

Abstract
Ryanodine receptors (RyRs) are the Ca2+ release channels in the sarcoplasmic reticulum in striated muscle which play an important role in excitation-contraction coupling and cardiac pacemaking. Single channel recordings have revealed a wealth of information about ligand regulation of RyRs from mammalian skeletal and cardiac muscle (RyR1 and RyR2, respectively). RyR subunit has a Ca2+ activation site located in the luminal and cytoplasmic domains of the RyR. These sites synergistically feed into a common gating mechanism for channel activation by luminal and cytoplasmic Ca2+. RyRs also possess two inhibitory sites in their cytoplasmic domains with Ca2+ affinities of the order of 1 μM and 1 mM. Magnesium competes with Ca2+ at these sites to inhibit RyRs and this plays an important role in modulating their Ca2+-dependent activity in muscle. This review focuses on how these sites lead to RyR modulation by Ca2+ and Mg2+ and how these mechanisms control Ca2+ release in excitation-contraction coupling and cardiac pacemaking.
Keywords: Ryanodine receptor, RyR1, RyR2, Excitation-contraction coupling, Cardiac pacemaking, Ca2+ activation, Mg2+ inhibition, Ca2+ release channels
Introduction
Muscle contracts in response to the electrical excitation of the surface membrane by a process called excitation-contraction (E-C) coupling whereby surface membrane depolarisation elicits calcium release from the sarcoplasmic reticulum (SR). The SR is the primary intracellular Ca2+ store and ryanodine receptors (RyR) are the Ca2+ release channels. Three RyR isoforms have been identified in mammals: RyR1 in skeletal muscle, RyR2 in cardiac and smooth muscle and RyR3 in many cell types (Ogawa 1994). Regulation of RyRs by intracellular Ca2+ and Mg2+ (Coronado et al. 1994; Meissner 1994) plays a major role in how the cell controls SR Ca2+ release. Single channel recordings have revealed a wealth of information about Ca2+ and Mg2+ regulation of RyR and have gone a long way to untangling their many interdependent RyR gating mechanisms. This article reviews the current general picture of RyR regulation by Ca2+ and Mg2+. A detailed review of functional models of RyR2 gating by Ca2+ and Mg2+ has been given elsewhere (Laver 2010).
RyR2 activation by cytoplasmic and luminal Ca2+
Both RyR1 (Hymel et al. 1988; Smith et al. 1986) and RyR2 isoforms (Meissner et al. 1986) exhibit activation by micrometre concentrations of cytoplasmic Ca2+. In the absence of any other channel activator, cytoplasmic Ca2+ can increase channel open probability (Po) from virtually zero up to Po ~ 0.6 with a half-maximal activation concentration (Ka) of 1–5 μM (Laver et al. 1995; Sitsapesan and Williams 1994a; Xu et al. 1996). RyR2 was shown to have a steep Po dependence on cytoplasmic [Ca2+], typically exhibiting Hill coefficients of 2–4 (Sitsapesan and Williams 1994a) as would be expected if RyR2 activation resulted from cooperative involvement of one high-affinity (~ 1 μM) Ca2+ binding site on each subunit of the homotetrameric channel (Zahradnik et al. 2005). The cytoplasmic Ca2+ activation site is referred to here as the A-site, a name originally coined by Balog et al. (2001) (A for Activation).
Early studies gained clues to the distance of the A-sites from the channel pore from their sensitivity to Ca2+ flowing from the luminal side of the membrane into a cytoplasmic milieu containing a fast Ca2+ chelator such as BAPTA (Laver 2007; Tripathy and Meissner 1996). The dilution and sequestering of Ca2+ emanating from the pore creates a concentration gradient such that the [Ca2+] at the A-site will depend on its distance from the pore. Analytic solutions of the Ca2+ diffusion/buffering equations (Bers and Peskoff 1991) predicted a distance of 11 nm between the A-site and the cytoplasmic pore mouth. More recent mutagenesis experiments revealed that the RyR1 mutation, E4032A and the corresponding mutation E3987A in RyR2 impeded channel activation by ~ 1000-fold, implicating this amino acid with the A-site (Fessenden et al. 2001; Li and Chen 2001). Recently, cryo-EM reconstructions of RyR1 identified the A-site Ca2+ binding pocket near, but not including, E4032. It is formed by the carboxylate side chains of E3893 and E3967 of the RyR1 core solenoid and the backbone carbonyl of T5001 in the C-terminal domain (des Georges et al. 2016). The approximate location of the A-site is shown in a RyR1 gating domain schematic in Fig. 1. Interestingly, the location of the site is ~ 9 nm from the pore gate, consistent with estimates from diffusion/chelation experiments (Laver 2007; Tripathy and Meissner 1996). Even though the nearby E4032 did not coordinate with Ca2+, it was essential for the stability of the Ca2+ binding pocket comprising the A-site.
Fig. 1.
