

SUMMARY
Intestinal immune homeostasis is preserved by commensal bacteria interacting with the host to generate a balanced array of cytokines that are essential for wound repair and for combatting infection. Inflammatory Bowel Disease (IBD), which can lead to colitis-associated cancer (CAC), is thought to involve chronic microbial irritation following a breach of the mucosal intestinal epithelium. However, the innate immune pathways responsible for regulating these inflammatory processes remain to be fully clarified. Here we show that commensal bacteria influence STING-signaling predominantly in mononuclear phagocytes to produce both pro-inflammatory cytokines as well as anti-inflammatory IL-10. Enterocolitis, manifested through loss of IL-10 was completely abrogated in the absence of STING. Intestinal inflammation was less severe in the absence of cGAS, possibly suggesting a role for cyclic dinucleotides (CDNs) indirectly regulating STING signaling. Our data sheds insight into the causes of inflammation and provides a potential therapeutic target for prevention of IBD.
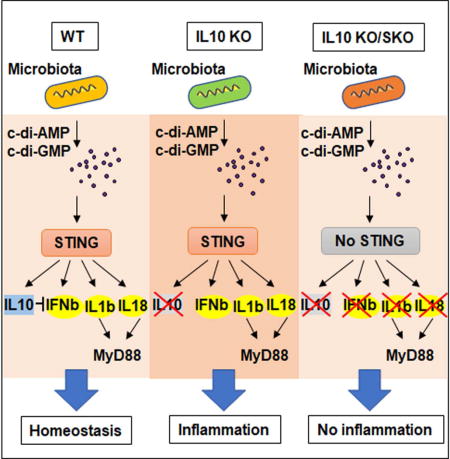
Graphical abstract
INTRODUCTION
The pro-inflammatory response, while essential for initiating wound repair and protection against pathogens, if uncontrolled, is known to drive a variety of maladies including rheumatoid arthritis, inflammatory bowel disease (IBD) and even cancer (de Souza and Fiocchi, 2016; Grivennikov et al., 2010; Nagata and Kawane, 2011; Saleh and Trinchieri, 2011; Trinchieri, 2012). Incidences of IBD, such as Crohn’s disease and ulcerative colitis are increasing although the mechanistic causes remain to be clarified (de Souza and Fiocchi, 2016; Loftus, 2004; Sartor, 2006). Gut inflammatory responses are capably circumvented even though the gastrointestinal tract contains trillions of microbes (Belkaid and Hand, 2014). Indeed, most intestinal bacteria are considered commensal to the host, generating nutritional metabolites and even contributing towards facilitating the homeostasis of the immune system (Honda and Littman, 2016). However, damage to the intestinal mucous membrane, comprising the lamina propria and epithelial cells enable microbes including non-commensal dysbiotic bacteria access to which respond by overproducing cytokines to generate an inflammatory state (Varol et al., 2010).
Antibiotics are known to subdue the immune system signifying that mucosal commensal bacteria possibly contribute towards priming the immune system in the gut through a balanced production of a variety of innate immune regulated proteins, including a key anti-inflammatory cytokine IL-10 (Arthur et al., 2014; Arthur et al., 2012; Honda and Littman, 2016; Uronis et al., 2009). Mice deficient in IL-10 can develop severe enterocolitis, resembling Crohn’s disease unless they are treated with antibiotics (Hoentjen et al., 2003; Kuhn et al., 1993; Madsen et al., 2000). The innate signaling pathway(s) mainly responsible for pro-inflammatory cytokine production, normally suppressed by IL-10, remains to be clarified. However, loss of the TLR adaptor protein, myeloid-differentiation primary response protein (MyD88) in MNP’s, can eliminate inflammation in IL-10-deficient mice suggesting a role for the TLR pathway or for pro-inflammatory cytokines that require MyD88 for signaling such as IL-1β or IL-18 (Hoshi et al., 2012; Rakoff-Nahoum et al., 2006; Salcedo et al., 2010). IL-10 production can also be initiated through TLR signaling or by type I interferon (IFN) which utilize IFN-regulatory transcription factors (IRF) and NF-ĸB to exert their affects (Ouyang et al., 2011). Following binding to the IL-10 receptor (IL-R1/2), IL-10 principally signals through STAT3 to prevent the pro-inflammatory effects of cytokines such as IL-12, IL-23 and IFN-λ (Chang et al., 2007; Hutchins et al., 2013; Manzanillo et al., 2015; Saraiva and O'Garra, 2010).
It has recently been shown that another key innate immune pathway, controlled by an endoplasmic reticulum (ER) associated protein referred to as STING (stimulator of interferon genes) may also be involved in controlling inflammation (Ishikawa and Barber, 2008; Ishikawa et al., 2009). STING is activated by cyclic dinucleotides (CDNs) such as cyclic di-AMP or GMP (c-di-AMP, c-di-GMP) directly exuded by certain bacteria, or by cGAMP (cyclic GMP–AMP) which is generated by the cellular synthase cGAS, following association with microbial or self-dsDNA species (Ablasser et al., 2013; Barber, 2013; Burdette et al., 2011). Here, we have evaluated the role of STING signaling in influencing intestinal inflammation and demonstrate that this innate immune pathway interacts with host commensal bacteria to play a key role in producing both pro- and anti-inflammatory cytokines that facilitate gut immune homeostasis.
