

Summary
Discriminating transcriptional changes that drive disease pathogenesis from nonpathogenic and compensatory responses is a daunting challenge. This is particularly true for neurodegenerative diseases, which affect the expression of thousands of genes in different brain regions at different disease stages. Here we integrate functional testing and network approaches to analyze previously reported transcriptional alterations in the brains of Huntington’s Disease (HD) patients. We selected 312 genes whose expression is dysregulated both in HD patients and in HD mice, and then replicated and/ or antagonized each alteration in a Drosophila HD model. High-throughput behavioral testing in this model and controls revealed that transcriptional changes in synaptic biology and calcium signaling are compensatory, whereas alterations involving the actin cytoskeleton and inflammation drive disease. Knockdown of disease-driving genes in HD patient-derived cells lowered mutant Huntingtin levels and activated macroauthophagy, suggesting a mechanism for mitigating pathogenesis. Our multilayered approach can thus untangle the wealth of information generated by transcriptomics and identify early therapeutic intervention points.
eTOC blurb
Various ‘omic’ methodologies produce voluminous amounts of data, but distinguishing the changes that drive a disease from those that represent compensatory mechanisms or secondary responses is exceedingly difficult, time-consuming, and costly. A combination of functional and network approaches can tackle the challenge on a large scale, as shown here using Huntington’s disease transcriptomics as a test case.
Introduction
As our understanding of the molecular pathogenesis of human disease grows, we are faced with a dizzying array of possible leads for future therapies. Even in the case of monogenic disorders, the number of alterations that occur at the transcriptional level—or proteomic, or metabolomic levels— is in the thousands. Undoubtedly some of this complexity arises from the body’s own compensatory responses. Yet we lack a reliable means to distinguish changes that are non-pathogenic or compensatory from those that drive disease pathogenesis, and it is no small matter to decipher which pathways offer the best therapeutic entry points.
Consider the case of Huntington’s Disease (HD). The underlying mutation is well-defined: expansion of a CAG repeat that then encodes an abnormally long polyglutamine tract in the Huntingtin (HTT) protein (The HD Collaborative Research Group, 1993). The pathogenic cascade has a clear origin: the expanded polyglutamine tract hinders mutant HTT (mHTT) clearance, causing a slow accumulation of the protein that leads to neurotoxicity. The anatomic derangement is relatively circumscribed, with neuronal loss being concentrated in the striatum and cortex (Jimenez-Sanchez et al., 2017). Yet, despite this seemingly straightforward account, mHTT is associated with disruptions of many cellular functions, from autophagy to mitochondrial respiration, synaptic transmission, calcium signaling, axonal transport, intra-cellular trafficking, transcription, spindle orientation, and DNA repair, among other processes (Harjes and Wanker, 2003; Jimenez-Sanchez et al., 2017; Ross and Tabrizi, 2011; Saudou and Humbert, 2016). The complexity of HD pathogenesis is even more overwhelming when assessed molecularly, with recent studies revealing expression changes in thousands of genes, depending on the age (disease duration), genetic background, and length of polyglutamine tract (Ament et al., 2017; Langfelder et al., 2016).
There have been few explicit attempts to isolate pathogenic from compensatory pathways in neurological disease, and fewer still to go beyond correlating gene expression changes with disease features to testing whether these correlations might reflect causal relationships in vivo. Several studies are noteworthy in this regard. Using a mouse model of tauopathy, Karsten et al. performed microarray analysis on unaffected as well as affected brain regions in a mouse model of tauopathy (the assumption being that the former might reveal neuroprotective compensatory mechanisms) and tested three of the candidates from the microarray analysis using functional assays (Karsten et al., 2006). Rosen et al., seeking to understand the molecular basis of progranulin deficiency leading to frontotemporal dementia, used weighted coexpression network analysis (WGCNA) to investigate transcriptional alterations in progranulin-deficient human cells and confirmed dysregulation of the Wnt receptor Fzd2 in GRN knockout mice (Rosen et al., 2011). Bando et al. applied complex network analysis to the transcriptomes of two subtypes of refractory temporal lobe epilepsy (Bando et al., 2014), though they did not test the results experimentally. Stroedicke et al. focused on HTT protein interaction partners whose abundance is abnormal in HD affected brain regions and applied sequential interaction network filtering to highlight 13 protein interactors, one of which was subsequently validated as a modifier in vivo (Stroedicke et al., 2015). More recently, Ingram et al. used WGCNA to reveal two gene networks correlating with Purkinje cell degeneration in a Spino-cerebellar ataxia type 1 mouse model, which led to the discovery that upregulation of the neurohormone cholecystokinin mitigates Purkinje cell pathology (Ingram et al., 2016). The main constraint on these kinds of studies is the difficulty, cost, and amount of time it takes to test candidate genes in mice or in human neurons, which usually means that researchers can reasonably examine the effects of only a few genes at a time.
