

Abstract
Most structural techniques provide averaged information or information about a single predominant conformational state. However, biological macromolecules typically function through series of conformations. Therefore, a complete understanding of macromolecular structures requires knowledge of the ensembles that represent probabilities on a conformational free energy landscape. Here we describe an emerging approach, X-ray scattering interferometry (XSI), a method that provides instantaneous distance distributions for molecules in solution. XSI uses gold nanocrystal labels site-specifically attached to a macromolecule and measures the scattering interference from pairs of heavy metal labels. The recorded signal can directly be transformed into a distance distribution between the two probes. We describe the underlying concepts, present a detailed protocol for preparing samples and recording XSI data, and provide a custom-written graphical user interface to facilitate XSI data analysis.
Keywords: X-ray scattering interferometry, small angle X-ray scattering, gold nanocrystals, molecular ruler, structure determination, ensemble determination, energy landscape
Introduction
Richard Feynman famously said “… everything that is living can be understood in terms of the jiggling and wiggling of atoms” (Feynman et al., 1989). Biological macromolecules such as unfolded or partially folded RNAs or intrinsically disordered proteins are especially dynamic, given the noncovalent forces that hold them together, their aqueous surroundings, and physiological temperature that provides thermal energy. Moreover, significant conformational changes of molecules can be triggered by external stimuli and are typically integrally involved in the functions of biomolecules (Fischer et al., 2010; Frauenfelder, 1989; Müller et al., 2016). Thus, conformational changes play an important role in understanding the basic mechanics and are key in reconstructing causes from the molecular level to macromolecular systems.
This unit describes an emerging molecular ruler, termed X-ray scattering interferometry (XSI) (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2014; Shi et al., 2015; Shi et al., 2013; Shi et al., 2016; Shi et al., 2017), which can be used to generate whole distance distributions at Ångström resolution. XSI measures the interference of scattered X-rays between two specifically attached gold nanocrystals (Fig. 1). The strengths of XSI are that it provides: i. distance information in solution; ii. the distance information that is unperturbed by temporal averaging because scattering is fast relative to atomic motions; iii. the direct mathematical relationship between scattering and distance, formally related by Fourier transformation, allows XSI data to be unambiguously converted into a calibrated distance distribution; and iv. while sample preparation is time-consuming, it is straightforward, highly reliable and highly reproducible.
Figure 1. Schematic of AuSAXS workflow to determine gold label-gold label distance distributions.

Scattering intensity equation for a single-labeled molecule. The scattering signal can be decomposed into a sum of the individual scattering contributions: double-labeled sample, the macromolecule only, two gold-macromolecule cross-terms, and the interference from the gold labels. Schematic of the workflow to determine the Au-Au distance distribution. The SAXS profiles of the shown samples are used to extract the gold-gold interference scattering profile. The interference pattern is Fourier transformed into a distance distribution using basic profiles generated with the size distribution of Au nanocrystals.
Over the past decade, XSI has been successfully applied to nucleic acids and nucleic acid/protein complexes (G. L. Hura et al., 2013; Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2014; Shi et al., 2015; Shi et al., 2013; Shi et al., 2016; Shi et al., 2017). Labelling strategies employing gold nanocrystals of various sizes, diverse macromolecules, and variable attachment positions have been reported: So far, demonstrated labeling strategies include: i. end labeled DNA nanocrystal conjugates (Ackerson et al., 2005; Alivisatos et al., 1996; Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2014; Shi et al., 2015; Shi et al., 2013), ii. end labeled RNA molecules (Shi et al., 2015; Shi et al., 2016; Shi et al., 2017), iii. gold labels positioned at defined internal sites of DNA or RNA helices (Mathew-Fenn et al., 2008a; Shi et al., 2014; Shi et al., 2015; Shi et al., 2013; Shi et al., 2016; Shi et al., 2017), and iv. protocols to form single-labeled protein constructs (Aubin-Tam & Hamad-Schifferli, 2005; Aubin-Tam et al., 2009; Azubel & Kornberg, 2016).
This unit covers the design and preparation of end-labeled nucleic acid samples. Below we present the procedures required to prepare samples, to acquire XSI data, and to generate ensemble distance distributions.
Basic Protocol 1 describes sample preparation for end-labeled nucleic acid gold conjugates and includes a protocol for gold nanocrystal synthesis.
Basic Protocol 2 describes the acquisition of a full data set at a synchrotron radiation facility for XSI analysis.
Basic Protocol 3 describes the data analysis of XSI data and the use of a custom-written graphical user interface (GUI) in MATLAB.
The detailed protocols and the user interface presented in this chapter will enable scientists interested in molecular distance measurements to perform and analyze XSI measurements easily.
Basic protocol 1, Sample preparation
In this unit, we focus on the preparation of end labeled nucleic acid samples for XSI. An additional protocol on labeling proteins will be forthcoming.
Briefly, thioglucose protected gold nanocrystals are synthesized using the method of Schaaff and coworkers (Schaaff et al., 1998). DNA or RNA oligonucleotides are ordered with a commercially available C3-thiol-modification for end labeling, or a C2 dT amino-modification for internal labeling. High-performance liquid chromatography (HPLC), performed either in-house or by the oligonucleotide vendor, is used to purify the oligonucleotides. In the case of the internal C2 dT amino-modification, the amino group is converted to a thiol group using the commercially-available SPDP (N-succinimidyl3-[2-pyridyldithio]-propionate) cross-linker. For a detailed protocol of internal label attachment see (Shi et al., 2015). The gold nanocrystals couple to thiol groups, forming stable conjugates. A second HPLC purification step is used to purify 1:1 nanocrystal-nucleic-acid conjugates, eliminating nanocrystals coupled to multiple oligonucleotides and excess gold particles. Finally, modified and unmodified single-stranded molecules are mixed in various combinations to form a sample quartet, which consists of one unmodified construct, two complementary single-labeled molecules with a single gold nanocrystal attached to one of the two labeling sites respectively, and one double-labeled construct. After HPLC purification and desalting, these duplexed constructs can be stored at −20 °C for several months. A full set of samples is required for the data analysis to work as explained in detail in basic protocol 3.
CAUTION: Some of the chemicals and reagents used are flammable. Refer to material safety data sheets prior to use. Some of the reactions should be conducted in a well-ventilated fume hood. Use of personal protection equipment is highly recommended.
Materials
Dithiothreitol (Thermo Fisher, cat. No. R0861)
Tris hydrochloride
FPLC cleaning solution (100 mM DTT, 20 mM Tris-HCl, pH 8.0)
Ammonium acetate (Sigma Aldrich, cat. No. A1542)
Size exclusion running buffer (150 mM ammonium acetate, pH 5.6)
Methanol
Acetic acid
Hydrogen tetrachloroaurate (III) hydrate (Sigma Aldrich, cat. No. 50790)
1-thio-β-D-glucose (Sigma Aldrich, cat. No. T6375)
Sodium borohydride
Wet ice
3′-thiol modified oligonucleotides
Oligonucleotides
2 M ammonium acetate, pH 5.6
Sodium chloride (NaCl)
Boric acid
Sodium hydroxid (NaOH)
Low salt borate buffer (10 mM NaCl, 20 mM Na-borate, pH 7.8)
High salt borate buffer (1.5 M NaCl, 20 mM Na-borate, pH 7.8)
Ethanol
Magnesium chloride (MgCl2)
Dry ice
Low salt acetate buffer (10 mM NaCl, 20 mM ammonium acetate, pH 5.6)
High salt acetate buffer (1.5 M NaCl, 20 mM ammonium acetate, pH 5.6)
G25 column, 26/10 Housing (Sigma Aldrich, cat. No. GE17-5087)
FPLC system (Dual Wavelength detector recommended)
Superdex 30 column, 16/600 Housing (Sigma Aldrich, cat. No. GE28-9893-31)
250-mL round-bottom flask
Parafilm
Magnetic stirrer/hotplate
Vortexer
0.22 μm syringe filter units
3 kDa and 10 kDa Amicon spin filtration units (Sigma Aldrich)
Dionex DNAPac Pa200 column, 9/250 Housing (Thermo Fisher, cat. No. 063421)
HPLC System (Dual Wavelength detector recommended)
Rotary evaporator equipped with a dry ice condenser and connected to an oil pump
−20 or −80 °C freezer
Nanodrop ND-1000 spectrophotometer or other UV spectrophotometer
The protocol below describes: i. the synthesis of monodisperse, thiol-passivated gold nanocrystals with 0.7 nm radius; ii. the preparation of end-labeled gold-oligonucleotides conjugates; and iii. the preparation of a sample quartet that is ready for XSI data acquisition. The concentrations cited below are based on ordering a 200 nmol scale nucleic-acid synthesis, and can be adjusted for alternate quantities of starting material.
