitney RT, Murakami H, Zucker IH. Electrophysiology of rabbit ventricular myocytes following sustained rapid ventricular pacing. J Mol Cell Cardiol. 1997;29(2):S. 721–S. 732. doi: 10.1006/jmcc.1996.0314. [DOI] [PubMed] [Google Scholar]
- Sanders P, Kistler PM, Morton JB, Spence SJ, Kalman JM. Remodeling of sinus node function in patients with congestive heart failure. Reduction in sinus node reserve. Circulation. 2004;110(8):S. 897–S. 903. doi: 10.1161/01.CIR.0000139336.69955.AB. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Wiedmann F, Kallenberger SM, Ratte A, Schulte JS, Scholz B, Müller FU, Voigt N, Zafeiriou MP, Ehrlich JR, Tochtermann U, Veres G, Ruhparwar A, Karck M, Katus HA, Thomas D. Stretch-activated two-pore-domain (K2P) potassium channels in the heart focus on atrial fibrillation and heart failure. Prog Biophys Mol Biol. 2017;130(Pt B):S. 233–S. 243. doi: 10.1016/j.pbiomolbio.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation a translational appraisal. Physiol Rev. 2011;91(1):S. 265–S. 325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- Schram G. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90(9):S. 939–S. 950. doi: 10.1161/01.res.0000018627.89528.6f. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome from genetics to management. Circ Arrhythm Electrophysiol. 2012;5(4):S. 868–S. 877. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer PA, Schröter J, Greiner S, Haas J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A, Koenen M, Meder B, Katus HA, Thomas D. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol. 2014;64(8):S. 757–S. 767. doi: 10.1016/j.jacc.2014.06.1155. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80(1):S. 9–S.19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res. 2000;48(2):S. 300–S. 309. doi: 10.1016/s0008-6363(00)00180-2. [DOI] [PubMed] [Google Scholar]
- Tsuji Y, Zicha S, Qi X-Y, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113(3):S. 345–S. 355. doi: 10.1161/CIRCULATIONAHA.105.552968. [DOI] [PubMed] [Google Scholar]
- Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001;276(30):S. 28197–S. 28203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, Bakker JM de (2004) Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 109 (8), S. 1048–S. 1055 [DOI] [PubMed]
- Vermeer AMC, Lodder EM, Thomas D, Duijkers FAM, Marcelis C, van Gorselen EOF, Fortner P, Buss SJ, Mereles D, Katus HA, Wilde AAM, Bezzina CR, Boekholdt SM, Schweizer PA, Christiaans I. Dilation of the aorta ascendens forms part of the clinical spectrum of HCN4 mutations. J Am Coll Cardiol. 2016;67(19):2313–2315. doi: 10.1016/j.jacc.2016.01.086. [DOI] [PubMed] [Google Scholar]
- Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klöpping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28(10):S. 1547–S. 1554. doi: 10.1093/cvr/28.10.1547. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 2008;5(6 Suppl):S. 1–S. 6. doi: 10.1016/j.hrthm.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Li G, Huang CLH, Lei M, Wu L. Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflugers Arch - Eur J Physiol. 2018;470(3):S. 461–S. 469. doi: 10.1007/s00424-017-2079-7. [DOI] [PubMed] [Google Scholar]
- Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561(Pt 3):S. 735–S. 748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha S, Fernández-Velasco M, Lonardo G, L'Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. 2005;66(3):S. 472–S. 481. doi: 10.1016/j.cardiores.2005.02.011. [DOI] [PubMed] [Google Scholar]