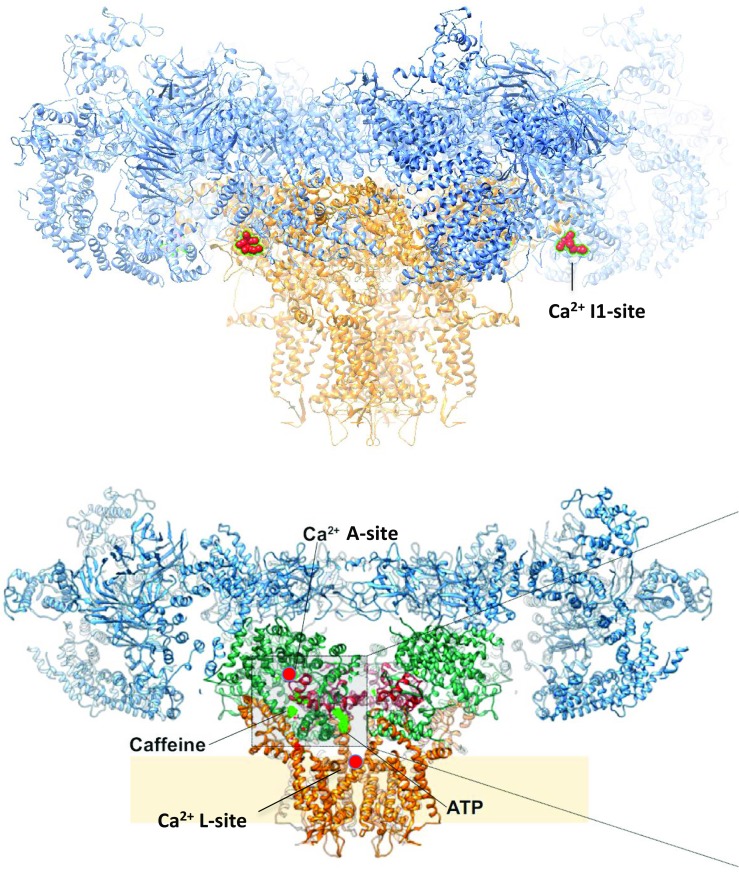
RyR1 structure reconstructed from cryo-EM images (des Georges et al. 2016) showing putative locations of key regulation sites on the RyR. Ca2+ sites are marked in red. Top—a distinctive sequence of 30 negative amino acids at positions 1873–1903 postulated to form the Ca2+/Mg2+ inhibitory; I1-site (Laver et al. 1997b) is shown on two of the four subunits. Bottom—a cutaway of the RyR1 structure showing activation sites for Ca2+ (A-site), caffeine and ATP (des Georges et al. 2016) and the L-site (Chen et al. 2014)
It has been long recognised that Ca2+ loading of the SR can trigger Ca2+ release (Fabiato 1985) but it was Sitsapesan and Williams (1994b) who first showed that luminal Ca2+ could directly activate RyR2. However, at that time, there was no consensus as to the location of the Ca2+ binding sites for this process. Based on the finding that luminal Ca2+ activation was abolished by tryptic digestion of the luminal domains of the RyR2 (Ching et al. 2000), Sitsapesan and Williams argued that luminal activation was due to luminal facing Ca2+ binding sites. Counter to this, Tripathy and Meisner (1996) provided evidence that luminal activation relies on the flow of Ca2+ through the pore to the cytoplasm (Ca2+ feedthrough) where it activates RyR1 via the A-site. By varying the electrochemical Ca2+ driving force (i.e., by varying both luminal [Ca2+] and transmembrane voltage), they showed that RyR1 activation increased with increasing Ca2+ flux through the channel. However, any firm assignment of luminal activation to either cytoplasmic or luminal domains of the RyR was confounded by the fact that the putative luminal and cytoplasmic Ca2+ sites were connected by a Ca2+ conducting pore. This problem was considerably simplified by performing separate analyses of channel open and closed durations associated with luminal activation (Laver 2007; Laver and Honen 2008; Laver et al. 2004). By studying channel closed events, it was possible to examine luminal activation of RyRs when no Ca2+ was flowing through the channel. Analysis of RyR2 opening rate (the reciprocal of mean closed time) demonstrated the existence of a luminal facing Ca2+ binding site with an affinity of 40 μM (L-site). Separate analysis of channel open events provided information about the effects of Ca2+ feedthrough on channel gating. It was found that upon the triggering of a channel opening by the L-site, subsequent Ca2+ feedthrough to the A-site further stabilised the open channel state, thus enhancing channel activation (Laver 2007). Analysis of RyR1 opening rates demonstrated an L-site that not only triggered channel openings, but also decreased A-site-mediated Mg2+ inhibition through an allosteric interaction (Laver et al. 2004) (see below).
The location of the L-site has not yet been determined in cryo-EM reconstructions. The fact that the Ca2+ affinity of the L-site does not depend on voltage (Laver 2007) suggests that the site lies outside the transmembrane portion of the RyR. Mutagenesis experiments show luminal Ca2+ activation of RyR2 can be abolished by point mutations at E4872A in the S6 helix bundle crossing with relatively minor changes in cytosolic Ca2+ activation (Chen et al. 2014). This places the luminal Ca2+ site at the proposed gate of the RyR. Moreover, introducing histidine at this site converts RyR2 into a luminal Ni2+-activated channel.