Synthesis and Purification of gold nanocrystals
Steps 1–3: Preparation of FPLC columns (start two days in advance)
-
1
Using a flow rate of 2 mL/min, wash the G25 column with two column volumes of water, followed by four column volumes of the FPLC cleaning solution (20 mM Tris-HCl, pH 8.0, and 100 mM DTT). Equilibrate the column with four column volumes of water.
-
2
Using a flow rate of 0.75 mL/min, wash the Superdex 30 column with two column volumes of water, followed by 5 column volumes of the FPLC cleaning solution (same cleaning solution as in step 1). Remove the FPLC cleaning solution with 5 volumes of water. Finally, equilibrate the Superdex 30 with two column volumes of the size exclusion running buffer (150 mM ammonium acetate, pH 5.6).
Note: The gold nanocrystals cause the column resin to turn brown. This coloration is reversed by the DTT in the cleaning solution. If the resin does not revert to off-white after 5 column volumes, apply additional cleaning solution before equilibrating the column. Start the cleaning and equilibration of both columns at least one day before the synthesis.
Steps 3–11: Synthesis of gold nanocrystals (4 hours)
-
3
Rinse a 250-mL round-bottom flask with isopropanol, dry in a heated oven and cool (to room temperature). Add a stir bar, cap the flask with parafilm and mount the flask above a magnetic stir plate.
-
4
Prepare 72 mL of a 5:1 v/v mixture of methanol-acetic acid (60 mL : 12 mL).
-
5
Weigh 0.544 g of hydrogen tetrachloroaurate (III) hydrate and immediately transfer it to the round bottom flask. Add 36 mL of the 5:1 methanol-acetic acid solution to the flask. The color should be a clear, bright orange.
-
6
Dissolve 1 g of 1-thio-β-D-glucose in 36 mL of the 5:1 methanol-acetic acid mixture and vortex the solution until the powder is fully dissolved.
-
7
Add the dissolved 1-thio-β-D-glucose to the 250-mL round-bottom flask. The solution should turn cloudy. Stir the mixture for 20 min at room temperature.
-
8
Weigh 0.9 g of sodium borohydride and dissolve it in 20 mL of ddH2O by vortexing.
-
9
Carefully add the sodium borohydride solution dropwise to the reaction flask over 12–15 min. Use a 1 mL pipette for this step, or alternatively an addition funnel with a metering valve (for example Chemglass, cat. No. CG-1714).
Important!: it is critical to keep the addition speed and droplet volume constant to ensure monodisperse and high-quality gold nanocrystals. -
10
Stir the mixture for 30 min at room temperature.
-
11
Concentrate the reacted solution (approximately 92 mL) to 12–20 mL using a rotary evaporator. The water bath needs to stay at room temperature.
Steps 12–16: Purification of gold nanocrystals (10 hours)
-
12
Filter the crude nanocrystal solution using a 0.22 μm filter unit, and store the solution on ice.
-
13
Desalt the filtered nanocrystals with the prepared G25 column. Split the total volume into 2–3 runs. Load one aliquot of the solution (at most 5 mL with a standard sample loop) while applying a flow rate of 2 mL/min using water as the running buffer. Set the FPLC system up to monitor 260 nm or 280 nm to detect the Au nanocrystals. The particles should elute in a window between 8–14 min (see Fig. 2). Also monitor the conductance of the solution to detect the salt peak, which elutes after the nanocrystals. Repeat the desalting procedure for the additional aliquots. The desalted Au nanocrystals are stable can be stored at 4 °C for up to a few days or at −20°C for months.
-
14
Concentrate the desalted Au nanocrystal solution to 10 mL with centrifugal filter units (3 kDa cutoff, 15mL, 3,000×g, 40 min) at 4 °C. The flow through should be clear.
-
15
Purify a monodisperse population of Au nanocrystals on the Superdex 30 size exclusion column (see Fig. 3). Set up the FPLC system to monitor 260 nm or 280 nm to detect the nanocrystals. You can inject up to 5 mL of sample per run. After loading an aliquot of the filtered, desalted and concentrated nanocrystal solution, apply the size exclusion running buffer at 0.75 mL/min for at least 210 minutes. Collect only the center of the largest Au nanocrystal elution peak (see Fig. 3), and discard the lower and upper shoulders. Immediately desalt the solution using ddH2O and centrifugal filter units (3 kDa cutoff, 15mL) at 3,000 × g and 4 °C. Repeat the centrifugal desalting three times and pool the concentrated particles after the final run.
-
16
Determine the final gold nanocrystal concentration by measuring the UV absorption. The extinction coefficient is 0.076 μm/cm at 360 nm. Store the solution at −20°C. Typically one synthesis yields 3–10 μmol of purified gold nanocrystals.
Figure 2. A sample FPLC chromatogram of Au nanocrystal desalting.
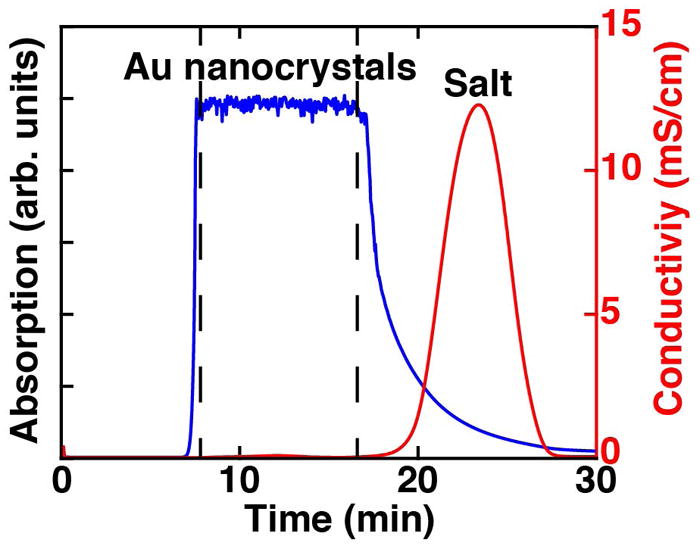
The absorption of the Au nanocrystals (blue) is monitored at 260 nm and separated from the salt front (red) observed by a peak in conductivity. The sample eluting between the two dashed lines was used.
Figure 3. A sample FPLC chromatogram of Au nanocrystal size exclusion.
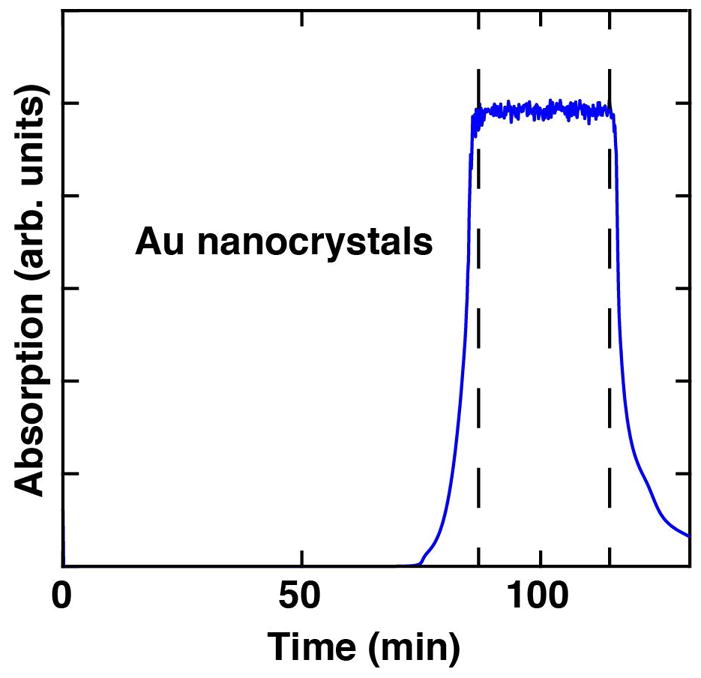
The absorption of the Au nanocrystals (blue) is monitored at 260 nm. The sample between the two dashed lines can be used for a highly uniform particle size distribution.
