

The Sydney Heart Bank (SHB) has contributed and continues to contribute samples of human left ventricles to our understanding of molecular mechanisms underlying heart failure (Li et al. 2013; Li and dos Remedios 2015). The great majority of the publications concern myofibrillar proteins. To date, the SHB has published about 120 research papers with about 60 collaborators around the world. About 20% of them involved genes and proteins that did not involve sarcomeric proteins.
Working with the SHB
The authors took the position that we should not expect to identify all of the causes of heart failure mechanisms, and that the best way forward would be to collaborate with the many laboratories in searching for an answer. The major obstacle to this aspiration was the need to convince these labs that your heart samples were unique in several ways. First we had to dispel the notion that our samples were degraded, or at least partially degraded due to the time between harvesting and preservation in liquid nitrogen. That obstacle would only be dispelled by sending out samples and wait for the results. This was particularly true for the donor hearts.
Minimal delay collecting donor hearts
Many cardiac research laboratories assumed that the non-failing donors (some wrongly called them “controls”) were obtained post-mortem, i.e., obtained 12–24 h after death. In fact, our donors come from healthy individuals on life support that are harvested by the heart transplant surgeons soon after the declaration of brain death by the transplant coordinators. They were rejected by the transplant surgeons for a range of reasons, commonly because no tissue-type match could be found in the heart failure clinic at St Vincent’s Hospital, but sometimes, the hearts were suspected of having coronary vascular disease, but usually this was dispelled once we began sectioning the heart before freezing. Sometimes, the hearts were simply too young.
All donor hearts were quickly perfused with ice-cold cardioplegia that rapidly chilled the heart and arrested the heart-beat. When the valves were required for transplantation, the hearts were rapidly transported to St Vincent’s Hospital in Sydney. When a donor was a juvenile or infant, the valves were not required and were delivered to the SHB team. Infant and juvenile hearts were obtained very precious and, thanks to the care and diligence of the clinical teams in participating hospitals particularly at the John Hunter Hospital in Newcastle. We are very grateful to those families of those very young donors who hoped some good would come from the death of their children.
This bank of non-diseased donors turned out to be more sought-after than the failing hearts. Today after nearly 30 years, we have assembled a substantial bank of hearts where the older heart groups were used as age-matched (and sex-matched) donors for the failing hearts. Figure 1 shows the age distribute of the donor hearts. The SHB currently has 120 of these hearts.
Fig. 1.

The age distribution of the non-diseased donor hearts in the SHB (5 years per group). A unique feature of these donor heart samples is they enable laboratories to investigate changes in expression of genes and proteins over a period of six decades
The failing hearts were also preserved in liquid nitrogen (− 194 °C) as close as possible to the time when they were removed from the transplant patient. They remain perfused by the cardiac by-pass pump until the moment the aorta was cross-clamped. Within 2–3 min, the patient aortic and pulmonary semilunar valved were removed by the Sydney Valve Clinic personnel and the first 10–20 1 g samples of left ventricle free wall were frozen within 5–15 min. This speed of collection is probably the single main factor responsible for the high quality of the SHB heart samples.
Non-sarcomeric causes of human heart failure
Figure 2 summarizes papers published by the SHB and its collaborators (Pleuss et al. 2015). Our major contribution was the selection and provision of high-quality failing and non-failing samples of left ventricle. When the research was carried out in laboratories outside of the Sydney, heart samples (usually were left ventricle) were transported to their final destination in a nitrogen dry shipper which maintained temperatures at about − 180 °C. The dewar shipped maintained temperature for about 2 weeks, which took the pressure of the shippers when deliveries took more than the usual few days.
Fig. 2.