Recent measurements of RyR2 gating have shown that activation of RyR2 by luminal and cytoplasmic Ca2+ cannot be thought of as independent processes (Laver and Honen 2008). Opening rate increased in proportion to the third power of cytoplasmic [Ca2+] (in the μM range) and the 2nd power of luminal [Ca2+], peaking at ~ 1000 openings per second. By applying combinations of various cytoplasmic and luminal [Ca2+], Laver and Honen (2008) reported a marked synergy between A- and L-site activation of the RyR2. Increasing cytoplasmic [Ca2+] from 1 to 100 nM increased luminal Ca2+ activation by 4-fold without altering the apparent affinity of the L-site. Likewise, increasing luminal [Ca2+] from 1 nM to 0.1 mM increased cytoplasmic Ca2+ activation without altering the apparent affinity of the A-site. Therefore, Ca2+ activation is not uniquely attributable to either the A- or L-sites but rather is a combination of both. However, this synergy was only apparent at cytoplasmic [Ca2+] lower than 10 μM. At higher concentrations, the RyR opening rate is insensitive to luminal [Ca2+]. This synergy between A- and L-sites is also apparent in mutagenesis studies (Chen et al. 2014) where loss of either A- or L-site activation was always accompanied by partial loss of activation (opening rate) by the other site.
Cytoplasmic Ca2+ inhibition of RyR1 and RyR2
Millimolar cytoplasmic [Ca2+] inhibits RyR1 (Bezprozvanny et al. 1991; Meissner 1986) and RyR2 (Laver et al. 1995). In RyR1, Po markedly declines when cytoplasmic [Ca2+] rises above 100 μM indicating the presence of low-affinity inhibitory sites on the RyR. RyR2 is 10-fold less sensitive to Ca2+ inhibition than RyR1 (Laver et al. 1995). In keeping with the nomenclature of Balog et al. (2001), this site is referred to here as the I1-site (I for inhibition and I1 to distinguish it from a recently identified high-affinity inhibition site, I2-site, see below). At physiological concentrations of monovalent ions, the apparent Ca2+ affinity of the I1-site in RyR1 was 100 μM and this increased as the 2nd power of monovalent ion concentration (Meissner et al. 1997; Shomer et al. 1993). Together, the A- and I1-sites generate the well-known, bell-shaped cytoplasmic [Ca2+] dependence of Po which characterises RyR activation at micrometre Ca2+ and inhibition at millimolar Ca2+. Since Ca2+ levels in the bulk cytoplasm only reach ~ 1 μM (Bers 2002c), this process was originally not seen as physiologically important (Bezprozvanny et al. 1991). Even taking into account the much higher levels of Ca2+ in the triadic cleft (200 μM (Soeller and Cannell 1997)), Mg2+ inhibition by the I1-site will be more significant to muscle function than Ca2+ inhibition (see below).
The location of the I1-site has not yet been determined in cryo-EM reconstructions. Mutated RyR1, which lacked amino acids 1641–2437 in the so-called junctional solenoid domain (des Georges et al. 2016) or Handle domain (Peng et al. 2016), required 10-fold higher Ca2+ than wt-RyR1 for inhibition but still had normal Ca2+ activation (Bhat et al. 1997). It has been noted (Laver et al. 1997b) that within this region of RyR1 is a distinctive sequence of 30 negative amino acids at positions 1873–1903, whereas in the corresponding region in RyR2 has far fewer negative amino acids. These would create a diffuse electrostatic binding region consistent with a low-affinity binding site that produces equal inhibition by Ca2+ and Mg2+ (Mg2+ is reviewed below). Also, the reduced negative charge in the corresponding region in RyR2 might explain their lower sensitivity Ca2+ inhibition.
Another cytoplasmic Ca2+ inhibition phenomenon has been identified in single channel recordings of RyR2 which has been attributed to a high-affinity, cytoplasmic facing site (I2-site) (Laver 2007). It is manifest as a cytoplasmic [Ca2+]-dependent reduction in channel open time in the sub micrometre range. Ca2+ diffusion/buffering experiments (described above for the A-site) estimate a 26-nm distance between the I2-site and the cytoplasmic mouth of the channel, somewhat further away than the A-site (Laver 2007). As yet, the physiological significance and precise location of this site are unknown.
Mg2+ inhibition of RyR1 and RyR2
Mg2+ is a strong inhibitor of RyRs that acts as a competitive Ca2+ antagonist at the A-site (Smith et al. 1986) with a binding affinity of ~ 50 μM (Laver and Honen 2008; Meissner et al. 1986; Meissner and Henderson 1987). The effect of physiological concentrations of Mg2+ (1 mM (Godt and Maughan 1988)) is to shift Ka for Ca2+ activation from 1 μM in the absence of Mg2+ to 50 μM (Cannell et al. 2013). Mg2+ binding at the A-site not only prevents Ca2+ activation but can also close the channels that are activated by luminal Ca2+ or ryanodine (Laver et al. 2004) which bind at other sites.
A biphasic [Ca2+] dependence of the Mg2+ IC50 revealed a second Mg2+ inhibitory mechanism due to the I1-site (Laver et al. 1997a). At low cytoplasmic [Ca2+], the IC50 for Mg2+ inhibition increased in proportion to [Ca2+] as would be expected for Ca2+/Mg2+ competition at the A-site. However, at higher concentrations, the IC50 for Mg2+ plateaued as if another non-competitive mechanism dominated. These IC50 values for Mg2+ and Ca2+ were identical in each individual channel in spite of substantial channel-to-channel variations indicating that Mg2+ is a surrogate for Ca2+, the I1-site and both Ca2+ and Mg2+ cause channel closures (Laver et al. 1997a). This finding provided an explanation for the physiological significance of the I1-site in skeletal muscle where the IC50 for the I1-site is ~ 0.1 mM. Even though physiological concentrations of Ca2+ are well below the IC50, Mg2+ levels are 10-fold higher than this and will strongly inhibit RyR1 in the cell. However, in RyR2, the IC50 of the I1-site is ~ 10 mM so it is unlikely that intracellular Mg2+ will have a sufficient inhibitory action at this site to make it a significant regulator in the heart.