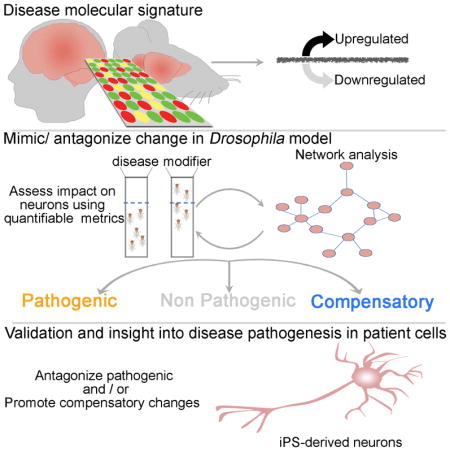
Here we demonstrate a high-throughput approach that can discriminate between changes that are primary drivers of HD and changes that are compensatory or simply non-consequential. We first queried transcriptional alterations observed in the brains of early-stage HD patients and several HD mouse models, to minimize confounding effects from downstream secondary events. Focusing on 312 genes that are affected in both species, we then replicated or antagonized these molecular alterations in a well-established Drosophila HD model (Branco et al., 2008; Langfelder et al., 2016; Lu et al., 2013) (testing hundreds of transcriptional alterations is not feasible in mice, and cellular models do not allow assessment of dysfunction in the context of a nervous system). We classified the transcriptional changes as likely pathogenic or compensatory according to whether they relieved or aggravated motor deficits of the HD Drosophila HD model, which provides a rapid, quantitative readout of nervous system function. We then applied network analysis to the data obtained in Drosophila to delineate functionally related genes and identify new candidate modifiers in the resulting networks. Finally, we used HD patient-derived fibro-blasts and iPSC-derived neurons to validate the Drosophila and network findings and to define the effects of key gene networks on mHTT protein stability and clearance.
Results
Functional analysis categorizes transcriptomic changes as likely compensatory or pathogenic
We began by selecting ~1000 genes that showed differential expression in the caudate nuclei of 32 patients with early-stage HD (Vonsattel grades 0–2) compared to caudate nuclei from 32 unaffected human controls (Hodges et al., 2006). We then compared these ~1000 human genes with ~3,000 differentially expressed genes from several HD mouse models in order to enrich for causal changes and filter out alterations due to individual variability, batch effects, and environmental influences. We used RNAseq data from HD knock-in mice with several different polyglutamine tract lengths (Langfelder et al., 2016) (see STAR Methods), and microarray data from both the HD knock-in mouse models (bearing Q111 and Q150; (Fossale et al., 2011) and (Kuhn et al., 2007), respectively) and the R6/2 transgenic mice, which express a truncated HTT with exon 1 and 150 CAG repeats (Strand et al., 2007). This comparison yielded 312 genes that are dysregulated early in the disease progression in both humans and mice (Table S1; see STAR Methods). We subjected these 312 genes to functional analysis to investigate their potential to modify HTT-induced neuronal dysfunction.
Because murine models are not practical for functional analysis of large numbers of genes, we used a Drosophila model of HD expressing human expanded N-terminal HTT (HTTN231Q128) exclusively in neurons (elavC155>GAL4 paneuronal driver). This fruit fly model develops a late-onset, progressive motor performance deficit that is readily quantifiable in a climbing assay (Langfelder et al., 2016; Lu et al., 2013; Lu et al., 2014; Shirasaki et al., 2012; Yao et al., 2015), enabling us to automate the identification of genetic modifiers that worsen or improve neuronal function in the context of the whole organism and before cell death takes place. We chose this model over our full-length HD fly model (Romero et al., 2008) for the primary readout because a screen as large as this would not have been possible with the much lower throughput and phenotypic sensitivity of the full-length model, but we conducted secondary assays in HD patient cells expressing full-length HTT (see below).
BLAST analysis revealed that 249 out of the 312 genes from human patients and mouse models have Drosophila homologs (cut-off was set at BLAST e-value=1E-10). In some cases we identified more than one Drosophila homolog, so there were a total of 268 genes to test in the HD fly model. To investigate whether the gene expression alterations identified in the mammalian HD transcriptome are likely to be compensatory or pathogenic, we mimicked or antagonized each gene expression alteration in the HD Drosophila model by using knockdown or overexpression alleles and constructs. We then compared the motor performance of these flies with three separate controls: HD flies (HTTN231Q128) with no candidate modifier gene as a positive control, flies with neither mHTT nor a candidate modifier gene as a negative control (these essentially wild-type animals provided a baseline for behavior), and flies bearing only the candidate modifier gene without mHTT as modifier controls.
We tested all the mutant alleles available in public Drosophila strain repositories corresponding to the 268 Drosophila genes for their ability to modify the motor impairment in the HD fruit fly model (Figure 1, Figure S1 and S2); results of testing each modifier in isolation, in the modifier control flies, are shown in Figures S3 and S4. We reasoned that replicating a pathogenic transcriptomic alteration should worsen performance on the climbing assay, whereas replicating a compensatory change should mitigate motor deficits in the Drosophila HD model. Similarly, antagonizing a pathogenic change should improve their motor function, whereas antagonizing a compensatory change should worsen their climbing assay performance (Figure 1). Applying these criteria to the selected ~312 gene expression changes, we were able classify 38 as likely compensatory (Figure 1A–B) and 44 as likely pathogenic (Figure 1C–D) (henceforth referred to as compensatory or pathogenic for the sake of simplicity; Tables S2 and S3).
Figure 1. Functional analysis of HD transcriptomic alterations.