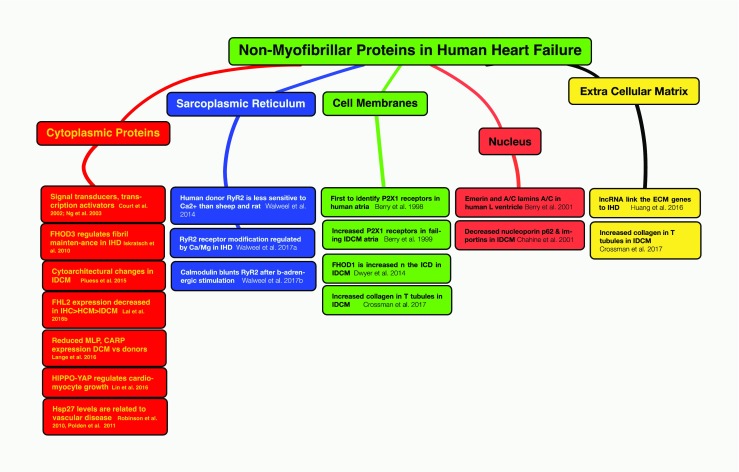
This figure summarizes the contributions of the SHB to our knowledge of the role of extra-sarcomeric proteins in human heart failure. These proteins are located in the cardiomyocyte cytoplasm, the internal membranes of the sarcoplasmic reticulum, the sarcolemma of cardiomyocytes including the intercalated disc membranes, the nucleus, and the proteins of the extracellular space
Signal transduction proteins (STAT and MAPK)
We were the first to report the role of signal transducers and activators of transcription (STATs) and mitogen-activated protein kinases (MAPKs) using LV samples from patients with ischemic heart disease (IHD, 11 samples), dilated cardiomyopathy (DCM, 6 samples), and non-diseased donor LV (4 samples). Significant expression was observed in cardiomyocytes that had a punctate distribution and was not associated with the sarcomeric repeats. MAPKs expression was also post-translationally modified (phosphorylated) only in IHD. Different members of the STAT family were also phosphorylated in both forms of heart failure (Court et al. 2002). In a slightly larger (IHD = 11, DCM = 9, age-matched donors = 9) study of STATs1, 3, 5, and 6, only STAT1 and STAT5 were phosphorylated in IHD. MAPK p38 was only phosphorylated in IHD. They concluded that only STAT1 and STAT5 are likely to play a role in regulating human heart failure (Ng et al. 2003).
FHOD3
Iskratsch et al. (2010) identified a novel striated muscle-specific splice variant of the formin, FHOD3 that introduces a casein kinase 2 phosphorylation site. The specific targeting of muscle FHOD3 to the cardiac myofibrils is abolished in phosphor-mutants or by the inhibition of CK2. FHOD3 efficiently promotes the polymerization of cardiomyocyte actin filaments and downregulation of its expression severely affects myofibril integrity. In murine and human cardiomyopathy, FHOD3 expression is reduced with a concomitant isoform switch and change of subcellular targeting. The data suggest that a muscle-specific isoform of FHOD3 is required for the maintenance of the contractile structures in heart muscle and that its function is regulated by post-translational modification.
FHOD1
Dwyer et al. characterized the expression and subcellular localization of FHOD3’s closest relative, FHOD1, in the heart. Confocal microscopy shows it is mainly located at the intercalated disc, the cell-cell contact between adjacent cardiomyocytes. It is also partially associated with the myofibrils. Subcellular targeting of FHOD1 is probably mediated by its N-terminal domain, since expression constructs lacking this domain show aberrant localization in primary cultures of neonatal rat cardiomyocytes. Finally, it was shown that in contrast to FHOD3, expression of FHOD1 is increased in DCM LV, suggesting that the two formins play distinct roles and are differentially regulated in cardiomyocytes (Dwyer et al. 2015).