Preparation of gold nanocrystal-nucleic acid conjugates
Step 17–22: Preparation of oligonucleotides (about 8 hours for 4 oligonucleotides)
-
17
Purchase 3′-thiol modified oligonucleotides (C3-S-S) and unmodified oligonucleotides with the same sequences. It is critical to use the short three-carbon linker for end labeled samples to ensure high-quality results for the measurements of the distance distributions. Design the construct such that the terminating bases are GC base pairs to minimize end fraying.
-
18
Purify the ordered oligonucleotides using the Dionex DNAPac 200 and anion exchange HPLC. Inject up to 100 nmol of oligonucleotide onto the column and apply a flow of 3 mL/min. The salt gradient is formed from low salt borate buffer (10 mM NaCl, 20 mM Na-borate, pH 7.8) and high salt borate buffer (1.5 M NaCl, 20 mM Na-borate, pH 7.8). Tune the period of the gradient according to the length of your oligonucleotide. Perform an analytical run before the preparative ones to determine the elution time of the product. For analytical runs adjust the injected sample according to the instrument detection sensitivity.
-
19
Desalt the purified oligonucleotides by buffer exchange into water using centrifugal filter units and ddH2O (3 kDa cutoff, 4 mL, 4,000×g in a swinging basket centrifuge or 7,500×g in a fixed angle rotor, 30 min, 4 °C). Repeat this step three times. Finally, reduce the volume to ~40 μL using centrifugal filter units (3 kDa cutoff, 0.5 mL, 14,000×g using a benchtop centrifuge, 30 min, 4 °C). Typically, ~60 nmol of oligonucleotide remain after purification of a 200 nmol scale synthesis. To protect against loss of oligonucleotide from a broken filter unit, keep the flow through and check its absorbance at 260 nm. The oligonucleotides can be stored in a freezer at −20°C.
Steps 20–26: These steps use the thiol-modified oligonucleotides (about 3 hours)
-
20
Immediately before coupling oligonucleotides to gold nanocrystals, ensure that the pendant thiols are fully reduced by adding 150 μL of 200 mM DTT, 50 mM Tris-HCl, pH 9.0 and incubating for 30 min at 60°C for DNA or 50°C for RNA.
Note: The DTT can be replaced by immobilized TCEP Reducing Gel (Thermo Fisher, cat. No. 77712). Follow the manual provided by the vendor to reduce and extract the oligonucleotides and proceed with step 23 afterwards. -
21
Purify the oligonucleotide by ethanol precipitation. Add 2 μL of 2 M MgCl2 and 1 mL of cold ethanol and mix the solution. Incubate the mixture on dry ice for 40 min. Spin the mixture (15,000×g, 30 min, 4 °C). Remove the supernatant and wash with 1 mL ethanol. Spin the mixture again (15,000×g, 15 min, 4 °C) and remove the supernatant. Be careful not to disturb the precipitated pellet on the bottom.
-
22
Dissolve the precipitated pellet in 500 μL ddH2O and add it to a filter unit (3 kDa, 0.5 mL). Spin the solution (14,000×g, 30 min, 4 °C) to remove residual DTT and determine the final concentration of the oligonucleotide by UV absorption at 260 nm. Use the extinction coefficient provided by the manufacturer or calculate it using the nucleic acid sequence (Internet resource: “OligoAnalyer 3.1”). If a strong absorbance at 230 nm and a shoulder peak above 300 nm is observed repeat this step to remove excess DTT. Again, keep the flow-through to test for broken filter units. Immediately proceed to step 23.
Steps 23–26: Conjugate oligonucleotides and nanocrystals (about 8 hours)
-
23
Add a six-fold molar excess of purified and desalted gold nanocrystals to the reduced oligonucleotide and vortex the mixture (i.e. add 300 nmol Au particles to 50 nmol of oligonucleotides). Add 20 μL of 1 M Tris-HCl, pH 9.0 and vortex again. Incubate the solution for 2 h at room temperature.
-
24
Add 15 μL of 2 M ammonium acetate, pH 5.6 to stop the reaction and store the mixture on ice.
-
25
Purify the solution using the Dionex DNAPac 200 and anion exchange HPLC. Elute the conjugates with a salt gradient using a low salt acetate buffer (10 mM NaCl, 20 mM ammonium acetate, pH 5.6) and a high salt acetate buffer (1.5 M NaCl, 20 mM ammonium acetate, pH 5.6). Again, tune the period of the gradient according to the length of your oligonucleotide. Typically, 15-mer DNA-gold nanocrystal conjugates elute around 40% of the high salt buffer. Set the flow to 3 mL/min and monitor the absorbance at 260 nm and 360 nm. The oligonucleotides only absorb at 260 nm whereas the gold nanocrystals absorb at both wavelengths. Typically, oligonucleotides with a single gold nanocrystal elute earlier than unlabeled oligonucleotides of the same length. Free gold nanocrystals elute very early and gold nanocrystals with multiple oligonucleotides elute later than 1:1 conjugates. For a detailed chromatogram see (Shi et al., 2015). Use a small amount of sample to perform an initial analytical run, so that you can to make adjustments to the salt gradient if required.
-
26
Desalt the gold-oligonucleotide conjugates using centrifugal filter units (3 kDa, 4,000×g, 35–40 min, 4 °C) and ddH2O. Repeat this step three times. Determine the concentration of the purified conjugate by measuring the absorption at 360 nm (0.076 μM/cm). The desalted conjugates are stable and can be stored at −20°C for months. Typically, 12 nmol of sample can be recovered.
Steps 27–29: Preparation of final duplex conjugates for a sample quartet (about 5 hours)
-
27
Mix pairs of complementary single-stranded oligonucleotides in equimolar ratio and incubate DNA at room temperature or RNA at 40°C for 30 min. A samples quartet consists of a double-labeled sample with two modified strands, two single-labeled samples with a single modified strand, and an unlabeled sample (see Fig. 1 and Fig. 4). Use unmodified oligonucleotides for the unlabeled sample or as the complementary strand for the single-labeled samples.
Note: Use desalted oligonucleotides from step 26. In case your structure does not form in ddH2O and room temperature only, add the required buffer and salt to the solution and perform thermal annealing. -
28
Purify the annealed constructs by anion exchange HPLC using the same approach as in step 25. Use a small amount of sample to perform an initial analytical run so that you can make adjustments to the salt gradient if required. The duplex constructs typically elute later then single-stranded conjugates.
-
29
Collect the desired HPLC fractions and immediately desalt them using centrifugal filter units (10 kDa, 4,000×g, 15 min, 4 °C) and ddH2O. Repeat this step three times. Determine the concentration of the purified conjugate by measuring the absorption at 360 nm (0.076 μM/cm for single-labeled samples and 0.152 μM/cm for double-labeled samples). Typically, 2–3 nmol of each final duplexed construct should be obtained. The desalted conjugates are stable and can be stored at −20°C for months.
Figure 4. Example scattering profiles of XSI samples.

a) Buffer-only scattering profile used for buffer subtraction (blue), scattering profile of an unlabeled sample without (brown) and with buffer subtraction (red). b) Ten individual exposures for a double-labeled sample recorded in one run. All profiles match and thus radiation damage can be excluded. These scattering profiles are buffer subtracted. c) One full set of samples including one double-labeled sample (red), two orthogonal single-labeled samples (green, grey), bare gold nanocrystals (yellow) and unlabeled sample (blue). All scattering profiles are buffer subtracted.