(A) Representative transcriptomic alterations categorized as likely compensatory (the rest of the compensatory genes are shown in Figure S1). Scatter plots on the left show expression levels of the indicated human gene in the caudate nucleus of HD patients with Vonsattel grades 0–2 (reanalyzed after (Hodges et al., 2006) and controls, and the charts on the right show motor performance as a function of age in Drosophila negative controls (blue dashed lines, elavC155>GAL4/w1118), positive controls expressing HTTN231Q128 in neurons (black dotted line, elavC155>GAL4/w1118; UAS-HTTN231Q128/+) and experimental animals (red line, elavC155>GAL4/w1118; UAS-HTTN231Q128/+; modifier/+). CACNB4 is downregulated in HD human brains and neuron-specific knockdown (LOF) of its Drosophila homolog improves the performance of the HD fly model on the climbing assay, so it is classified as compensatory. MAN1A1 is downregulated in HD human brains and neuron-specific overexpression (OE) of its Drosophila homolog worsens HTTN231Q128-induced motor deficits, so it is also classified as compensatory. (B) Bar graph summarizing the integration of the human transcriptomic data (grey bars), Drosophila allele class (arrowhead pointing down denotes reduced function and arrowhead pointing up denotes increased function), and the average % effect of the mHTT-induced motor deficits in Drosophila (blue bars) for the all gene expression changes classified as compensatory. (C) Representative transcriptomic changes we categorized as likely pathogenic (the rest of the pathogenic genes are shown in Figure S2). Neuron-specific knockdown (shRNA) of the Drosophila homolog for ACTN, which is downregulated in human HD brains, worsens the motor deficit. PRPF40A is upregulated in HD human brains, and neuron-specific overexpression (OE) of its Drosophila homolog markedly worsens motor performance. LIMK2 is upregulated in HD human brains and neuron-specific downregulation of its Drosophila homologue improves motor performance in HD flies. (D) Bar graph parallels that in B, except that it is for gene expression changes classified as pathogenic. Green error bars in gene expression scatter plots: average and standard deviation. Error bars in motor performance charts: s.e.m. Significant differences identified using Anova followed by Tukey’s post hoc test for each time point (α=0.05).
Network analysis confirms interactions within the pathogenic and compensatory categories and increasing dysregulation with more severe disease
We next constructed protein-protein interaction networks using the Inweb database (Rossin et al., 2011) and analyzed the properties of the compensatory and pathogenic subnetworks. The modifier genes within each subnetwork had a greater weight than would be expected at random (using as background either the full genome or genes expressed in the striatum), with higher values for node degree (number of connections per gene) and betweeness (number of paths per gene) (Figure 2A–D). Network analysis also showed lower average shortest path length within the compensatory and pathogenic subnetworks compared to the background (Figure 2E–F), indicating that the genes in each subnetwork are more functionally connected to one another than to background genes. Because node and network properties can vary depending on the database used, we also calculated these same attributes using the STRING network (Franceschini et al., 2013), again with both striatal genes and the whole genome as backgrounds; the node and network properties showed similar trends as with Inweb (Figure 2G–J).
Figure 2. Network analysis of the compensatory and pathogenic subnetworks.

(A and B) Subnetworks of the potentially compensatory (A) and pathogenic (B) genes from the HD transcriptome showing their primary interactors. (C) Average node degree (number of edges per node) among the compensatory and pathogenic genes compared to the striatum (Wilcoxon rank sum test, p=5.41e-4 and 6.85e-3, respectively) and whole genome (p=1.38e-4 and 1.81e-3, respectively) backgrounds indicates that the compensatory and pathogenic modifiers are more tightly connected than randomly expected. This is also supported in (D) by the average betweenness (number of shortest paths crossing each node) of compensatory and pathogenic genes compared to striatum (p= 7.58e-5 and 3.33e-4, respectively) or whole genome (p= 1.79e-5 and 7.26e-5, respectively). (E-F) Network properties also show a higher connectivity among the genes in the compensatory and pathogenic subnetworks than would be randomly expected. (E) Shortest path length distribution among the compensatory and pathogenic genes compared to the striatum background and average shortest path length within the compensatory and pathogenic subnetworks compared to the striatal background (p=0 and 9.6e-4 respectively). P calculated by performing a background probability distribution (black line) corresponding to the 1.0e5 randomized samplings we ran using the striatal background genes. (F) Same as E but compared to the whole genome background (p= 0 and 1.3e-4 respectively). P calculated by performing a background probability distribution (black line) corresponding to the 1.0e5 randomized samplings we ran using the striatal background genes from the Inweb network. (G–J) Network properties of compensatory and pathogenic genes using the STRING network. Compensatory changes show the same behavior as with Inweb. Betweenness centrality is significantly increased (Wilcoxon rank sum p= 2.19e-6 for striatum and p= 4.72e-6 for whole genome). Node degree is significantly increased compared to striatum (Wilcoxon rank sum p= 6.42e-6) and whole genome (p= 3.37e-6) and average shortest path is significantly decreased (p=0). For the pathogenic subnetwork the average shortest path is significantly decreased (p=2.6e-4 striatum and 3e-5 whole genome). The betweenness centrality and degree indicators also show a trend to increase, but the Wilcoxon rank sum test does not return statistical significance.
We also investigated the relationship between HD progression and gene expression changes in the compensatory and pathogenic networks. We found that, for most genes, the extent of transcriptomic change correlated with the patient Vonsattel grade, with overall expression changes becoming more prominent with more severe disease (Figure 3). This observation further supports the idea that the 82 modifier genes (38 compensatory, 44 pathogenic) we identified among genes dysregulated in human HD striatum are central to disease progression. Our next step was to investigate the biological processes regulated by genes in the compensatory and pathogenic networks.
Figure 3. Reanalysis of human microarray data for compensatory and pathogenic changes and disease correlation analysis.

(A) Heat map of the gene expression changes in the compensatory network. Gene probes in each panel are organized by increasing degree of correlation with disease stage from bottom to top. Disease stage is indicated at the top, donor identifier at the bottom, and the gene symbol targeted by each probe on the right side. Genes in red font belong to the PLC cascade (PRKCB, PLCB1 and ITPR1). (B) Heat map of the GEP changes in the pathogenic network. Data arranged as in (A). (C and D) Trend lines representing the correlation between aggregate gene expression changes (per individual) with HD stages for each of the panels shown in A and B respectively. Note the tendency of the transcriptomic changes to become more pronounced in each group as the HD grade becomes more severe. Green line represents loess regression and pink shade indicates confidence interval. Each point is the average transcriptomic change for all the probes (in each group, indicated in the title) corresponding to a specific donor (indicated in the x axis). Chart in C corresponds to panel in A, and the same correspondence exists between charts in D and panels B. A–D (reanalyzed after (Hodges et al., 2006)). Correlation of gene expression changes with disease progression was calculated using Spearman’s correlation.