LIM domain proteins
The role of the LIM domain family of proteins and heart failure is an ongoing interest at the SHB (Li et al. 2012). Bovill et al. (2008) first reported that Four-and-a-half LIM-protein 2 (FHL2) levels were preserved in non-failing human left ventricle samples, but levels were reduced by 53% in DCM hearts with gross dilatation. FHL2 was localized in the perinuclear Golgi apparatus complex. This change is associated with a reduction in the levels of several kinases, which might disrupt myocardial energy metabolism in heart failure. Lu et al. (2012) and later Lal et al. (2016) reported that FHL1 was significantly upregulated in rat models of HCM. Subsequently, Schreckenbach et al. (2013) reported mutations in the FHL1 gene where four members of a family died of heart failure under 14 years of age. Another LIM domain protein, muscle LIM protein (MLP) is a well-established cause of human heart failure and is known to cause both HCM and DCM in humans (Bos et al. 2006). It is the loss of MLP from the cytoskeleton that may be the cause (Boateng et al. 2007). More recently, Lange et al. (2016) concluded that the main role of MLP is to inhibit a kinase (PKCa) located at the intercalated disc and that this inhibition leads to heart failure.
Hippo-Yes-associated protein
The transcriptional co-activator Yes-associated protein (YAP) is a key driver of heart growth. It binds to several transcription factors to activate transcription of cell-cycle and cell-survival genes, thus promoting organ growth. YAP and Hippo are essential for normal heart development. YAP activation diminishes with age and Lin et al.’s paper shows that regulation of YAP activity is crucial for normal cardiac growth control. Moreover, they suggest that unknown mechanisms may suppress YAP mitogenic activity in the post-natal heart (Lin et al. 2016).
Coronary artery disease and Hsp27
Robinson et al. (2010) compared non-diseased human coronary arteries with those from IHD to determine the levels of phosphorylated heat-shock-protein, Hsp27, using Western blots. They reported that its expression is specifically decreased in IHD vessels, but not in non-diseased vessels. Hsp27 is expressed in both smooth muscle and endothelial cells. Two-dimensional difference gel electrophoresis detected proteins that were identified by mass spectrometry. The results are consistent with the hypothesis that phospho-Hsp27 protects against vascular disease possibly by stabilizing the actin cytoskeleton within endothelial and/or smooth muscle cells. In another report, we undertook the identification of basic proteins (pH 6–11) of the human LV using 2-DE. Tissue was lysed and proteins separated on pH 6–11 IPG strips followed by separation on 12% SDS polyacrylamide gels in the second dimension. Proteins were then identified by mass spectrometry and analyzed using in-house analysis. The proteome map contains 176 identified spots with 151 unique proteins and is available on the UCD-2DPAGE database at http://proteomics-portal.ucd.ie:8082. This reference map and others will aid future studies of heart diseases such as IDCM and IHD (Polden et al. 2011).
Sarcoplasmic reticulum
Small samples of LV tissue were used to isolate SR vesicles and measure their sensitivity to Ca2+ and Mg2+. The performance of the membrane RyR2 Ca2+ channels revealed they are sensitive to post-translational modifications including hyperphosphorylation of key serines residues and a reduction in free thiols. These changes did not depend on the nature of the heart failure Walweel et al. 2014, 2017a, b.
P2X receptors, intercalated discs, and sarcolemma
Berry et al. (1998) were the first to identify ATP-specific P2X1 receptors in the human heart. Using Western blots, they noted changes in sub-populations of these channels that were related to the LV ejection fraction of IDCM patients. In 1999, we reported (Berry et al. 1999) Western blots showing increases in the atria (but no significant increase in the ventricles) in DCM samples compared to healthy donor samples. In the next issue of the same journal, Hou et al. (1999) reported that P2X1 mRNA levels were significant increased in both atria and ventricles, and that P2X1 receptors were upregulated in congestive heart failure. However, by the following year (Berry et al. 2000), we had developed more specific antibodies, and, with the increase in the number of SHB hearts, we were compared P2X1 expression (Western blots) in 14 IDCM and 11 healthy donor hearts and observed a significantly increased (~ 80%) expression in both atria ventricles in DCM patients. The importance of these observations was highlighted in a recent review (Ralevic 2015) who pointed out the possible relevance of these receptors as therapeutic targets.
