

Abstract
Increasing usage of next-generation sequencing techniques pushed during the last decade cardiogenetic diagnostics leading to the identification of a huge number of genetic variants in about 170 genes associated with cardiomyopathies, channelopathies, or syndromes with cardiac involvement. Because of the biochemical and cellular complexity, it is challenging to understand the clinical meaning or even the relevant pathomechanisms of the majority of genetic sequence variants. However, detailed knowledge about the associated molecular pathomechanism is essential for the development of efficient therapeutic strategies in future and genetic counseling. Mutations in DES, encoding the muscle-specific intermediate filament protein desmin, have been identified in different kinds of cardiac and skeletal myopathies. Here, we review the functions of desmin in health and disease with a focus on cardiomyopathies. In addition, we will summarize the genetic and clinical literature about DES mutations and will explain relevant cell and animal models. Moreover, we discuss upcoming perspectives and consequences of novel experimental approaches like genome editing technology, which might open a novel research field contributing to the development of efficient and mutation-specific treatment options.
Keywords: Desmin, Cardiomyopathy, Desminopathy, Cardiovascular genetics, Intermediate filaments
Introduction
In clinical practice and historically, cardiomyopathies are classified according to their morphological and clinical symptoms into dilated (DCM), hypertrophic (HCM), restrictive (RCM), arrhythmogenic (ACM), and noncompaction (NCM) cardiomyopathy (McKenna et al. 2017). All these different forms of cardiomyopathies can be caused by genetic or nongenetic factors (Maron et al. 2006). The first cardiomyopathy causing genetic mutation was identified in 1990 in MYH7, encoding myosin heavy chain, by the group of Geisterfer-Lowrance et al. (1990). Pushed by the human genome project in the 1990s and by the development of efficient next-generation sequencing technology in the 2000s, a large number of different mutations in over 170 different genes were discovered, which are associated with cardiomyopathies, isolated channelopathies, or syndromes with cardiac involvement (Cahill et al. 2013).
In the last years, it became more and more evident that different cardiomyopathies can be caused by mutations in the same gene(s). DES, encoding the intermediate filament (IF) protein desmin (Figs. 1 and 2), is such a gene, where mutations are associated with DCM (Taylor et al. 2007; Brodehl et al. 2016a), HCM (Harada et al. 2018), RCM (Ojrzynska et al. 2017), ACM (Klauke et al. 2010), or left ventricular noncompaction cardiomyopathy (LVNC) (Miszalski-Jamka et al. 2017) (Figs. 3 and 4). In addition, DES mutations might cause different isolated or combined skeletal myopathies (Schirmer et al. 2018; Cetin et al. 2013; Goldfarb et al. 1998; Dalakas et al. 2000) (for details, see Table 1).
Fig. 1.

a Schematic overview about the localization of desmin filaments (red) in cardiomyocytes. Of note, desmin filaments connect several protein-protein complexes (e.g., desmosomes) and cell organelles (mitochondria, nuclei) to the cytoskeletal network. b Transmission electron microscopy image of murine cardiac tissue. A typical desmosome connecting two cardiomyocytes is marked by the red box. c Schematic illustration of the cardiac desmosomes, which are connected to the desmin filaments. DSC2 = desmocollin-2, DSG2 = desmoglein-2, PKP2 = plakophilin-2, PG = plakoglobin, PM = plasma membrane, DSP = desmoplakin
Fig. 2.
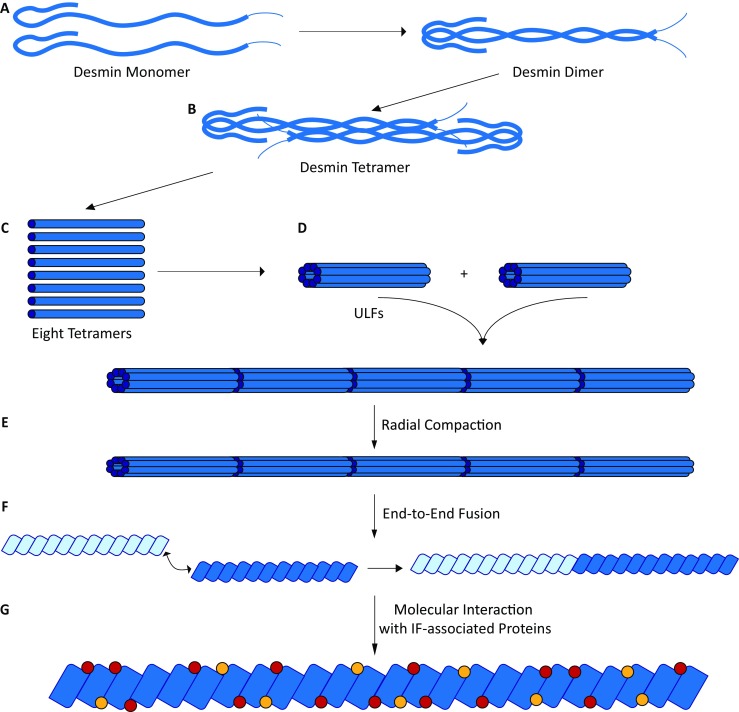
Schematic overview about the desmin filament assembly. a Desmin dimers are formed as coiled-coils by the rod domains. b Two desmin dimers anneal into anti-parallel tetramers. Based on a ‘divide and conquer’ strategy, the molecular tetrameric structure of the homologous protein vimentin has been proposed (Chernyatina et al. 2015). c Eight tetramers associate laterally into unit length filaments (ULFs). ULFs are the essential building blocks of the desmin filaments, which are formed by longitudinal elongation (d) followed by a radial compaction step (e). Desmin filaments could also fuse end-to-end to elongate into longer filamentous structures (f). In addition, several IF-associated proteins (shown in red or yellow) bind to desmin filaments (g)
Fig. 3.
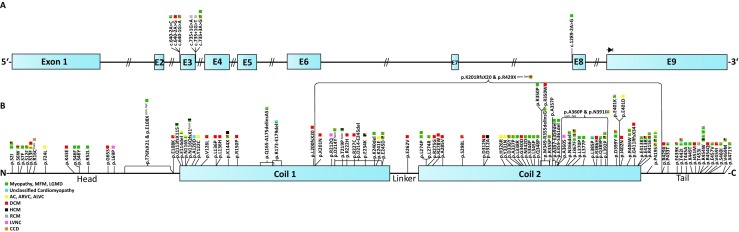
a Schematic overview of the DES gene and known splice site mutations. b Schematic overview of the desmin protein including the positions of known missense and small deletion mutations. Colored squares indicate the clinical phenotypes of DES mutation carriers
Fig. 4.
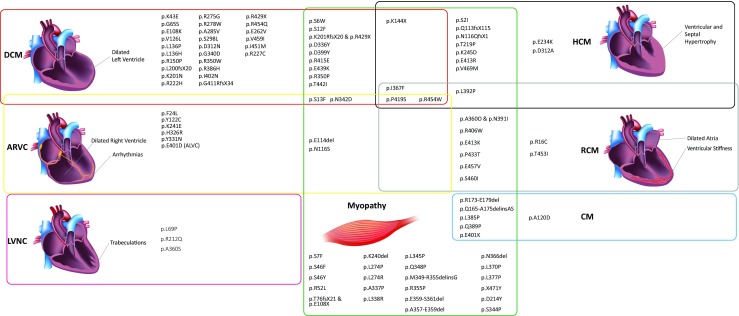
Venn diagram indicating the different cardiac and skeletal phenotypes of DES mutation carriers. Of note, the associated clinical phenotypes of different mutations overlap significantly. DCM = dilated cardiomyopathy, HCM = hypertrophic cardiomyopathy, ARVC = arrhythmogenic right ventricular cardiomyopathy, RCM = restrictive cardiomyopathy, LVNC = left ventricular cardiomyopathy, CM = atypical or unknown cardiomyopathy. (Images for DCM and HCM are licensed from shutterstock.de)
Table 1.
Overview about the known missense and deletion mutations
| Mutation | Comments | Family anamnesis | MAF (gnomAD, 11th May 2018) (Lek et al. 2016) | Categorization | Clinical symptoms | References |
|---|---|---|---|---|---|---|
| p.S2I | NO (Selcen et al. 2004); 2 mutation carriers (Wahbi et al. 2012) | – | Pathogenic | MFM (Selcen et al. 2004); HCM, bifascicular block, SM (Wahbi et al. 2012) | Sharma et al. (2009), Selcen et al. (2004), Wahbi et al. (2012) | |
| p.S6W | 2 mutation carriers | – | Pathogenic | DCM, atrial arrhythmias, SM | Weihl et al. (2015) | |
| p.S7F | 2 mutation carriers | – | Pathogenic | MFM | Vattemi et al. (2011) | |
| p.S12F | 4 mutation carriers | – | Pathogenic | DCM, AVB, SCD, SM, LW, dysphagia | Hong et al. (2011), Hong et al. (2010) | |
| p.S13F | Eight families with several mutation carriers (van Spaendonck-Zwarts et al. 2012; Bergman et al. 2007; van Tintelen et al. 2009) | – | Pathogenic | DCM (Walsh et al. 2017) | Brodehl et al. (2012a), van Spaendonck-Zwarts et al. (2012), McCormick et al. (2015), Bergman et al. (2007), Sharma et al. (2009), van Tintelen et al. (2009), Walsh et al. (2017), van Spaendonck-Zwarts et al. (2013) | |
| p.R16C | Homozygous | 1 (homo.) mutation carrier (Arbustini et al. 2006) | – | Pathogenic | RCM, biatrial dilation, AVB, HTx | Sharma et al. (2009), Arbustini et al. (2006) |
| p.F24L | NO | – | VUS | ARVC | Walsh et al. (2017) | |
| p.K43E | NO | – | VUS | DCM | Walsh et al. (2017), Pugh et al. (2014) | |
| p.S46Y | NO | – | Pathogenic | MFM | Sharma et al. (2009), Selcen et al. (2004) | |
| p.S46F | NO | – | Pathogenic | MFM | Sharma et al. (2009), Selcen et al. (2004), Baker et al. (2013) | |
| p.R52L | De novo | – | Pathogenic | LGMD | Yu et al. (2017) | |
| p.G65S | NO | 0.00002139 | VUS | DCM | Walsh et al. (2017), Pugh et al. (2014) | |
| p.L69P | NO | 0.00001067 | VUS | LVNC | Miszalski-Jamka et al. (2017) | |
| p.T76fsX21 and p.E108X | Compound heterozygous | 2 (c. h.) mutation carriers | – | Pathogenic | SM | Henderson et al. (2013) |
| p.E108K | NO | – | Pathogenic | DCM, LAFB | Taylor et al. (2007) | |
| p.Q113RfsX115 | Sporadic case | – | Pathogenic | HCM, arrhythmias, SM, LW | Hong et al. (2011) | |
| p.E114del | 3 mutation carriers | – | Pathogenic | ARCV, atrial dilation, arrhythmias, SCD, SM | Brodehl et al. (2012a), Vernengo et al. (2010), Hedde et al. (2012) | |
| p.N116S | De novo | – | Pathogenic | ARVC, HTx, SM | Klauke et al. (2010), Brodehl et al. (2012a), Hedde et al. (2012), Maerkens et al. (2013), Brodehl et al. (2012b) | |
| p.N116QfsX2 | Homozygous | 2 (homo.) mutation carriers | – | Pathogenic | HCM, respiratory failure, SM, LW | Durmus et al. (2016) |
| p.A120D | 3 mutation carrier + 3 obligate mutation carriers | – | Pathogenic | CM, biatrial dilation, arrhythmias, HTx, Ebstein’s anomaly, SCD | Brodehl et al. (2013a) | |
| p.Y122C | NO | – | Likely pathogenic | ARVC | Walsh et al. (2017) | |
| p.V126L | NO | – | VUS | DCM | Haas et al. (2015) | |
| p.R127P | NO | – | Pathogenic | DCM, SCD | Golbus et al. (2014) | |
| p.L136P | NO | – | Likely pathogenic | DCM | Brodehl et al. (2016a) | |
| p.L136H | NO | 0.00006302 | VUS | DCM | Pugh et al. (2014), Wilson et al. (2015) | |
| p.K144X | 2 mutation carriers | – | Pathogenic | DCM, LBBB, SM; HCM | Wahbi et al. (2012) | |
| p.R150P | NO | – | VUS | DCM | Walsh et al. (2017) | |
| p.Q165-A174delinsAS | NO | – | Likely pathogenic | CM, AVB, SM | Schirmer et al. (2018) | |
| p.E173-E179del | Yes | – | Pathogenic | CM, SM, SMD, respiratory failure | Munoz-Marmol et al. (1998), Pinol-Ripoll et al. (2009) | |
| p.L200fsX20 | NO | – | Likely pathogenic | DCM | Walsh et al. (2017) | |
| p.K201fsX20 and p.R429X | Compound heterozygous | 2 (homo.) mutation carriers | – | Pathogenic | DCM, arrhythmias, SM | McLaughlin et al. (2013) |
| p.K201N | NO | 0.000007214 | Likely pathogenic | DCM | Dal Ferro et al. (2017) | |
| p.R212Q | NO | 0.0002092 | VUS | LVNC | Miszalski-Jamka et al. (2017) | |
| p.D214Y | Homozygous | NO | – | Pathogenic | Arrhythmias, conduction disease, SM | Monies et al. (2017) |
| p.D214-E245del | NO | – | Pathogenic | Walsh et al. (2017) | ||
| p.T219P | Homozygous | – | Pathogenic | HCM, SM | Harada et al. (2018) | |
| p.R222H | NO | 0.0004004 | Likely pathogenic | DCM | Dal Ferro et al. (2017) | |
| p.R227C | 6 mutation carriers | 0.000008122 | Pathogenic | DCM | Liu et al. (2017) | |
| p.E234K | MYPN-p.R989H CACNA1C-p.R1973P | 2 mutation carriers | – | Likely pathogenic | HCM, AVB, SQTS | Chen et al. (2017) |
| p.K240del | Mutation corrected (Schroder et al. 2007) | NO | – | Pathogenic | SM, ventricular arrhythmias | Schroder et al. (2007), Schroder et al. (2003) |
| p.K241E | PKP2-p.T816RfsX10 | NO | – | VUS | ARVC | Lorenzon et al. (2013) |
| p.E245D | 2 mutation carriers (Wahbi et al. 2012); 5 mutation carriers (Strach et al. 2008) | – | Pathogenic | HCM, AVB, atrial flutter, SM (Wahbi et al. 2012); AVB, RBBB, SM (Strach et al. 2008) | Conover et al. (2009), Wahbi et al. (2012), Baker et al. (2013), Maerkens et al. (2013), Strach et al. (2008) | |
| p.E262V | NO | 0.0002472 | VUS | DCM | Pugh et al. (2014) | |
| p.L274R | 6 mutation carriers | – | Pathogenic | AVB, SM, LW | Hong et al. (2011) | |
| p.L274P | 6 mutation carriers (Hong et al. 2011); de novo (Yu et al. 2017) | – | Pathogenic | AVB, SCD, SM, LW (Hong et al. 2011); LGMD (Yu et al. 2017) | Hong et al. (2011), Hong et al. (2010), Yu et al. (2017) | |
| p.R275G | NO | – | VUS | DCM | Haas et al. (2015) | |
| p.R278W | NO | 0.000004077 | VUS | DCM | Walsh et al. (2017) | |
| p.A285V | NO | – | Pathogenic | DCM, arrhythmias, SCD | Tse et al. (2013) | |
| p.S298L | NO (Taylor et al. 2007) | 0.00007944 | Likely pathogenic | DCM, LBBB (Taylor et al. 2007) | Taylor et al. (2007), Andreasen et al. (2013), Ng et al. (2013) | |
| p.D312N | NO (Taylor et al. 2007; Pugh et al. 2014) | 0.0001879 | VUS | DCM, SCD | Taylor et al. (2007), Pugh et al. (2014), Andreasen et al. (2013) | |
| p.D312A | MYBPC3-p.R1002W MYH7-p.D43N | NO | 0.0003974 | VUS | HCM, SCD | Mook et al. (2013) |
| p.H326R | 2 mutation carriers + 1 obligate carrier | – | VUS | ARVC | Brodehl et al. (2013a) | |
| p.Y331N | NO | – | VUS | ARVC | Walsh et al. (2017) | |
| p.D336Y | NO | – | Pathogenic | DCM, trifascicular block, SM | Wahbi et al. (2012) | |
| p.A337P | 3 mutation carriers | – | Pathogenic | RBBB, SM | Goldfarb et al. (1998), Goudeau et al. (2006) | |
| p.L338R | (Goudeau et al. 2006) | – | Pathogenic | SM, LW, respiratory insufficiency | Goudeau et al. (2006) | |
| p.G340D | NO | – | VUS | DCM | Walsh et al. (2017) | |
| p.N342D | NO (Wahbi et al. 2012); 2 affected and 1 unaffected mutation carrier (Dalakas et al. 2003) | – | Pathogenic | AVB, SM (Wahbi et al. 2012) | Dalakas et al. (2000), Brodehl et al. (2012a), van Spaendonck-Zwarts et al. (2012), Wahbi et al. (2012), Brodehl et al. (2012b), Dalakas et al. (2003) | |
| p.S344P | De novo | – | Pathogenic | LGMD | Yu et al. (2017) | |
| p.L345P | NO | – | Pathogenic | RBBB, SM (Wahbi et al. 2012) | Sjoberg et al. (1999) | |