Luminal Mg2+ was found to inhibit RyR2 by two mechanisms (Laver and Honen 2008). Firstly, Mg2+ flows through the channel and binds to the A-site and closes the channel (RyRs have the same permeability for both Mg2+ and Ca2+). Secondly, luminal Mg2+ is a competitive antagonist for Ca2+ at the luminal L-site where Ca2+ and Mg2+ have the same affinity. Hence, Mg2+ shifts the Ka for luminal Ca2+ activation from 40 μM in the absence of Mg2+ to 1 mM at physiological [Mg2+]. This is likely to be very important for the way luminal [Ca2+] regulates SR Ca2+ release. The physiological [Ca2+] in the junctional SR cycles between 0.3 and 1 mM (Bers 2002b) which, in the absence of Mg2+, would be well above the Ka so that Ca2+ release would be virtually unregulated by SR Ca2+ loading. However, in the presence of 1 mM Mg2+, the Ka lies within the physiological range will have a substantial effect on RyR2 activity and Ca2+ release.
RyR1 regulation during excitation-contraction coupling in skeletal muscle
In striated muscle, the L-type Ca2+ channels (DHPRs) located in the sarcolemma and transverse tubules are the voltage sensor. In skeletal muscle, the DHPRs directly interact with RyR1 and their depolarisation by an action potential induces conformational changes that are relayed to RyR1 to cause it to open (Tanabe et al. 1990). E-C coupling in skeletal muscle does not rely on the influx of Ca2+ so that DHPRs can activate RyRs even though cytoplasmic [Ca2+] is 100 nM. Once RyR1 opens, SR Ca2+ release increases cytoplasmic [Ca2+] to 10 μM.
The properties of cytoplasmic Ca2+, Mg2+ and ATP regulation of RyR1 give insight into the way DHPRs control Ca2+ release in skeletal muscle. In skeletal muscle, the I1- and A-sites both have Mg2+ IC50 values of 50–100 μM so that both sites make similar contributions to Mg2+ inhibition. The fact that Mg2+ inhibition at the I1-site is not alleviated by elevated cytoplasmic [Ca2+] (unlike the A-site) means that I1-site-mediated Mg2+ inhibition provides an extremely strong brake on Ca2+ release. In fact, without some way of bypassing this Mg2+ inhibition, RyR1 would not release Ca2+ in skeletal muscle at all. However, experiments on skinned muscle fibres have shown that depolarisation of DHPRs leads to a loss of Mg2+ inhibition of RyR1 (Lamb and Stephenson 1994). Though the particular Mg2+ inhibition mechanism being modulated has not been determined, it would need to involve the I1-site for it to be effective in de-repressing RyR1.
ATP can substantially activate RyR1 even when cytoplasmic [Ca2+] is at resting levels (~ 100 nM). This explains why RyR1 can be opened without there being any influx of extracellular Ca2+ to act as a trigger. Therefore, in resting muscle, RyR1 is primed for activation and once the activation of DHPRs alleviates Mg2+ inhibition, the RyR1 will open. Removal of ATP stimulation of RyR1 ‘deprimes’ RyR1 resulting in a loss of E-C coupling, i.e. the E-C coupling mechanism in skeletal muscle absolutely requires upregulation of RyR1 by ATP (Blazev and Lamb 1999a, 1999b). Adenosine, a non-agonist competitor for the ATP site, inhibited voltage sensor control of Ca2+ release in rat skinned muscle fibres. Therefore, it is likely that the DHPR triggers SR Ca2+ release simply by removing the inhibitory effect of Mg2+ exerted at the I1-site, possibly by lowering its affinity for Mg2+ as suggested previously (Lamb 2000). ATP would then be allowed to activate RyR1 and the released Ca2+ could then reinforce further activation.
Decreasing SR luminal Ca2+ from resting levels of 1 to 0.1 mM decreases the Ki for Mg2+ at the A-site in RyR1 from 72 to 20 μM without altering the Ki for the I1-site (Laver et al. 2004). Therefore, depletion of the SR will impose a stronger Mg2+ inhibition by the A-site which could explain why chemical stimulation of RyR1 (i.e. lowering Mg2+ and application of agonists such as caffeine) is unable to fully deplete the SR (Fryer and Stephenson 1996; Lamb and Stephenson 1994). However, activation of DHPRs can completely empty the SR of Ca2+, even in the presence of physiological [Mg2+] (Kurebayashi and Ogawa 2001; Posterino and Lamb 2003) which has led to the proposal that DHPRs alleviate Mg2+ inhibition by ‘commandeering’ the L-site/A-site allosteric interaction (Laver et al. 2004).