Downregulation of genes involved in synaptic biology and Ca2+ homeostasis are compensatory re- sponses to mitigate the harmful effects of mHTT
Using the DAVID/KEGG database (http://david.abcc.ncifcrf.gov/) we found that neuronal excitation (calcium signaling) is the most enriched category among the changes we classified as likely compensatory (Figure S5A). Ingenuity Pathway Analysis (http://www.ingenuity.com/) revealed that many of these genes form a network involved in synaptic biology and Ca2+ homeostasis (Figure 4A). Although it has been well recognized that impairments in calcium homeostasis and synaptic function are important pathogenic mechanisms in HD (Pchitskaya et al., 2017; Raymond, 2017), it has never been shown that neurons respond by downregulating the expression of genes involved in these cellular processes. Yet the genes involved in synaptic biology and Ca2+ homeostasis show decreased expression in HD brains, and decreasing the activity of the corresponding orthologs clearly improved the motor performance of HD flies (Figure 4B and S1, S3). Note that this motor performance reflects neuronal, not muscle, function (or dysfunction), since mHTT is expressed exclusively in Drosophila neurons.
Figure 4. Compensatory and pathogenic networks.

(A) Functional interaction network of potentially compensatory genes (blue triangles) and modifiers identified through pathway extension (faded green circles) generated using Ingenuity Pathway Analysis. (B) Motor assays of the GPCR cascade genes identified by pathway extension show that when knocked down, they too relieve motor impairments caused by HTTN231Q128-induced neuronal dysfunction. (C) Functional interaction network of 19 genes (orange triangles) in the pathogenic network together with additional modifiers identified through pathway extension (faded green circles). Interactions generated using Ingenuity Pathway Analysis. A more detailed interaction map is shown in (Figure S5F). All genes in bold font also modulate mHTT protein levels in fibroblasts from HD patients (Figure 5). (D) Effect on mHTT-induced motor deficits of additional modifiers identified by pathway extension of genes categorized as pathogenic. In B and D, charts show motor performance as a function of age in Drosophila negative controls (blue dashed lines, elavC155>GAL4/w1118), positive controls expressing HTTN231Q128 in the nervous system (black dotted line, elavC155>GAL4/w1118; UAS-HTTN231Q128/+) and experimental animals (red line, elavC155>GAL4/w1118; UAS-HTTN231Q128/+; modifier/+). For B and D sh: shRNA; LOF: loss of function. Error bars in motor performance charts: s.e.m. Significant differences identified using Anova followed by Tukey’s post hoc test for each time point (α=0.05). Table S5 lists the specific modifier alleles identified through pathway expansion.
Compensatory expression changes were observed in genes with roles in vesicle formation, release and reuptake (i.e., DNM1, PACSIN1, STXBP1 and RAB3A; Figure 4A, area encircled with dotted line), as well as Ca2+ channels and glutamate receptors (e.g., CACNA1, CACNA4 and GRIA) and downstream cascade components (PRKC, PLCB and ITPR1; Figure 4A, shaded oval area). These genes, which are implicated in membrane depolarization, Ca2+ signaling and homeostasis, are among the top genes correlating with disease progression (Figure 3A, red font).
Having identified this network of compensatory alterations in Ca2+ homeostasis and synaptic biology, we performed pathway extension analysis to uncover additional modifiers. We identified three candidate modifiers (Figure 4A, faded green circles)—GNAQ (G- protein-qα), DGKI/Z (diacylglycerol kinase) and TRPC4/5 (Trp type Ca2+ channel)—and found that reducing the expression of their homologs improved the motor performance of the HD Drosophila model (Figure 4B, controls in Figure S5B). Together these data suggest that neurons downregulate the expression levels of glutamate receptors, Ca2+ channels, and downstream signaling genes in the human HD transcriptome to counteract the harmful effects of excitotoxicity.
Alterations in the expression of actin cytoskeleton and inflammation genes drive pathogenesis
Functional enrichment analysis of the modifier genes and their shared primary interactors in the pathogenic network using the DAVID/KEGG database (http://david.abcc.ncifcrf.gov/) revealed enrichment in genes involved in “regulation of actin cytoskeleton,” “inflammation mediated by chemokine and cytokine receptors” and “MAPK signaling pathway.” These pathways, all intricately connected to the inflammatory response, were enriched regardless of whether we used the Inweb or STRING identified in- teractors (Figure 4C and Figure S5E–F).
We again used pathway extension analysis to further investigate these networks and identified the Drosophila homologs of RAC2, PAK1/7, RHOA, IKBKB (catalytic subunit of the IKK complex), NFKB2 and IRAK1 as additional suppressors of mutant HTT-induced behavioral impairments (Figure 4D, Figure S5C; modifier controls in Figure S5D). These additional modifiers form a functional network together with 19 of the genes whose expression changes in the human HD transcriptome were categorized as pathogenic (Figure 4C and Figure S5E–F). At the core of this network are genes involved in the inflammatory response, downstream of the cytokine/chemokine receptor cascade; the network has two interconnected branches, one involved in regulation of the actin cytoskeleton (Figure 4C, shaded area) and one involved in NFKB activation (Figure 4C, area encircled by dotted line, also see http://www.genome.jp/kegg-bin/show_pathway?hsa04062).