Changes in the nucleus
Lamins are nuclear protein that came to prominence when it was discovered that mutations in the rod domain of both lamin A and lamin C are a known cause of DCM (Fatkin et al. 1999), but are they homodimers or heterodimers? In 2001, we used Western blots to addressed this question. We analyzed LV samples from 10 DCM LV samples and non-diseased donors and unexpectedly their evidence led them to conclude the nuclei carried lamin A and lamin C as well as lamin C2, a short lamin associated with the nuclear envelope (Berry et al. 2001). During the hypertrophy of cardiomyocytes, there is an increase in the size and expression of cytoskeletal proteins and a reactivation of a fetal gene program. We investigated the role of a range of nuclear import and export molecules that control of hypertrophy in rat and human heart failure. In cardiomyocytes from patients with DCM, nuclear size was increased and there were changes in nuclear transport molecules related to increased export and decreased import activity. These changes occur early in hypertrophic development, suggesting that hypertrophy can be prevented, or even reversed, by targeting import/export, which may open new therapeutic opportunities (Chahine et al. 2014).
Extracellular matrix proteins
The SHB provided 15 failing (IHD) and 15 non-failing LV to Bill Pu’s laboratory at Harvard Medical School to test his hypothesis that the expression and function of long non-coding RNAs (lncRNAs) are differentially regulated in diseased hearts. They performed RNA deep sequencing of protein-coding and non-coding RNAs and found changes in 35 lncRNAs. Expression correlation coefficient analyses revealed a strong association between lncRNAs and extracellular matrix (ECM) protein-coding genes. Overexpressed or knocked-down experiments suggest that lncRNAs are important regulators of fibrosis and the expression of ECM synthesis genes. Thus, lncRNAs may be novel regulators of heart function and cardiac disorders (Huang et al. 2016). Considering the recent observation by David Crossman and his colleagues, these changes in the ECM are consistent with increased collagen around the openings of the T tubules in the sarcolemma, which particularly included type IV collagen (Crossman et al. 2017).
Access to SHB heart samples?
If the above discussion has stimulated readers to attempt using human heart samples to test important hypotheses, they should contact the Sydney Heart Bank via any of the first three authors. We will work with you to design experiments that are statistically relevant, and perhaps avoid a decision to employ an animal model, at least as a first step. You will need (1) a clear set of aims, (2) a rationale for the amounts of tissue required, and (3) current human ethics approval (or a waiver) for the project. The independent SHB Board will make the final decision and the University of Sydney Research office will arrange for a Material Transfer agreement with the receiving institution. There are no charges for the tissue or the associated clinical data, but the recipient is asked to actual cover the FedEx cost of transporting the nitrogen-vapor dewar to the recipient’s laboratory and returning the empty dewar to the SHB. The process typically takes 3–4 months.
Compliance with ethical standards
C. G. dos Remedios declares that he has no conflicts of interest. A. Li declares that she has no conflicts of interest. S. Lal declares that he has no conflicts of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
This article is part of a Special Issue on ‘Heart Failure Due to Non-Myofibrillar Defects’ edited by Elisabeth Ehler and Katja Gehmlich.