RyR2 regulation during excitation-contraction coupling in cardiac muscle
In the presence of diastolic concentrations of Mg2+ (1 mM) and Ca2+ (100 nM), the open probability of RyR2 has been measured at ~ 10−6 (Laver unpublished). Hence, during diastole, RyR2 are inactive because of insufficient activation by the A-site. RyR2 is not as strongly activated by ATP as RyR1 and so ATP is not the primary RyR2 agonist. Instead, Ca2+ influx through DHPRs increases [Ca2+] in the dyadic cleft to ~ 100 μM (Soeller and Cannell 1997) which provides the activating stimulus to RyR2 via their A-sites, even in the presence of 1 mM Mg2+. The subsequent release of Ca2+ from the SR further increases dyadic [Ca2+] to ~ 200 μM (Laver et al. 2013) to cause further RyR2 activation. This process, known as Ca2+-induced Ca2+ release (CICR (Fabiato 1983)), provides a strong positive feedback that should sustain RyR2 activation until the SR is substantially depleted of Ca2+ (Zima et al. 2008). However, in myocytes, Ca2+ release can undergo rapid termination of CICR in spite of only moderate SR depletion as measured by fluo-5N (Picht et al. 2011; Shannon et al. 2003). This is clearly seen in localised SR Ca2+ release events (Ca2+ sparks) that exhibit CICR lasting only 10 ms, after which the positive feedback cycle of CICR is broken and SR Ca2+ release ceases (Cheng et al. 1993). It was a long-standing problem that single channel studies have not found any RyR2 regulation mechanisms that could terminate CICR this rapidly (see reviews by Cannell and Kong 2012; Stern and Cheng 2004). Recently, two theoretical studies and one experimental study have provided a plausible explanation for a negative feedback mechanism (Cannell et al. 2013; Guo et al. 2012; Laver et al. 2013). Modelling of RyR2-mediated Ca2+ release by simulating Ca2+diffusion and buffering within a 3D spatial model of a dyad accurately reproduced the experimental values of Ca2+ spark duration, refractory period and SR depletion. The model also produced reliable and spontaneous RyR2 closure without any form of RyR2 inactivation (Cannell et al. 2013; Laver et al. 2013), by a mechanism called ‘induction decay’ (Laver et al. 2013). In this process, depletion of the junctional SR leads to a decay of CICR and subsequent deactivation of RyR2. The junctional SR depleted by ~ 90% with virtually no depletion of the longitudinal SR due to the limited connection between the two SR compartments. Model simulations of confocal images revealed that the inability of confocal microscopes to resolve fluo-5N fluorescence in junctional and longitudinal SR leads to an underestimation of junctional SR depletion and so reconciled model and experimental estimates of SR depletion. Experimental support for the idea of ‘induction decay’ came from the observation that reducing RyR2 conductance increased the rate of Ca2+ spark termination (Guo et al. 2012).
RyR2 regulation during cardiac pacemaking
In cardiomyocytes, SERCA2a and RyR2 carry Ca2+ uptake and release, respectively, across the SR membrane while DHPRs and Na/Ca exchangers (NCXs) carry Ca2+ uptake and release across the sarcolemma, respectively (Dibb et al. 2007). These transporters generate the large oscillations in free [Ca2+] in the cytoplasm ([Ca2+] ~ 0.1 to 1 μM) and SR lumen ([Ca2+] ~ 1 to 0.3 mM) between diastole and systole, respectively (Bers 2002b). Spontaneously, cyclic SR Ca2+ uptake and release have been observed as Ca2+ waves in intact cardiomyocytes under conditions of Ca2+ overload (Bers 2002a). This process was first shown to generate a ‘clock cycle’ that could entrain pacemaker action potentials in lymphatic smooth muscle (Van Helden 1993) and more recently in cardiac pacemaking in the sinoatrial node (Ju and Allen 1998; Ju and Allen 2007; Lakatta and DiFrancesco 2009; Rigg and Terrar 1996). Although this process depends on many ion transport and buffering mechanisms (Bers 2002a), the role of the RyR2 luminal and cytoplasmic activation sites in cyclic Ca2+ uptake and release is envisaged as follows. First, SERCA2a sequesters cytoplasmic Ca2+, loading the SR to a point where elevated luminal [Ca2+] causes opening of RyR2 via the L-site, a process dubbed store overload-induced Ca2+ release (SOICR (Jiang et al. 2005)). Second, during Ca2+ release, CICR provides positive reinforcement of RyR2 activity via the A-site which continues until the SR lumen depletes and the decaying CICR can no longer support RyR2 activation and RyR2 close. Subsequently, the SR reloads and the cycle is repeated, producing a cyclic, bistable oscillation analogous to the motion of a tipping urn (Fig. 2). By this analogy, one can see how cycling rate is influenced by L-site sensitivity of RyR2 (pivot point), SERCA2a activity (filling rate) and Ca2+ capacity/buffering of the SR (urn capacity). SR Ca2+ cycling is coupled to sarcolemmal pacemaking by the NCX. Ca2+ released during RyR2 activation phase of each cycle promotes Ca2+ extrusion from the cell via NCX leading to depolarization of the sarcolemma (i.e. 3 Na+ enter for every Ca2+ extruded) and enhanced sarcolemmal excitability with the effect of entraining pacemaker action potentials (Bers 2002a).
Fig. 2.