| p.Q348P | 2 mutation carriers | – | Pathogenic | SM, LW | Fichna et al. (2014) | |
| p.M349-R355delinsG | 6 affected and 3 unaffected mutation carriers (Cao et al. 2013) | – | Pathogenic | AVB, SCD, SM | Cao et al. (2013) | |
| p.R350P | 6 mutation carriers (Strach et al. 2008) | – | Pathogenic | SM (Strach et al. 2008) | Clemen et al. (2015), Winter et al. (2016), Durmus et al. (2016), Bar et al. (2005b), Strach et al. (2008), Bonakdar et al. (2012), Levin et al. (2010), Walter et al. (2007) | |
| p.R350W | NO (Taylor et al. 2007) | 0.00002437 | Likely pathogenic | DCM | Taylor et al. (2007), Andreasen et al. (2013) | |
| p.R355P | NO (Wahbi et al. 2012) | – | Pathogenic | AF, SCD, SM, atrial dilation, bifascicular block, SM (Wahbi et al. 2012) | Wahbi et al. (2012), Fidzianska et al. (2005) | |
| p.A357P | 2 mutation carriers (Dagvadorj et al. 2003) | – | Pathogenic | Chourbagi et al. (2011), Dagvadorj et al. (2003), Fischer et al. (2006) | ||
| p.A357-E359del | 13 mutation carriers (3 families) | – | Pathogenic | SM, LW | Fichna et al. (2014) | |
| p.E359-S361del | 4 mutation carriers (2 families) | – | Pathogenic | SM, LW | Kaminska et al. (2004) | |
| p.A360S | LDB3-p.I615N | NO | – | Pathogenic | LVNC | Miszalski-Jamka et al. (2017) |
| p.A360P and p.N393I | Compound heterozygous | 3 (c. h.) mutation carriers | – | Pathogenic | RCM, AVB, respiratory insufficiency, SM | Goldfarb et al. (1998), Goudeau et al. (2006) |
| p.N366del | NO | – | Pathogenic | Left anterior hemi-block, SCD, SM | Kaminska et al. (2004) | |
| p.I367F | NO (Olive et al. 2007); 11 mutation carriers (Ripoll-Vera et al. 2015) | – | Pathogenic | HCM, RCM, AVB, SCD, SM (Olive et al. 2007); RCM, SCD, SM (Kreplak and Bar 2009) | Olive et al. (2007), Ripoll-Vera et al. (2015) | |
| p.L370P | NO (Dagvadorj et al. 2003); de novo (Yu et al. 2017) | – | Pathogenic | LGMD (Yu et al. 2017) | Chourbagi et al. (2011), Yu et al. (2017), Dagvadorj et al. (2003), Arias et al. (2006), Olive et al. (2011) | |
| p.L377P | Sporadic | 0.000004061 | Pathogenic | SM | Strach et al. (2008) | |
| p.L385P | De novo | – | Pathogenic | CM, SM, LW | Sugawara et al. (2000) | |
| p.R386H | NO | 0.000004061 | Pathogenic | DCM | Zhao et al. (2015) | |
| p.Q389P | Sporadic | – | Pathogenic | CM, RBBB, SM | Chourbagi et al. (2011), Goudeau et al. (2001) | |
| p.L392P | NO | – | Pathogenic | HCM, RCM, CCD, SCD, SM, LW, respiratory insufficiency (Olive et al. 2007) | Maerkens et al. (2013), Olive et al. (2007), Olive et al. (2011) | |
| p.D399Y | NO (Strach et al. 2008; Goudeau et al. 2006) | – | Pathogenic | SM (Strach et al. 2008); DCM, AVB, SCD, SM, LW (Goudeau et al. 2006) | Chourbagi et al. (2011), Maerkens et al. (2013), Strach et al. (2008), Goudeau et al. (2006), Fokstuen et al. (2016) | |
| p.E401D | 23 mutation carriers + 2 obligate carriers | – | Pathogenic | ALVC | Bermudez-Jimenez et al. (2017) | |
| p.E401K | NO | – | Pathogenic | CM, heart block, SM, LW (Goudeau et al. 2006) | Chourbagi et al. (2011), Goudeau et al. (2006) | |
| p.I402N | 2 mutation carriers | – | Pathogenic | DCM, arrhythmias, SCD, respiratory insufficiency, dysphagia | Weihl et al. (2015) | |
| p.R406W | 3 mutation carriers (Arbustini et al. 2006); NO (Wahbi et al. 2012) | – | Pathogenic | RCM, biatrial dilation, AVB (Arbustini et al. 2006); AVB, AF, SM (Wahbi et al. 2012) | Dalakas et al. (2000), Chourbagi et al. (2011), Wahbi et al. (2012), Arbustini et al. (2006), Punetha et al. (2016) | |
| p.G411RfsX34 | NO | – | Likely pathogenic | DCM | Dal Ferro et al. (2017) | |
| p.E413K | 3 mutation carriers + 1 obligate carrier | – | Pathogenic | RCM, AVB, AF, SCD (Pruszczyk et al. 2007) | Chourbagi et al. (2011), Levin et al. (2010), Pruszczyk et al. (2007), Bar et al. (2007) | |
| p.E413R | 2 mutation carriers (Wahbi et al. 2012) | – | Pathogenic | HCM, LBBB, SM | Wahbi et al. (2012) | |
| p.R415E | 5 mutation carriers | – | Pathogenic | LVRC, SCD, DCM, SM, RCM, SM | Ripoll-Vera et al. (2015) | |
| p.P419S | 3 mutation carriers (Wahbi et al. 2012); 2 mutation carriers (Olive et al. 2007); 2 mutation carriers (Ripoll-Vera et al. 2015); 7 mutation carriers (Hedberg et al. 2012) | – | Pathogenic | DCM, HCM, bifascicular block, SM (Wahbi et al. 2012); HCM, left atrial dilation, heart block, SM (Olive et al. 2007); RCM, SM (Ripoll-Vera et al. 2015) | Hedberg et al. (2012), Brodehl et al. (2013b), Wahbi et al. (2012), Maerkens et al. (2013), Olive et al. (2007), Ripoll-Vera et al. (2015), Olive et al. (2011), Hedberg et al. (2013) | |
| p.R429X | NO (Walsh et al. 2017) | 0.000008122 | Likely pathogenic | DCM (Walsh et al. 2017) | McLaughlin et al. (2013), Walsh et al. (2017), Pugh et al. (2014), Zhu et al. (2015) | |
| p.P433T | NO | – | Pathogenic | RCM, SM | Jurcu et al. (2017) | |
| p.E439K | 2 mutation carriers | – | Pathogenic | DCM, AF, RBBB, respiratory insufficiency, SM | Wahbi et al. (2012) | |
| p.T442I | 3 mutation carriers (Wahbi et al. 2012) | – | Pathogenic | CM, arrhythmia, SM; DCM, SCD, RBBB, LBBB, respiratory insufficiency, SM (Wahbi et al. 2012) | Chourbagi et al. (2011), Wahbi et al. (2012), Bar et al. (2007) | |
| p.T445A | Sporadic case | 0.00003243 | Pathogenic | SM, LW, respiratory insufficiency | Hong et al. (2011) | |
| p.K449T | NO | – | Pathogenic | MFM | Chourbagi et al. (2011), Selcen et al. (2004), Bar et al. (2007), Maddison et al. (2012) | |
| p.I451M | 3 affected and 3 unaffected mutation carriers (Dalakas et al. 2003) | 0.00006598 | VUS | DCM | Chourbagi et al. (2011), Dalakas et al. (2003), Bar et al. (2007), Li et al. (1999) | |
| p.T453I | Sporadic case | – | Pathogenic | RCM, left atrial dilation, AVB | Chourbagi et al. (2011), Arbustini et al. (2006), Baker et al. (2013) | |
| p.R454W | MYOT-p.Q74K (Bar et al. 2007) | De novo (Bar et al. 2007); 2 mutation carriers (Weihl et al. 2015); NO (Wahbi et al. 2012; Vattemi et al. 2011) | – | Pathogenic | HOCM, SM (Bar et al. 2007); CM, biatrial dilation, SCD, arrhythmias, SM (Weihl et al. 2015); MFM (Vattemi et al. 2011); RCM, AF, AVB, HTx, SM (Wahbi et al. 2012) | Brodehl et al. (2012a), Wahbi et al. (2012), Weihl et al. (2015), van Spaendonck-Zwarts et al. (2013), Hedde et al. (2012), Levin et al. (2010), Punetha et al. (2016), Bar et al. (2007), Cerino et al. (2017), Haskell et al. (2017), Shanks et al. (2017), Ackerman et al. (2016) |
| p.R454Q | NO | – | VUS | DCM | Haas et al. (2015) | |
| p.E457V | 5 mutation carriers | – | Pathogenic | RCM, AVB, AF, SCD, SM, LW | Hong et al. (2011) | |
| p.V459I | 2 unrelated mutation carriers (Taylor et al. 2007) | 0.003191 | VUS | DCM, AVB (Taylor et al. 2007) | Taylor et al. (2007), Weihl et al. (2015), Andreasen et al. (2013), Nouhravesh et al. (2016) | |
| p.S460I | NO (Bar et al. 2007) | – | Pathogenic | RCM, AVB, SCD, SM | Chourbagi et al. (2011), Bar et al. (2007) | |
| p.V469M | LMNA-p.R644C | NO | – | VUS | HCM, heart block, SM (Muntoni et al. 2006) | Chourbagi et al. (2011), Bar et al. (2007), Muntoni et al. (2006) |
| p.X471Y | NO | – | Pathogenic | AVB, SM | Wahbi et al. (2012) |
AF = atrial fibrillation, ALVC = arrhythmogenic left ventricular cardiomyopathy, ARVC = arrhythmogenic right ventricular cardiomyopathy, AVB = atrioventricular block, CM = cardiomyopathy, DCM = dilated cardiomyopathy, VUS = genetic variant of unknown significance, HCM = hypertrophic cardiomyopathy, HOCM = hypertrophic obstructive cardiomyopathy, HTx = heart transplantation, LAFB = left anterior fascicular block, LBBB = left bundle-branched block, LGMD = limb-girdle muscular dystrophy, LW = limp weakness, MAF = minor allele frequency, MFM = myofibrillar myopathy, NMD = nonsense-mediated RNA decay, NO = not observed, RBBB = right bundle-branched block, RCM = restrictive cardiomyopathy, SCD = sudden cardiac death, SM = skeletal myopathy, SMD = smooth muscle defect, SQTS = short QT syndrome
In this article, we review the genetic, cellular, molecular, and biophysical pathomechanisms of DES mutations with a focus on cardiomyopathies. In addition, we summarize relevant in vitro experiments using recombinant desmin as well as existing cell culture and animal models, which were used in combination with biochemical and biophysical methods to investigate the underlying pathomechanism of DES mutations.
Cellular functions of desmin
Three filamentous systems build the eukaryotic cytoskeleton. Actin filaments are important for the structural integrity and stability of nearly all mammalian cells and are essential components of the thin sarcomeric filaments in (cardio)myocytes. Microtubules are composed of α- and β-microtubulin and are necessary for the kinesin-mediated vesicle transport. In contrast to these filament types, IFs are formed by nonglobular, highly flexible protein units, which consist mostly of α-helices. Therefore, IF proteins are nanomolecular springs, which provide enormous structural flexibility. In humans, 70 different genes including the DES gene encode members of the IF protein family (Szeverenyi et al. 2008). Desmin is expressed in cardiac, skeletal, and smooth muscle cells (Hnia et al. 2015; The Human Protein Atlas, https://www.proteinatlas.org; Uhlen et al. 2015). Due to their specific cellular expression pattern, several diverse genetic diseases like cardiomyopathies (Brayson and Shanahan 2017) and cutaneous (Coulombe 2017) or neurological diseases (Brenner et al. 2001) might be caused by mutations in genes encoding IF proteins.
Due to their assembling mechanism, IF proteins can be categorized into different groups, which were previously reviewed in detail (Herrmann and Aebi 2004). Desmin belongs to assembly group type III, which contains also vimentin and glial fibrillary acidic protein (GFAP). In general, desmin filaments connect and anchor different cell structures like desmosomes, mitochondria, and costameres or Z-bands to the cytoskeleton.
Desmosomes are cell-cell junctions, which mediate the cell-cell adhesion in a Ca2+-dependent way (Patel and Green 2014) (Fig. 1). They are localized at the intercalated disc in myocardial tissue. Members of the cadherin family (desmocollins and desmogleins) bind via trans interaction to their counterparts from the neighboring cells (Harrison et al. 2016; Lapouge et al. 2006; Dieding et al. 2017). The cytoplasmic domains of these desmosomal cadherins are bound from proteins of the Armadillo family (plakophilins and plakoglobin) (Kami et al. 2009; Chen et al. 2002; Chitaev et al. 1998). These Armadillo proteins are connected to the cytolinker protein desmoplakin (Hofmann et al. 2000). Desmin filaments are linked via desmoplakin to the desmosomes (Choi et al. 2002), which reached a special interest since it became evident that mutations within the genes encoding for desmosomal structural proteins are the main cause of ACM (Gerull et al. 2004; Heuser et al. 2006; Bauce et al. 2005; Bhuiyan et al. 2009; McKoy et al. 2000).
Costameres are multiprotein complexes, which mediate the interactions of (cardio)myocytes with the extracellular matrix and which are localized at the sarcolemma. The dystrophin-glycoprotein and the integrin-vinculin-talin systems are the main components of the costameres (Jaka et al. 2015). Desmin forms heteropolymers with the IF-protein synemin (encoded by SYNM) (Granger and Lazarides 1980; Price and Lazarides 1983; Bellin et al. 1999). Synemin is expressed in all muscle cell types (Olive et al. 2003; https://www.proteinatlas.org) and co-localizes with desmin at the Z-bands in striated muscle (Bellin et al. 1999; Bilak et al. 1998; Hirako et al. 2003). In addition to the characteristic rod domain, synemin has a short N-terminal head and a large C-terminal tail domain (Becker et al. 1995). Synemin links the IFs via binding to α-actinin within the Z-bands (Bellin et al. 1999, 2001; Lund et al. 2012). In addition, synemin binds to different costameric proteins like vinculin, dystrophin, and talin and mediates the interaction of IFs with costameres (Bellin et al. 2001; Bhosle et al. 2006; Sun et al. 2008). Interestingly, DES missense mutations leading to an abnormal cytoplasmic desmin aggregation cause also a co-aggregation of the binding partner synemin (Chourbagi et al. 2011). Although no human SYNM mutations are described so far, Synm knock-out mice develop cardiomyopathies (Garcia-Pelagio et al. 2018).