Basic protocol 2, Collecting X-ray scattering interferometry data
To date, XSI data has been successfully recorded at beamline 4-2 of the Stanford Synchrotron Radiation Lightsource (SSRL) (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2014; Shi et al., 2013; Shi et al., 2016; Shi et al., 2017), beamline 12-ID of the Advanced Photon Source (APS) (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b), the SIBLYS beamline of the Advanced Light Source (ALS) (G. L. Hura et al., 2013), and the BM29 beamline of the European Synchrotron Radiation Facility (ESRF) (see Fig. 8). In general, measurements can be carried out at any synchrotron with beam lines set up for SAXS measurements that meet the following requirements:
Figure 8. Maximum entropy analysis of XSI data.

a) Radius distribution of gold nanocrystals used to generate basis profiles I(S,D). b) Calculated interference pattern for the example data set and c) final distance distribution averaged over 10 maximum entropy fitting runs.
S-range: 0.0015 – 0.11 Å−1 (optimal, for details see below) corresponding to a q-range 0.01 – 0.7 Å−1
X-ray energy: 9 keV – 15 keV (9 – 11 keV is optimal). This is the tested energy range used in experiments to date, for details see below.
Sample volumes: 16–40 μL (typical amounts used at state-of-the-art synchrotrons (Lipfert et al., 2006)). This amount allows for 10 independent exposures without requiring large quantities of sample.
It is important to pay attention to the definition of the magnitude of the momentum transfer vector S as two different conventions are used. In this protocol S is defined as S = 2sin(θ/λ) (q is defined as 4·π·sin(θ/λ)), where λ is the X-ray wavelength and θ is half the total scattering angle. S is alternatively reported in units of Å−1 and nm−1. We report S in Å−1 in this protocol. Set up a sample-to-detector distance that covers an S-range from 0.0015 – 0.11 Å−1 (for example, this corresponds to a sample-to-detector distance of 1.1 m for the Pilatus 300K detector at 11 keV on Stanford beamline 4-2). Typically, the sample and detector configuration must be arranged with beamline scientists well in advance of data collection, since it requires hardware alignment and calibration. If the beamline cannot cover the full “optimal” S-range (0.0015 – 0.11 Å−1), Smax should not be less than 0.095 Å−1 as the labeled samples and the bare gold particles contribute scattering intensity up to 0.085 Å−1. This is crucial for obtaining a valid interference profile by the analysis procedure described below. If the chosen beamline cannot reach this Smax for lower X-ray energies, one solution is to extend the S-range by selecting higher energies (for example, using 15 keV instead of 11 keV). However, X-ray energies close to gold absorption edges (L-III at 11.92 keV, L-II at 13.73 keV and L-I at 14.35 keV) should be avoided and energies below L-III are preferable to minimize X-ray fluorescence from these edges. Be aware that important details of the scattering profile can be lost in the low S-range for X-ray energies chosen too low or depleted by low signal-to-noise at X-ray energies set too high. For general protocols on SAXS sample preparation, data collection, and data analysis see Unit 7.14, Unit 17.18 and (Doniach & Lipfert, 2012; Dyer et al., 2014; Greg L. Hura et al., 2013; Jeffries et al., 2016; Lipfert & Doniach, 2007; Tuukkanen et al., 2017).
Materials
Scattering standard sample (e.g. cytochrome c)
Tris-HCl buffer, pH 7.4
Sodium ascorbate
Sodium chloride (NaCl)
Purified gold nanocrystal sample for titration series (10 nmol)
Purified double-labeled sample (1 nmol per buffer condition)
Purified 2x orthogonal single-labeled samples (1 nmol per buffer condition, each)
Purified unlabeled sample (1 nmol per buffer condition)
Sample buffer
MilliQ water
UV/Vis spectrometer
Vortex mixer
−20 or −80 °C freezer
Dry ice
Microfuge
0.5 ml centrifugal filters (Amicon Ultra)
The protocol below is a suggested workflow for data acquisition at a synchrotron facility. It may vary based on the instrument setup at the facility.
Sample preparation and setup initialization
-
1
If you are not familiar with the beamline and the settings, record data for a standard, e.g. cytochrome c (Lipfert et al., 2006), and compare it to benchmark profiles (Internet resource: “Small Angle Scattering Biological Data Bank”).
-
2
Prepare a 10x buffer mixture containing Tris-HCl and sodium ascorbate together with the desired amount of additional salt and other components (e.g. ligands) required for the experiment. An example 10x buffer solution for near-physiological conditions is 700 mM Tris-HCl, pH 7.4, 100 mM sodium ascorbate, 1.5 M NaCl, and 10 mM MgCl2. Use a Vortex mixer to ensure proper mixing of the components. It is important to use sodium ascorbate and Tris-HCl as radical scavengers in the buffer solution to capture free radicals and thus to reduce radiation damage to your sample during X-ray exposure; this allows longer total exposure times and therefore better signal-to-noise. Replace the sodium ascorbate stock solution every 3 h to ensure good scavenger capability. Cover the sodium ascorbate with aluminum foil or store it at a dark place.
-
3
The total exposure time depends on the flux at the synchrotron beam line used. A typical scheme, used at beam line 4-2, is to set the total exposure time to 30 s as a series of 10 independent repeats of 3 s each for data collection. Screen each trace for radiation damage, which can be detected by a gradual change in scattering intensity especially in the low S-range region in subsequent X-ray exposures. Exclude scattering profiles with oxidative damage determined from subsequent analysis. Furthermore, do not reuse samples that have been exposed to the X-ray beam. If the photon flux is much less than 1012 photons/s, extend the total exposure time.
-
4
If you have not done so previously (see step 29 in Preparation of gold nanocrystal-nucleic acid conjugates), determine the concentration of your sample using a UV/Vis spectrometer. The extinction coefficient of the gold nanocrystals is 0.076 μM/cm at 360 nm.
-
5
Record a titration series of gold nanocrystal for every beam time as a scattering standard and to obtain the nanocrystal size distribution required for the further analysis. A typical concentration series is 200, 100, 50, and 25 μM of gold particles (include higher concentrations if they are necessary for your experiment). The shape of the scattering profile should not change with concentration, and the scattering profiles should be superimposable after normalization. Interparticle scattering should be avoided. It can be detected by a concentration-dependent change in the scattering profile at low S.
-
6
Store gold-labeled samples on dry ice or in a freezer at −20°C until the beam line is set up for the experiments. During data acquisition, store the sample stock solutions on ice or in a fridge.
-
7
Thaw the required amount of sample to room temperature and vortex before the measurements.
-
8
Combine 10x buffer, water and the concentrated sample to achieve a final sample concentration of 30 μM (for example 0.9 nmol of sample in 30 μL). If it is not possible to prepare the buffer as a 10x stock solution (for example due to solubility limitations), or if the concentration of charge of the macromolecule is comparable to the concentration of counterions in solution at very low ionic strength, prepare the sample by buffer exchange using centrifugal filter units with a suitable molecular weight cutoff (e.g., 10 kDa Amicon, three repeats, 35 min each).
-
9
Spin the final sample mixture for 2 min at 10,000 × g at 4 °C to sediment out any large contaminant particles. Large particles strongly perturb the scattering signal, as the forward scattering intensity of an object grows quadratically with its molecular mass.
Data recording
-
10
Prepare five samples for data collection. The full set of samples consists of an unlabeled molecule, two complementary single-labeled samples, one double-labeled macromolecule and a buffer-only sample (Fig. 4). Again, the concentration of the macromolecule should be at least 30 μM in 1x buffer to provide good signal-to-noise. Measure the scattering profiles of the five samples on the same set up, in direct succession, to keep the conditions as similar as possible. Always record the buffer-only scattering profile (at least) twice, once at the beginning of the acquisition sequence and once at the end (for example: buffer, unlabeled molecule, two complementary single-labeled samples, double-labeled macromolecule and buffer again). An automated sample changer installed at the beamline can aid data collection for such a series.