RESULTS
Commensal Bacteria-Host Interactions Influences STING-Signaling
To evaluate the role of STING in influencing colitis, we orally treated mice containing (WT) or lacking STING (SKO) with dextran sodium sulfate (DSS) which can trigger intestinal inflammation (Ahn et al., 2015). Principally, the experiments were conducted with mice housed in a barrier facility, containing Helicobacter spp, (helicon+ barrier) which is known to influence the outcome of colitis (Fox et al., 2011; Oliveira et al., 2004). We observed that mice lacking STING modestly lost weight in response to DSS treatment over an 8 day period, compared to similarly treated WT mice (day 5 p=0.005, day6 p=0.007, day7 p=0.027) (Figure. 1A). Correspondingly modest differences in histology were noted, which nevertheless indicated that mice lacking STING may play a role in early innate immune responses to colonic irritation, under these conditions (Figures. S1A-S1C). However, similar treatment of mice housed in a Helicobacter spp negative environment (helicon− SPF) indicated no significant differences in inflammatory responses to DSS treatment (Figures. 1B and S1D). A similar observation, namely lack of notable inflammation in response to DSS treatment, was observed using mice deficient in the cyclic GMP-AMP synthase (cGAS) (Figures. S1D-F). This data allude that intestinal flora may play a role in influencing STING–dependent inflammatory responses. That STING signaling may be important in intestinal immunity has been demonstrated by observing that SKO mice, treated with Azoxymethane/dextran sodium sulfate (AOM/DSS), which can induce colitis associated cancer (CAC), develop increased polyp formations compared to control mice (Ahn et al., 2015). To further investigate the importance of STING signaling in intestinal immunity, we treated mice housed in a helico+ barrier room with AOM/DSS for approximately 4 months, and confirmed that SKO mice develop higher numbers of polyps compared to similarly treated WT mice (Figure. 1C). To appraise the role of commensal bacteria in influencing this outcome, we correspondingly treated SKO mice with AOM/DSS in helico+ barrier housing conditions in the presence or absence of antibiotics. Our results demonstrated that antibiotic-treated SKO mice developed less polyps compared to untreated SKO mice, inferring a key role for bacteria in manipulating this event (Figure. 1D). 16S ribosomal RNA sequence analysis of commensal microbial populations within the SKO mice housed under helico+ barrier housing conditions indicated, by principal coordinate analysis (PCoA) using Bray-Curtis dissimilarity at the operational taxonomic unit (OTU) level, significant bacterial differences to that of healthy WT mice (Figure. S2). This included key differences in Turicibacter and Odoribacter species, implying that loss of STING-signaling can influence commensal bacteria portfolios (Figure. S2). Of interest was that cGAS-deficient mice (cGASKO) did not appear to exhibit an increased amount of polyp formation compared to WT mice, suggesting that bacteria produced CDNs rather than genomic DNA may play a role in triggering STING activity (Figure. 1C). This data indicates that STING signaling is required to recognize AOM/DSS-induced DNA damage and initiate wound repair processes, which if not instigated may enable microbial-influenced inflammatory events to facilitate CAC. This possibility was enforced by observing that AOM/DSS treated SKO mice housed in a Helicobacter spp negative environment did not exhibit increased polyp formation (Figure. 1E). Rather, we observed that SKO mice housed in Helicobacter spp free conditions had a significantly higher inflammation and mortality rates compared to WT or cGASKO mice (Figure. 1F-H). This may be explained in part, by noting that SKO mice failed to generate anti-inflammatory IL-10 in response to AOM/DSS treatment (Figure. 1I). Collectively, our observations suggest that STING may interact with commensal bacteria to generate anti-inflammatory cytokines such as IL-10 and play a role in maintaining gut immune homeostasis.
Figure 1. Commensal bacteria-host interactions influence colonic polyp formation.
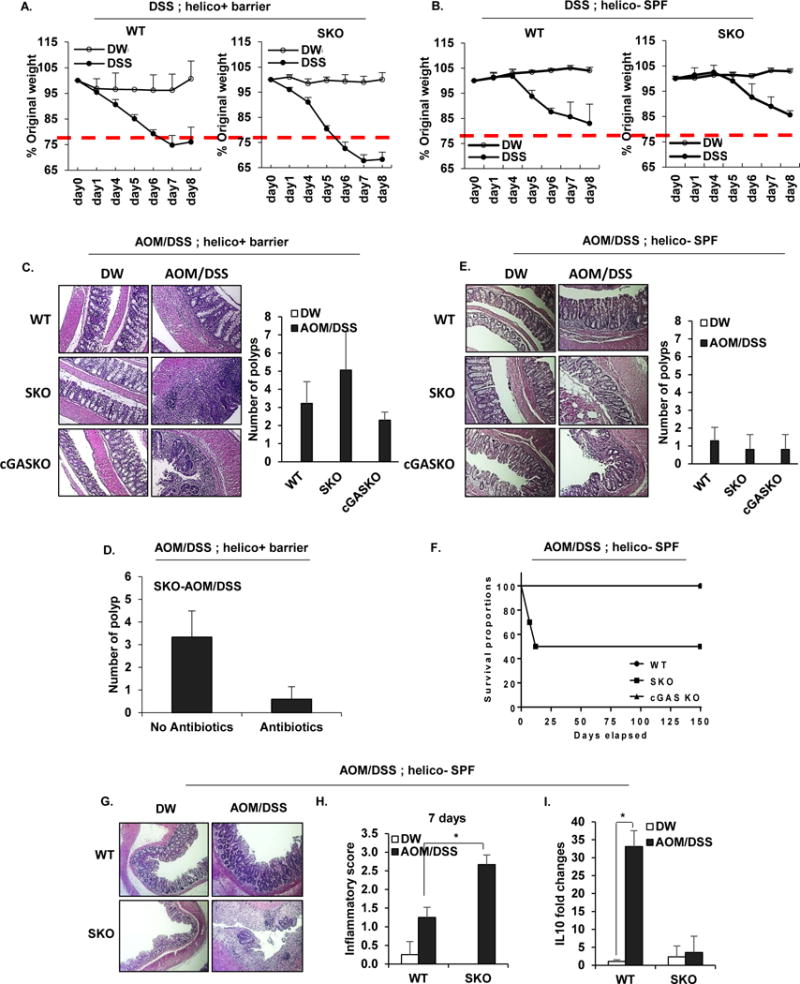
Body weight assessment of B6 background of Wild-type (WT) and Sting Knockout (SKO) mice received 5% of Dextran sodium sulfate (DSS) in drinking water for 7 days in (A) helico+ barrier room or helico− SPF room (B). The data are representative of at least two independent experiments. (C) Representative photographs of Hematoxylin and eosin staining (H&E) of colon tissues and the number of polyps in colon tissue of WT and SKO mice either AOM/DSS-treated or normal water treated in the helico+ barrier room. (D) The number of polyps in AOM/DSS-treated SKO mice either antibiotics treated or not. An antibiotic cocktail was administered in the drinking water of a separate cohort of mice for 1 month prior to AOM/DSS treat. (E) Representative photographs of H&E staining and the number of polyps in colon tissue of WT and SKO mice either AOM/DSS-treated or normal water treated in the helico− SPF room. (F) Mortality rates of AOM/DSS-treated WT, SKO, and cGASKO mice in helico− SPF room. (G) Representative photographs of H&E staining and (H) Inflammation score (0: Normal to 3: most severe) in either AOM/DSS-treated or normal water treated in helico− SPF room for 7 days. (I) quantitative PCR (qPCR) analysis of IL10 expression in colon from the mice same as (H). Error bars indicated s. d. Statistical analysis was performed using Student’s t-test. *; p ≤ 0.05
IL-10 Suppresses STING-aggravated Inflammatory Colitis
That commensal bacteria can influence intestinal inflammation has been observed using other models of chronic colitis. For example, loss of IL-10, a major immunosuppressive cytokine, induces spontaneous colitis in mice because the effects of concomitant pro-inflammatory cytokine production including interferon-λ (IFN-λ) and IL-12 are not blocked (Davidson et al., 1998). Since antibiotics can eliminate the observed colitis and CAC in this model, commensal bacteria may constitutively stimulate innate immune signaling pathways resulting in the production of both pro-inflammatory cytokines as well as counter-balancing anti-inflammatory IL-10. However, the innate immune pathways that control host interactions with intestinal flora remain to be clarified. Given that antibiotics can also eliminate STING-dependent CAC and our observations of reduced levels of IL-10 in SKO mice treated with AOM/DSS (Figure. 1D and 1I), we thus examined the importance of STING in the development of IL-10-controlled colitis and polyp formation. To achieve this we generated IL-10−/−STING−/−double deficient mice (IL10KO/SKO). Our results indicated that while IL-10−/− mice (IL10KO) developed severe colitis within 10 weeks, IL-10−/−STING−/−mice did not exhibit any significant intestinal inflammatory disease for over 19 weeks (Figure. 2A). The pronounced thickening of the bowel wall and slightly shortened colon length characteristic for IL-10 deficient mice was also reduced in IL-10−/−STING−/−mice (Figures. 2B-D and Figure. S3A). The incidence of spontaneous polyp formation in IL-10-deficient mice was also completely eliminated in the absence of STING (Figure. 2E and Figure. S3B). Gene expression profiles were measured on the various mice, using PCR and microarray analysis, which indicated that high levels of pro-inflammatory cytokine production including IL-1b, IL-22 and IL-12 as well as members of the Regenerating Family (Reg3b/g) typically detected in the colon of IL10KO mice, were similarly greatly repressed in the absence of STING signaling (Figures. 2F, G). A similar effect was seen crossing cGAS-deficient mice with IL-10 deficient mice (IL10KO/cGASKO) (Figure. S4A). However, we observed that 10% of the examined IL10KO/cGASKO mice developed polyps perhaps again suggesting a direct role for CDN’s on influencing STING signaling (Figure. S4B and C). Our data thus indicates that STING-signaling may play a significant role in the development of colitis in the absence of IL-10, plausibly by interacting with microbes to generate pro-inflammatory cytokines.