Structural organization of desmin filaments
Nearly all IF proteins including desmin are built by a central homologous rod domain flanked by head and tail domains of different sizes and sequences (Carlsson and Thornell 2001). Linker sequences divide the rod domain into two coil subdomains. Because of the high oligomerization of IF proteins, it is difficult to crystallize and determine their molecular structure. Therefore, a ‘divide and conquer’ strategy was applied leading to a tetrameric detailed molecular model of the IF-protein vimentin (Chernyatina et al. 2012, 2015). However, the structure and architecture of the completely assembled IFs remains still widely unknown.
Desmin filaments are assembled in a stepwise process (Herrmann et al. 2009) (Fig. 2). A repeated heptade sequence with hydrophobic amino acids at positions A and D is characteristic for IF proteins (Chernyatina et al. 2015). These amino acids form a hydrophobic seam, which is essential for the dimerization into coiled-coils (Herrmann et al. 2000). Two highly conserved motifs at the beginning and the end of the rod domain are essential for the assembly process (Hatzfeld and Weber 1992; Albers and Fuchs 1992). In the second step, desmin dimers anneal into anti-parallel tetramers (Herrmann et al. 2009; Potschka et al. 1990; Kooijman et al. 1995; Geisler et al. 1985). Because the anti-parallel tetramers have no orientation, IFs have in contrast to actin filaments or microtubules no polarity. Eight of these tetramers anneal laterally into unit length filaments (ULFs), which are the essential building blocks of desmin filaments (Herrmann et al. 2009). ULFs are longitudinally elongated and compacted into regular IFs (Herrmann et al. 2009). Ando and colleagues demonstrated a helical right-handed twist of the homologous vimentin filaments (Ando et al. 2004), which was previously also shown for desmin filaments by atomic force microscopy (Brodehl et al. 2012a, 2016a). In addition, IFs can fuse end-to-end to elongate longitudinally (Colakoglu and Brown 2009; Winheim et al. 2011). Interestingly, different disease-causing DES mutations interfere at different stages within this assembly process (Brodehl et al. 2012a; Bar et al. 2005a). The sequences of the head and rod domains are highly variable. Site-directed spin labeling in combination with electron paramagnetic resonance (EPR) spectroscopy revealed that the head domain of type III IF proteins interacts at specific sites with the rod domain (Aziz et al. 2009, 2010). Therefore, the head domain is essential for IF assembly, whereas deletion studies revealed that the tail domain of desmin and vimentin is not essential for the formation of IFs (Herrmann et al. 1996; Kaufmann et al. 1985). Thus, the exact molecular function of the desmin tail domain is unknown. However, it was suggested that the tail domain is involved in width control of ULFs (Herrmann et al. 1996) and mediates Ca2+- or Mg2+-dependent cross-linking (Lin et al. 2010).
Biochemical and biophysical experimental approaches
Originally, desmin was purified from muscle tissue (Izant and Lazarides 1977). Human monomeric desmin has a molecular mass of about 55 kDa and consists of 470 amino acids. Because posttranslational modifications (PTMs) are not essential for the assembly process, recombinant desmin can be efficiently expressed in bacterial cells (Escherichia coli). It can be isolated from inclusion bodies and purified under denaturing conditions (8 M urea) by ionic exchange and immobilized metal affinity chromatography followed by refolding through a stepwise dialysis, to reduce the urea concentration (Brodehl et al. 2012a; Kreplak and Bar 2009).
The assembly of recombinant desmin can be initiated in vitro by adding sodium chloride (Kreplak and Bar 2009). Transmission electron microscopy (TEM) or atomic force microscopy (AFM) can be applied for visualization of the assembly process (Brodehl et al. 2012a; Harder et al. 2013; Bar et al. 2006). Of note, desmin contains one cysteine residue (p.C333), which can be used for site-specific labeling with chemical fluorescent dyes (Harder et al. 2013). Apertureless scanning near-field microscopy (aSNOM) was previously used in combination with Atto740-conjugated recombinant desmin to investigate the co-assembly of mutant and wild-type desmin (Brodehl et al. 2016a; Harder et al. 2013) (Fig. 5e).
Fig. 5.
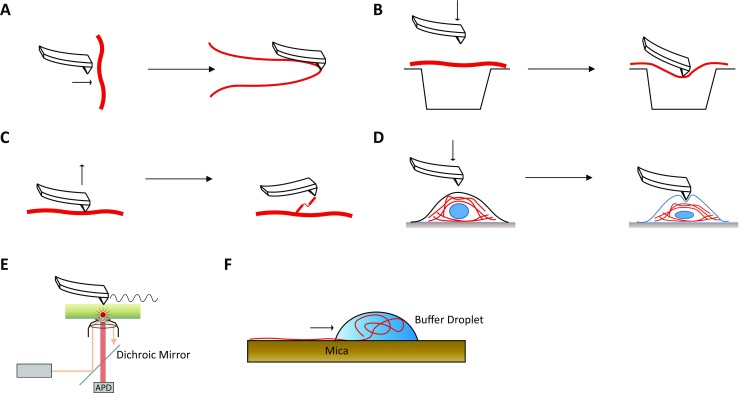
Overview of biophysical approaches to investigate the nanomechanical and structural properties of different desmin filaments. a Longitudinal stretching of isolated desmin filaments using the tip of an atomic force microscopy (Kreplak and Bar 2009). b Lateral stretching of IFs by pushing desmin filaments into small holes using the tip of an atomic force microscope (Guzman et al. 2006). c Molecule force spectroscopy using an atomic force microscope (Kiss et al. 2006). d Cellular stretching of transfected cells expressing mutant desmin (Plodinec et al. 2011). e Schematic overview of apertureless scanning near-field optical microscopy (aSNOM) (Harder et al. 2013). f Longitudinal stretching of desmin filaments using the withdrawing meniscus of a buffer droplet. Centrifugation is used to apply centrifugal forces (Kiss and Kellermayer 2014)
Because IF proteins including desmin are important for the cellular stability and integrity, several studies developed techniques to investigate the nanomechanical properties of IFs. Kreplak and Bär used the cantilever tip of an AFM to stretch single desmin filaments in lateral orientation (Kreplak and Bar 2009) (Fig. 5a). Guzman et al. (2006) used a tip of an AFM to push single IFs into holes of a porous membrane to determine elasticity and Young’s module (Fig. 5b). Kiss et al. (2006) used also the AFM tip to lift coiled-coil desmin dimers from assembled filaments (Fig. 5c). This approach is useful to investigate the stretching and sliding of the dimers from single IFs. A further approach published by Kiss et al. used the centrifugal force of a centrifuge to displace a droplet containing preassembled desmin filaments on mica surface (Kiss and Kellermayer 2014). In this approach, the desmin filaments are stretched longitudinally by the withdrawing meniscus of the buffer droplet (Kiss and Kellermayer 2014) (Fig. 5f). The cellular stiffness of transfected cells can also be investigated using AFM (Fig. 5d). Plodinec et al. (2011) revealed that the molecular changes caused by DES missense mutations are associated with altered nanomechanical properties of the cells, which might contribute to disease progression. A similar approach consisting of two microplates, which are used as a stretching device for single cells, revealed that traction forces of myoblasts expressing mutant desmin are altered (Charrier et al. 2016).
Animal models
Besides some rare exceptions (Mencarelli et al. 2011), insects express only nuclear IF proteins but do not express cytoplasmic IF proteins (Herrmann and Strelkov 2011). Therefore, the model organism Drosophila melanogaster (fruit fly) has limited value for the investigation of DES mutations.
Li et al. (1996) and Capetanaki et al. (1997) generated independently Des knock-out mouse models by homologous recombination. Surprisingly, Des knock-out mice were viable and fertile but developed defects of all three muscle types. Magnetic resonance imaging (MRI) revealed a biventricular reduced ejection fraction and a decreased cardiac output (Sprinkart et al. 2012). Homozygous Des knock-out mice develop a severe cardiomyopathy including hemorrhaging, extensive fibrosis, and calcification in the septum and the ventricular walls, whereas heterozygous mice were not affected (Li et al. 1996; Capetanaki et al. 1997). Degeneration of cardiomyocytes, accumulation of macrophages, and severe fibrosis were also found in these Des knock-out mice (Thornell et al. 1997). Of note, calcified lesions were mainly present in the right ventricular wall and the septum (Thornell et al. 1997), which is in good agreement with the identification of human DES mutations associated with predominantly right ventricular cardiomyopathy (Klauke et al. 2010). ARVC is mainly caused by mutations in genes, encoding desmosomal structural proteins (Gerull et al. 2004; Rampazzo et al. 2002; Gehmlich et al. 2011, 2012). Interestingly, gene expression analysis revealed a remarkable overlap of differentially expressed gene networks between mouse models for desmosomal genes and the Des knock-out mice suggesting comparable molecular pathomechanisms (Brodehl et al. 2017a; Psarras et al. 2012). For example, genes encoding matricellular proteins like osteopontin (Spp1) are highly upregulated in both mouse models, which might explain the extensive fibrotic remodeling of the extracellular matrix (Brodehl et al. 2017a; Psarras et al. 2012). Ventricular arrhythmias are frequently observed in human patients with DES mutations. However, the Des knock-out mice present atrial fibrillation but do not develop severe ventricular tachycardia (Schrickel et al. 2010). Interestingly, the phenotype of Des knock-out mice can be decreased by adeno-associated viruses, encoding the cDNA of wild-type desmin (Heckmann et al. 2016) suggesting a compensatory therapeutic approach for DES null mutations. However, the Des knock-out mice have some limitations, because only a few DES null mutations have been described in humans at all (McLaughlin et al. 2013) and the majority of DES mutations induce a dominant aggregation by functional alterations.
Therefore, different transgenic and knock-in mouse models expressing mutant desmin have been developed and characterized (Raats et al. 1996; Kostareva et al. 2008; Joanne et al. 2013; Diermeier et al. 2017a; Clemen et al. 2015) (Fig. 6). However, some of these mice develop only a mild or even no obvious phenotype (Kostareva et al. 2008). In contrast, the Des-p.R345P knock-in mice develop DCM in combination with cardiac arrhythmias and conduction disease, which is quite comparable to the clinical phenotype of human patients with the corresponding DES mutation (Clemen et al. 2015). Only homozygous animals develop, in addition, skeletal myopathy. These mice present mitochondrial abnormalities (Winter et al. 2016), increased stiffness of myoblasts (Diermeier et al. 2017a), and age-dependent myofibrillar changes (Diermeier et al. 2017b). Using optical voltage mapping in a transgenic mouse model with the cardiac-specific expression of mutant desmin (seven amino acid deletion), Gard et al. (2005) demonstrated the impairment of ventricular conduction by this mutation.
Fig. 6.
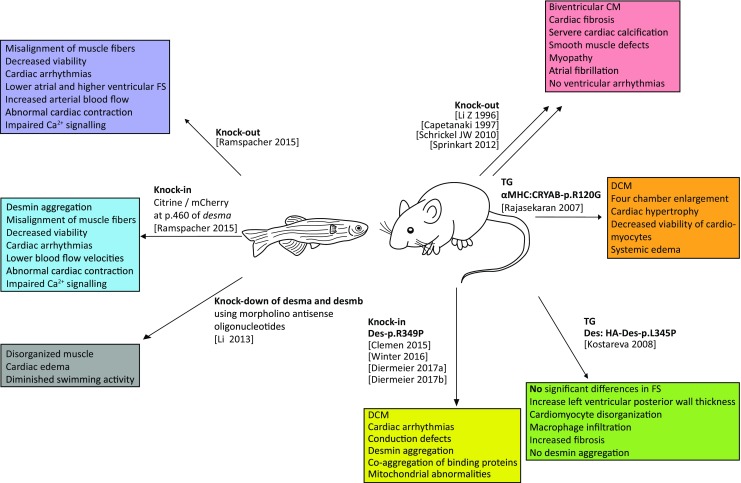
Overview of animal models used for modeling desminopathies. CM = cardiomyopathy, DCM = dilated cardiomyopathy
Recently, Rainer et al. (2018) applied transverse aortic constriction (TAC) in mice and demonstrated that toxic preamyloid oligomers containing cleaved desmin species accumulate in this heart failure model. This report indicates that in addition to DES mutations, also environmental factors can contribute to altered desmin structural and functional properties leading as a consequence to heart failure (Rainer et al. 2018).
Besides mutations in DES, DRM could be also caused by mutations in CRYAB, encoding the small heat shock protein αB-crystallin (Vicart et al. 1998). αB-crystallin is a binding partner of desmin filaments (Elliott et al. 2013). Mutations in both genes cause an abnormal co-aggregation of both proteins. Because mutations in CRYAB (αB-crystallin) cause also DRM, a transgenic mouse model with a cardiac-specific overexpression of mutant αB-crystallin (p.R120G) has been developed to investigate DRM. These mice are characterized by high mortality in early adulthood (Wang et al. 2001), toxic cardiac aggresomes containing desmin and αB-crystallin (Sanbe et al. 2004), increased apoptosis of cardiomyocytes (Maloyan et al. 2005), and oxido-reductive stress (Rajasekaran et al. 2007). However, it is currently unclear if all findings might be also relevant for DES mutations.
The zebrafish (Danio rerio) has two different desmin genes desma and desmb, which are 81 or 83% homologous to human DES (Ramspacher et al. 2015), respectively. Using morpholino antisense oligonucleotides, Li et al. (2013) knocked down by 50% the expression desma and desmb (Fig. 6). These embryos developed cardiac edema and presented a diminished swimming activity through disorganized muscles (Li et al. 2013). Ramspacher et al. generated different zebrafish lines to compare desma null lines with aggregate-forming lines. Both models developed embryonic cardiac defects like altered cardiac fractional shortening, perturbed heart biomechanics, and impaired Ca2+ signaling but showed also specific functional alterations (Ramspacher et al. 2015).
Genetic overview about human DES mutations
The American College of Medical Genetics and Genomics (ACMG) suggested a classification system for the interpretation of genetic sequence variants according to several criteria like co-segregation within the family, absence in controls, in silico prediction, or functional analysis (Richards et al. 2015). According to these guidelines, genetic sequence variants can be categorized into five classes: benign, likely benign, genetic variants with unknown significance (VUS), likely pathogenic, or pathogenic, respectively. The interpretation of novel DES sequence variants should follow these guidelines to increase the quality of genetic counseling of affected families and to prevent overinterpretation of rare sequence variants. Based on the minor allele frequency (MAF) in controls, Kostareva et al. (2011) demonstrated for example that DES-p.A213V is rather a benign single nucleotide polymorphism (SNP) than a pathogenic mutation.