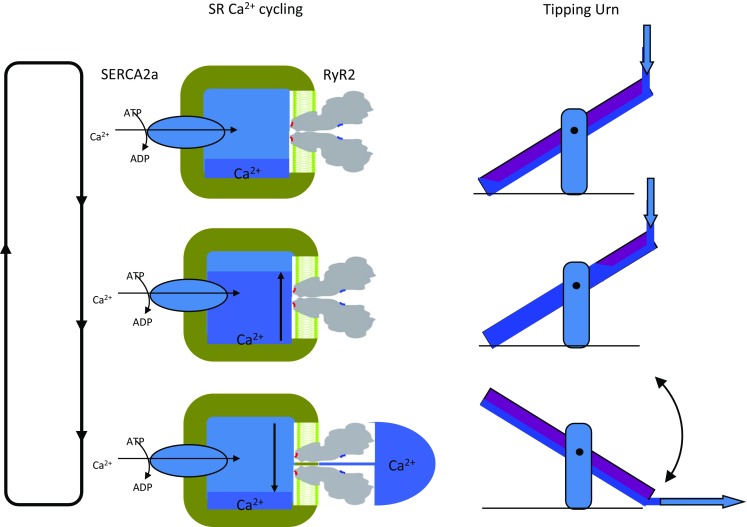
Two schematics illustrating how SERCA2a and RyR2 produce cycling of Ca2+ uptake and release in cardiac SR. The left shows an idealised SR with SERCA2a driven uptake and RyR2-mediated release which is controlled by luminal and cytoplasmic facing Ca2+ activation sites (L-sites—red; A-sites—blue). Store filling with Ca2+ is signified by dark blue. As the store fills (middle left), the L-sites trigger the opening of RyR2 (bottom left) and the resulting Ca2+ flow to the A-sites reinforce channel opening until the stores are nearly depleted SERCA2a (top left). The right side shows the analogy of a ‘tipping urn’ that undergoes bistable oscillations by similar mechanisms. The thick arrows indicate water flow. Analogous features are (1) SERCA2a transport rate and water flow rate; (2) SR capacity and urn capacity; and (3) effective Ca2+ affinity of the L-site and the pivot point
An example of how this would work within a physiological setting is the case of the sympathetic ‘fight or flight’ response that increases heart rate and contractility. Increased catecholamine concentrations stimulate cardiac β-adrenergic receptors (β-AR), resulting in phosphorylation and increased activity of key Ca2+ transporters including SERCA2a and RyR2 (Bers 2002b). Many studies have demonstrated increased phosphorylation of RyR2 at S2808, S2814 and S2030 (Benkusky et al. 2007; Carter et al. 2006; Currie et al. 2004; Ferrero et al. 2007; Huke and Bers 2008; Li et al. 2013; Rodriguez et al. 2003; Xiao et al. 2006). Detailed analysis of RyR2 regulation of channels isolated from adrenergic-stimulated hearts showed that adrenergic stimulation increased their L-site-mediated luminal Ca2+ activation without affecting A-site RyR2 activation (Li et al. 2013). Such an increase in L-site sensitivity would cause earlier onset of the Ca2+ release phase in the cardiac cycle which would increase Ca2+ cycling rate and so provide a driver for increasing pacemaking frequency.
Concluding remarks
Thus far, four Ca2+regulation sites have been identified in RyRs. They have two Ca2+ activation sites located in the luminal and cytoplasmic domains of the RyR (L- and A-sites). These sites feed into a common gating mechanism and produce synergistic activation by luminal and cytoplasmic Ca2+. The cytoplasmic domains also possess two inhibitory sites (I1- and I2- sites) with Ca2+ affinities of the order of millimolar and micromolar, respectively. Magnesium competes with Ca2+ at these sites and plays an important role in inhibiting RyRs and shaping the Ca2+-dependent activation of RyRs in muscle. Recent analysis of RyR1 structure has identified one A-site in each subunit at the interfaces between the C-terminal domain and the core solenoid, at a distance from the cytoplasmic pore mouth that was predicted by functional studies.
RyR2 and RyR1 share common Ca2+/Mg2+ regulation mechanisms albeit with different sensitivities and these differences lead to quite different types of cellular control over SR Ca2+ release in skeletal and cardiac muscle (Lamb 2000; Laver et al. 2001). In resting skeletal muscle, RyR1 would be activated by ATP if it were not for Mg2+ which strongly inhibits the channels via the I1-site. RyR1 activation by voltage sensor depolarisation is due to a conformational coupling between the voltage sensor and RyR1 that alleviates this Mg2+ inhibition thereby de-repressing RyR1. Once the voltage sensor deactivated upon membrane repolarisation, Mg2+ inhibition is restored and this will close RyR1 even in the presence of high [Ca2+]. Thus, cessation of Ca2+ release is under voltage sensor control. In cardiac muscle during diastole, RyR2 inhibition by Mg2+ is weak because the I1-site is ~ 100-fold less sensitive than in RyR1 and Mg2+ inhibition that occurs at the A-site is competitively overcome by the Ca2+ stimulus. Hence, RyR2 inactivity in diastole is due to a lack of RyR2 stimulation rather than inhibition. During E-C coupling, Ca2+ entry through the voltage sensor is sufficient to stimulate SR Ca2+ release but cessation of Ca2+ release is not under voltage sensor control. Rather, it is controlled by restricted Ca2+ diffusion between the junctional and longitudinal SR leading to ‘induction decay’ of RyR2.
Finally, RyR2 activation by L- and A-sites serves different roles in SR Ca2+ release. The A-site stimulates RyR2 opening during cardiac E-C coupling where the trigger for Ca2+ release is the Ca2+ influx through voltage sensors, whereas the L-site stimulates RyR2 opening in response to store loading which forms part of the cardiac pacemaking mechanism.
Acknowledgments
Thanks to Dirk van Helden for suggesting the ‘Tipping Urn’ analogy in Fig. 2 and to Oliver Clarke for locating amino acids on the RyR1 structure in Fig. 1.