The genetic interactions delineated here dovetail nicely with reports of abnormal immune activation and elevated levels of IL-6, TNF-α, and other proinflammatory cytokines in HD mouse models as well as postmortem striatum and plasma from HD patients (Andre et al., 2016; Bjorkqvist et al., 2008; Wild et al., 2011). The present data provide new insight into these previous studies and support the notion that abnormally increased signaling through the NFKB/ actin cytoskeleton regulation pathways drives HD pathogenesis.
Reducing the expression of genes in the inflammatory/ actin cytoskeleton network lowers mHTT levels in patient fibroblast cells and suppresses mHTT toxicity in iPS-derived HD neurons
Next we turned to a human HD cell model to test the validity of the Drosophila data, to further define primary disease drivers, and to gain mechanistic insight. We were particularly interested in the effects of altered expression of the various genes on the accumulation of mHTT, since this is the fundamental driver of pathogenesis. These modifier genes would be particularly interesting from a therapeutic point of view, since reducing mHTT levels in mouse HD models reverses neurodegeneration even at relatively advanced stages (Yamamoto et al., 2000).
We transfected HD patient fibroblasts (HTT[Q68]) with siRNAs targeting each of the 82 human genes (8 individual siRNAs per gene) that are altered in the HD transcriptome and whose Drosophila homologue we defined as a modifier using the behavioral assay. We then monitored mHTT levels in these cells using Homogeneous Time Resolved Fluorescence (HTRF). We used HTRF instead of western blot or ELISA because it provides a more sensitive and quantitative assay of protein levels and lacks the artifacts associated with signal amplification or protein isolation inherent to these techniques (Lotz and Weiss, 2013; Lu et al., 2013; Weiss et al., 2009; Weiss et al., 2012). The HTRF secondary screen was performed in duplicate, and we used scrambled siRNAs as well as siRNAs targeting HTT as negative and positive controls, respectively. Genes affecting cell viability or cell cycle were filtered out (see STAR Methods).
The initial screen in human fibroblasts (HTT[Q68]) identified several hits that consistently reduced mHTT levels (Figure 5A). The hits were then re-tested using a different HD fibroblast line (HTT[45Q]) to minimize the possibility of genetic background effects (Figure 5B). We identified four genes whose knockdown decreased mHTT levels (DACH1, DOCK5, DPYSL3 and NFKBIA), and two that led to increased mHTT (PREPL and GNB5) (Figure 5B). Five of these six genes belong to the pathogenic network (Figures 4C and S5E–F), which suggested that other pathogenic modifiers we identified through network analysis (faded green circles in Figure 4C) might also modulate mHTT levels. Accordingly, we studied these additional genes by using siRNAs followed by HTRF and found that decreasing the levels of PAK1, RAC2/3, NFKB2, the IKK adaptor protein IKBKAP, and the upstream activator IRAK1 reduced mHTT levels in both patient fibroblast lines (Figure 5C; see also (Thompson et al., 2009) for IKK role on HTT clearance).
Figure 5. Knockdown of genes involved in inflammation/ regulation of actin cytoskeleton decreases mHTT protein levels in HD fibroblasts; pathway validation in HD iPS-derived neurons.

(A–B) Analysis of mHTT protein levels in fibroblasts from HD patients. Fibroblasts were transfected with siRNAs targeting the human homologs of the Drosophila genetic modifiers identified among genes altered in the HD transcriptome. (A) Scatter plot summarizing the HTRF screen in HTT[Q68] fibroblasts for genes modulating mHTT protein levels. All 82 human genes whose Drosophila homologs modified motor impairments caused by HTTN231Q128-induced neuronal dysfunction were targeted using 8 siR-NAs per gene. Screen was done in duplicate (experiment-1 and -2). (B) Effect of the hit genes on mHTT levels in HTT[Q68] and HTT[Q45] patient fibroblast lines normalized to negative control. Error bars: standard deviations. (C) Analysis of mHTT protein levels in HD fibroblasts transfected with siRNAs targeting the additional genetic modifiers identified by pathway extension analysis. Data is shown for both HTT[Q68] and HTT[Q45] fibroblast lines. (D) Charts showing average caspase-3 activation following BDNF deprivation as a function of time. Black line: iPS-derived neuron like cells from a HTT[Q47] patient. Red line: HTT[Q47]-derived neurons transfected with siRNAs targeting the indicated gene. Blue line: control iPS-derived neuron like cells. The donor was the sibling of the patient that donated the HTT[Q47] cells. Error bars indicate standard deviation. All the differences shown in B and C where significantly different compared to the corresponding negative controls (using Anova followed by Student’s t test, α=0.05).
To more rigorously assess the relevance of these findings to HD, we tested whether genes in the inflammatory/ actin cytoskeleton network suppress mHTT-induced toxicity in iPS-derived neurons from HD patients, using the previously reported sensitivity of HD iPS-derived neurons to withdrawal of BDNF as a benchmark (HD iPSC Consortium, 2012; Yao et al., 2015). iPS cells were differentiated into Tuj1-positive neuronal-like populations (Figure S6) and then transfected with pools of siRNAs. We specifically targeted PAK1, RAC2, NFKB2, IKBKA and IRAK1 (Figure 5D). Knocking down any of these genes suppressed caspase-3 activation following BDNF withdrawal (Figure 5D and Figure S6B).
The genetic modifiers identified in the Drosophila model system have thus been validated in three different human cell lines (HTTFLQ45, HTTFL68 fibroblasts, and HTTFLQ47 iPS-derived neurons). The results suggest that the genes involved in actin cytoskeleton and NFKB activation form a functional network that drives HD pathogenesis by increasing the levels of mHTT protein.