During the 1980s and 1990s, several reports described an abnormal cytoplasmic desmin accumulation in muscle tissue of patients with cardiac and/or skeletal myopathies without identifying the relevant molecular trigger (Stoeckel et al. 1981; Goebel 1997; Osborn and Goebel 1983; Schroder et al. 1990). Several different terms like desmin-related myopathy (DRM), desminopathy, or inclusion body myopathy were used to describe this disease (Goebel 1997). The term DRM is mainly used to describe an abnormal accumulation of desmin and associated proteins leading to skeletal and cardiac myopathies. Although DRM and desminopathy are sometimes used as synonyms, most authors want to underline by using the term desminopathy that a pathogenic mutation in DES is the most likely genetic factor. However, specific cases with an abnormal desmin aggregation caused by mutations in further genes like CRYAB are also known (Vicart et al. 1998). The genetic trigger for DRM remained unknown until the end of the 1990s. Vicart et al. (1998) identified in a French family with DRM a pathogenic mutation in CRYAB (p.R120D). CRYAB encodes the small heat shock protein αB-crystallin, which is a binding partner of desmin filaments (Elliott et al. 2013). Although it was discovered as a structural component of the eye lenses, it is also highly expressed in (cardio)myocytes (Dubin et al. 1989). Small heat shock proteins are chaperones preventing the aggregation of misfolded proteins (Garrido et al. 2012). Interestingly, CRYAB mutations cause a co-aggregation of desmin and αB-crystallin (Vicart et al. 1998). Biochemical studies revealed later on that the protein-protein interaction between αB-crystallin and desmin is affected by mutations in both genes leading to an abnormal cytoplasmic co-aggregation of both proteins and in consequence to comparable clinical symptoms (Elliott et al. 2013; Rajasekaran et al. 2007; Brodehl et al. 2017b).
Shortly after the identification of the CRYAB mutation, two independent research groups described in parallel the first DES mutations causing DRM (Goldfarb et al. 1998; Munoz-Marmol et al. 1998). The human DES gene contains nine exons and has been mapped to chromosome 2 (2q35) (Li et al. 1989). In the last decades, it became more and more evident that DES mutations can cause different forms of skeletal and cardiac myopathies or variable combinations of both. Most of the known DES mutations are missense mutations or small in-frame deletions (Fig. 1). Many missense mutations introduce prolines (Brodehl et al. 2016a; Harada et al. 2018; Clemen et al. 2015; Fichna et al. 2014). Because of the cyclic imidic residue, prolines are incompatible with the formation of hydrogen bonds within the peptide bonds of α-helices and destabilize therefore the desmin structure.
The majority of DES mutations are heterozygously inherited indicating a dominant negative genetic mechanism or putative haploinsufficiency (Hedberg et al. 2012). This is in good agreement with the findings that mutant and wild-type desmin partially or completely co-aggregate (Brodehl et al. 2012a). However, some rare cases with compound heterozygous or homozygous DES truncating mutations were also described indicating that in specific cases the inheritance can be also recessive (Cetin et al. 2013; McLaughlin et al. 2013; Henderson et al. 2013; Tian et al. 2016; Pinol-Ripoll et al. 2009; Durmus et al. 2016). Frequently, mRNA molecules of genes with premature termination codons (PTCs) are degraded by nonsense-mediated mRNA decay or the truncated proteins are instable and consequently degraded (Alonso 2005). Consequently, patients carrying compound heterozygous DES truncating mutations do not express any desmin (McLaughlin et al. 2013). Heterozygous family members with one wild-type allele and a DES truncating mutation did not develop a phenotype excluding haploinsufficiency as the main molecular mechanism (McLaughlin et al. 2013; Henderson et al. 2013; Durmus et al. 2016). This is in agreement with heterozygous Des knock-out mice, which develop also no obvious phenotype (Li et al. 1996).
Of note, DES mutations might also occur de novo, and in these specific cases, it is difficult to recognize the genetic etiology based on the family anamnesis alone (Klauke et al. 2010; Park et al. 2000a; Dagvadorj et al. 2004; Sugawara et al. 2000). Furthermore, some DES splice site mutations were described (Ojrzynska et al. 2017; Khudiakov et al. 2017; Park et al. 2000b; Dunand et al. 2009; Kostareva et al. 2006; Gudkova et al. 2013) (Fig. 1a). However, it is challenging to predict the molecular consequences of splice site mutations at the mRNA and protein level because multiple unknown cryptic splice sites might be used or because exons can be completely skipped.
Clinical phenotypes associated with DES mutations
The clinical phenotypes associated with DES mutations are heterogeneous and range from isolated myopathies to different kinds of isolated cardiomyopathies and/or cardiac conduction disease. Most of the patients with DES mutations present a combined skeletal and cardiac myopathy. A meta-analysis published by van Spaendonck-Zwarts et al. (2011) revealed that about 75% of the patients with DES mutations present cardiac symptoms and only 22% of them have an isolated cardiac phenotype. However, it cannot be excluded that these patients might develop later also a phenotype of skeletal muscles since the onset of the disease appears to be independent in the different muscle systems and not predictable from the mutation. In addition, there is no evidence that the smooth muscle is concerned in desminopathies.
Some DES mutations are associated with an incomplete penetrance and diverse expressivity (Brodehl et al. 2016a). Even within the same family expressivity and severity of the associated clinical phenotypes of different mutation carriers might be remarkably heterogeneous (Palmio et al. 2013; van Spaendonck-Zwarts et al. 2012; McCormick et al. 2015; Bergman et al. 2007; Pica et al. 2008). Of note, the specific clinical phenotype can develop progressively and might change age dependently. In this context, data from clinical follow-up studies over longer periods are missing for most DES mutation carriers. Typically, DRM is clinically diagnosed during the third decade of life (van Spaendonck-Zwarts et al. 2011). However, the onset of disease is also highly variable and juvenile and even infantile onsets were reported (Klauke et al. 2010; Pinol-Ripoll et al. 2009).
Remarkably, there is no clear correlation between the position of the DES mutation and the associated clinical entities affecting the cardiac and skeletal muscle to a different degree (Figs. 3 and 4).
Furthermore, there is currently no molecular explanation for the broad spectrum of clinical phenotypes associated with DES mutations. Therefore, it has to be assumed that further genetic, epigenetic, and environmental factors modulate the clinical phenotype.
Cellular and molecular pathomechanisms caused by DES mutations
The histopathological hallmark of many but not of all DES mutations is an abnormal cytoplasmic desmin aggregation in (cardio)myocytes (Brodehl et al. 2012a; Goldfarb and Dalakas 2009; Herrmann et al. 2007). This desmin aggregation can be observed in cell transfection studies (Bar et al. 2005b; Brodehl et al. 2013a), in animal models (Clemen et al. 2015), and also in explanted myocardial tissue from DES mutation carriers (Brodehl et al. 2013a; Goebel and Muller 2006). However, there is a controversial on-going debate in what respect these desmin aggregates are toxic or whether the disturbed IF network is the molecular trigger for the degeneration of the cardiomyocytes (Goldfarb and Dalakas 2009; McLendon and Robbins 2011; Capetanaki et al. 2015). It has been suggested that mutant desmin inhibits the ubiquitin-proteasome system (Liu et al. 2006a, b). Several further secondary and tertiary molecular and cellular pathomechanisms have been reported, which contribute consequently to the disease progression. Conover et al. (2009) demonstrated that in addition to the IF system, also the actin filaments are affected by DES-p.E245D mutation. This can be explained by the finding that the IF system is cross-linked with several cytoskeletal components. In consequence, the force generation of myoblast and the complete cell elasticity can be altered by DES mutations (Charrier et al. 2016; Even et al. 2017). Different protein-protein interactions of desmin and IF-associated proteins can also be affected by DES mutations leading to a co-aggregation of these binding proteins (Chourbagi et al. 2011; Elliott et al. 2013). Interestingly, several studies reported a structural or functional impairment of the mitochondria by DES mutations (Winter et al. 2016; Henderson et al. 2013; McCormick et al. 2015; Smolina et al. 2014). The interplay between IFs and mitochondria was previously reviewed in detail (Schwarz and Leube 2016).
It has to be mentioned that not every pathogenic DES mutation causes desmin aggregation. Especially, mutations localized in the tail domain do not cause abnormal aggregation (Sharma et al. 2009; Brodehl et al. 2013b; Bar et al. 2010), which is in agreement with the finding that the deletion of the tail domain has no obvious effect on filament assembly (Kaufmann et al. 1985). However, different studies indicate that the nanomechanical properties and the network formation can nevertheless be changed by these mutations (Kreplak and Bar 2009; Bar et al. 2010).
Because desmin filaments are cellular scaffolds connecting different cellular components with the cytoskeleton, it is not surprising that DES mutations cause multiple pathomechanisms leading to death of cardiomyocytes and contributing accordingly to disease progression. Although the detailed molecular pathways triggered by the abnormal desmin aggregates are currently unknown, it was shown that desmin filaments are also substrates of caspases promoting apoptosis (Chen et al. 2003). Comparable abnormal protein aggregates caused by the CRYAB mutation p.R120G lead to an increased apoptosis including activation of caspase-3 in transgenic mice (Maloyan et al. 2005; Maloyan et al. 2010). However, it was also suggested that necrosis contributes to the pathogenicity in desmin knock-out mice (Sprinkart et al. 2012).
Therapeutic approaches
Currently, there is no specific molecular treatment available for desminopathies or DRM, respectively. However, some experimental reports using different mouse or cell culture models describe first putative therapeutic approaches, which will we summarized in the following paragraph. Sanbe et al. used oral administration of geranylgeranylacetone (GGA) to induce the expression of small heat shock proteins in a transgenic mouse model expressing mutant αB-crystallin (CRYAB-p.R120G), which is a model for DRM (Rajasekaran et al. 2007). Small heat shock proteins are adenosine triphosphate (ATP)-independent chaperones, which bind unfolded proteins and prevent protein accumulation and aggregation (Garrido et al. 2012). Remarkably, GGA protected CRYAB-p.R120G transgenic mice significantly against cardiac death by inducing the expression of small heat shock proteins and inhibiting protein aggregation (Sanbe et al. 2009). Nicorandil is a small compound, which is cardioprotective (Zhao et al. 2014). Recently, it was demonstrated that the administration of nicorandil improves the fractional shortening and reverses cardiac electrical remodeling in the CRYAB-p.R120G transgenic mouse model (Matsushita et al. 2014; Sanbe et al. 2011). Pharmacological analysis using C2C12 cells transiently expressing mutant desmin revealed that inhibition of the Rac1 pathway, activation of autophagy pathways using PP242, and further inducers of autophagy and antioxidant treatment significantly reduce the desmin aggregation (Cabet et al. 2015). However, it is unclear how or if these preclinical data can be translated to the treatment of patients with DES mutations.
Despite these pilot experiments, there is currently no molecular gene therapy available. In general, gene therapy suffered in the last decades several setbacks (Yla-Herttuala and Baker 2017), and before applicable under standard clinical conditions, several ethical and technical issues have to be solved.
Future perspectives
The majority of DES mutations are missense or small deletion mutations, which might be classified into two groups: aggregate-forming and filament-forming mutations. For most of the filament-forming mutations, the exact pathomechanisms are widely unknown and future molecular studies are necessary to elucidate them. Several mutations in further genes like CRYAB (Brodehl et al. 2017b), FLNC (Brodehl et al. 2016b), BAG3 (Schanzer et al. 2018), and MYOT (Maerkens et al. 2016) cause also an abnormal protein aggregation leading in consequence to cardiac and skeletal myopathies.
However, for the majority of DES mutations, the aggregate formation seems to be a first direct trigger of the disease. However, the downstream effects are diverse and heterogeneous. Hopefully, new developments in molecular and cell biology will help to develop molecular therapies for desminopathies. Because the aggregate formation is a direct consequence of DES mutations, it can be suggested to focus primarily on the prevention of aggregate formation. Targeting DES gene regulation leading to a decreased expression of the mutant DES allele might be a promising strategy. Several putative approaches based on DNA genome editing using CRISPR-Cas9 or TALENs (Jinek et al. 2012), RNA-targeted therapeutics (Crooke et al. 2018), or the modulation of protein degradation (Clift et al. 2017) or folding are relevant for preclinical proof-of-concept studies to specifically treat DES mutation carriers. However, at present, these novel technologies are far away from a transfer to clinical application.
Summary
Different forms of cardiomyopathies and skeletal myopathies can be caused by DES mutations. Novel DES mutations should be carefully interpreted according to ACMG guidelines to improve genetic counseling. The majority of pathogenic DES mutations cause an abnormal cytoplasmic desmin aggregation, which can be verified by cell transfection experiments (Brodehl et al. 2013a). Because desmin is a scaffolding protein connecting different cell organelles, the secondary and tertiary molecular and cellular pathomechanism in vitro and in vivo are diverse and affect different cellular compartments. Currently, there is no specific therapy for desminopathies available. Therefore, there is a strong need for the development of efficient molecular therapies in the future.
Acknowledgements
The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. We thank Dr. Volker Walhorn (Experimental Biophysics and Applied Nanoscience, Faculty of Physics and Bielefeld Institute for Biophysics and Nanoscience (BINAS), Bielefeld University, Germany) for providing Fig. 5e.
Abbreviations
- ACM
Arrhythmogenic cardiomyopathy
- ACMG
American College of Medical Genetics and Genomics
- AF
Atrial fibrillation
- AFM
Atomic force microscopy
- ALVC
Arrhythmogenic left ventricular cardiomyopathy
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- aSNOM
Apertureless scanning near-field microscopy
- ATP
Adenosine triphosphate
- AVB
Atrioventricular block
- CM
Cardiomyopathy
- DCM
Dilated cardiomyopathy
- DRM
Desmin-related myopathy
- DSC2
Desmocollin-2
- DSG2
Desmoglein-2
- DSP
Desmoplakin
- EPR
Electron paramagnetic resonance
- GFAP
Glial fibrillary acidic protein
- GGA
Geranylgeranylacetone
- HCM
Hypertrophic cardiomyopathy
- HOCM
Hypertrophic obstructive cardiomyopathy
- HTx
Heart transplantation
- IF
Intermediate filament
- LAFB
Left anterior fascicular block
- LBBB
Left bundle-branched block
- LGMD
Limb-girdle muscular dystrophy
- LVNC
Left ventricular noncompaction cardiomyopathy
- LW
Limp weakness
- MAF
Minor allele frequency
- MFM
Myofibrillar myopathy
- MRI
Magnetic resonance imaging
- NCM
Noncompaction cardiomyopathy
- NMD
Nonsense-mediated RNA decay
- PG
Plakoglobin
- PKP2
Plakophilin-2
- PTC
Premature termination codon
- PTM
Posttranslational modification
- RBBB
Right bundle-branched block
- RCM
Restrictive cardiomyopathy
- SCD
Sudden cardiac death
- SM
Skeletal myopathy
- SMD
Smooth muscle defect
- SNP
Single nucleotide polymorphism
- SQTS
Short QT syndrome
- TAC
Transverse aortic constriction
- TEM
Transmission electron microscopy
- ULF
Unit length filament
- VUS
Variant of unknown significance
Compliance with ethical standards
AB received a grant of the German Society of Heart Research (DSHF, F/07/17) and a grant of the University Bielefeld (Reseach fond OWL). AGR is supported by the Medical Faculty of the Ruhr-University Bochum (FoRUM). HM is thankful for funding of the German Research Foundation (DFG, MI 1146/2-1) and the Erich and Hanna Klessmann Foundation (Gütersloh, Germany).
Andreas Brodehl declares that he has no conflicts of interest. Anna Gaertner-Rommel declares that she has no conflicts of interest. Hendrik Milting declares that he has no conflicts of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
This article is part of a Special Issue on ‘Heart Failure Due to Non-Myofibrillar Defects’ edited by Elisabeth Ehler and Katja Gehmlich.
Additional databases
1. Human Intermediate Filament Database, www.interfil.org (Szeverenyi et al. 2008).
2. The Human Protein Atlas, https://www.proteinatlas.org (Uhlen et al. 2015).
3. ClinVar, https://www.ncbi.nlm.nih.gov/clinvar (Landrum et al. 2016).
4. Leiden Open Variation Database, http://www.dmd.nl.
5. Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org/ (Lek et al. 2016).
6. Genome Aggregation Database (gnomAD), http://gnomad.broadinstitute.org/ (Lek et al. 2016).
Contributor Information
Andreas Brodehl, Phone: +49-(0)5731-973530, Email: abrodehl@hdz-nrw.de.