Funding
This work was supported by an infrastructure grant from NSW Health through Hunter Medical Research Institute.
Compliance with ethical standards
Derek R. Laver declares that he has no conflict of interest.
This article does not contain any studies with human participants or animals performed by the author.
Footnotes
This article is part of a Special Issue on ‘Heart Failure Due to Non-Myofibrillar Defects’ edited by Elisabeth Ehler and Katja Gehmlich.
References
- Balog EM, Fruen BR, Shomer NH, Louis CF. Divergent effects of the malignant hyperthermia-susceptible arg(615)cys mutation on the Ca2+ and Mg2+ dependence of the RyR1. Biophys J. 2001;81:2050–2058. doi: 10.1016/S0006-3495(01)75854-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkusky NA, et al. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium and cardiac rhythms: physiological and pathophysiological. Circ Res. 2002;90:14–17. [PubMed] [Google Scholar]
- Bers DM (2002b) Cardiac excitation-contraction coupling. Nature 415 [DOI] [PubMed]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM, Peskoff A. Diffusion around a cardiac calcium channel and the role of surface bound calcium. Biophys J. 1991;59:703–721. doi: 10.1016/S0006-3495(91)82284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bhat MB, Zhao J, Hayek S, Freeman EC, Takeshima H, Ma J. Deletion of amino acids 1641-2437 from the foot region of skeletal muscle ryanodine receptor alters the conduction properties of the Ca release channel. Biophys J. 1997;73:1320–1328. doi: 10.1016/S0006-3495(97)78165-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazev R, Lamb GD. Adenosine inhibits depolarization-induced Ca2+ release in mammalian skeletal muscle. Muscle Nerve. 1999;22:1674–1683. doi: 10.1002/(SICI)1097-4598(199912)22:12<1674::AID-MUS9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Blazev R, Lamb GD. Low [ATP] and elevated [Mg2+] reduce depolarization-induced Ca2+ release in rat skinned skeletal muscle fibres. JPhysiolLond. 1999;520:203–215. doi: 10.1111/j.1469-7793.1999.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Kong CH. Local control in cardiac E-C coupling. J Mol Cell Cardiol. 2012;52:298–303. doi: 10.1016/j.yjmcc.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Kong CH, Imtiaz MS, Laver DR. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J. 2013;104:2149–2159. doi: 10.1016/j.bpj.2013.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–1513. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- Chen W, et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat Med. 2014;20:184–192. doi: 10.1038/nm.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca2+ activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circ Res. 2000;87:201–206. doi: 10.1161/01.RES.87.3.201. [DOI] [PubMed] [Google Scholar]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM. Structure and function of ryanodine receptors. Am J Physiol. 1994;266:C1485–C1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- Currie S, Loughrey CM, Craig MA, Smith GL. Calcium/calmodulin-dependent protein kinase II delta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem J. 2004;377:357–366. doi: 10.1042/bj20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Georges A, et al. Structural basis for gating and activation of RyR1. Cell. 2016;167:145–157. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb KM, Graham HK, Venetucci LA, Eisner DA, Trafford AW. Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium. 2007;42:503–512. doi: 10.1016/j.ceca.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Phys. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. JGenPhysiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero P, Said M, Sanchez G, Vittone L, Valverde C, Donoso P, Mattiazzi A, Mundina-Weilenmann C. Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during beta-adrenergic stimulation. J Mol Cell Cardiol. 2007;43:281–291. doi: 10.1016/j.yjmcc.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessenden JD, Chen L, Wang Y, Paolini C, Franzini-Armstrong C, Allen PD, Pessah IN. Ryanodine receptor point mutant E4032A reveals an allosteric interaction with ryanodine. Proc Natl Acad Sci U S A. 2001;98:2865–2870. doi: 10.1073/pnas.041608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. JPhysiolLond. 1996;493:357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godt RE, Maughan DW. On the composition of the cytosol of relaxed skeletal muscle of the frog. AmJPhysiol. 1988;254:C591–C604. doi: 10.1152/ajpcell.1988.254.5.C591. [DOI] [PubMed] [Google Scholar]
- Guo T, Gillespie D, Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res. 2012;111:28–36. doi: 10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke S, Bers DM. Ryanodine receptor phosphorylation at serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hymel L, Inui M, Fleischer S, Schindler H. Purified ryanodine receptor of skeletal muscle sarcoplasmic reticulum forms Ca2+-activated oligomeric Ca2+ channels in planar bilayers. Proc Natl Acad Sci U S A. 1988;85:441–445. doi: 10.1073/pnas.85.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- Ju YK, Allen DG. Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. JPhysiolLond. 1998;508:153–166. doi: 10.1111/j.1469-7793.1998.153br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YK, Allen DG. Store-operated Ca2+ entry and TRPC expression; possible roles in cardiac pacemaker tissue. Heart Lung Circ. 2007;16:349–355. doi: 10.1016/j.hlc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. JPhysiolLond. 2001;533:185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009;47:157–170. doi: 10.1016/j.yjmcc.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb GD. Excitation-contraction coupling in skeletal muscle: comparisons with cardiac muscle. Clin Exp Pharmacol Physiol. 2000;27:216–224. doi: 10.1046/j.1440-1681.2000.03224.x. [DOI] [PubMed] [Google Scholar]