Decreasing the activity of NFKB2 and RAC2 results in activation of macroautophagy
To ascertain the mechanism by which the identified modifier genes affect mHTT clearance, we investigated the status of the autophagic pathway, which degrades mHTT (Koga et al., 2011; Ravikumar et al., 2002; Sarkar and Rubinsztein, 2008; Yamamoto et al., 2006). There is also abundant evidence that mHTT impairs selective macroautophagy (Cortes and La Spada, 2014; Cuervo and Zhang, 2015; Martin et al., 2014b; Rui et al., 2015).
We used a high-content microscopy approach to assess macroautophagy in cells by monitoring the levels of the autophagy marker LC3. First, we monitored LC3-positive vesicles in HeLa cells after testing siRNAs targeting each of the 12 genes that are able to modulate mHTT levels in the extended pathogenic network (genes in Figure 5B and C). We found that knocking down RAC2 and NFKB2 increased the number of LC3-positive vesicles compared to scrambled siRNA controls (Figure 6A). We validated these results in human fibroblasts from HD patients and control individuals and again found that suppressing NFKB2 and RAC2 increased the number of LC3-positive vesicles (Figure 6B). The levels of the autophagosome-associated LC3II were also monitored by western blot analysis. Consistent with cell immunofluorescence, we found that knocking down NFKB2 and RAC2 led to increased LC3II levels in HTT[Q68] fibroblasts (Figure 6C). Similar results were observed with LC3 upon down-regulation of NFKB1 (Figure 6). Genes that block autophagic degradation can also lead to increased LC3II levels; this was not the case in our assays, since adding the lysosomal blocker bafilomycin resulted in a further increase in the levels of LC3II under all conditions, which suggests that knockdown of target genes leads to induction of basal autophagy (Figure 6C). These observations indicate that at least some of the pathogenesis-driving genes in the cytoskeletal remodeling and NFKB network (Figure 4C) raise mHTT protein levels by hindering autophagic activity in cells.
Figure 6. Knockdown of NFKB2 and RAC2 results in activation of autophagy.

(A) Immunofluorescence (IF) staining showing LC3-positive vesicles and quantification in HeLa cells with decreased levels of NFKB1/2 and RAC2. (B) IF staining and quantification in control (WT) and HD patient (HTT[Q68]) fibroblasts showing the number of LC3-positive vesicles. (C) Western blot (WB) analysis and quantification showing increased levels of the autophagic vesicle bound LC3II in HD patient (HTT[Q68]) fibroblasts with decreased levels of NFKB1/2 and RAC2 in normal conditions (“-” in WB and charts). Also shown is the effect of bafilomycin treatment (“+” in WB and charts). Significant differences were identified using Anova followed by Student’s t test (α=0.05).
Discussion
The main goal of this work was to develop an approach capable of distinguishing compensatory responses from disease-driving alterations, using the multitude of gene expression changes that take place during relatively early stages of HD pathogenesis as a test case. We reasoned that identifying these two distinct classes of molecular changes could provide a framework for interpreting findings from previous and ongoing studies as well as designing effective therapeutic interventions. For example, interventions that antagonize pathogenic gene expression alterations (e.g., actin cytoskeleton/ inflammation) could be rendered ineffectual if they also antagonize compensatory gene expression changes (e.g., Ca2+ homeostasis/ synaptic). Understanding specific pathogenic and compensatory changes should also enable us to develop molecular biomarkers that can monitor the efficacy of treatment interventions.
Admittedly, the 249 genes shared between human and mouse HD microarrays analyzed here represent only a small portion of the transcriptomic alterations present in HD. Much more data, including longitudinal datasets from human tissues and a variety of model systems, will be required to define the HD transcriptome. Nevertheless, our functional analyses demonstrate the power of interrogating even partial transcriptomic data. We had previously shown, for example, that partial loss of function of voltage-gated Ca2+ channels decreases abnormally high Ca2+ levels in the synaptic boutons and mitigates mHTT-induced neurodegeneration (Romero et al., 2008), but the data in the present study strongly indicate that HD neurons reduce the transcription of genes required for Ca2+ entry and Ca2+ signaling to compensate for high intracellular Ca2+ levels. Moreover, given the genetic and pharmacological evidence that excitotoxicity can impair autophagy (Williams et al., 2008), it seems likely that reducing elevated intracytosolic Ca2+ levels would not only reduce excitotoxicity but relieve the impairment of autophagy, thereby retarding HD progression.
It is worth noting that some of the compensatory changes we identified may exert their effect in part by modulating the function of genes in the pathogenic network. For example, the PLCB/PRKC hub, which can be activated downstream of chemokine receptors, can also activate the IKK kinase (Moscat, et al., 2003; Shinohara et al., 2005; Macdonald et al., 1999; Su et al., 2002; see PRKC and PLCB in Figures 4A and 4C). In this scenario, the observed decreases in PLCB/PRKC levels in HD patients would lead to the attenuation of IKK/NFKB signaling in the pathogenic module. Likewise, the observed decreases in ROCK2 in HD brains may lead to the attenuation of LIMK signaling (Govek et al., 2005), another component of the pathogenic network. In sum, understanding the countermeasures neurons deploy against HD pathogenesis will be useful in devising possible therapies.
It is also worth noting that certain inflammation/ actin cytoskeleton network-related genes are primary disease drivers and that reducing their expression suppresses mHTT-induced pathogenesis in several model systems. It is particularly interesting that this suppression takes place by reducing mHTT protein levels, the accumulation of which is a central driver of disease (Jimenez-Sanchez et al., 2017). Therapeutic targets that reduce mHTT levels should influence the myriad downstream pathogenic events that underlie the progression of cognitive, psychiatric and motor symptoms in HD. We predict that targets that can modestly reduce mHTT levels without severely compromising the neuroprotective functions of wild-type HTT hold the greatest therapeutic promise.