-
11
Repeat the 5-sample data acquisition sequence with each macromolecule construct and/or condition in your experiment (for example at varying salt concentrations, with and without ligand binding partners, etc.).
Basic protocol 3, Analyzing X-ray scattering interferometry data
The data obtained at the beamline can be processed either by applying individual scripts step-by-step or using the custom written GUI in Matlab (“au_saxs_gui.m”, see Materials). The underlying principles are described in detail by Mathew-Fenn et al. (2008b) and a brief summary is as follows. After standard SAXS data processing, as outlined below, the radius distribution of the spherical gold nanocrystals is determined first, from scattering data of the unconjugated gold labels. To accomplish this, the recorded scattering profile of the gold nanocrystals is decomposed into a volume-weighted sum of scattering profiles of spheres with radii ranging from 1 to 100 Å. Using the nanocrystal synthesis protocol described above, (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2015) the nanocrystal size distribution should have a radius centered at 6–7 Å. The obtained radius distribution is then used to calculate the precise basis scattering functions I(S,D), which are the scattering interference patterns for two nanocrystal separated by a fixed center-to-center distance D, where D is varied from 1 to 200 Å. These basis functions will be used to decompose IAu-Au(S), which is the experimentally-determined scattering interference pattern for the two gold nanocrystals attached to the macromolecule. Importantly, the measured scattering profile from the double-labeled macromolecule includes contributions from the macromolecule itself and from the cross-scattering terms between the gold labels and the macromolecule, in addition to IAu-Au. To extract IAu-Au, the scattering profiles for the quartet of samples must be summed, and the summation requires that the profile intensities are accurately scaled relative to each other. Finding the correct scaling factors, denoted cU, cA+B and cBuf (see Eqn. 2 and 3), is the most difficult part of the data processing. To do this, the measured scattering profiles of the double-, single-, and unlabeled constructs and of the buffer (IAB(S), IA(S), IB(S), IU(S) and IBuf(S)) are first transformed into interatomic Patterson distributions PAB(D), PA(D), PB(D), PU(D) and PBuf(D) using point-scatterer basis functions (see Eqn. 1). Using the measured scattering profiles and the Patterson distributions, the scaling factors are optimized to satisfy two constraints: i. that the integral of the sinusoidal function S•IAu-Au sums to zero (Eqn. 4), and ii. that none of the computed interatomic distances between gold labels are negative, which should not be possible (Eqn. 5). Deviations from these constraints are summed together in a target function T (see Critical Parameters and Troubleshooting), and the scaling factors that minimize T are determined. Using the optimized scaling factors, the measured profiles are summed to obtain IAu-Au.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Finally, IAu-Au is decomposed into a sum of the I(S,D) basis functions using a maximum entropy algorithm, resulting in a center-to-center distance distribution between the two gold nanocrystals of the double-labeled sample (Eqn. 1). Alternatively, the IAu-Au decomposition can be performed using non-negative least squares algorithms that are available in most scientific programming languages.
Some beamlines, such as the beamline 4-2 at SSRL and beamline BM29 at ESRF, provide beamline software packages that perform radial averaging and buffer subtraction of scattering profiles, which allows for immediate detection of radiation damage or other technical problems.
Materials
-
Data set including scattering profiles from:
Gold nanocrystal sample
Double-labeled sample
Orthogonal single-labeled samples (two individual scattering profiles)
Unlabeled sample
Sample buffer
Computer (Minimum requirements: Any Intel or AMD x86-64 processor, 2.5 GB Disk space, 2 GB RAM)
MATLAB license (GUI support from version 2015b guaranteed)
Au-SAXS GUI (https://gitlab.physik.uni-muenchen.de/Jan.Lipfert/AuSAXSGUI.git)
Example files (https://gitlab.physik.uni-muenchen.de/Jan.Lipfert/AuSAXSGUI.git)
A step-by-step guide on how to obtain the scattering interference pattern IAu-Au including an example set of data (see exemplary files) is given below.
Data preparation
-
1
If it has not already been done automatically by the beamline software, reduce the 2D scattering matrix into a one-dimensional scattering profile by radial averaging. The output should be a matrix with three columns for the scattering momentum transfer vector S, the corresponding scattering intensity, and the variance/standard deviation in scattering intensity at different radial positions.
Note: If the incident X-ray beam is polarized, simple radial averaging cannot be performed. See (Pauw, 2014; Svergun & Koch, 2003) for further instruction on how to process the raw data into one-dimensional scattering profiles. -
2
Before starting the custom written au_saxs_gui.m GUI and loading the data, rename the data files ‘*_i.dat’ where * is any name for the sample and i is the ith exposure, i.e., ranging from 01 to 10 for 10 exposures per molecule (‘AB_01.dat’, ‘AB_02.dat’, ‘AB_03.dat’, …, for double-labeled samples). Structure the data files so that the scattering momentum vector occupies the first column, the corresponding recorded scattering intensity occupies the second column and the variance/standard deviation occupies the third column, and the column entries are separated by a single blank space (see example files for comparison).
-
3
Initialize the GUI by executing the ‘au_saxs_gui.m’ script. For proper execution the files ‘au_saxs_gui.m’, ‘au_saxs_gui.fig’ and the folder ‘subroutines’ have to be stored in the same directory to allow the main script to find the required subroutines.
Data initialization
-
4
Enter the full path into the field ‘Data path’ (Fig. 5.1).
-
5
Optional: Manipulate the scattering momentum by setting the lower (qmin) (Fig. 5.2) and the upper (qmax) (Fig. 5.3) limit for the scattering angle. The default input is the 35th data point of the initial data up to data point 500; however, this strongly depends on the settings of the beamline and the type of sample.
-
6
Optional: Untick the ‘Data is in q’ box (Fig. 5.4) to switch the scattering momentum vector to S (2sin(θ/λ) in Å−1).
Note: The scattering momentum vector is set to q per default (4π sin(θ/λ), in Å−1) since q is the common output format at synchrotrons. -
7
Optional: Modify the number of samples (Fig. 5.5) if required. (Default is 5 for one full set of samples.)
-
8
Optional: Change the number of exposures (Fig. 5.6) the number of files read per sample. (Default is 10 as described in the protocol.)
-
9
Press ‘Ok’ to initialize the script. After doing so new buttons and a table will appear on the left side of the window (see Fig. 6).
-
10
Enter all sample names in the first column of the table (see Fig. 6).
-
11
Enter the corresponding buffer in the second column.
-
12
Enter the determined sample concentration (in μM) in the third column.
-
13
Optional: Enter save as filenames for all samples in the fourth column (see Fig. 6.1).
-
14
Load the data files by pressing the ‘Load data files’ button (Fig. 6.2). After successfully doing so, a window indicating ‘All data was successfully imported!’ will be displayed. In case a file could not be found or a wrong sample name was entered a warning will be generated noting to the incorrect position. If the concentration was not entered properly a similar error message will be produced.
Figure 5. GUI to analyze XSI data.

Default values can be modified to adjust the data analysis. The GUI contains panels to specify the following: (1) storage path of the data; (2) adjust qmin 2); (3) adjust qmax; (4) specify the momentum transfer convention according to the scattering data; (5) specify the number of samples; and (6) specify the number of exposures per sample.
Figure 6. GUI to analyze XSI data.

Sample and corresponding buffer filenames, sample concentration and save as filename (optional) are entered for a full set of data (1). Initial files are loaded (2) and can be plotted (4–6) (optional). Unified and truncated scattering profiles can be saved (3).
Data testing
-
15
Optional: Inspect the scattering profiles for all samples either by plotting all exposures per sample as individual traces into one figure per sample (Fig. 6.4) or by plotting the averaged profile over all exposures (Fig. 6.5).
-
16
Optional: Overlay these plots by pressing the ‘Plot sample data’ first and the ‘Plot mean data’ second.
-
17
Optional: Plot the averaged buffer for each sample using the ‘Plot buffer’ button (Fig. 6.6).