Figure 2. IL10 suppresses STING-induced inflammatory colitis and CAC.
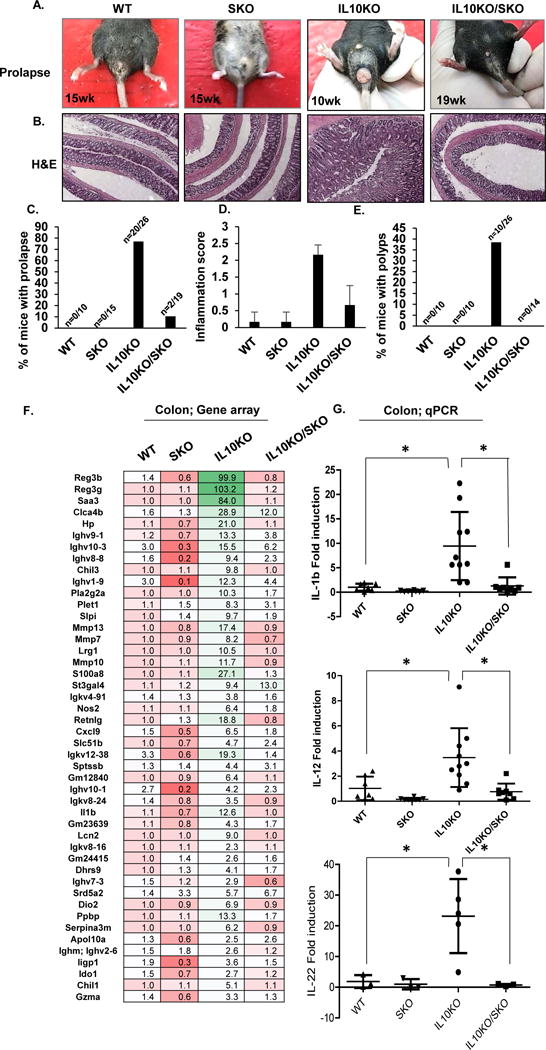
(A) Representative photographs of rectal prolapse, (B) representative photographs of H&E staining, (C) % of mice with prolapse (D) inflammation score, and (E) the number of polyps in 10~19 weeks old WT (n=10), SKO (n=15), IL10KO (n=26) and IL10KO/SKO mice (n=19). (F) Gene array analysis of colon tissue from 8 weeks old age of WT, SKO, IL10KO and IL10KO/SKO mice (n=5). Fold changes were estimated by WT mice and the highest variable genes are shown. Pseudo-colors indicate transcript levels equal to below (red) or above (green). (G) qPCR of IL1β, IL12, and IL22 mRNA level in in each genotype of colon tissue. All data are the mean of at least 7 mice. Error bars indicated s. d. Statistical analysis was performed using Student’s t-test. *; p ≤ 0.05
STING-signaling does not require the adaptor MyD88
It is thus conceivable that STING-signaling is stimulated following interaction with commensal bacteria. STING-activation may invoke the generation of pro-inflammatory cytokines, which are normally suppressed by IL-10, to induce colitis. However, it is known that loss of MyD88 can also eliminate inflammatory colitis induced by IL-10 deficiency (Hoshi et al., 2012; Rakoff-Nahoum et al., 2006). Thus, it is acceptable that MyD88 may play a direct role in STING-dependent signaling, or alternatively that STING-dependent pro-inflammatory cytokines may require downstream MyD88-dependent signaling to exert their effect. To determine this, we treated STING or MyD88-deficient murine embryonic fibroblasts (MEFs), bone marrow derived macrophages (BMDM) or dendritic cells (BMDC) with exogenous CDN’s or cytosolic dsDNA (ISD) which triggers STING-signaling and type I IFN production. This data confirmed that STING but not MyD88 was required for CDNs or cytosolic dsDNA-dependent type I IFN production as determined by microarray analysis and PCR (Figures. 3A-D). We further confirmed that STING was not required for lipopolysaccharide (LPS) driven innate immune signaling which requires TLR4 and the adaptors MyD88 or TRIF to drive cytokine production such as IL-1b, at least in BMDM or BMDC (Figures. 3E-H). However, loss of STING was seen to somewhat abrogate IL1β production in response to LPS in MEFs, for reasons that presently remain unclear (Figure. 3E, F). Of interest was that we observed that STING-signaling could trigger STAT3 phosphorylation as well as STAT1 via cytosolic DNA stimulation. This was particularly noticeable in BMDC’s compared to BMDM’s (Figures. 4A and Figure. S5). Cells retrieved from SKO mice were confirmed to retain sensitivity to LPS treatment, unlike those analyzed from MyD88-deficient mice (Figure 4B and Figure. S6). cGAS was seen to be required to activate STAT1 and STAT3 by ISD but not by exogenous CDNs which acted on STING-directly (Figure. S5). Given this data, it is possible that commensal bacteria may stimulate STING-signaling to induce cytokines that bind to receptors requiring MyD88 to exert their downstream pro-inflammatory effects. Thus, at least in part, loss of MyD88 may abrogate colitis manifested by IL-10 deficiency by preventing the action of STING-dependent pro-inflammatory cytokines.
Figure 3. STING-signaling does not require the adaptor MyD88.