Some of the suppressors that reduced mHTT protein levels did so by a mechanism involving macroautophagy. mHTT has been shown to be degraded by both selective macroautophagy (Ravikumar et al., 2002; Sarkar and Rubinsztein, 2008; Yamamoto et al., 2006) and chaperone-mediated autophagy (Koga et al., 2011), and among mHTT-expressing neurons those showing greater autophagy flux tend to live longer (Tsvetkov et al., 2013). Wild-type HTT functions as a scaffold for selective autophagy (Martin et al., 2014b; Ochaba et al., 2014; Rui et al., 2015), whereas mHTT impairs autophagy (Cortes and La Spada, 2014; Martin et al., 2014a). mutant HTT sequesters mTOR, which leads to induction of autophagy (Ravikumar et al., 2004) and a rise in autophagosome numbers (Kegel et al., 2000; Martinez-Vicente et al., 2010; Petersen et al., 2001), but the autophagosomes in HD neurons cannot efficiently trap cargo for degradation (Martinez-Vicente et al., 2010). Moreover, mHTT impairs vesicle trafficking, disrupting au-tophagosome and lysosome dynamics (del Toro et al., 2009; Wong and Holzbaur, 2014). The suppressors identified here that reduce mHTT protein levels may thus work by alleviating mHTT-induced autophagy impairments.
We were surprised, in fact, to find that knockdown of specific inflammation/ actin cytoskeleton genes reduced mHTT levels by inducing autophagy. It is possible that these genes have other, unknown, functions directly related to protein turnover, but there is abundant evidence linking autophagy and inflammation—in both directions. Autophagy is suspected to be defective in a number of inflammatory diseases, such as Crohn’s disease, systemic lupus erythematosus, arthritis and sepsis (reviewed in (Choi and Ryter, 2011; Jones et al., 2013)); it could be that chronic inflammation eventually leads to impairment of autophagy. On the other hand, autophagy seems to be one cellular mechanism for controlling inflammation, insofar as it can degrade cytokines (Harris et al., 2011; Shi et al., 2012), inflammasome components (Shi et al., 2012), and cellular factors causing inflammasome activation (Nakahira et al., 2011; Zhou et al., 2011). The work presented here suggests that dampening the response of specific genes in the inflammatory network could, in fact, enhance autophagy. Given that much of the very large literature on neuroinflammation has focused on glia (Crotti and Glass, 2015; Andre, 2016), many questions remain to be answered regarding the role of inflammation genes in neurons themselves.
The wide variance observed in age of onset for any given polyglutamine repeat length strongly suggests the existence of HD modifier genes (Wexler et al., 2004). Conceivably, some human modifiers could affect the expression or activity of the same genes that we showed here as modifier genes. Our data overall indicate that antagonizing pathogenic variants (which in human could be identified as being associated with earlier age of onset) could prove to be as or even more beneficial than mimicking compensatory variants (i.e., human modifiers associated with later age of onset).
The analysis of HD transcriptomic alterations described here also suggests that functional querying of disease gene expression datasets should expedite the identification of therapeutic targets for many other disorders for which such data are available. The ability to distinguish between compensatory and pathogenic transcriptomic alterations should also facilitate the search for candidate therapeutic compounds through network pharmacology (Hopkins, 2008; Lamb et al., 2006; Pang et al., 2014), because drugs reversing pathogenic but not compensatory alterations should be most efficacious. The same approach to classifying compensatory and pathogenic alterations could be used for querying the significance of proteomic or metabolomic alterations during disease.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Dr. Juan Botas (jbotas@bcm.edu).
Experimental model and subject details
Drosophila melanogaster
The HTTN231Q128 Drosophila HD model, which pan-neurally expresses an N-terminal fragment of mHTT with 128Q under the elavC155>GAL4 driver, has been previously described (Branco et al., 2008). Mutant strains for screening the HD-transcriptional changes were obtained from Bloomington Drosophila Stock Center at Indiana University and the Vienna Drosophila RNAi Center at Vienna, Austria. Strains were maintained at 18°C in standard molasses, yeast extract, agar media until their experimental use. All experiments behavioral experiments were done on females raised at 28°C.
Cultured cells
We used Human fibroblasts obtained from Coriell Cell Repositories (GM02153-female for wt, GM03868-female HTT[Q45] and GM21757-male HTT[Q68]) and immortalized by SV40 large T-antigen. Cells were cultured in MEM (Life Technologies, #10370) with 15% FBS (Life Technologies, #10082-147) plus 1X GlutaMax (Life Technologies, #35050079) and transfected with each siRNA at a final concentration of 20nM. (See Table S6 for all siRNAs used here). We also used for the high-content assay to quantify LC3-positive vesicles, HeLa Cells (female).
iPSC-derived neuron-like cells
We generated HD iPSC lines and iPSC-derived neuron-like cells as previously described (Yao et al., 2015). Briefly, iPSCs were differentiated to Pax6-expressing primitive neuroepithelia (NE) for 10–12 days in a neural induction medium. For neuronal differentiation, neural progenitor clusters were dissociated and placed onto laminin-coated coverslips at day 26 in Neurobasal medium, with a set of trophic factors, including brain-derived neurotrophic factor (BDNF, 20 ng/ml, Protech, cat. no. 450-02), glial-derived neurotrophic factor (GDNF, 10 ng/ml, Protech, cat. no. 450-10), insulin-like growth factor 1 (IGF1, 10 ng/ml, Protech, cat. no. 100-11), and Vitamin C (Sigma cat. no. D-0260, 200 ng/ml). All the cells were maintained at 37 °C in an incubator with 5% CO2. All the cells were tested for mycoplasma contamination and confirmed to be clear. iPSC-derived neuron-like cells from HD patients and controls (non-HD sibling) were cultured in NIM (1% N2 in DMEM:F12).