Hendrik Milting, Phone: +49-(0)5731-973510, Email: hmilting@hdz-nrw.de.
References
- Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, and Ackerman MJ (2016) The promise and peril of precision medicine: phenotyping still matters most. Mayo Clinic proceedings [DOI] [PMC free article] [PubMed]
- Albers K, Fuchs E. The molecular biology of intermediate filament proteins. Int Rev Cytol. 1992;134:243–279. doi: 10.1016/s0074-7696(08)62030-6. [DOI] [PubMed] [Google Scholar]
- Alonso CR. Nonsense-mediated RNA decay: a molecular system micromanaging individual gene activities and suppressing genomic noise. BioEssays. 2005;27:463–466. doi: 10.1002/bies.20227. [DOI] [PubMed] [Google Scholar]
- Ando S, Nakao K, Gohara R, Takasaki Y, Suehiro K, Oishi Y. Morphological analysis of glutaraldehyde-fixed vimentin intermediate filaments and assembly-intermediates by atomic force microscopy. Biochim Biophys Acta. 2004;1702:53–65. doi: 10.1016/j.bbapap.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunso S, Svendsen JH, Olesen MS. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013;21:918–928. doi: 10.1038/ejhg.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, Repetto A, Bellini O, Azan G, Scaffino M, Campana C, Piccolo G, Vigano M, Tavazzi L. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8:477–483. doi: 10.1016/j.ejheart.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Arias M, Pardo J, Blanco-Arias P, Sobrido MJ, Arias S, Dapena D, Carracedo A, Goldfarb LG, Navarro C. Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord. 2006;16:498–503. doi: 10.1016/j.nmd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Aziz A, Hess JF, Budamagunta MS, FitzGerald PG, Voss JC. Head and rod 1 interactions in vimentin: identification of contact sites, structure, and changes with phosphorylation using site-directed spin labeling and electron paramagnetic resonance. J Biol Chem. 2009;284:7330–7338. doi: 10.1074/jbc.M809029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz A, Hess JF, Budamagunta MS, Voss JC, Fitzgerald PG. Site-directed spin labeling and electron paramagnetic resonance determination of vimentin head domain structure. J Biol Chem. 2010;285:15278–15285. doi: 10.1074/jbc.M109.075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LK, Gillis DC, Sharma S, Ambrus A, Herrmann H, Conover GM. Nebulin binding impedes mutant desmin filament assembly. Mol Biol Cell. 2013;24:1918–1932. doi: 10.1091/mbc.E12-11-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar H, Mucke N, Kostareva A, Sjoberg G, Aebi U, Herrmann H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc Natl Acad Sci U S A. 2005;102:15099–15104. doi: 10.1073/pnas.0504568102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar H, Fischer D, Goudeau B, Kley RA, Clemen CS, Vicart P, Herrmann H, Vorgerd M, Schroder R. Pathogenic effects of a novel heterozygous R350P desmin mutation on the assembly of desmin intermediate filaments in vivo and in vitro. Hum Mol Genet. 2005;14:1251–1260. doi: 10.1093/hmg/ddi136. [DOI] [PubMed] [Google Scholar]
- Bar H, Mucke N, Ringler P, Muller SA, Kreplak L, Katus HA, Aebi U, Herrmann H. Impact of disease mutations on the desmin filament assembly process. J Mol Biol. 2006;360:1031–1042. doi: 10.1016/j.jmb.2006.05.068. [DOI] [PubMed] [Google Scholar]
- Bar H, Goudeau B, Walde S, Casteras-Simon M, Mucke N, Shatunov A, Goldberg YP, Clarke C, Holton JL, Eymard B, Katus HA, Fardeau M, Goldfarb L, Vicart P, Herrmann H. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat. 2007;28:374–386. doi: 10.1002/humu.20459. [DOI] [PubMed] [Google Scholar]
- Bar H, Schopferer M, Sharma S, Hochstein B, Mucke N, Herrmann H, Willenbacher N. Mutations in desmin's carboxy-terminal “tail” domain severely modify filament and network mechanics. J Mol Biol. 2010;397:1188–1198. doi: 10.1016/j.jmb.2010.02.024. [DOI] [PubMed] [Google Scholar]
- Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26:1666–1675. doi: 10.1093/eurheartj/ehi341. [DOI] [PubMed] [Google Scholar]
- Becker B, Bellin RM, Sernett SW, Huiatt TW, Robson RM. Synemin contains the rod domain of intermediate filaments. Biochem Biophys Res Commun. 1995;213:796–802. doi: 10.1006/bbrc.1995.2200. [DOI] [PubMed] [Google Scholar]
- Bellin RM, Sernett SW, Becker B, Ip W, Huiatt TW, Robson RM. Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J Biol Chem. 1999;274:29493–29499. doi: 10.1074/jbc.274.41.29493. [DOI] [PubMed] [Google Scholar]
- Bellin RM, Huiatt TW, Critchley DR, Robson RM. Synemin may function to directly link muscle cell intermediate filaments to both myofibrillar Z-lines and costameres. J Biol Chem. 2001;276:32330–32337. doi: 10.1074/jbc.M104005200. [DOI] [PubMed] [Google Scholar]
- Bergman JE, Veenstra-Knol HE, van Essen AJ, van Ravenswaaij CM, den Dunnen WF, van den Wijngaard A, van Tintelen JP. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet. 2007;50:355–366. doi: 10.1016/j.ejmg.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Bermudez-Jimenez, F. J., V. Carriel, A. Brodehl, M. Alaminos, A. Campos, I. Schirmer, H. Milting, B. Alvarez Abril, M. Alvarez, S. Lopez-Fernandez, D. Garcia-Giustiniani, L. Monserrat, L. Tercedor, and J. Jimenez-Jaimez. 2017. The novel desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation [DOI] [PubMed]
- Bhosle RC, Michele DE, Campbell KP, Li Z, Robson RM. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun. 2006;346:768–777. doi: 10.1016/j.bbrc.2006.05.192. [DOI] [PubMed] [Google Scholar]
- Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, Schouten M, Jongbloed R, Cox MG, van Wolferen M, Rodriguez LM, van Gelder IC, Bikker H, Suurmeijer AJ, van den Berg MP, Mannens MM, Hauer RN, Wilde AA, van Tintelen JP. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- Bilak SR, Sernett SW, Bilak MM, Bellin RM, Stromer MH, Huiatt TW, Robson RM. Properties of the novel intermediate filament protein synemin and its identification in mammalian muscle. Arch Biochem Biophys. 1998;355:63–76. doi: 10.1006/abbi.1998.0702. [DOI] [PubMed] [Google Scholar]
- Bonakdar N, Luczak J, Lautscham L, Czonstke M, Koch TM, Mainka A, Jungbauer T, Goldmann WH, Schroder R, Fabry B. Biomechanical characterization of a desminopathy in primary human myoblasts. Biochem Biophys Res Commun. 2012;419:703–707. doi: 10.1016/j.bbrc.2012.02.083. [DOI] [PubMed] [Google Scholar]
- Brayson D, Shanahan CM. Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus. 2017;8:17–33. doi: 10.1080/19491034.2016.1260798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Brodehl A, Hedde PN, Dieding M, Fatima A, Walhorn V, Gayda S, Saric T, Klauke B, Gummert J, Anselmetti D, Heilemann M, Nienhaus GU, Milting H. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J Biol Chem. 2012;287:16047–16057. doi: 10.1074/jbc.M111.313841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Schmidt T, Gummert J, Milting H. Molecular insights into filament assembly defects of ARVC-related desmin mutations. Cardiovasc Res. 2012;93:S38–S38. [Google Scholar]
- Brodehl A, Dieding M, Klauke B, Dec E, Madaan S, Huang T, Gargus J, Fatima A, Saric T, Cakar H, Walhorn V, Tonsing K, Skrzipczyk T, Cebulla R, Gerdes D, Schulz U, Gummert J, Svendsen JH, Olesen MS, Anselmetti D, Christensen AH, Kimonis V, Milting H. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ Cardiovasc Genet. 2013;6:615–623. doi: 10.1161/CIRCGENETICS.113.000103. [DOI] [PubMed] [Google Scholar]
- Brodehl A, Dieding M, Cakar H, Klauke B, Walhorn V, Gummert J, Anselmetti D, Milting H. Functional characterization of desmin mutant p.P419S. Eur J Hum Genet. 2013;21:589–590. doi: 10.1038/ejhg.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Dieding M, Biere N, Unger A, Klauke B, Walhorn V, Gummert J, Schulz U, Linke WA, Gerull B, Vorgert M, Anselmetti D, Milting H. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J Mol Cell Cardiol. 2016;91:207–214. doi: 10.1016/j.yjmcc.2015.12.015. [DOI] [PubMed] [Google Scholar]
- Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, Gibson WT, McKinnon ML, McGillivray B, Alvarez N, Giuffre M, Schwartzentruber J, Consortium FC, Gerull B. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat. 2016;37:269–279. doi: 10.1002/humu.22942. [DOI] [PubMed] [Google Scholar]
- Brodehl A, Belke DD, Garnett L, Martens K, Abdelfatah N, Rodriguez M, Diao C, Chen YX, Gordon PM, Nygren A, Gerull B. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS One. 2017;12:e0174019. doi: 10.1371/journal.pone.0174019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodehl A, Gaertner-Rommel A, Klauke B, Grewe SA, Schirmer I, Peterschroder A, Faber L, Vorgerd M, Gummert J, Anselmetti D, Schulz U, Paluszkiewicz L, Milting H. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum Mutat. 2017;38:947–952. doi: 10.1002/humu.23248. [DOI] [PubMed] [Google Scholar]
- Cabet E, Batonnet-Pichon S, Delort F, Gausseres B, Vicart P, Lilienbaum A. Antioxidant treatment and induction of autophagy cooperate to reduce desmin aggregation in a cellular model of desminopathy. PLoS One. 2015;10:e0137009. doi: 10.1371/journal.pone.0137009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res. 2013;113:660–675. doi: 10.1161/CIRCRESAHA.113.300282. [DOI] [PubMed] [Google Scholar]
- Cao L, Hong D, Zhu M, Li X, Wan H, Hong K. A novel heterozygous deletion-insertion mutation in the desmin gene causes complete atrioventricular block and mild myopathy. Clin Neuropathol. 2013;32:9–15. doi: 10.5414/NP300514. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y, Milner DJ, Weitzer G. Desmin in muscle formation and maintenance: knockouts and consequences. Cell Struct Funct. 1997;22:103–116. doi: 10.1247/csf.22.103. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatsellas G, Tsikitis M. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol. 2015;32:113–120. doi: 10.1016/j.ceb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson L, Thornell LE. Desmin-related myopathies in mice and man. Acta Physiol Scand. 2001;171:341–348. doi: 10.1046/j.1365-201x.2001.00837.x. [DOI] [PubMed] [Google Scholar]
- Cerino M, Gorokhova S, Laforet P, Ben Yaou R, Salort-Campana E, Pouget J, Attarian S, Eymard B, Deleuze JF, Boland A, Behin A, Stojkovic T, Bonne G, Levy N, Bartoli M, Krahn M. Genetic characterization of a French cohort of GNE-mutation negative inclusion body myopathy patients with exome sequencing. Muscle Nerve. 2017;56:993–997. doi: 10.1002/mus.25638. [DOI] [PubMed] [Google Scholar]
- Cetin N, Balci-Hayta B, Gundesli H, Korkusuz P, Purali N, Talim B, Tan E, Selcen D, Erdem-Ozdamar S, Dincer P. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet. 2013;50:437–443. doi: 10.1136/jmedgenet-2012-101487. [DOI] [PubMed] [Google Scholar]
- Charrier EE, Asnacios A, Milloud R, De Mets R, Balland M, Delort F, Cardoso O, Vicart P, Batonnet-Pichon S, Henon S. Desmin mutation in the C-terminal domain impairs traction force generation in myoblasts. Biophys J. 2016;110:470–480. doi: 10.1016/j.bpj.2015.11.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J Biol Chem. 2002;277:10512–10522. doi: 10.1074/jbc.M108765200. [DOI] [PubMed] [Google Scholar]
- Chen F, Chang R, Trivedi M, Capetanaki Y, Cryns VL. Caspase proteolysis of desmin produces a dominant-negative inhibitor of intermediate filaments and promotes apoptosis. J Biol Chem. 2003;278:6848–6853. doi: 10.1074/jbc.M212021200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Barajas-Martinez H, Zhu D, Wang X, Chen C, Zhuang R, Shi J, Wu X, Tao Y, Jin W, Wang X, Hu D. Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J Transl Med. 2017;15:78. doi: 10.1186/s12967-017-1180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyatina AA, Nicolet S, Aebi U, Herrmann H, Strelkov SV. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc Natl Acad Sci U S A. 2012;109:13620–13625. doi: 10.1073/pnas.1206836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyatina AA, Guzenko D, Strelkov SV. Intermediate filament structure: the bottom-up approach. Curr Opin Cell Biol. 2015;32:65–72. doi: 10.1016/j.ceb.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Chitaev NA, Averbakh AZ, Troyanovsky RB, Troyanovsky SM. Molecular organization of the desmoglein-plakoglobin complex. J Cell Sci. 1998;111(Pt 14):1941–1949. doi: 10.1242/jcs.111.14.1941. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Park-Snyder S, Pascoe LT, Green KJ, Weis WI. Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat Struct Biol. 2002;9:612–620. doi: 10.1038/nsb818. [DOI] [PubMed] [Google Scholar]
- Chourbagi O, Bruston F, Carinci M, Xue Z, Vicart P, Paulin D, Agbulut O. Desmin mutations in the terminal consensus motif prevent synemin-desmin heteropolymer filament assembly. Exp Cell Res. 2011;317:886–897. doi: 10.1016/j.yexcr.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Clemen CS, Stockigt F, Strucksberg KH, Chevessier F, Winter L, Schutz J, Bauer R, Thorweihe JM, Wenzel D, Schlotzer-Schrehardt U, Rasche V, Krsmanovic P, Katus HA, Rottbauer W, Just S, Muller OJ, Friedrich O, Meyer R, Herrmann H, Schrickel JW, Schroder R. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015;129:297–315. doi: 10.1007/s00401-014-1363-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC, Schuh M. A method for the acute and rapid degradation of endogenous proteins. Cell. 2017;171(1692–1706):e1618. doi: 10.1016/j.cell.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colakoglu G, Brown A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J Cell Biol. 2009;185:769–777. doi: 10.1083/jcb.200809166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover GM, Henderson SN, Gregorio CC. A myopathy-linked desmin mutation perturbs striated muscle actin filament architecture. Mol Biol Cell. 2009;20:834–845. doi: 10.1091/mbc.E08-07-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe PA. The molecular revolution in cutaneous biology: keratin genes and their associated disease: diversity, opportunities, and challenges. J Invest Dermatol. 2017;137:e67–e71. doi: 10.1016/j.jid.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-targeted therapeutics. Cell Metab. 2018;27:714–739. doi: 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Goudeau B, Hilton-Jones D, Blancato JK, Shatunov A, Simon-Casteras M, Squier W, Nagle JW, Goldfarb LG, Vicart P. Respiratory insufficiency in desminopathy patients caused by introduction of proline residues in desmin C-terminal alpha-helical segment. Muscle Nerve. 2003;27:669–675. doi: 10.1002/mus.10370. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Olive M, Urtizberea JA, Halle M, Shatunov A, Bonnemann C, Park KY, Goebel HH, Ferrer I, Vicart P, Dalakas MC, Goldfarb LG. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol. 2004;251:143–149. doi: 10.1007/s00415-004-0289-3. [DOI] [PubMed] [Google Scholar]