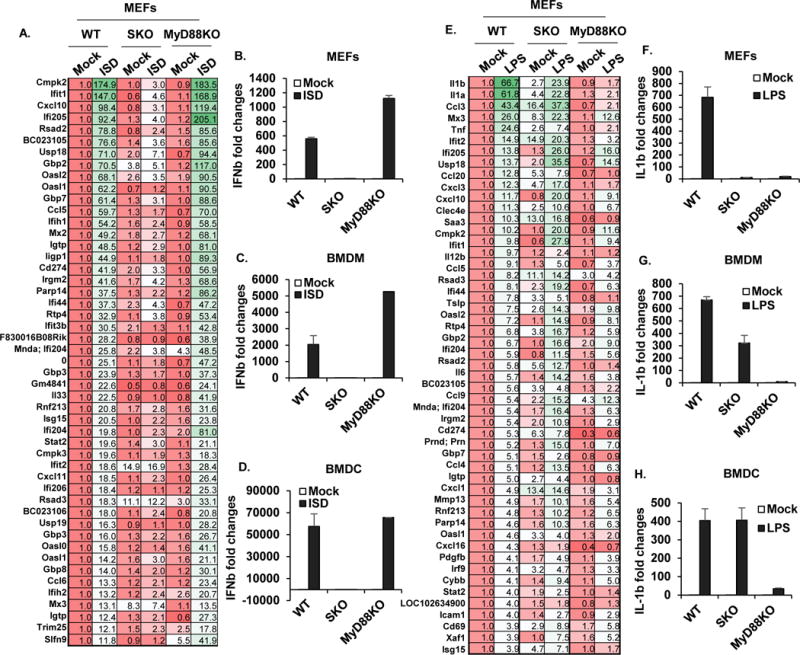
(A, E) Gene array analysis from total RNA purified in WT, SKO and MyD88KO mouse embryonic fibroblasts (MEFs) treated with 3ug/ml of ISD (A) or 1 ug/ml of LPS (E) for 6 hours. Fold changes were estimated by WT mice and the highest variable genes are shown. Pseudo-colors indicate transcript levels equal to below (red) and above (green). qPCR analysis of IFNβ mRNA level in MEFs (B), Bone marrow derived macrophages (BMDM) (C) and Bone marrow derived dendritic cells (BMDC) (D) treated same as (A). qPCR analysis of IL1β mRNA level in MEFs (F), BMDM (G) and BMDC (H) treated same as (E).
Figure 4. STING-signaling drives pro-inflammatory, MyD88-dependent gene induction.
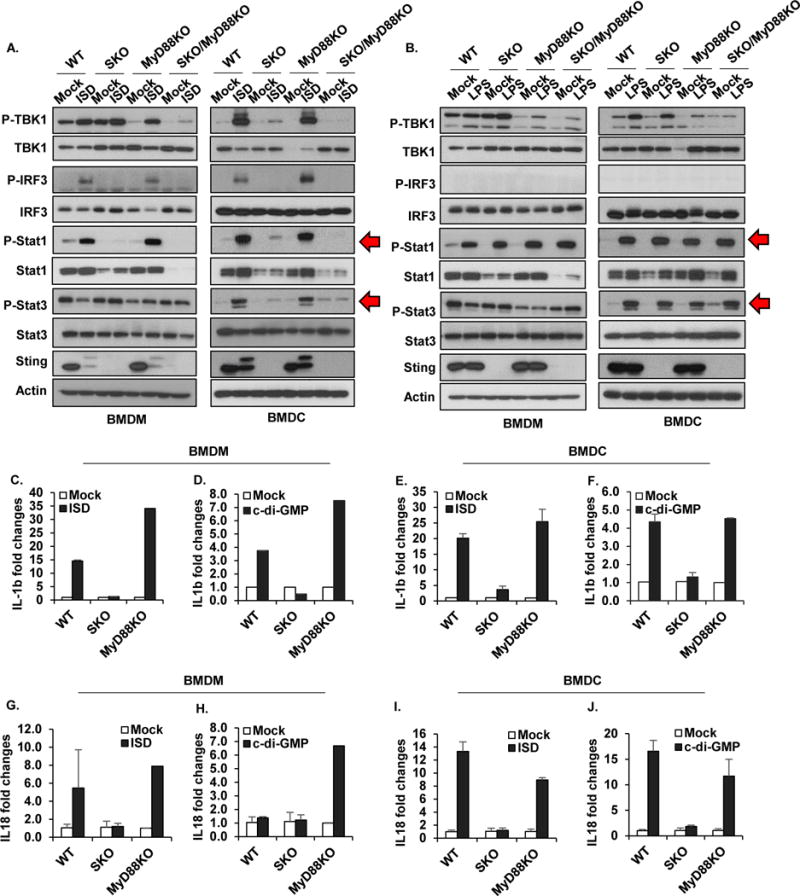
(A, B) Immunoblot analysis to determine the levels of pTBK1, pIRF3, pStat1, pStat3, and STING in BMDM or BMDC treated with 3ug/ml of ISD (A) or 3ug/ml of LPS (B) for 6 hours. qPCR analysis of IL1β mRNA level in BMDM (C, D) or BMDC (E, F) treated with 3ug/ml of ISD (C, E) or 3ug/ml of c-di-GMP (E, F) for 6 hours. qPCR analysis of IL18 mRNA level in BMDM (G, H) or BMDC (I, J) treated with 3ug/ml of ISD (G, I) or 3ug/ml of c-di-GMP (H, J) for 6 hours. Data are representative of at least 2 independent experiments. Error bars indicated s. d.
STING-signaling drives pro-inflammatory, MyD88-dependent gene induction
Key pro-inflammatory cytokines that bind to receptors requiring MyD88 to exert pro-inflammatory responses include IL-1β and IL-18 (Salcedo et al., 2013; Salcedo et al., 2010). The promoter region of both these cytokines are known to harbor NF-κB and STAT3 transcription factor binding sites which, in turn, are stimulated directly or indirectly by STING signaling (Ahn et al., 2015; Ahn et al., 2014; Barber, 2015). To confirm whether these cytokines can be induced in a STING-dependent manner, we treated BMDM or BMDC’s with CDN’s or cytosolic DNA and first measured IL-1β induction by PCR. This data indicated that IL-1β production was indeed detectable in response to CDN’s and dsDNA in a STING dependent manner at least in these cell types (Figures. 4C-F). A similar study indicated that IL-18 production could also be augmented by STING signaling in BMDM or BMDC’s (Figures. 4G-J). Thus, the stimulation of STING signaling by CDN’s/dsDNA can help increase the production of pro-inflammatory cytokines, IL-1β and IL-18 that require MyD88 to exert their influence in select cells. Therefore, the enhancement of colitis driven by loss of IL-10, may be partially explained by STING-dependent genes such as IL-1β and IL-18 exerting an inflammatory effect.
The expression of anti-inflammatory IL-10 can be modulated by STING signaling
The observation that loss of STING can rescue colitis manifested by loss of IL-10 implies not only that STING signaling may contribute towards the production of pro-inflammatory cytokines whose function is generally suppressed by IL-10 signaling but also that IL-10 expression itself may be regulated by STING as our preliminary data suggests (Figure. 1I). IL-10 is expressed in a wide variety of immune related cells including mononuclear phagocytes (MNP) in response to a variety of stimuli including the LPS-triggered TLR4 pathway, requiring MyD88, as well as type I IFN (Saraiva and O'Garra, 2010). Indeed, the promoter region of IL-10 is known to harbor sites for STAT1, 3, NF-κB and members of the interferon regulatory factor (IRF) family. IL-10 binds to IL-10 receptors (IL-10R1/R2) and triggers the activation of STAT3 signaling to downregulate proteins involved in inflammation such as IL-12 and IL-23 and IFN-λ. Upon analysis of BMDM as well as BMDC, we confirmed that IL-10 can also be induced by cytosolic DNA, in a STING-dependent manner (Figures. 5A-D). An additional member of the IL-10 family is IL-22, which is also inducible by NF-κB, STAT3-dependent signaling (Hutchins et al., 2013; Manzanillo et al., 2015). IL-22 is known to induce proinflammatory cytokines in the gut such as the S100 family, IL-6 and IL-8 (Andoh et al., 2005; Kolls et al., 2008; Zenewicz et al., 2008). Examination of the colon retrieved from IL-10 deficient mice indicated the presence of significant levels of IL-22 was eliminated in the absence of STING, as described (Figure. 2G). Thus, STING signaling can influence both the production of pro-inflammatory cytokines such as IL-1 and IL18 and directly or indirectly IL-22, in vitro and in vivo to influence the outcome of colitis.