C. G. dos Remedios, A. Li and S. Lal contributed equally to this work.
References
- Berry D, Yao M, Barden JA, Balcar VJ, Hansen MA. Alterations in the expression of P2X1 receptors in failing and non-diseased human atria. Electrophoresis. 1998;19:856–859. doi: 10.1002/elps.1150190542. [DOI] [PubMed] [Google Scholar]
- Berry D, Barden JA, Balcar VJ, Keogh A, dos Remedios CG. Increase in expression of P2X1 receptors in the atria of patients suffering from dilated cardiomyopathy. Electrophoresis. 1999;20:2059–2064. doi: 10.1002/(SICI)1522-2683(19990701)20:10<2059::AID-ELPS2059>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Berry D, Balcar VJ, Barden JA, Keogh A, dos Remedios CG. Determination of P2X1α-sarcoglycan (adhalin) expression levels in failing human dilated cardiomyopathic left ventricles changes in P2X1 receptors in human left ventricles and their relationship to the acto-ATPase α-sarcoglycan (adhalin) Electrophoresis. 2000;21:3857–3862. doi: 10.1002/1522-2683(200011)21:17<3857::AID-ELPS3857>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Berry DA, Keogh A, dos Remedios CG. Nuclear membrane proteins in failing human dilated cardiomyopathy. Proteomics. 2001;1:1507–1512. doi: 10.1002/1615-9861(200111)1:12<1507::AID-PROT1507>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Boateng SY, Belin RJ, Geenen DL, Margulies KB, Martin JL, et al. Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am J Physiol Heart Circ Physiol. 2007;292:H259–H269. doi: 10.1152/ajpheart.00766.2006. [DOI] [PubMed] [Google Scholar]
- Bos JM, Poley RN, Ny M, Tester DJ, Xu X, et al. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78–85. doi: 10.1016/j.ymgme.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovill E, Westaby S, Crisp A, Jacobs S, Shaw T. Reduction of four-and-a-half LIM-protein 2 expression occurs in human left ventricular failure and leads to altered localization and reduced activity of metabolic enzymes. J Thorac Cardiovasc Surg. 2008;137:853–861. doi: 10.1016/j.jtcvs.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Chahine MN, Mioulane M, Sikkel MB, O’Gara P, dos Remedios CG, et al. Nuclear pore rearrangements and nuclear trafficking in cardiomyocytes from rat and human failing hearts. Cardiovasc Res. 2014;105:31–43. doi: 10.1093/cvr/cvu218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court NW, dos Remedios CG, Cordell J, Bogoyevitch MA. Cardiac expression and subcellular localization of the p38 mitogen-activated protein kinase member, stress-activated protein kinase-3 (SAPK3) J Mol Cell Cardiol. 2002;34:413–426. doi: 10.1006/jmcc.2001.1523. [DOI] [PubMed] [Google Scholar]
- Crossman DJ, Shen X, Jüllig M, Munro M, Hou Y, et al. Increased collagen within the transverse tubules in human heart failure. Cardiovasc Res. 2017;113:879–891. doi: 10.1093/cvr/cvx055. [DOI] [PubMed] [Google Scholar]
- Dwyer J, Pluess M, Iskratsch T, dos Remedios CG, Ehler E. The formin FHOD1 in cardiomyocytes. Anat Rec. 2015;297:1560–1570. doi: 10.1002/ar.22984. [DOI] [PubMed] [Google Scholar]
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, et al. Misspence mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- Hou M, Malmsjö M, Möller S, Pantev E, Bergdahl A, Zhao XH, Sun XY, Hedner T, Edvinsson L, Erling D. Increase in cardiac P2X1- and P2Xy2-receptor mRNA levels in congestive heart failure. Life Sci. 1999;65:1195–1206. doi: 10.1016/S0024-3205(99)00353-7. [DOI] [PubMed] [Google Scholar]
- Huang Z-P, Ding Y, Chen J, Wu G, Kataoka M, Wang D-Z, et al. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc Res. 2016;112:543–554. doi: 10.1093/cvr/cvw201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskratsch T, Lange S, Kho AL, dos Remedios CG, Ehler E. Formin follows function: a muscle specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol. 2010;191:1159–1172. doi: 10.1083/jcb.201005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal S, Li A, dos Remedios C. Limitations in translating animal studies to humans in cardiovascular disease. J Cardiovasc Transl Res. 2016;9:165–166. doi: 10.1007/s12265-016-9676-2. [DOI] [PubMed] [Google Scholar]