- Dal Ferro M, Stolfo D, Altinier A, Gigli M, Perrieri M, Ramani F, Barbati G, Pivetta A, Brun F, Monserrat L, Giacca M, Mestroni L, Merlo M, Sinagra G. Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart. 2017;103:1704–1710. doi: 10.1136/heartjnl-2016-311017. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Dagvadorj A, Goudeau B, Park KY, Takeda K, Simon-Casteras M, Vasconcelos O, Sambuughin N, Shatunov A, Nagle JW, Sivakumar K, Vicart P, Goldfarb LG. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 2003;13:252–258. doi: 10.1016/s0960-8966(02)00271-7. [DOI] [PubMed] [Google Scholar]
- Dieding M, Debus JD, Kerkhoff R, Gaertner-Rommel A, Walhorn V, Milting H, Anselmetti D. Arrhythmogenic cardiomyopathy related DSG2 mutations affect desmosomal cadherin binding kinetics. Sci Rep. 2017;7:13791. doi: 10.1038/s41598-017-13737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diermeier S, Iberl J, Vetter K, Haug M, Pollmann C, Reischl B, Buttgereit A, Schurmann S, Sporrer M, Goldmann WH, Fabry B, Elhamine F, Stehle R, Pfitzer G, Winter L, Clemen CS, Herrmann H, Schroder R, Friedrich O. Early signs of architectural and biomechanical failure in isolated myofibers and immortalized myoblasts from desmin-mutant knock-in mice. Sci Rep. 2017;7:1391. doi: 10.1038/s41598-017-01485-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diermeier S, Buttgereit A, Schurmann S, Winter L, Xu H, Murphy RM, Clemen CS, Schroder R, Friedrich O. Preaged remodeling of myofibrillar cytoarchitecture in skeletal muscle expressing R349P mutant desmin. Neurobiol Aging. 2017;58:77–87. doi: 10.1016/j.neurobiolaging.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Dubin RA, Wawrousek EF, Piatigorsky J. Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol Cell Biol. 1989;9:1083–1091. doi: 10.1128/mcb.9.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunand M, Lobrinus JA, Jeannet PY, Behin A, Claeys KG, Selcen D, Kuntzer T. Confirmation that abnormal desmin accumulation and migration are due to a desmin gene mutation in a familial cardiomyopathy and distal myopathy. Neuromuscul Disord. 2009;19:802. doi: 10.1016/j.nmd.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Durmus H, Ayhan O, Cirak S, Deymeer F, Parman Y, Franke A, Eiber N, Chevessier F, Schlotzer-Schrehardt U, Clemen CS, Hashemolhosseini S, Schroder R, Hemmrich-Stanisak G, Tolun A, Serdaroglu-Oflazer P. Neuromuscular endplate pathology in recessive desminopathies: lessons from man and mice. Neurology. 2016;87:799–805. doi: 10.1212/WNL.0000000000003004. [DOI] [PubMed] [Google Scholar]
- Elliott JL, Der Perng M, Prescott AR, Jansen KA, Koenderink GH, Quinlan RA. The specificity of the interaction between alphaB-crystallin and desmin filaments and its impact on filament aggregation and cell viability. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20120375. doi: 10.1098/rstb.2012.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even C, Abramovici G, Delort F, Rigato AF, Bailleux V, de Sousa Moreira A, Vicart P, Rico F, Batonnet-Pichon S, Briki F. Mutation in the core structure of desmin intermediate filaments affects myoblast elasticity. Biophys J. 2017;113:627–636. doi: 10.1016/j.bpj.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichna JP, Karolczak J, Potulska-Chromik A, Miszta P, Berdynski M, Sikorska A, Filipek S, Redowicz MJ, Kaminska A, Zekanowski C. Two desmin gene mutations associated with myofibrillar myopathies in Polish families. PLoS One. 2014;9:e115470. doi: 10.1371/journal.pone.0115470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidzianska A, Kotowicz J, Sadowska M, Goudeau B, Walczak E, Vicart P, Hausmanowa-Petrusewicz I. A novel desmin R355P mutation causes cardiac and skeletal myopathy. Neuromuscul Disord. 2005;15:525–531. doi: 10.1016/j.nmd.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Fischer D, Clemen CS, Olive M, Ferrer I, Goudeau B, Roth U, Badorf P, Wattjes MP, Lutterbey G, Kral T, van der Ven PF, Furst DO, Vicart P, Goldfarb LG, Moza M, Carpen O, Reichelt J, Schroder R. Different early pathogenesis in myotilinopathy compared to primary desminopathy. Neuromuscul Disord. 2006;16:361–367. doi: 10.1016/j.nmd.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Fokstuen S, Makrythanasis P, Hammar E, Guipponi M, Ranza E, Varvagiannis K, Santoni FA, Albarca-Aguilera M, Poleggi ME, Couchepin F, Brockmann C, Mauron A, Hurst SA, Moret C, Gehrig C, Vannier A, Bevillard J, Araud T, Gimelli S, Stathaki E, Paoloni-Giacobino A, Bottani A, Sloan-Bena F, Sizonenko LD, Mostafavi M, Hamamy H, Nouspikel T, Blouin JL, Antonarakis SE. Experience of a multidisciplinary task force with exome sequencing for Mendelian disorders. Hum Genom. 2016;10:24. doi: 10.1186/s40246-016-0080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pelagio KP, Chen L, Joca HC, Ward C, Jonathan Lederer W, Bloch RJ. Absence of synemin in mice causes structural and functional abnormalities in heart. J Mol Cell Cardiol. 2018;114:354–363. doi: 10.1016/j.yjmcc.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard JJ, Yamada K, Green KG, Eloff BC, Rosenbaum DS, Wang X, Robbins J, Schuessler RB, Yamada KA, Saffitz JE. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res. 2005;67:539–547. doi: 10.1016/j.cardiores.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Garrido C, Paul C, Seigneuric R, Kampinga HH. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44:1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Gehmlich K, Lambiase PD, Asimaki A, Ciaccio EJ, Ehler E, Syrris P, Saffitz JE, McKenna WJ. A novel desmocollin-2 mutation reveals insights into the molecular link between desmosomes and gap junctions. Heart Rhythm. 2011;8:711–718. doi: 10.1016/j.hrthm.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehmlich K, Syrris P, Reimann M, Asimaki A, Ehler E, Evans A, Quarta G, Pantazis A, Saffitz JE, McKenna WJ. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc Pathol. 2012;21:275–282. doi: 10.1016/j.carpath.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Geisler N, Kaufmann E, Weber K. Antiparallel orientation of the two double-stranded coiled-coils in the tetrameric protofilament unit of intermediate filaments. J Mol Biol. 1985;182:173–177. doi: 10.1016/0022-2836(85)90035-x. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- Goebel HH. Desmin-related myopathies. Curr Opin Neurol. 1997;10:426–429. doi: 10.1097/00019052-199710000-00012. [DOI] [PubMed] [Google Scholar]
- Goebel HH, Muller HD. Protein aggregate myopathies. Semin Pediatr Neurol. 2006;13:96–103. doi: 10.1016/j.spen.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. doi: 10.1161/CIRCGENETICS.113.000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- Goudeau B, Dagvadorj A, Rodrigues-Lima F, Nedellec P, Casteras-Simon M, Perret E, Langlois S, Goldfarb L, Vicart P. Structural and functional analysis of a new desmin variant causing desmin-related myopathy. Hum Mutat. 2001;18:388–396. doi: 10.1002/humu.1210. [DOI] [PubMed] [Google Scholar]
- Goudeau B, Rodrigues-Lima F, Fischer D, Casteras-Simon M, Sambuughin N, de Visser M, Laforet P, Ferrer X, Chapon F, Sjoberg G, Kostareva A, Sejersen T, Dalakas MC, Goldfarb LG, Vicart P. Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum Mutat. 2006;27:906–913. doi: 10.1002/humu.20351. [DOI] [PubMed] [Google Scholar]
- Granger BL, Lazarides E. Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell. 1980;22:727–738. doi: 10.1016/0092-8674(80)90549-8. [DOI] [PubMed] [Google Scholar]
- Gudkova A, Kostareva A, Sjoberg G, Smolina N, Turalchuk M, Kuznetsova I, Rybakova M, Edstrom L, Shlyakhto E, Sejersen T. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr Cardiol. 2013;34:467–470. doi: 10.1007/s00246-012-0312-x. [DOI] [PubMed] [Google Scholar]
- Guzman C, Jeney S, Kreplak L, Kasas S, Kulik AJ, Aebi U, Forro L. Exploring the mechanical properties of single vimentin intermediate filaments by atomic force microscopy. J Mol Biol. 2006;360:623–630. doi: 10.1016/j.jmb.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim WK, Zhao X, Fradkin D, Kohler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjaer H, Jorgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Morner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135a. doi: 10.1093/eurheartj/ehu301. [DOI] [PubMed] [Google Scholar]
- Harada H, Hayashi T, Nishi H, Kusaba K, Koga Y, Koga Y, Nonaka I, Kimura A. Phenotypic expression of a novel desmin gene mutation: hypertrophic cardiomyopathy followed by systemic myopathy. J Hum Genet. 2018;63:249–254. doi: 10.1038/s10038-017-0383-x. [DOI] [PubMed] [Google Scholar]
- Harder A, Dieding M, Walhorn V, Degenhard S, Brodehl A, Wege C, Milting H, Anselmetti D. Apertureless scanning near-field optical microscopy of sparsely labeled tobacco mosaic viruses and the intermediate filament desmin. Beilstein J Nanotechnol. 2013;4:510–516. doi: 10.3762/bjnano.4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, Shapiro L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci U S A. 2016;113:7160–7165. doi: 10.1073/pnas.1606272113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell GT, Jensen BC, Samsa LA, Marchuk D, Huang W, Skrzynia C, Tilley C, Seifert BA, Rivera-Munoz EA, Koller B, Wilhelmsen KC, Liu JD, Alhosaini H, Weck KE, Evans JP, Berg JS (2017) Whole exome sequencing identifies truncating variants in nuclear envelope genes in patients with cardiovascular disease. Circ Cardiovasc Genet 10 [DOI] [PMC free article] [PubMed]
- Hatzfeld M, Weber K. A synthetic peptide representing the consensus sequence motif at the carboxy-terminal end of the rod domain inhibits intermediate filament assembly and disassembles preformed filaments. J Cell Biol. 1992;116:157–166. doi: 10.1083/jcb.116.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann MB, Bauer R, Jungmann A, Winter L, Rapti K, Strucksberg KH, Clemen CS, Li Z, Schroder R, Katus HA, Muller OJ. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther. 2016;23:673–679. doi: 10.1038/gt.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg C, Melberg A, Kuhl A, Jenne D, Oldfors A. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur J Human Genet. 2012;20:984–985. doi: 10.1038/ejhg.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg C, Melberg A, Kuhl A, Jenne D, Oldfors A. Reply to Brodehl et al. Eur J Hum Genet. 2013;21:590. doi: 10.1038/ejhg.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedde PN, Gayda S, Brodehl A, Gummert J, Milting H, Nienhaus GU. Colocalization analysis of mutant and wildtype desmin using dual color super-resolution microscopy. Biophys J. 2012;102:722a–722a. [Google Scholar]
- Henderson M, De Waele L, Hudson J, Eagle M, Sewry C, Marsh J, Charlton R, He L, Blakely EL, Horrocks I, Stewart W, Taylor RW, Longman C, Bushby K, Barresi R. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. 2013;125:917–919. doi: 10.1007/s00401-013-1113-x. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Aebi U. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu Rev Biochem. 2004;73:749–789. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Strelkov SV. History and phylogeny of intermediate filaments: now in insects. BMC Biol. 2011;9:16. doi: 10.1186/1741-7007-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H, Haner M, Brettel M, Muller SA, Goldie KN, Fedtke B, Lustig A, Franke WW, Aebi U. Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J Mol Biol. 1996;264:933–953. doi: 10.1006/jmbi.1996.0688. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Strelkov SV, Feja B, Rogers KR, Brettel M, Lustig A, Haner M, Parry DA, Steinert PM, Burkhard P, Aebi U. The intermediate filament protein consensus motif of helix 2B: its atomic structure and contribution to assembly. J Mol Biol. 2000;298:817–832. doi: 10.1006/jmbi.2000.3719. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Strelkov SV, Burkhard P, Aebi U. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119:1772–1783. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, MacRae CA, Gerull B. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirako Y, Yamakawa H, Tsujimura Y, Nishizawa Y, Okumura M, Usukura J, Matsumoto H, Jackson KW, Owaribe K, Ohara O. Characterization of mammalian synemin, an intermediate filament protein present in all four classes of muscle cells and some neuroglial cells: co-localization and interaction with type III intermediate filament proteins and keratins. Cell Tissue Res. 2003;313:195–207. doi: 10.1007/s00441-003-0732-2. [DOI] [PubMed] [Google Scholar]
- Hnia K, Ramspacher C, Vermot J, Laporte J. Desmin in muscle and associated diseases: beyond the structural function. Cell Tissue Res. 2015;360:591–608. doi: 10.1007/s00441-014-2016-4. [DOI] [PubMed] [Google Scholar]
- Hofmann I, Mertens C, Brettel M, Nimmrich V, Schnolzer M, Herrmann H. Interaction of plakophilins with desmoplakin and intermediate filament proteins: an in vitro analysis. J Cell Sci. 2000;113(Pt 13):2471–2483. doi: 10.1242/jcs.113.13.2471. [DOI] [PubMed] [Google Scholar]
- Hong DJ, Zhang W, Jiang TY, Feng L, Wang ZX, Yuan Y. Clinical characteristics and desmin mutations in patients with desminopathy associated cardiomyopathy from 5 Chinese families. Zhonghua xin xue guan bing za zhi. 2010;38:420–424. [PubMed] [Google Scholar]
- Hong D, Wang Z, Zhang W, Xi J, Lu J, Luan X, Yuan Y. A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol Appl Neurobiol. 2011;37:257–270. doi: 10.1111/j.1365-2990.2010.01112.x. [DOI] [PubMed] [Google Scholar]
- Izant JG, Lazarides E. Invariance and heterogeneity in the major structural and regulatory proteins of chick muscle cells revealed by two-dimensional gel electrophoresis. Proc Natl Acad Sci U S A. 1977;74:1450–1454. doi: 10.1073/pnas.74.4.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaka O, Casas-Fraile L, Lopez de Munain A, Saenz A. Costamere proteins and their involvement in myopathic processes. Expert Rev Mol Med. 2015;17:e12. doi: 10.1017/erm.2015.9. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joanne P, Chourbagi O, Hourde C, Ferry A, Butler-Browne G, Vicart P, Dumonceaux J, Agbulut O. Viral-mediated expression of desmin mutants to create mouse models of myofibrillar myopathy. Skelet Muscle. 2013;3:4. doi: 10.1186/2044-5040-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurcu TR, Bastian AE, Militaru S, Popa A, Manole E, Popescu BA, Tallila J, Popescu BO, Ginghina CD. Discovery of a new mutation in the desmin gene in a young patient with cardiomyopathy and muscular weakness. Rom J Morphol Embryol. 2017;58:225–230. [PubMed] [Google Scholar]
- Kami K, Chidgey M, Dafforn T, Overduin M. The desmoglein-specific cytoplasmic region is intrinsically disordered in solution and interacts with multiple desmosomal protein partners. J Mol Biol. 2009;386:531–543. doi: 10.1016/j.jmb.2008.12.054. [DOI] [PubMed] [Google Scholar]