Figure 5. IL10 can be modulated by STING signaling.
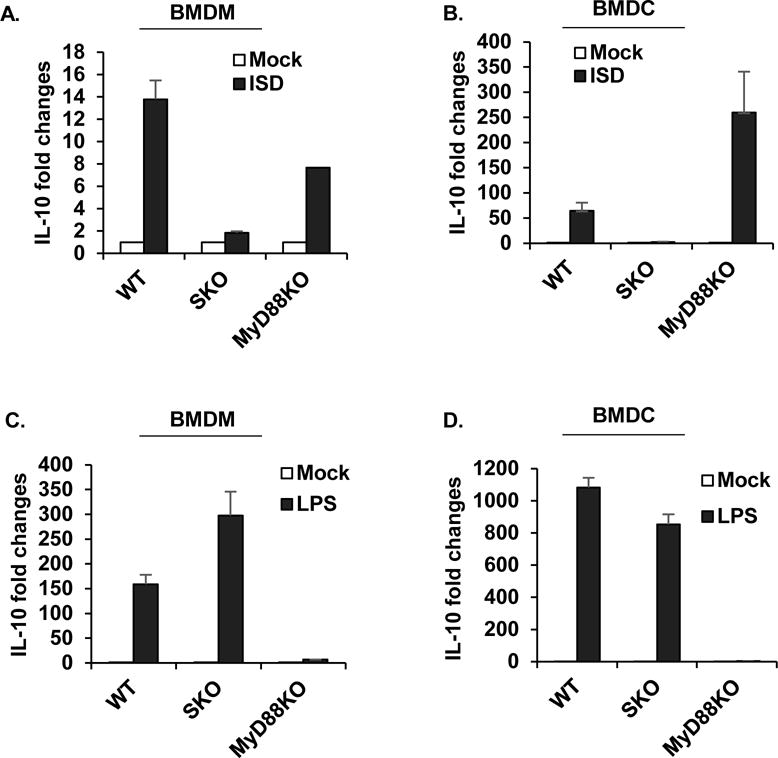
qPCR analysis of IL10 mRNA level in BMDM (A, C) or BMDC (B, D) treated with 3ug/ml of ISD (A, B) or 1ug/ml of LPS (C, D) for 6 hours. Data are representative of at least 2 independent experiments. Error bars indicated s. d.
Monocyte lineages are predominantly responsible for STING-mediated pro-inflammatory responses
Our data indicates that STING signaling is required to initiate wound healing processes in response to AOM/DSS injury. Loss of STING facilitates polyp formation perhaps as a result of dysbiotic microbial infiltration of the lamina propria which initiates STING-independent inflammatory responses. Further, loss of STING may also reduce anti-inflammatory IL-10 production. To start to delineate the cells including MNP’s responsible for STING’s role in maintaining gut immune homeostasis, mice with STING floxed allele(s) were crossed with different cre-recombinase expressing mouse lines (LysM-SKO, which deleted STING from macrophages and neutrophils and CD11c-SKO, which deleted STING from dendritic cells)(Bouabe and Okkenhaug, 2013; Caton et al., 2007; Clausen et al., 1999). To examine the role of STING-signaling in the macrophage and dendritic cell subsets, the LysM-SKO and CD11c-SKO mice were treated with AOM/DSS as described. Body weight was monitored and effects on the colon including polyp formation observed. This analysis indicated that WT, SKO, LysM-SKO, and CD11c-SKO mice all exhibited modest weight reduction as a result of treatment (Figures. 6A-D). However, the LysM-SKO, and CD11c-SKO mice, however, were noted to lose slightly less weight (Figures. 6C-D). In addition, both SKO-LysM and SKO-CD11C mice exhibited less inflammation and polyp formation in response to AOM/DSS treatment and compared to SKO mice (SKO-LysM: p=0.0005, SKO-CD11C: p=0.0019) (Figures. 6E, F). Our data would suggest that in vivo, STING-signaling in MNP’s representing both macrophages and DC’s lineages may play an important role in recognizing microbes, and/or DNA damaging events, to generate cytokines that can pre-dominantly aggravate inflammatory colitis. However, STING-signaling in other cell types may also contribute towards maintaining gut homeostasis since SKO mice, which do not express STING in any tissue, exhibit different responses to carcinogenic events in vivo compared to WT and LysM-SKO/CD11c-SKO mice (Figures. 6E, F)(Ahn et al., 2015). Our data underscores the importance of STING-innate immune signaling in interacting with commensal bacteria and controlling inflammatory colitis.
Figure 6. Monocytes are predominantly responsible for STING-mediated pro-inflammatory responses.