- Lange S, Gehmlich K, Lun AS, Blondelle J, Hooper C, et al. MLP and CARP are linked to chronic PKCa signalling in dilated cardiomyopathy. Nat Commun. 2016;7:12120. doi: 10.1038/ncomms12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, dos Remedios CG. Special issue on human heart failure. Biophys Rev. 2015;7:1–3. doi: 10.1007/s12551-014-0161-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Ponten F, dos Remedios CG. The interactome of Lim domain proteins: the contributions of LIM domain proteins to heart failure and heart development. Proteomics. 2012;12:203–225. doi: 10.1002/pmic.201100492. [DOI] [PubMed] [Google Scholar]
- Li A, Estigoy C, Raftery M, Cameron D, Odeberg J, et al. Heart research advances using database search engines, human protein atlas and the Sydney Heart Bank. Heart Lung Circ. 2013;22:819–826. doi: 10.1016/j.hlc.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Lin Z, Guo H, Cao Y, Zohrabian S, Zhou P, et al. Acetylation of VGLL4 regulates hippo-YAP regulates growth-YAP signaling and postnatal growth. Dev Cell. 2016;39:466–479. doi: 10.1016/j.devcel.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Yu H, Zwartbol M, Ruifrok WP, van Gilst WH, et al. Identification of hypertrophy- and heart failure-associated genes by combining in vitro and in vivo models. Physiol Genomics. 2012;44:443–454. doi: 10.1152/physiolgenomics.00148.2011. [DOI] [PubMed] [Google Scholar]
- Ng DCH, Court NW, dos Remedios CG, Bogoyevitch MA. Activation of signal transducer and activator of transcription (STAT) pathways in failing human hearts. Cardiovasc Res. 2003;57:333–346. doi: 10.1016/S0008-6363(02)00664-8. [DOI] [PubMed] [Google Scholar]
- Pleuss M, Daeubler G, dos Remedios CG, Ehler E. Adaptations of cytoarchitecture in human dilated cardiomyopathy. Biophys Rev. 2015;7:25–32. doi: 10.1007/s12551-014-0146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polden J, McManus CA, dos Remedios C, Dunn MJ. A 2-D gel reference map of the basic human heart proteome. Proteomics. 2011;11:3582–3586. doi: 10.1002/pmic.201000182. [DOI] [PubMed] [Google Scholar]
- Ralevic C. P2X receptors in the cardiovascular system and their potential as therapeutic targets in disease. Curr Med Chem. 2015;22:851–865. doi: 10.2174/0929867321666141215094050. [DOI] [PubMed] [Google Scholar]
- Robinson AA, Dunn MJ, McCormack A, dos Remedios CG, Rose ML. Protective effect of phosphorylated Hsp27 in coronary arteries through actin stabilization. J Mol Cell Cardiol. 2010;49:370–379. doi: 10.1016/j.yjmcc.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Schreckenbach T, Henn W, Kress W, Roos A, Maschke M, et al. Novel FHL1 mutation in a family with reducing body myopathy. Muscle Nerve. 2013;47:127–134. doi: 10.1002/mus.23500. [DOI] [PubMed] [Google Scholar]
- Walweel K, Li J, Molenaar P, Imtiaz MS, Quail A, et al. Regulation of human RyR2 by Ca2+ and Mg2+ in the cytoplasm and in the lumen of the sarcoplasmic reticulum. J Gen Physiol. 2014;144:263–271. doi: 10.1085/jgp.201311157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walweel K, Gomez-Hurtado N, Oo YW, Beard NA, dos Remedios C, et al. Calmodulin mutants linked to catecholaminergic polymorphic ventricular tachycardia fail to inhibit human RyR2 channels during adrenergic stress. J Am Coll Cardiol. 2017;70:115–117. doi: 10.1016/j.jacc.2017.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walweel K, Molenaar P, Imtiaz MS, Denniss A, dos Remedios C, et al. Ryanodine receptor modification and regulation by intracellular Ca2+ and Mg2+ in healthy and failing human hearts. J Mol Cell Cardiol. 2017;104:263–271. doi: 10.1016/j.yjmcc.2017.01.016. [DOI] [PubMed] [Google Scholar]