- Kaminska A, Strelkov SV, Goudeau B, Olive M, Dagvadorj A, Fidzianska A, Simon-Casteras M, Shatunov A, Dalakas MC, Ferrer I, Kwiecinski H, Vicart P, Goldfarb LG. Small deletions disturb desmin architecture leading to breakdown of muscle cells and development of skeletal or cardioskeletal myopathy. Hum Genet. 2004;114:306–313. doi: 10.1007/s00439-003-1057-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann E, Weber K, Geisler N. Intermediate filament forming ability of desmin derivatives lacking either the amino-terminal 67 or the carboxy-terminal 27 residues. J Mol Biol. 1985;185:733–742. doi: 10.1016/0022-2836(85)90058-0. [DOI] [PubMed] [Google Scholar]
- Khudiakov A, Kostina D, Zlotina A, Nikulina T, Sergushichev A, Gudkova A, Tomilin A, Malashicheva A, Kostareva A. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. 2017;24:77–80. doi: 10.1016/j.scr.2017.08.015. [DOI] [PubMed] [Google Scholar]
- Kiss B, Kellermayer MS. Stretching desmin filaments with receding meniscus reveals large axial tensile strength. J Struct Biol. 2014;186:472–480. doi: 10.1016/j.jsb.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Kiss B, Karsai A, Kellermayer MS. Nanomechanical properties of desmin intermediate filaments. J Struct Biol. 2006;155:327–339. doi: 10.1016/j.jsb.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, Bohms B, Schulz U, Knyphausen EZ, Vorgerd M, Gummert J, Milting H. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19:4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- Kooijman M, van Amerongen H, Traub P, van Grondelle R, Bloemendal M. The assembly state of the intermediate filament proteins desmin and glial fibrillary acidic protein at low ionic strength. FEBS Lett. 1995;358:185–188. doi: 10.1016/0014-5793(94)01419-2. [DOI] [PubMed] [Google Scholar]
- Kostareva A, Gudkova A, Sjoberg G, Kiselev I, Moiseeva O, Karelkina E, Goldfarb L, Schlyakhto E, Sejersen T. Desmin mutations in a St. Petersburg cohort of cardiomyopathies. Acta Myologica Myopathies Cardiomyopathies. 2006;25:109–115. [PubMed] [Google Scholar]
- Kostareva A, Sjoberg G, Bruton J, Zhang SJ, Balogh J, Gudkova A, Hedberg B, Edstrom L, Westerblad H, Sejersen T. Mice expressing L345P mutant desmin exhibit morphological and functional changes of skeletal and cardiac mitochondria. J Muscle Res Cell Motil. 2008;29:25–36. doi: 10.1007/s10974-008-9139-8. [DOI] [PubMed] [Google Scholar]
- Kostareva A, Sjoberg G, Gudkova A, Smolina N, Semernin E, Shlyakhto E, Sejersen T. Desmin A213V substitution represents a rare polymorphism but not a mutation and is more prevalent in patients with heart dilation of various origins. Acta Myologica. 2011;30:42–45. [PMC free article] [PubMed] [Google Scholar]
- Kreplak L, Bar H. Severe myopathy mutations modify the nanomechanics of desmin intermediate filaments. J Mol Biol. 2009;385:1043–1051. doi: 10.1016/j.jmb.2008.10.095. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K, Fontao L, Champliaud MF, Jaunin F, Frias MA, Favre B, Paulin D, Green KJ, Borradori L. New insights into the molecular basis of desmoplakin- and desmin-related cardiomyopathies. J Cell Sci. 2006;119:4974–4985. doi: 10.1242/jcs.03255. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, C. Exome Aggregation Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin J, Bulst S, Thirion C, Schmidt F, Botzel K, Krause S, Pertl C, Kretzschmar H, Walter MC, Giese A, Lochmuller H. Divergent molecular effects of desmin mutations on protein assembly in myofibrillar myopathy. J Neuropathol Exp Neurol. 2010;69:415–424. doi: 10.1097/NEN.0b013e3181d71305. [DOI] [PubMed] [Google Scholar]
- Li ZL, Lilienbaum A, Butler-Browne G, Paulin D. Human desmin-coding gene: complete nucleotide sequence, characterization and regulation of expression during myogenesis and development. Gene. 1989;78:243–254. doi: 10.1016/0378-1119(89)90227-8. [DOI] [PubMed] [Google Scholar]
- Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 1996;175:362–366. doi: 10.1006/dbio.1996.0122. [DOI] [PubMed] [Google Scholar]
- Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100:461–464. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- Li M, Andersson-Lendahl M, Sejersen T, Arner A. Knockdown of desmin in zebrafish larvae affects interfilament spacing and mechanical properties of skeletal muscle. J Gen Physiol. 2013;141:335–345. doi: 10.1085/jgp.201210915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Broedersz CP, Rowat AC, Wedig T, Herrmann H, Mackintosh FC, Weitz DA. Divalent cations crosslink vimentin intermediate filament tail domains to regulate network mechanics. J Mol Biol. 2010;399:637–644. doi: 10.1016/j.jmb.2010.04.054. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 2006;20:362–364. doi: 10.1096/fj.05-4869fje. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–454. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G, Rampazzo A. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400–405. doi: 10.1016/j.amjcard.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund LM, Kerr JP, Lupinetti J, Zhang Y, Russell MA, Bloch RJ, Bond M. Synemin isoforms differentially organize cell junctions and desmin filaments in neonatal cardiomyocytes. FASEB J. 2012;26:137–148. doi: 10.1096/fj.10-179408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison P, Damian MS, Sewry C, McGorrian C, Winer JB, Odgerel Z, Shatunov A, Lee HS, Goldfarb LG. Clinical and myopathological characteristics of desminopathy caused by a mutation in desmin tail domain. Eur Neurol. 2012;68:279–286. doi: 10.1159/000341617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkens A, Kley RA, Olive M, Theis V, van der Ven PF, Reimann J, Milting H, Schreiner A, Uszkoreit J, Eisenacher M, Barkovits K, Guttsches AK, Tonillo J, Kuhlmann K, Meyer HE, Schroder R, Tegenthoff M, Furst DO, Muller T, Goldfarb LG, Vorgerd M, Marcus K. Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J Proteome. 2013;90:14–27. doi: 10.1016/j.jprot.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkens A, Olive M, Schreiner A, Feldkirchner S, Schessl J, Uszkoreit J, Barkovits K, Guttsches AK, Theis V, Eisenacher M, Tegenthoff M, Goldfarb LG, Schroder R, Schoser B, van der Ven PF, Furst DO, Vorgerd M, Marcus K, Kley RA. New insights into the protein aggregation pathology in myotilinopathy by combined proteomic and immunolocalization analyses. Acta Neuropathol Commun. 2016;4:8. doi: 10.1186/s40478-016-0280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloyan A, Sayegh J, Osinska H, Chua BH, Robbins J. Manipulation of death pathways in desmin-related cardiomyopathy. Circ Res. 2010;106:1524–1532. doi: 10.1161/CIRCRESAHA.109.212639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, A. American Heart, H. F. Council on Clinical Cardiology, C. Transplantation, C. Quality of, R. Outcomes, G. Functional, G. Translational Biology Interdisciplinary Working, E. Council on, and Prevention Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Hirose M, Sanbe A, Kondo Y, Irie Y, Taira E. Nicorandil improves electrical remodelling, leading to the prevention of electrically induced ventricular tachyarrhythmia in a mouse model of desmin-related cardiomyopathy. Clin Exp Pharmacol Physiol. 2014;41:89–97. doi: 10.1111/1440-1681.12185. [DOI] [PubMed] [Google Scholar]
- McCormick EM, Kenyon L, Falk MJ. Desmin common mutation is associated with multi-systemic disease manifestations and depletion of mitochondria and mitochondrial DNA. Front Genet. 2015;6:199. doi: 10.3389/fgene.2015.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121:722–730. doi: 10.1161/CIRCRESAHA.117.309711. [DOI] [PubMed] [Google Scholar]
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- McLaughlin HM, Kelly MA, Hawley PP, Darras BT, Funke B, Picker J. Compound heterozygosity of predicted loss-of-function DES variants in a family with recessive desminopathy. BMC Med Genet. 2013;14:68. doi: 10.1186/1471-2350-14-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon PM, Robbins J. Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol. 2011;301:H1220–H1228. doi: 10.1152/ajpheart.00601.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencarelli C, Ciolfi S, Caroti D, Lupetti P, Dallai R. Isomin: a novel cytoplasmic intermediate filament protein from an arthropod species. BMC Biol. 2011;9:17. doi: 10.1186/1741-7007-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miszalski-Jamka K, Jefferies JL, Mazur W, Glowacki J, Hu J, Lazar M, Gibbs RA, Liczko J, Klys J, Venner E, Muzny DM, Rycaj J, Bialkowski J, Kluczewska E, Kalarus Z, Jhangiani S, Al-Khalidi H, Kukulski T, Lupski JR, Craigen WJ, Bainbridge MN (2017) Novel genetic triggers and genotype-phenotype correlations in patients with left ventricular noncompaction. Circ Cardiovasc Genet 10 [DOI] [PMC free article] [PubMed]
- Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, Al-Owain M, Shah A, Rahbeeni Z, Al-Muhaizea MA, Alzaidan HI, Cupler E, Bohlega S, Faqeih E, Faden M, Alyounes B, Jaroudi D, Goljan E, Elbardisy H, Akilan A, Albar R, Aldhalaan H, Gulab S, Chedrawi A, Al Saud BK, Kurdi W, Makhseed N, Alqasim T, El Khashab HY, Al-Mousa H, Alhashem A, Kanaan I, Algoufi T, Alsaleem K, Basha TA, Al-Murshedi F, Khan S, Al-Kindy A, Alnemer M, Al-Hajjar S, Alyamani S, Aldhekri H, Al-Mehaidib A, Arnaout R, Dabbagh O, Shagrani M, Broering D, Tulbah M, Alqassmi A, Almugbel M, AlQuaiz M, Alsaman A, Al-Thihli K, Sulaiman RA, Al-Dekhail W, Alsaegh A, Bashiri FA, Qari A, Alhomadi S, Alkuraya H, Alsebayel M, Hamad MH, Szonyi L, Abaalkhail F, Al-Mayouf SM, Almojalli H, Alqadi KS, Elsiesy H, Shuaib TM, Seidahmed MZ, Abosoudah I, Akleh H, AlGhonaium A, Alkharfy TM, Al Mutairi F, Eyaid W, Alshanbary A, Sheikh FR, Alsohaibani FI, Alsonbul A, Al Tala S, Balkhy S, Bassiouni R, Alenizi AS, Hussein MH, Hassan S, Khalil M, Tabarki B, Alshahwan S, Oshi A, Sabr Y, Alsaadoun S, Salih MA, Mohamed S, Sultana H, Tamim A, El-Haj M, Alshahrani S, Bubshait DK, Alfadhel M, Faquih T, El-Kalioby M, Subhani S, Shah Z, Moghrabi N, Meyer BF, Alkuraya FS. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–939. doi: 10.1007/s00439-017-1821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook OR, Haagmans MA, Soucy JF, van de Meerakker JB, Baas F, Jakobs ME, Hofman N, Christiaans I, Lekanne Deprez RH, Mannens MM. Targeted sequence capture and GS-FLX titanium sequencing of 23 hypertrophic and dilated cardiomyopathy genes: implementation into diagnostics. J Med Genet. 2013;50:614–626. doi: 10.1136/jmedgenet-2012-101231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez-Figueras MT, Navas-Palacios JJ, Ariza A, Fuchs E. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci U S A. 1998;95:11312–11317. doi: 10.1073/pnas.95.19.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Bonne G, Goldfarb LG, Mercuri E, Piercy RJ, Burke M, Yaou RB, Richard P, Recan D, Shatunov A, Sewry CA, Brown SC. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain J Neurol. 2006;129:1260–1268. doi: 10.1093/brain/awl062. [DOI] [PubMed] [Google Scholar]
- Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, Lewis KL, Cooper DN, Stenson PD, Mullikin JC, Biesecker LG, N. I. H. I. S. C. C. S. Program Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. 2013;6:337–346. doi: 10.1161/CIRCGENETICS.113.000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouhravesh N, Ahlberg G, Ghouse J, Andreasen C, Svendsen JH, Haunso S, Bundgaard H, Weeke PE, Olesen MS. Analyses of more than 60,000 exomes questions the role of numerous genes previously associated with dilated cardiomyopathy. Mol Genet Genomic Med. 2016;4:617–623. doi: 10.1002/mgg3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojrzynska N, Bilinska ZT, Franaszczyk M, Ploski R, Grzybowski J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol Pol. 2017;75:723. doi: 10.5603/KP.2017.0129. [DOI] [PubMed] [Google Scholar]
- Olive M, Goldfarb L, Dagvadorj A, Sambuughin N, Paulin D, Li Z, Goudeau B, Vicart P, Ferrer I. Expression of the intermediate filament protein synemin in myofibrillar myopathies and other muscle diseases. Acta Neuropathol. 2003;106:1–7. doi: 10.1007/s00401-003-0695-0. [DOI] [PubMed] [Google Scholar]
- Olive M, Armstrong J, Miralles F, Pou A, Fardeau M, Gonzalez L, Martinez F, Fischer D, Martinez Matos JA, Shatunov A, Goldfarb L, Ferrer I. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord. 2007;17:443–450. doi: 10.1016/j.nmd.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive M, Odgerel Z, Martinez A, Poza JJ, Bragado FG, Zabalza RJ, Jerico I, Gonzalez-Mera L, Shatunov A, Lee HS, Armstrong J, Maravi E, Arroyo MR, Pascual-Calvet J, Navarro C, Paradas C, Huerta M, Marquez F, Rivas EG, Pou A, Ferrer I, Goldfarb LG. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord. 2011;21:533–542. doi: 10.1016/j.nmd.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M, Goebel HH. The cytoplasmic bodies in a congenital myopathy can be stained with antibodies to desmin, the muscle-specific intermediate filament protein. Acta Neuropathol. 1983;62:149–152. doi: 10.1007/BF00684933. [DOI] [PubMed] [Google Scholar]
- Palmio J, Penttila S, Huovinen S, Haapasalo H, Udd B. An unusual phenotype of late-onset desminopathy. Neuromuscul Disord. 2013;23:922–923. doi: 10.1016/j.nmd.2013.06.374. [DOI] [PubMed] [Google Scholar]
- Park KY, Dalakas MC, Semino-Mora C, Lee HS, Litvak S, Takeda K, Ferrans VJ, Goldfarb LG. Sporadic cardiac and skeletal myopathy caused by a de novo desmin mutation. Clin Genet. 2000;57:423–429. doi: 10.1034/j.1399-0004.2000.570604.x. [DOI] [PubMed] [Google Scholar]
- Park KY, Dalakas MC, Goebel HH, Ferrans VJ, Semino-Mora C, Litvak S, Takeda K, Goldfarb LG. Desmin splice variants causing cardiac and skeletal myopathy. J Med Genet. 2000;37:851–857. doi: 10.1136/jmg.37.11.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DM, Green KJ. Desmosomes in the heart: a review of clinical and mechanistic analyses. Cell Commun Adhes. 2014;21:109–128. doi: 10.3109/15419061.2014.906533. [DOI] [PubMed] [Google Scholar]
- Pica EC, Kathirvel P, Pramono ZA, Lai PS, Yee WC. Characterization of a novel S13F desmin mutation associated with desmin myopathy and heart block in a Chinese family. Neuromuscul Disord. 2008;18:178–182. doi: 10.1016/j.nmd.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Pinol-Ripoll G, Shatunov A, Cabello A, Larrode P, de la Puerta I, Pelegrin J, Ramos FJ, Olive M, Goldfarb LG. Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul Disord. 2009;19:418–422. doi: 10.1016/j.nmd.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plodinec M, Loparic M, Suetterlin R, Herrmann H, Aebi U, Schoenenberger CA. The nanomechanical properties of rat fibroblasts are modulated by interfering with the vimentin intermediate filament system. J Struct Biol. 2011;174:476–484. doi: 10.1016/j.jsb.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Potschka M, Nave R, Weber K, Geisler N. The two coiled coils in the isolated rod domain of the intermediate filament protein desmin are staggered. A hydrodynamic analysis of tetramers and dimers. Eur J Biochem. 1990;190:503–508. doi: 10.1111/j.1432-1033.1990.tb15602.x. [DOI] [PubMed] [Google Scholar]