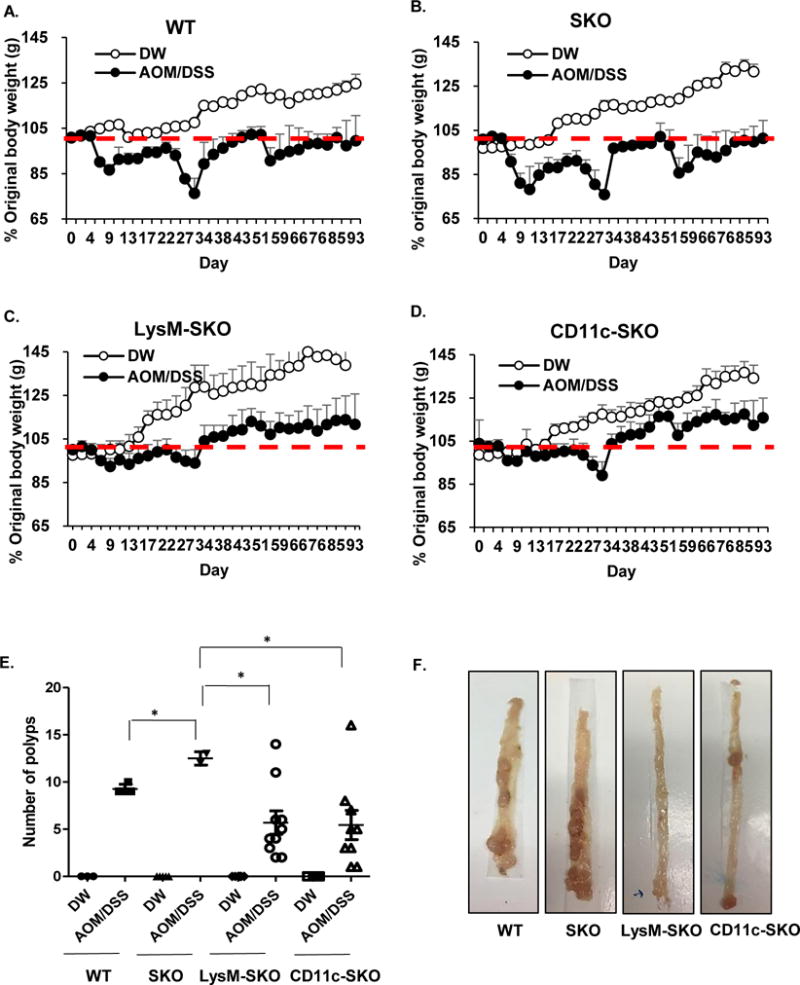
Body weight assessment in WT (A), SKO (B), LysM-SKO (C) and CD11c-SKO (D) mice treated with AOM/DSS- or normal water as control. LysM-SKO for AOM/DSS (n=10), LysM-SKO for DW control (n=6), CD11c-SKO for AOM/DSS (n=9), CD11c-SKO for DW control (n=5), WT for AOM/DSS (n=4), WT for DW control (n=3), SKO for AOM/DSS (n=4), and SKO mice for DW control (n=5). (E) Number of polys (F) and representative photographs of polyps in colon. All data are the mean of at least 3 mice. Error bars indicated s. d. Statistical analysis was performed using Student’s t-test. *; p ≤ 0.05
DISCUSSION
Our data demonstrates a key role for STING signaling in interacting with commensal bacteria and influencing gut immune homeostasis. STING-deficient mice (SKO) were found to harbor altered portfolios of commensal bacteria compared to similarly housed wild type mice, possibly due to loss of an important innate immune pathway enabling the dominance of select species for reasons that presently remain unclear. Significantly, loss of STING-signaling was found to reverse the severe form of colitis that ensues in mice in the absence of the key anti-inflammatory cytokine IL-10. Thus, it is possible that CDNs produced from intracellular bacteria, as well as microbial DNA, may constitutively stimulate STING-dependent signaling. Plausibly, under normal conditions, intracellular microbes may obtain access to scavenging intestinal epithelial cells and/or MNP’s and activate STING signaling, or conversely that exogenous CDNs produced from extracellular microbes may gain access to such immune cells that harbor STING. It is also possible that self-DNA may trigger the production of CDNs in immune cells, although the observation that treatment of STING-deficient mice with antibiotics eliminated the incidence polyp formation would argue against this. While it is known that different housing conditions that contain varying bacteria species, as well as the background of the mice themselves can affect the outcome of inflammatory stimuli, our data would nevertheless suggest an important role for STING signaling in interacting with the microbiome (De Robertis et al., 2011; Laukens et al., 2016; Mahler et al., 1998).
The production of IL-10 requires NF-κB or IRF transcription factor activation which can be triggered by TLR stimulation or by interferon (Chang et al., 2007; Ouyang et al., 2011). Since STING-signaling utilizes these same pathways it is perhaps unsurprising that STING can also stimulate the production of IL-10. Our data indicates that through interaction with commensal bacteria, STING-signaling may play a major role maintaining an appropriate amount of immunosuppressive IL-10. Without STING-dependent IL-10 production, levels of STING-dependent pro-inflammatory cytokines may increase and trigger an inflammatory state. These aggravating cytokines may be predominantly generated by MNP’s, as our data suggests since LysM-SKO/CD11c-SKO exhibited less inflammation in response to carcinogens. The MNP population implicated here include both P1 (CD11c+ CD11b−, CD103+ CX3CR1− DEC205+ F4/80−) and P2 (CD11c+CD11b+,CD11c+CD103−CX3CR1+ macrophage [MP2] subsets as determined using CD11c-Cre and LysM-Cre mice respectively. However, the cytokine profile triggered in response to STING activation may vary depending on the cell-type. In the complete absence of STING, SKO mice were more prone to inflammation and polyp formation in response to DSS and AOM/DSS treatment respectively. We postulate that DSS-induced inflammation may enable microbes access to STING-containing immune cells such as CD11c+ CD11b+ (P2). STING-signaling may be important for facilitating rapid wound healing and antimicrobial processes, that if not enabled allow microbes access to the lamina propria where they can aggravate STING-independent inflammatory responses, such as through the TLR pathway (Salcedo et al., 2010). AOM-instigated DNA damage events also likely trigger intrinsic STING-dependent cytokine production, and loss of STING may enable DNA-damaged cells to escape the immune system and proliferate (Ahn et al., 2015). Loss of extrinsic STING signaling in APC’s may also affect anti-tumor T-cells responses, and additionally enable pre-cancerous cells to escape (Woo et al., 2014). It is presently unclear why cGAS-deficient (cGASKO) mice exhibited less pronounced inflammatory responses or polyp formation following DSS or AOM/DSS treatment, compared to SKO mice. However, in these transient early-immediate circumstances, it may be that CDNs generated by bacteria exert a more important affect upon STING signaling than self or microbial cytosolic dsDNA species (Danilchanka and Mekalanos, 2013).
That the elimination of STING signaling prevents colitis-associated with IL-10 deficiency suggests that STING-signaling may be responsible for the generation of liable pro-inflammatory cytokines in this model (Figure. 7). STING-dependent signaling certainly seems capable of influencing the production of pro-inflammatory cytokines IL1b, IL-18 and possible IL-22 at least indirectly, that after binding to appropriate receptors require the adaptor MyD88 to stimulate additional pro-inflammatory production (Ahn et al., 2015; Salcedo et al., 2013). In this light it is well know that loss of MyD88 can also eliminate colitis associated with loss of IL-10 (Hoshi et al., 2012; Rakoff-Nahoum et al., 2006). This would suggest that MyD88 plays a role in STING signaling or facilitates the downstream effects of STING-triggered pro-inflammatory cytokines such as IL1b and IL18. Our data would favor the latter model since Myd88 did not affect cytosolic dsDNA dependent signaling. Similarly, STING was not directly required for LPS signaling in BMDM or BMDC, which predominantly required MyD88 or TRIF. However, it is also likely that the interaction of commensal bacteria with members of the TLRs may also play a key role in IL-10 production and that both the STING and the TLR pathways synergistically function to maintain immune homeostasis of the gut (Hoshi et al., 2012; Uronis et al., 2009). Since loss of TLR4 does not eliminate IL-10 mediated suppression, this may suggest that at least this TLR may not be a major cause of pro-inflammatory cytokines that drive colitis (Gonzalez-Navajas et al., 2010). Collectively, our data indicates that STING-may play a key role in maintaining intestinal immune homeostasis and be a key producer of anti-inflammatory IL-10 as well as pro-inflammatory cytokines and type I IFN. Thus, deregulation of the STING-pathway, which is being more commonly associated with a variety of autoinflammatory disease such as Aicardi-Goutieres Syndrome (AGS) and STING-associated vasculopathy within onset in infancy (SAVI), may also play a role in IBD. Therefore, the development of therapeutics that may target the STING pathway may have benefit in the treatment of such malaise.