-
18
Optional: In case too many MATLAB figures are open, close all except for the main GUI by pressing the ‘Close Figures’ button (Fig. 6.7).
-
19
Optional: Save the scattering data for all loaded samples as ‘YOURFILENAMEHERE_scattering_data.mat’ files using ‘Save data’ (Fig. 6.3).
Maximum entropy fitting
-
20
Set the options by specifying the sample positions according to the row number in the table (see Fig. 7.1), i.e., Gold sample position = 1, A-label sample position = 2, B-label sample position = 3, Double-label sample position = 4 and Unlabeled sample position = 5.
-
21
Optional: Change the number of runs for the maximum entropy fit (default is 10, Fig. 7.3). The output is one high-resolution distance distribution per run. Average the distributions over all runs to obtain the final distribution. Lower the number to shorten the required computational time for test purposes.
-
22
Optional: Change the minimization function option to extract the gold-gold interference pattern IAu-Au (default is 5 ranging from 1 to 7, see Critical Parameters and Troubleshooting for more details, Fig. 7.2).
-
23
Start the maximum entropy fit by pressing ‘Max Entropy fit’ (Fig. 7.4. A progress bar in the lower left corner of the main GUI will display the progress and vanish as soon as the calculations are finished. Three new figures showing the gold nanocrystal radius distribution, the gold-gold scattering interference signal IAu-Au, and the final distance distribution determined via the maximum entropy fit will appear (Fig. 8).
-
24
Enter a save as filename (e.g. ‘YOURSAMPLENAME’) for the distance distribution (Save Max Entropy Data, right side, Fig. 7.5). This file has one column per maximum entropy run (10 as default) and a 1 Å spaced distance distribution ranging from 1 Å up to 200 Å.
-
25
After setting the name, press ‘Save data’ (Save Max Entropy Data, right side) a file in the format of ‘YOURSAMPLENAME_Distance_Distribution.mat’ will be saved in your current folder.
Optional: Save the gold nanocrystal radius distribution and/or the Au-Au interference pattern IAu-Au checking the individual boxes.
Figure 7. GUI to analyze XSI data.

Panels to set the sample type (1) according to the position in the sample loading table (2). The choice of minimization function (3) and the number of individual maximum entropy fitting runs (4) can be specified prior to starting the routine (5). Final distance distributions (Au radius distribution and interference pattern I(S), optional) can be saved (6).
REAGENTS AND SOLUTIONS
-
FPLC cleaning solution:
100 mM DTT, 20 mM Tris-HCl, pH 8.0 (prepare fresh)
-
Size exclusion running buffer:
150 mM ammonium acetate, pH 5.6 (prepare fresh)
-
Ammonium acetate stock buffer:
2 M ammonium acetate, pH 5.6 (store up to one month at 4 °C)
-
Low salt borate buffer:
10 mM NaCl, 20 mM Na-borate, pH 7.8 (store up to one month at room temperature)
-
High salt borate buffer:
1.5 M NaCl, 20 mM Na-borate, pH 7.8 (store up to one month at room temperature)
-
Low salt ammonium acetate buffer:
10 mM NaCl, 20 mM ammonium acetate, pH 5.6 (store up to one month at room temperature)
-
High salt ammonium acetate buffer:
1.5 M NaCl, 20 mM ammonium acetate, pH 5.6 (store up to one month at room temperature)
COMMENTARY
Background Information
Basic principle
XSI measures the interference between X-rays scattered by two gold nanocrystals attached to a macromolecule or macromolecular complex. In addition to the desired nanocrystal-nanocrystal interference pattern, the recorded scattering profile includes signals from scattering interference between pairs of atoms in the macromolecule, between pairs of atoms in a single gold nanocrystal, and between pairs of atoms in the macromolecule and the nanocrystal. It is critical to isolate the nanocrystal-nanocrystal interference pattern from the other signals, since it alone contains direct information about the distribution of center-to-center distances between the nanocrystal probes. This distance distribution is obtained by Fourier transformation of the nanocrystal-nanocrystal interference pattern. A limitation of XSI is the time-consuming sample preparation, which only allows low throughput. The strength of XSI is that it provides accurate ensemble distance distributions even for dynamic and rapidly interconverting macromolecules, with precise external distance calibration from the wavelength of the incident X-ray radiation. These absolute distances can be directly compared to high-resolution atomic models from NMR spectroscopy, X-ray crystallography, electron microscopy or molecular simulations. XSI is very complementary to spectroscopic rulers such as FRET, and combined application of the two types of ruler presents interesting possibilities.
Application of XSI to nucleic acids and nucleic acid-protein complexes
Over the last decade, XSI has been successfully applied to dsDNA (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2013), nucleic acid two-way junctions (Shi et al., 2014; Shi et al., 2017) and nucleic acid-protein complexes (G. L. Hura et al., 2013; Shi et al., 2016). It has been used to characterize the stretching (Mathew-Fenn et al., 2008a), twisting, and bending elasticity (Shi et al., 2013) of short DNA helices. Recently, DNAs and RNAs containing bulge sequences have been studied, revealing a complex multistate behavior that responds to the solution conditions (Shi et al., 2014; Shi et al., 2017). In addition, an RNA-protein complex has been investigated (Shi et al., 2016). The use of metal clusters as structural probes makes XSI scalable. Clusters of a single or few metal atoms are suitable for smaller systems (Miake-Lye et al., 1983; Vainshtein et al., 1980), while clusters containing thousands of atoms can be applied to large complexes (G. L. Hura et al., 2013). While small labels are desirable for accurate position determination and to minimize the perturbation of the molecular structures of interest, for large macromolecules noise from the macromolecule signal can prevent accurate Au-Au distance determination and larger gold nanocrystals are needed.
A variant of the XSI interference technique has recently been demonstrated that uses anomalous SAXS to directly extract the IAu-Au interference pattern from double-labeled samples without the need to record any single- or unlabeled samples (Zettl et al., 2016). This approach will be in particular relevant for samples where it is difficult to prepare matching single labeled constructs.
Future directions
The future challenges and possible applications of XSI are numerous. Using the labeling and measuring protocols described here, the conformational ensembles of diverse nucleic-acid motifs and nucleic acid-protein complexes can be determined, including their dependence on environmental conditions. The application of XSI to nanocrystal-labeled proteins will provide a new window into protein conformational ensembles, especially for folding intermediates and intrinsically disordered proteins (IDPs). Finally, the application of next-generation free electron lasers with very high brilliance could allow measurement of correlated and time resolved distances between multiple sites in a macromolecule, with a time resolution of tens of femtoseconds (Arnlund et al., 2014; Bada et al., 2000; Ball, 2017; Doniach, 2000; Mendez et al., 2014; Mendez et al., 2016; Schenk et al., 2016; SLAC & LCLS, 2010; Smolsky et al., 2007).
Critical Parameters and Troubleshooting
A key aspect for successful determination of intramolecular distance distributions using XSI is sample preparation and sample quality. The samples have to be highly homogeneous (e.g., no free gold nanocrystals in solution) and free of degradation. Furthermore, buffer matching should be as precise as possible. Given that XSI requires very pure samples it is crucial to monitor and adjust the sample quality in advance. To assess sample quality, non-denaturing polyacrylamide gel electrophoresis (PAGE) (UNIT 10.1) or ion exchange chromatography (UNIT 10.5) can be used. In addition, we advise the user to perform additional experiments (e.g., UV melting curve analysis and circular dichroism spectroscopy, see (Mathew-Fenn et al., 2008b)) to test whether the attached gold nanocrystals perturb the macromolecular structure and, ideally, to establish the absence of such perturbations. In addition, G/C base pairs at the blunt ends of nucleic acids are recommended to avoid fraying of nucleic acid helices.