- Lamb GD, Stephenson DG. Effects of intracellular pH and [Mg2+] on excitation-contraction coupling in skeletal muscle fibres of the rat. JPhysiolLond. 1994;478:331–339. doi: 10.1113/jphysiol.1994.sp020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver D (2010) Regulation of the RyR channel gating by Ca, Mg and ATP. In structure-function of calcium release channels. In: Serysheva I (ed) Current topics in membranes, vol 66. Elsevier, pp 69–89 [DOI] [PubMed]
- Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys J. 2007;92:3541–3555. doi: 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, Baynes TM, Dulhunty AF. Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J Memb Biol. 1997;156:213–229. doi: 10.1007/s002329900202. [DOI] [PubMed] [Google Scholar]
- Laver DR, Honen BN. Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: cytoplasmic and luminal regulation modeled in a tetrameric channel. JGenPhysiol. 2008;132:429–446. doi: 10.1085/jgp.200810001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, Kong CHT, Imtiaz MS, Cannell MB. Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J Mol Cell Cardiol. 2013;54:98–100. doi: 10.1016/j.yjmcc.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Laver DR, Lenz GK, Lamb GD. Regulation of the calcium release channel from rabbit skeletal muscle by the nucleotides ATP, AMP, IMP and adenosine. JPhysiolLond. 2001;537:763–778. doi: 10.1111/j.1469-7793.2001.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, O’Neill ER, Lamb GD. Luminal Ca2+-regulated Mg2+ inhibition of skeletal RyRs reconstituted as isolated channels or coupled clusters. JGenPhysiol. 2004;124:741–758. doi: 10.1085/jgp.200409092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, Owen VJ, Junankar PR, Taske NL, Dulhunty AF, Lamb GD. Reduced inhibitory effect of Mg2+ on ryanodine receptor-Ca2+ release channels in malignant hyperthermia. Biophys J. 1997;73:1913–1924. doi: 10.1016/S0006-3495(97)78222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, Roden LD, Ahern GP, Eager KR, Junankar PR, Dulhunty AF. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J Memb Biol. 1995;147:7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- Li J, Imtiaz MS, Beard NA, Dulhunty AF, Thorne R, vanHelden DF, Laver DR. ss-Adrenergic stimulation increases RyR2 activity via intracellular Ca2+ and Mg2+ regulation. PLoS One. 2013;8:e58334. doi: 10.1371/journal.pone.0058334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Chen SR. Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor) J Gen Physiol. 2001;118:33–44. doi: 10.1085/jgp.118.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annual Reviews in Physiology. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Meissner G, Darling E, Eveleth J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry. 1986;25:236–244. doi: 10.1021/bi00349a033. [DOI] [PubMed] [Google Scholar]
- Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem. 1987;262:3065–3073. [PubMed] [Google Scholar]
- Meissner G, Rios E, Tripathy A, Pasek DA. Regulation of skeletal muscle Ca2+ release channel (ryanodine receptor) by Ca2+ and monovalent cations and anions. J Biol Chem. 1997;272:1628–1638. doi: 10.1074/jbc.272.3.1628. [DOI] [PubMed] [Google Scholar]
- Ogawa Y. Role of ryanodine receptors. Crit Rev Biochem Mol Biol. 1994;29:229–274. doi: 10.3109/10409239409083482. [DOI] [PubMed] [Google Scholar]
- Peng W et al (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354. 10.1126/science.aah5324 [DOI] [PubMed]
- Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posterino GS, Lamb GD. Effect of sarcoplasmic reticulum Ca2+ content on action potential-induced Ca2+ release in rat skeletal muscle fibres. JPhysiolLond. 2003;551:219–237. doi: 10.1113/jphysiol.2003.040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase a. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.M301180200. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- Shomer NH, Louis CF, Fill M, Litterer LA, Mickelson JR. Reconstitution of abnormalities in the malignant hyperthermia-susceptible pig ryanodine receptor. AmJPhysiol. 1993;264:C125–C135. doi: 10.1152/ajpcell.1993.264.1.C125. [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Gating of the native and purified cardiac SR Ca2+-release channels with monovalent cations as permeant species. Biophys J. 1994;67:1484–1494. doi: 10.1016/S0006-3495(94)80622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by luminal Ca2+ J Memb Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Smith JS, Coronado R, Meissner G. Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum. Activation by Ca2+ and ATP and modulation by Mg2+ JGenPhysiol. 1986;88:573–588. doi: 10.1085/jgp.88.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD, Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium. 2004;35:591–601. doi: 10.1016/j.ceca.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- Tripathy A, Meissner G. Sarcoplasmic reticulum lumenal Ca2+ has access to cytosolic activation and inactivation sites of skeletal muscle Ca2+ release channel. Biophys J. 1996;70:2600–2615. doi: 10.1016/S0006-3495(96)79831-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Helden DF. Pacemaker potentials in lymphatic smooth muscle of the guinea-pig mesentery. J Physiol. 1993;471:465–479. doi: 10.1113/jphysiol.1993.sp019910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, et al. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res. 1996;79:1100–1109. doi: 10.1161/01.RES.79.6.1100. [DOI] [PubMed] [Google Scholar]
- Zahradnik I, Gyorke S, Zahradnikova A. Calcium activation of ryanodine receptor channels—reconciling RyR gating models with tetrameric channel structure. J Gen Physiol. 2005;126:515–527. doi: 10.1085/jgp.200509328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA (2008) Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion Circ Res 103:e105–e115 doi:CIRCRESAHA.107.183236 [DOI] [PMC free article] [PubMed]