Figure 7.
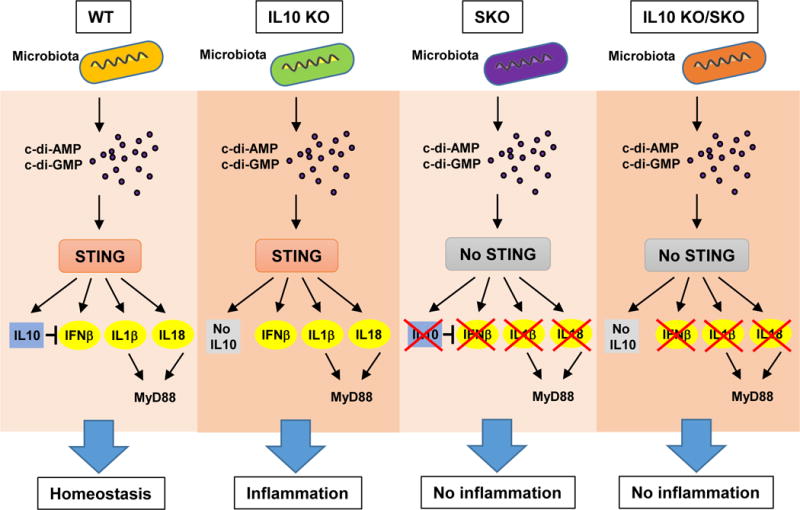
A schematic illustration of STING signaling responsible for the generation of pro-inflammatory cytokines to prevents colitis-associated with IL-10 deficiency.
METHODS
Mice
STING knockout mice (Sting−/−; SKO) were generated in our laboratory (Ishikawa 2008). We have generated B6 background wild type (WT) and SKO from original clone of B6/129 background SKO mice. Our original SKO mice in 129/B6 mixed background were backcrossed onto a WT B16 background more than 7 times until the hetero mice (STING+/−) were > 97% B16 in our conventional mice room (Helico spp +). To generate Helicobacter free STING KO mice, we utilized an in vitro fertilization method and the oocytes were transferred to Helicobacter free recipient females. We bred the hetero mice (STING+/−) to generate WT and SKO mice in a specific pathogen free room (Helico spp −). Both Helico+ and Hlico− rooms were inspected the same Rodent Health Surveillance authorities, quarterly by Division of Veterinary Resources in University of Miami. All use autoclaved caging, supplies, water and irradiated food. cGAS Knockout mice (cGASKO) were kindly provided by Dr. Herbert W. Virgin IV (Washington University School of Medicine). MyD88 knockout mice (MyD88 KO) and IL10 knockout mice (IL10KO) were purchased from Jackson Laboratory. To generate SKO mice on IL10 KO background, B6/129 background SKO mice were crossed to B6/129 background IL10 KO mice. The IL10 Ko mice and IL10KO/SKO mice were bred in a Helico+ room. We used Helico+ WT and SKO mice as controls. To generate the conditioning Sting knockout mice, we developed animals with the STING gene floxed. In brief, the exons 1-5 were flanked with loxp sites in C57/BL6 derived embryonic stem (ES) cells in order to render STING susceptible to Cre-mediated recombination. The floxed STING mice were crossed to mice expressing Cre under a cell specific promoter (LysM-Cre, CD11C-Cre) to generate LysM-SKO and CD11c-SKO mice. Mice care and study were conducted under approval from the Institutional Animal Care and Use Committee (IACUC) of the University of Miami. Mouse genotypes from tail biopsies were determined by real-time PCR with specific probes designed for each gene by commercial vendor (Transnetyx).
Acute DSS colitis
WT and SKO mice 6-8 weeks of ages were divided into experimental and control groups. Mice in experimental group were received 5% Dextran sodium sulfate (DSS, MP 160110; MW 36000-5000) for 7 days. The following day, the mice were sacrificed and colon was removed to proceed histology. Distilled water was administered into control group mice. The mice were monitored on every day to evaluate disease activity index.
AOM/DSS induced colitis-associated tumor induction
WT, SKO, and cGASKO mice were injected intraperitoneally with Azoxymethane (AOM; MP 180139, Sigma-Aldrich A5486) at a dose of 10mg/kg. DSS at 5% was administered in the drinking water for 7 days every 3 weeks. DSS cycle was repeated 4 times. On 125 days, the mice were sacrificed and colon was resected, flushed with PBS to count polyps. Colon were fixed in formalin for histology and frozen for RNA expression analysis. For antibiotic treatment, mice were treated with an antibiotic cocktail of Ampicillin (1g/L), Neomycin (1/L), Metronidazole (1g/L), and Vancomycin (500mg/L) in their drinking water for 4 weeks prior to AOM/DSS administration.
V4 16S rRNA gene sequencing
Stool was collected from WT and SKO mice and the commensal microbiota composition was evaluated by V4 hypervariable region of the 16S rRNA gene sequencing by Second Genome (The microbiome company). Second Genome performed nucleic isolation with the MoBio PowerMag® Microbiome kit (Carlsbad, CA) according to manufacturer’s guidlines and optimized for high-throughput processing. All samples were quantified via the Qubit® Quant-iT dsDNA High Sensitivity Kit (Invitrogen, Life Technologies, Grand Island, NY). To enrich the sample for bacterial 16S V4 rDNA region, DNA was amplified utilizing fusion primers designed against the surrounding conserved regions which are tailed with sequences to incorporate Illumina (San Diego, CA) adapters and indexing barcodes. Each sample was PCR amplified with two differently bar coded V4 fusion primers. Samples that met the post-PCR quantification minimum and were advanced for pooling and sequencing. For each sample, amplified products were concentrated using a solid-phase reversible immobilization method for the purification of PCR products and quantified by qPCR. A pool containing 16S V4 enriched, amplified, barcoded samples were loaded into a MiSeq® reagent cartridge, and then onto the instrument along with the flow cell. After cluster formation on the MiSeq instrument, the amplicons were sequenced for 250 cycles with custom primers designed for paired-end sequencing. Samples are processed in a Good Laboratory Practices (GLP) compliant service laboratory running Quality Management Systems for sample and data tracking. The laboratory implements detailed SOPs, equipment and process validation, training, audits and document control measures. QC and QA metrics are maintained for all sample handling, processing and storage procedures.
Primary cell culture
Mouse embryonic fibroblasts (MEFs) were obtained from e15 embryos by a standard procedure as described (Ishikawa 2008). Bone marrow derived dendritic cells were isolated from hind-limb femurs of 8-10 weeks old mice. Briefly, the marrow cells were flushed from the bones with Dulbecco’s modified eagle medium (DMEM, Invitrogen), 10% heat-inactivated fetal calf serum (FCS, Invitrogen) with a 23 gauge needle and incubated at 37°C for 4 hours. After harvesting floating cells, approximately 2×107 cells were seeded in 10 cm dish with complete DMEM including 10 ng/ml of Recombinant mouse GM-CSF (GM-CSF, R&D systems) or 10 ng/ml of Recombinant mouse M-CSF (M-CSF, R&D systems) for dendritic cells (BMDC) or macrophages (BMDM).