Another requirement of the XSI technique is high signal-to-noise SAXS measurements of the set of double-, single-, and unlabeled samples. We recommend testing the beam line set up with readily available and characterized samples, such as cytochrome c, bovine serum albumin (BSA), or unlabeled double-stranded DNA (Internet resource: “Small Angle Scattering Biological Data Bank”), prior to an XSI experiment. The measurements of the scattering standard samples should give the expected scattering profiles and radii of gyration. Such control measurements should be repeated every time the SAXS setup is reconfigured and can help to detect misalignment of the X-ray beam, problems with parasitic scattering, and issues in detector calibration and conversion of the detector image to the 1D scattering profile.
Sometimes, it is not possible to obtain a high-quality distance distribution even when the sample preparation and recording are performed correctly. Potential issues can be the scattering intensity ratio between the molecule and the gold nanocrystals resulting in poor signal-to-noise for larger nucleic acid structures or highly flexible molecules. Additionally, larger metal clusters should be considered in cases of insufficient signal-to-noise.
Some additional factors that can affect the data analysis and the final distance distribution and how to address them are listed below:
-
Parasitic X-ray scattering in the low S-range as well as high noise levels in the high S-range can occur and influence the normalization and calculation of the interference pattern.
Solution: We recommend measuring scattering standards to test and optimize the X-ray set up before running XSI samples. In the post-processing of recorded data sets, one should vary Smin and/or Smax multiple times and test the impact on the resulting distance distribution.
-
The scattering profile can be distorted by various effects such as radiation damage or bubbles passing through the X-ray beam.
Solution: Carefully compare scattering profiles from subsequent exposures of the same sample and exclude all traces differing from the majority.
-
Choice of minimization function T to extract to gold-gold scattering interference Patterson IAu-Au where S is the scattering momentum vector, IU the scattering intensity vector of the unlabeled sample and PAu-Au the label-label interatomic radial Patterson. The gold-gold scattering interference Patterson IAu-Au looks like a sinc (i.e. a decaying oscillation) function (see Fig. 8B). If that is not the case redo the analysis with a different choice for T. As guidance and for comparison, multiple interference pattern of static and dynamic systems can be found in the published literature (Mathew-Fenn et al., 2008a; Mathew-Fenn et al., 2008b; Shi et al., 2014; Shi et al., 2015; Shi et al., 2013; Shi et al., 2016; Shi et al., 2017) for comparison.
Choice 1: Original function described by Mathew-Fenn, Das and coworkers (Mathew-Fenn et al., 2008b)Minimization function similar to Kratky analysis dividing out the decay of the scattering intensity by weighting the interference pattern IAu-Au by S2.
Choice 2:Au-Au interference pattern is only weighted by S, thus IAu-Au minimization is dominated by values in the low S-region.
Choice 3:The low S-region of IAu-Au is weighted more compared to choice 1, but less than choice 2 and the high S-region is weighted less compared to choice 1 and more than choice 2.
Choice 4:Very similar to choice 3 in the high S-range, however the high S-range is weighted slightly less.
Choice 5:Very similar to choice 3 in the high S-range, however the high S-range does contribute very little.
Choice 6:Only PAu-Au(D) is minimized such that it does not include negative values, however the oscillations in IAu-Au are not minimized.
Choice 7:Only the high S-region of IAu-Au is minimized similar to a base line correction. PAu-Au(D) is allowed to include negative values.
Anticipated Results
The protocols presented here guide experimenters to synthesize gold nanocrystals, prepare and purify gold-labeled nucleic acid constructs, measure a complete set of SAXS profiles for the labeled samples, and to extract intra-molecular distance distributions from the data.
Following the steps presented in Basic Protocol 1, the following yields are typically achieved: the gold nanocrystal synthesis yields 3–10 μmol after final purification and desalting; the gold nanocrystal attachment protocol results in a yield of 2–3 nmol of each labeled and purified duplexed construct for XSI measurements.
Basic Protocol 2 and 3 describe a reliable procedure to obtain SAXS scattering profiles and a step-by-step guide on how to use the provided GUI to extract high-resolution distance distributions on an absolute scale (Fig. 8C). To study more complex macromolecules that can undergo conformational changes or to disentangle complex geometries and motions, multiple label pairs attached to different positions can be prepared and analyzed to increase the information content.
Time Considerations
The sample preparation should be performed about one month in advance. The final purification (step 28 and 29 in Basic Protocol 1) should be carried out shortly before data collection to ensure the highest sample quality. The overall data acquisition time varies depending on the synchrotron facility. Typically, data collection for several samples can take up to 1 day. The run time of the custom-written Matlab routine to compute distance distributions that is provided with this protocol is on the order of minutes on a standard personal computer. The total time required for data analysis, validation, and interpretation is variable and can take several days.
Significance Statement.
Most techniques in structural biology provide information on averaged structures or predominant states of biological macromolecules and their complexes. However, considering the importance of the entire conformational ensemble to understanding folding, recognition, and function, tools are needed to probe and quantify structural ensembles. Here we present X-ray scattering interferometry (XSI), an emerging approach that makes use of gold nanocrystal-nucleic acid conjugates to provide accurate ensemble information. The scattering signal can be transformed into a distance distribution between pairs of gold nanocrystal labels and, therefore, provides direct information about the structural ensemble. XSI data can be directly compared to atomic coordinates from additional experimental approaches or molecular dynamics simulations and be used to reveal hidden substates.
Acknowledgments
We thank T. Matsui and T. Weiss at beamline 4-2 of the Stanford Synchrotron Radiation Lab (SSRL) and Gabriele Giachin at beamline BM29 of the European Synchrotron Radiation Facility (ESRF) for technical support in synchrotron small-angle X-ray scattering experiments, and Steffen Sedlak for helpful discussions and comments on the manuscript. This work was supported by Sonderforschungsbereich (SFB) 1032 and the N.I.H. to D.H. (P01 GM066275 to D.H. and DP-OD000429 to P.H.).
Footnotes
NOTE: Use ultrapure water in all solutions and protocol steps.
INTERNET RESOURCES
https://eu.idtdna.com/calc/analyzer
Online tool to calculate melting temperature, extinction coefficient for a given DNA sequence.
Small Angle Scattering Biological Data Bank providing experimental measured SAXS profiles.
https://gitlab.physik.uni-muenchen.de/Jan.Lipfert/AuSAXSGUI.git
Git repository providing the Matlab AuSAXS Gui and example data files.