- Price MG, Lazarides E. Expression of intermediate filament-associated proteins paranemin and synemin in chicken development. J Cell Biol. 1983;97:1860–1874. doi: 10.1083/jcb.97.6.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruszczyk P, Kostera-Pruszczyk A, Shatunov A, Goudeau B, Draminska A, Takeda K, Sambuughin N, Vicart P, Strelkov SV, Goldfarb LG, Kaminska A. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int J Cardiol. 2007;117:244–253. doi: 10.1016/j.ijcard.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Psarras S, Mavroidis M, Sanoudou D, Davos CH, Xanthou G, Varela AE, Panoutsakopoulou V, Capetanaki Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur Heart J. 2012;33:1954–1963. doi: 10.1093/eurheartj/ehr119. [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–608. doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- Punetha J, Kesari A, Uapinyoying P, Giri M, Clarke NF, Waddell LB, North KN, Ghaoui R, O'Grady GL, Oates EC, Sandaradura SA, Bonnemann CG, Donkervoort S, Plotz PH, Smith EC, Tesi-Rocha C, Bertorini TE, Tarnopolsky MA, Reitter B, Hausmanowa-Petrusewicz I, Hoffman EP. Targeted re-sequencing emulsion PCR panel for myopathies: results in 94 cases. J Neuromuscul Dis. 2016;3:209–225. doi: 10.3233/JND-160151. [DOI] [PubMed] [Google Scholar]
- Raats JM, Schaart G, Henderik JB, van der Kemp A, Dunia I, Benedetti EL, Pieper FR, Ramaekers FC, Bloemendal H. Muscle-specific expression of a dominant negative desmin mutant in transgenic mice. Eur J Cell Biol. 1996;71:221–236. [PubMed] [Google Scholar]
- Rainer PP, Dong P, Sorge M, Fert-Bober J, Holewinski RJ, Wang Y, Foss C, An SS, Baracca A, Solaini G, Glabe C, Pomper MG, Van Eyk JE, Tomaselli GF, Paolocci N, Agnetti G (2018) Desmin phosphorylation triggers preamyloid oligomers formation and myocyte dysfunction in acquired heart failure. Circ Res [DOI] [PMC free article] [PubMed]
- Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramspacher C, Steed E, Boselli F, Ferreira R, Faggianelli N, Roth S, Spiegelhalter C, Messaddeq N, Trinh L, Liebling M, Chacko N, Tessadori F, Bakkers J, Laporte J, Hnia K, Vermot J. Developmental alterations in heart biomechanics and skeletal muscle function in desmin mutants suggest an early pathological root for desminopathies. Cell Rep. 2015;11:1564–1576. doi: 10.1016/j.celrep.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll-Vera T, Zorio E, Gamez JM, Molina P, Govea N, Cremer D. Phenotypic patterns of cardiomyopathy caused by mutations in the desmin gene. A clinical and genetic study in two inherited heart disease units. Rev Esp Cardiol. 2015;68:1027–1029. doi: 10.1016/j.rec.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, Yamauchi J, Tanonaka K, Glabe C, Tanoue A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One. 2009;4:e5351. doi: 10.1371/journal.pone.0005351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe A, Marunouchi T, Yamauchi J, Tanonaka K, Nishigori H, Tanoue A. Cardioprotective effect of nicorandil, a mitochondrial ATP-sensitive potassium channel opener, prolongs survival in HSPB5 R120G transgenic mice. PLoS One. 2011;6:e18922. doi: 10.1371/journal.pone.0018922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanzer A, Rupp S, Graf S, Zengeler D, Jux C, Akinturk H, Gulatz L, Mazhari N, Acker T, Van Coster R, Garvalov BK, Hahn A. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol Genet Metab. 2018;123:388–399. doi: 10.1016/j.ymgme.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Schirmer I, Dieding M, Klauke B, Brodehl A, Gaertner-Rommel A, Walhorn V, Gummert J, Schulz U, Paluszkiewicz L, Anselmetti D, Milting H. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol Genet Genomic Med. 2018;6(2):288–293. doi: 10.1002/mgg3.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrickel JW, Stockigt F, Krzyzak W, Paulin D, Li Z, Lubkemeier I, Fleischmann B, Sasse P, Linhart M, Lewalter T, Nickenig G, Lickfett L, Schroder R, Clemen CS. Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J Interv Card Electrophysiol. 2010;28:71–80. doi: 10.1007/s10840-010-9482-8. [DOI] [PubMed] [Google Scholar]
- Schroder JM, Sommer C, Schmidt B. Desmin and actin associated with cytoplasmic bodies in skeletal muscle fibers: immunocytochemical and fine structural studies, with a note on unusual 18- to 20-nm filaments. Acta Neuropathol. 1990;80:406–414. doi: 10.1007/BF00307695. [DOI] [PubMed] [Google Scholar]
- Schroder R, Goudeau B, Simon MC, Fischer D, Eggermann T, Clemen CS, Li ZL, Reimann J, Xue ZG, Rudnik-Schoneborn S, Zerres K, van der Ven PFM, Furst DO, Kunz WS, Vicart P. On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum Mol Genet. 2003;12:657–669. doi: 10.1093/hmg/ddg060. [DOI] [PubMed] [Google Scholar]
- Schroder R, Goudeau B, Simon MC, Fischer D, Eggermann T, Clemen CS, Li ZL, Reimann J, Xue ZG, Rudnik-Schoneborn S, Zerres K, van der Ven PFM, Furst DO, Kunz WS, Vicart P. On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria (vol 12, pg 657, 2003) Hum Mol Genet. 2007;16:2989–2990. doi: 10.1093/hmg/ddg060. [DOI] [PubMed] [Google Scholar]
- Schwarz N, Leube RE (2016) Intermediate filaments as organizers of cellular space: how they affect mitochondrial structure and function. Cells 5 [DOI] [PMC free article] [PubMed]
- Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain J Neurol. 2004;127:439–451. doi: 10.1093/brain/awh052. [DOI] [PubMed] [Google Scholar]
- Shanks GW, Tester DJ, Nishtala S, Evans JM, Ackerman MJ (2017) Genomic triangulation and coverage analysis in whole-exome sequencing-based molecular autopsies. Circ Cardiovasc Genet 10 [DOI] [PubMed]
- Sharma S, Mucke N, Katus HA, Herrmann H, Bar H. Disease mutations in the “head” domain of the extra-sarcomeric protein desmin distinctly alter its assembly and network-forming properties. J Mol Med. 2009;87:1207–1219. doi: 10.1007/s00109-009-0521-9. [DOI] [PubMed] [Google Scholar]
- Sjoberg G, Saavedra-Matiz CA, Rosen DR, Wijsman EM, Borg K, Horowitz SH, Sejersen T. A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet. 1999;8:2191–2198. doi: 10.1093/hmg/8.12.2191. [DOI] [PubMed] [Google Scholar]
- Smolina N, Bruton J, Sjoberg G, Kostareva A, Sejersen T. Aggregate-prone desmin mutations impair mitochondrial calcium uptake in primary myotubes. Cell Calcium. 2014;56:269–275. doi: 10.1016/j.ceca.2014.08.001. [DOI] [PubMed] [Google Scholar]
- van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP. Desmin-related myopathy. Clin Genet. 2011;80:354–366. doi: 10.1111/j.1399-0004.2010.01512.x. [DOI] [PubMed] [Google Scholar]
- van Spaendonck-Zwarts KY, van der Kooi AJ, van den Berg MP, Ippel EF, Boven LG, Yee WC, van den Wijngaard A, Brusse E, Hoogendijk JE, Doevendans PA, de Visser M, Jongbloed JD, van Tintelen JP. Recurrent and founder mutations in the Netherlands: the cardiac phenotype of DES founder mutations p.S13F and p.N342D. Neth Hear J. 2012;20:219–228. doi: 10.1007/s12471-011-0233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, de Boer RA, Jongbloed JD, Pinto YM, van Tintelen JP. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail. 2013;15:628–636. doi: 10.1093/eurjhf/hft013. [DOI] [PubMed] [Google Scholar]
- Sprinkart AM, Block W, Traber F, Meyer R, Paulin D, Clemen CS, Schroder R, Gieseke J, Schild H, Thomas D. Characterization of the failing murine heart in a desmin knock-out model using a clinical 3 T MRI scanner. Int J Card Imaging. 2012;28:1699–1705. doi: 10.1007/s10554-011-9990-3. [DOI] [PubMed] [Google Scholar]
- Stoeckel ME, Osborn M, Porte A, Sacrez A, Batzenschlager A, Weber K. An unusual familial cardiomyopathy characterized by aberrant accumulations of desmin-type intermediate filaments. Virchows Archiv A Pathological Anat Histol. 1981;393:53–60. doi: 10.1007/BF00430870. [DOI] [PubMed] [Google Scholar]
- Strach K, Sommer T, Grohe C, Meyer C, Fischer D, Walter MC, Vorgerd M, Reilich P, Bar H, Reimann J, Reuner U, Germing A, Goebel HH, Lochmuller H, Wintersperger B, Schroder R. Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul Disord. 2008;18:475–482. doi: 10.1016/j.nmd.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Sugawara M, Kato K, Komatsu M, Wada C, Kawamura K, Shindo PS, Yoshioka PN, Tanaka K, Watanabe S, Toyoshima I. A novel de novo mutation in the desmin gene causes desmin myopathy with toxic aggregates. Neurology. 2000;55:986–990. doi: 10.1212/wnl.55.7.986. [DOI] [PubMed] [Google Scholar]
- Sun N, Critchley DR, Paulin D, Li Z, Robson RM. Human alpha-synemin interacts directly with vinculin and metavinculin. Biochem J. 2008;409:657–667. doi: 10.1042/BJ20071188. [DOI] [PubMed] [Google Scholar]
- Szeverenyi I, Cassidy AJ, Chung CW, Lee BT, Common JE, Ogg SC, Chen H, Sim SY, Goh WL, Ng KW, Simpson JA, Chee LL, Eng GH, Li B, Lunny DP, Chuon D, Venkatesh A, Khoo KH, McLean WH, Lim YP, Lane EB. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29:351–360. doi: 10.1002/humu.20652. [DOI] [PubMed] [Google Scholar]
- Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, Zhu X, Cavanaugh J, Sucharov CC, Long CS, Bristow MR, Lavori P, Mestroni L, R. Familial Cardiomyopathy. B. D. Bank Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115:1244–1251. doi: 10.1161/CIRCULATIONAHA.106.646778. [DOI] [PubMed] [Google Scholar]
- Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol. 1997;29:2107–2124. doi: 10.1006/jmcc.1997.0446. [DOI] [PubMed] [Google Scholar]
- Tian C, Fuller C, Miles L, Jefferies J, Ryan T, Sawnani H, Bolger A, Wong B. A novel homozygous desmin nonsense mutation causes pediatric onset autosomal recessive desminopathy with severe cardiomyopathy. Neuromuscul Disord. 2016;26:S114–S115. [Google Scholar]
- van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, Boven L, van den Heuvel F, Veenstra-Knol HE, Saffitz JE, Hofstra RM, van den Berg MP. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6:1574–1583. doi: 10.1016/j.hrthm.2009.07.041. [DOI] [PubMed] [Google Scholar]
- Tse HF, Ho JC, Choi SW, Lee YK, Butler AW, Ng KM, Siu CW, Simpson MA, Lai WH, Chan YC, Au KW, Zhang J, Lay KW, Esteban MA, Nicholls JM, Colman A, Sham PC. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet. 2013;22:1395–1403. doi: 10.1093/hmg/dds556. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vattemi G, Neri M, Piffer S, Vicart P, Gualandi F, Marini M, Guglielmi V, Filosto M, Tonin P, Ferlini A, Tomelleri G. Clinical, morphological and genetic studies in a cohort of 21 patients with myofibrillar myopathy. Acta Myologica. 2011;30:121–126. [PMC free article] [PubMed] [Google Scholar]
- Vernengo L, Chourbagi O, Panuncio A, Lilienbaum A, Batonnet-Pichon S, Bruston F, Rodrigues-Lima F, Mesa R, Pizzarossa C, Demay L, Richard P, Vicart P, Rodriguez MM. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul Disord. 2010;20:178–187. doi: 10.1016/j.nmd.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- Wahbi K, Behin A, Charron P, Dunand M, Richard P, Meune C, Vicart P, Laforet P, Stojkovic T, Becane HM, Kuntzer T, Duboc D. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: a 10-year longitudinal study. Neuromuscul Disord. 2012;22:211–218. doi: 10.1016/j.nmd.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, C. Exome Aggregation. MacArthur DG, Farrall M, Cook SA, Watkins H. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MC, Reilich P, Huebner A, Fischer D, Schroder R, Vorgerd M, Kress W, Born C, Schoser BG, Krause KH, Klutzny U, Bulst S, Frey JR, Lochmuller H. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain J Neurol. 2007;130:1485–1496. doi: 10.1093/brain/awm039. [DOI] [PubMed] [Google Scholar]
- Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Iyadurai S, Baloh RH, Pittman SK, Schmidt RE, Lopate G, Pestronk A, Harms MB. Autophagic vacuolar pathology in desminopathies. Neuromuscul Disord. 2015;25:199–206. doi: 10.1016/j.nmd.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KD, Shen P, Fung E, Karakikes I, Zhang A, InanlooRahatloo K, Odegaard J, Sallam K, Davis RW, Lui GK, Ashley EA, Scharfe C, Wu JC. A rapid, high-quality, cost-effective, comprehensive and expandable targeted next-generation sequencing assay for inherited heart diseases. Circ Res. 2015;117:603–611. doi: 10.1161/CIRCRESAHA.115.306723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winheim S, Hieb AR, Silbermann M, Surmann EM, Wedig T, Herrmann H, Langowski J, Mucke N. Deconstructing the late phase of vimentin assembly by total internal reflection fluorescence microscopy (TIRFM) PLoS One. 2011;6:e19202. doi: 10.1371/journal.pone.0019202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter L, Wittig I, Peeva V, Eggers B, Heidler J, Chevessier F, Kley RA, Barkovits K, Strecker V, Berwanger C, Herrmann H, Marcus K, Kornblum C, Kunz WS, Schroder R, Clemen CS. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol. 2016;132:453–473. doi: 10.1007/s00401-016-1592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yla-Herttuala S, Baker AH. Cardiovascular gene therapy: past, present, and future. Mol Ther. 2017;25:1095–1106. doi: 10.1016/j.ymthe.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Zheng Y, Jin S, Gang Q, Wang Q, Yu P, Lv H, Zhang W, Yuan Y, Wang Z. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS One. 2017;12:e0175343. doi: 10.1371/journal.pone.0175343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Liu L, Chen C, Shen JM. Exome sequencing identifies a novel DES mutation (R227C) in a Chinese dilated cardiomyopathy family. Cardiology. 2017;137:78–82. doi: 10.1159/000455181. [DOI] [PubMed] [Google Scholar]
- Zhao F, Chaugai S, Chen P, Wang Y, Wang DW. Effect of nicorandil in patients with heart failure: a systematic review and meta-analysis. Cardiovasc Ther. 2014;32:283–296. doi: 10.1111/1755-5922.12097. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Feng Y, Zhang YM, Ding XX, Song YZ, Zhang AM, Liu L, Zhang H, Ding JH, Xia XS. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med. 2015;36:1479–1486. doi: 10.3892/ijmm.2015.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, Dhindsa RS, Hitomi Y, Schoch K, Spillmann RC, Heimer G, Marek-Yagel D, Tzadok M, Han Y, Worley G, Goldstein J, Jiang YH, Lancet D, Pras E, Shashi V, McHale D, Need AC, Goldstein DB. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–781. doi: 10.1038/gim.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]