Histopathology
Mice are sacrificed and the colon tissues were fixed in 10% formalin for 48 hours. All processes for paraffin block and Hematoxylin and Eosin staining (H&E) was performed at the pathology research resources histology laboratory in University of Miami.
Gene array analysis
Total RNA was isolated from cells or tissues with RNeasy Mini kit (74104, Qiagen, Valencia, CA). RNA quality was analyzed by Bionalyzer RNA 6000 Nano (Agilent Technologies, Santa Clara CA). Gene array analysis was examined by Illumina Sentrix BeadChip Array (Mouse WG6 version 2) (Affymetrix, Santa Clara CA) at the Oncogenomics Core Facility, University of Miami. Raw intensity values from Illumina array are uploaded on GeneSpring™ software from Agilent. Values are Quantile normalized and log2 transformed to the median of all samples. Significantly differential expressed genes are computed using the Student’s t-test and selected using threshold of P-value ≤ 0.05. Hierarchical Clustering and visualization of selected differentially expressed genes is performed on GeneSpring using Pearson Correlation distance method and linkage was computed using the Ward method. Gene expression profiles were processed and statistical analysis was performed at the Sylvester Comprehensive Cancer Center Bioinformatics Core facility University of Miami.
Quantitative Real time PCR (qPCR)
Total RNA were reverse-transcribed using M-MLV Reverse Transcriptase (Promega). Real-time PCR was performed using Taqman Gene Expression Assay (Applied Biosystems) for innate immune genes and inflammatory cytokines (IFNb: Mm00439552, IL1b: Mm01336189, IL18: Mm00434225, IL-10: Mm01288386, Life Technologies)
Immunoblot analysis
Equal amounts of proteins were resolved on sodium dodecyl sulfate (SDS)-Polyacrylamide gels and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). After blocking with 5% Blocking Reagent, membranes were incubated with various primary antibodies (and appropriate secondary antibodies). The image was resolved using an enhanced chemiluminescence system ECL (Thermo Scientific) and detected by autoradiography. Antibodies: rabbit poyclonal antibody against STING was developed in our laboratory as described previously in Ishikawa et al, 2008; other antibodies were obtained from following sources: β-actin (Sigma-Aldrich, A2228), p-IRF3 (Cell Signaling, CST4947), IRF3 (Cell Signaling, CST4302), p-TBK1(Cell Signaling, CST15483), TBK1(abcam, ab12116), p p65(Cell Signaling, CST3033), p65(Cell Signaling, CST), p-Stat3 (Cell Signaling, CST9145), Stat3 (Cell Signaling, CST4904), p-Stat1 (Cell Signaling, CST9167), Stat1(Cell Signaling,CST14994).
Statistical Analysis
All statistical analysis was performed by Student’s t test unless specified. The data were considered to be significantly different when P < 0.05.
Supplementary Material
Highlights.
Commensal bacteria stimulate STING-signaling to control gut homeostasis.
Both pro- and anti-inflammatory (IL-10) cytokines expression is stimulated by STING.
STING-dependent pro-inflammatory cytokine activity is balanced by IL-10 production.
Monocyte lineages are primarily accountable for STING-mediated cytokine expression.
Acknowledgments
We thank Ms. Delia Gutman and Ms. Auristela Rivera for mice maintaining and genotyping; Dr. Tianli Xia for generating the STINGFL mice; Dr. William F Hulme for gene array analysis. This study was supported by the NIH grants (R01-AI079336-06A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.A. designed and carried out most of the experiments; S.S. performed animal experiments; S.C.O advised on experimental design; and G.N.B designed the experiments and wrote the manuscript.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Konno H, Barber GN. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene. 2015;34:5302–5308. doi: 10.1038/onc.2014.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol. 2014;193:4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- Arthur JC, Gharaibeh RZ, Muhlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2013 doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouabe H, Okkenhaug K. Gene targeting in mice: a review. Methods Mol Biol. 2013;1064:315–336. doi: 10.1007/978-1-62703-601-6_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EY, Guo B, Doyle SE, Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol. 2007;178:6705–6709. doi: 10.4049/jimmunol.178.11.6705. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–970. doi: 10.1016/j.cell.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson NJ, Hudak SA, Lesley RE, Menon S, Leach MW, Rennick DM. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J Immunol. 1998;161:3143–3149. [PubMed] [Google Scholar]
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. Journal of carcinogenesis. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13:13–27. doi: 10.1038/nrgastro.2015.186. [DOI] [PubMed] [Google Scholar]
- Fox JG, Ge Z, Whary MT, Erdman SE, Horwitz BH. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011;4:22–30. doi: 10.1038/mi.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, Raz E. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120:570–581. doi: 10.1172/JCI40055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoentjen F, Harmsen HJ, Braat H, Torrice CD, Mann BA, Sartor RB, Dieleman LA. Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin 10 gene deficient mice. Gut. 2003;52:1721–1727. doi: 10.1136/gut.52.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535:75–84. doi: 10.1038/nature18848. [DOI] [PubMed] [Google Scholar]
- Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, Reizis B, Shen Z, Fox JG, Iwasaki A, Medzhitov R. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun. 2012;3:1120. doi: 10.1038/ncomms2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. 2013;12:489–498. doi: 10.1093/bfgp/elt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Laukens D, Brinkman BM, Raes J, De Vos M, Vandenabeele P. Heterogeneity of the gut microbiome in mice: guidelines for optimizing experimental design. FEMS Microbiol Rev. 2016;40:117–132. doi: 10.1093/femsre/fuv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- Madsen KL, Doyle JS, Tavernini MM, Jewell LD, Rennie RP, Fedorak RN. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology. 2000;118:1094–1105. doi: 10.1016/s0016-5085(00)70362-3. [DOI] [PubMed] [Google Scholar]
- Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- Manzanillo P, Eidenschenk C, Ouyang W. Deciphering the crosstalk among IL-1 and IL-10 family cytokines in intestinal immunity. Trends Immunol. 2015;36:471–478. doi: 10.1016/j.it.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Nagata S, Kawane K. Autoinflammation by endogenous DNA. Advances in immunology. 2011;110:139–161. doi: 10.1016/B978-0-12-387663-8.00004-1. [DOI] [PubMed] [Google Scholar]
- Oliveira AG, das Gracas Pimenta Sanna M, Rocha GA, Rocha AM, Santos A, Dani R, Marinho FP, Moreira LS, de Lourdes Abreu Ferrari M, Moura SB, et al. Helicobacter species in the intestinal mucosa of patients with ulcerative colitis. J Clin Microbiol. 2004;42:384–386. doi: 10.1128/JCM.42.1.384-386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annual review of immunology. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Salcedo R, Cataisson C, Hasan U, Yuspa SH, Trinchieri G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol. 2013;34:379–389. doi: 10.1016/j.it.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O'HUigin C, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annual review of immunology. 2012;30:677–706. doi: 10.1146/annurev-immunol-020711-075008. [DOI] [PubMed] [Google Scholar]
- Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol. 2010;10:415–426. doi: 10.1038/nri2778. [DOI] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.