References
- Ackerson CJ, Sykes MT, Kornberg RD. Defined DNA nanoparticle conjugates. Proc Natl Acad Sci U S A. 2005;102:13383–13385. doi: 10.1073/pnas.0506290102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alivisatos AP, Johnsson KP, Peng X, Wilson TE, Loweth CJ, Bruchez MP, Schultz PG. Organization of ‘nanocrystal molecules’ using DNA. Nature. 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- Arnlund D, Johansson LC, Wickstrand C, Barty A, Williams GJ, Malmerberg E, Davidsson J, Milathianaki D, DePonte DP, Shoeman RL, Wang D, James D, Katona G, Westenhoff S, White TA, Aquila A, Bari S, Berntsen P, Bogan M, van Driel TB, Doak RB, Kjaer KS, Frank M, Fromme R, Grotjohann I, Henning R, Hunter MS, Kirian RA, Kosheleva I, Kupitz C, Liang M, Martin AV, Nielsen MM, Messerschmidt M, Seibert MM, Sjohamn J, Stellato F, Weierstall U, Zatsepin NA, Spence JC, Fromme P, Schlichting I, Boutet S, Groenhof G, Chapman HN, Neutze R. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat Methods. 2014;11(9):923–926. doi: 10.1038/nmeth.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin-Tam ME, Hamad-Schifferli K. Gold nanoparticle-cytochrome c complexes: The effect of nanoparticle ligand charge on protein structure. Langmuir. 2005;21:12080–12084. doi: 10.1021/la052102e. [DOI] [PubMed] [Google Scholar]
- Aubin-Tam ME, Hwang W, Hamad-Schifferli K. Site-directed nanoparticle labeling of cytochrome c. Proc Natl Acad Sci U S A. 2009;106:4095–4100. doi: 10.1073/pnas.0807299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azubel M, Kornberg RD. Synthesis of Water-Soluble, Thiolate-Protected Gold Nanoparticles Uniform in Size. Nano Lett. 2016;16(5):3348–3351. doi: 10.1021/acs.nanolett.6b00981. [DOI] [PubMed] [Google Scholar]
- Bada M, Walther D, Arcangioli B, Doniach S, Delarue M. Solution structural studies and low-resolution model of the Schizosaccharomyces pombe sap1 protein. Journal of Molecular Biology. 2000;300(3):563–574. doi: 10.1006/jmbi.2000.3854. S0022-2836(00)93854-3 [pii] [DOI] [PubMed] [Google Scholar]
- Ball P. Europe’s X-ray laser fires up. Nature. 2017;548:507–508. doi: 10.1038/548507a. [DOI] [PubMed] [Google Scholar]
- Doniach S. Fourth-generation X-ray sources: some possible applications to biology. J Synchrotron Radiat. 2000;7(Pt 3):116–120. doi: 10.1107/S0909049500004143. S0909049500004143 [pii] [DOI] [PubMed] [Google Scholar]
- Doniach S, Lipfert J. Small and wide angle x-ray scattering from biological macromolecules and their complexes in solution. Comprehensive Biophysics. 2012;1:376–397. doi: 10.1016/B978-0-12-374920-8.00122-3. [DOI] [Google Scholar]
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL. High-Throughput SAXS for the Characterization of Biomolecules in Solution: A Practical Approach. Methods in molecular biology (Clifton, NJ) 2014;1091:245–258. doi: 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feynman RP, Leighton RB, Sands M. The Feynman Lectures on Physics. The Feynman Lectures on Physics 1989 [Google Scholar]
- Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466(7304):329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H. New looks at protein motions. Nature. 1989:338. [Google Scholar]
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nature Methods. 2013;10:453. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Tsai CL, Claridge SA, Mendillo ML, Smith JM, Williams GJ, Mastroianni AJ, Alivisatos AP, Putnam CD, Kolodner RD, Tainer JA. DNA conformations in mismatch repair probed in solution by X-ray scattering from gold nanocrystals. Proc Natl Acad Sci U S A. 2013;110(43):17308–17313. doi: 10.1073/pnas.1308595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries CM, Graewert MA, Blanchet CE, Langley DB, Whitten AE, Svergun DI. Preparing Monodisperse Macromolecular Samples for Successful Biological Small-Angle X-ray and Neutron Scattering Experiments. Nat Protoc. 2016;11(11):2122–2153. doi: 10.1038/nprot.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Biophys Biomol Struct. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- Lipfert J, Millett IS, Seifert S, Doniach S. Sample holder for small-angle x-ray scattering static and flow cell measurements. Review of Scientific Instruments. 2006;77(4):26–28. [Google Scholar]
- Mathew-Fenn RS, Das R, Harbury PA. Remeasuring the double helix. Science. 2008a;322:446–449. doi: 10.1126/science.1158881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew-Fenn RS, Das R, Silverman JA, Walker PA, Harbury PA. A molecular ruler for measuring quantitative distance distributions. PLoS One. 2008b;3(10):e3229. doi: 10.1371/journal.pone.0003229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D, Lane TJ, Sung J, Sellberg J, Levard C, Watkins H, Cohen AE, Soltis M, Sutton S, Spudich J, Pande V, Ratner D, Doniach S. Observation of correlated X-ray scattering at atomic resolution. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369(1647):20130315. doi: 10.1098/rstb.2013.0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D, Watkins H, Qiao S, Raines KS, Lane TJ, Schenk G, Nelson G, Subramanian G, Tono K, Joti Y, Yabashi M, Ratner D, Doniach S. Angular correlations of photons from solution diffraction at a free-electron laser encode molecular structure. IUCrJ. 2016;3(Pt 6):420–429. doi: 10.1107/S2052252516013956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miake-Lye RC, Doniach S, Hodgson KO. Anomalous x-ray scattering from terbium-labeled parvalbumin in solution. Biophys J. 1983;41(3):287–292. doi: 10.1016/S0006-3495(83)84440-3. S0006-3495(83)84440-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller JP, Löf A, Mielke S, Obser T, Brützel LK, Vanderlinden W, Lipfert J, Schneppenheim R, Benoit M. pH-Dependent Interactions in Dimers Govern the Mechanics and Structure of von Willebrand Factor. Biophys J. 2016;111(2):312–322. doi: 10.1016/j.bpj.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauw BR. Everything SAXS: small-angle scattering pattern collection and correction. Journal of Physics: Condensed Matter. 2014;26:239501. doi: 10.1088/0953-8984/26/23/239501. [DOI] [PubMed] [Google Scholar]
- Schaaff TG, Knight G, Shafigullin MN, Borkman RF, Whetten RL. Isolation and Selected Properties of a 10.4 kDa Gold:Glutathione Cluster Compound. J Phys Chem B. 1998;102(52):10643–10646. doi: 10.1021/jp9830528. [DOI] [Google Scholar]
- Schenk G, Krajina B, Spakowitz A, Doniach S. Potential for measurement of the distribution of DNA folds in complex environments using Correlated X-ray Scattering. Modern physics letters B, Condensed matter physics, statistical physics, applied physics. 2016;30(8):1650117. doi: 10.1142/S0217984916501177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Beauchamp KA, Harbury PB, Herschlag D. From a structural average to the conformational ensemble of a DNA bulge. Proc Natl Acad Sci U S A. 2014;111(15):E1473–E1480. doi: 10.1073/pnas.1317032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Bonilla S, Herschlag D, Harbury P. Quantifying Nucleic Acid Ensembles with X-ray Scattering Interferometry. Methods Enzymol. 2015;558:75–97. doi: 10.1016/bs.mie.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Shi X, Herschlag D, Harbury PA. Structural ensemble and microscopic elasticity of freely diffusing DNA by direct measurement of fluctuations. Proc Natl Acad Sci U S A. 2013;110(16):E1444–E1451. doi: 10.1073/pnas.1218830110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Huang L, Lilley DM, Harbury PB, Herschlag D. The solution structural ensembles of RNA kink-turn motifs and their protein complexes. Nat Chem Biol. 2016 Jan;12:146–152. doi: 10.1038/nchembio.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Walker P, Harbury PA, Herschlag D. Determination of the conformational ensemble of the TAR RNA by X-ray scattering interferometry. Nucleic Acids Res. 2017;45(8):e64. doi: 10.1093/nar/gkw1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLAC & LCLS. Building X-ray lasers. Nat Photonics. 2010;4:0–1. doi: 10.1038/nphoton.2010.276. [DOI] [Google Scholar]
- Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. Biological small-angle x-ray scattering facility at the Stanford synchrotron radiation laboratory. J Appl Crystallogr. 2007;40:S453–S458. doi: 10.1107/s0021889807009624. [DOI] [Google Scholar]
- Svergun D, Koch M. Small-angle scattering studies of biological macromolecules in solution. Reports on Progress in Physics. 2003;66:1735–1782. doi: 10.1088/0034-4885/66/10/R05. [DOI] [Google Scholar]
- Tuukkanen AT, Spilotros A, Svergun DI. Progress in small-angle scattering from biological solutions at high-brilliance synchrotrons. IUCrJ. 2017;4(Pt 5):518–528. doi: 10.1107/S2052252517008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein BK, Feigin LA, Lvov YM, Gvozdev RI, Marakushev SA, Likhtenshtein GI. Determination of the distance between heavy-atom markers in haemoglobin and histidine decarboxylase in solution by small-angle X-ray scattering. Febs Letters. 1980;116(1):107–110. doi: 10.1016/0014-5793(80)80539-4. [DOI] [PubMed] [Google Scholar]
- Zettl T, Mathew RS, Seifert S, Doniach S, Harbury PA, Lipfert J. Absolute Intra-Molecular Distance Measurements with Ångström-Resolution using Anomalous Small-Angle X-ray Scattering. Nano Lett. 2016;16(9):5353–5357. doi: 10.1021/acs.nanolett.6b01160. [DOI] [PubMed] [Google Scholar]