Method details
Drosophila genetics and motor performance analysis
For motor performance tests, we used 15 age-matched virgin females per assay. Animals were tapped to the bottom of a 20cm long tube and the number of animals climbing past 9cm in 16 seconds was scored. For each genotype we used 4 replicates of 15 females, and we performed 10 consecutive trials/replica/day as shown in the charts. The specific alleles that caused modification for each hit gene are listed in Tables S2 and S3.
There were three controls for motor performance tests: 1) animals with no modifier gene (elavC155>GAL4/w1118; UAS-HTTN231Q128/+, positive controls); 2) animals not expressing mHTT (elavC155>GAL4/w1118, negative controls); 3) animals carrying the possible modifier but not expressing mHTT (elavC155>GAL4/w1118; modifier allele/+, modifier controls). The latter controls allowed us to better evaluate possible modifier genes and assess their potential compensatory or pathogenic effects in isolation from mHTT. Table S2 lists the human genes whose expression alterations we categorized as compensatory; Table S3 lists the human genes whose changes in HD we categorized as pathogenic.
HTRF analysis for mutant HTT levels in patient fibroblasts
Human fibroblasts were transfected with each siRNA at a final concentration of 20nM. (See Table S6 for all siRNAs used here.) For HTRF assays, the immortalized patient fibroblasts were cultured and transfected in 384-well plates. The medium was then aspirated with plate washer so that ~6 μl medium remained in each well of the 384-well plates. The antibody mixes were then prepared by adding the donor and acceptor antibodies into the assay buffer containing 50 mM NaH2PO4, 400 mM NaF, 0.1% bovine serum albumin (BSA), and 0.05% Tween 20, with an additional 1% Triton X-100 and 2X EDTA-free protease inhibitor (Calbiochem, #535140) to lyse the cells. For all the antibody pairs used for this study, the donor antibody 2B7-Tb concentration is 0.023 ng/μl and the acceptor antibody MW1-D2 concentration is 1.4 ng/μl in the antibody mix. 6 μl of the antibody mix were then added into each well of the 384-well plates and incubated at 4°C overnight using the Envision 2104 (PerkinElmer) HTRF reader using the HTRF protocol. This protocol gives an excitation pulse at 337 nM and then measures the fluorescent intensities at both 665 nM and 615 nM in a 400 μs time window after 150 μs delay.
For each sample, 30 repeated cycles of the excitation measurement (flashes) were performed with an interval of 16600 μs (the default setting by the HTRF reader), and the average intensities at each wavelength of 30 flashes were recorded by the HTRF reader for calculations. The ratio of the averaged 665 nm signals and 615 nm signals were calculated to obtain the raw HTRF signals. The signals were then blank-corrected by subtracting the average raw HTRF signals from the well without any cells. The blank-corrected signals were then normalized to Z scores based on the negative siRNA control samples: Z = (sample signal – median signal of negative siRNA samples on the same plate) / s.d. of negative siRNA samples in the library). Z > 2 or Z < 2 (typically more than 15 % changes of the blanked corrected HTRF signals) were used as the threshold. siRNAs that led to consistently above-threshold changes were selected. Paralleled transfections were performed and all the wells were tested with the Cell titer-glo (CTG) assays to measure the cell number in each well. To ensure that the HTRF signal change was not due to the cell number changes, we excluded the siRNAs that changed the CTG signals by more than 5% in the same direction as the change of the HTRF signals. The genes with at least two different siRNAs that met the above standard and showed consistent change were considered to be primary hits.
Immunofluorescence for LC3 in HeLa cells and human fibroblasts
For the high-content assay to quantify LC3-positive vesicles, HeLa cells were reverse transfected with pools of 4 siRNA oligos (against modifier genes) using Hamilton STARTLet liquid handling platform and plated in 96-well plates for 72 h. Then, cells were washed and fixed in methanol. Standard immunofluorescence experiments to detect LC3 protein were performed using specific LC3 antibodies (Novus Biologicals). The nuclei of cells were stained with Hoeshct solution for ten minutes. Images were taken by using the Opera high content system (Perkin Elmer). The images were analyzed by using a developed script in the analysis software Harmony (Perkin Elmer). Plots represent number of spots (LC3-positive vesicles) per cell. The data were analyzed and plotted using Prism software. A similar protocol was used for human fibroblasts, and nuclei were stained with DAPI.
Western blot in fibroblasts
After 72h of silencing with the selected siRNA oligonucleotides, human WT and HD [Q68] fibroblast cells were either left untreated or treated for 4 hours with 100nM Bafilomycin-A1 (Sigma). The cells were lysed with RIPA or NP40 buffer containing protease inhibitors (Roche). The samples were collected in 5x SDS-sample buffer and separated by 13% SDS-PAGE. Immunoblotting analysis was performed by transferring proteins onto Immobilon-P PVDF (Millipore) membrane (for LC3) or Nitrocellulose (Biorad) for HTT. The membrane was blocked with 5% non-fat milk in TBS-T and incubated for 1 hour with primary antibody anti-actin (Sigma; 1:2000) and then over-night with anti-LC3 (Covance; 1:1000). Membranes were washed three times with TBS-T and incubated with HRP-conjugated secondary antibodies (Pierce) at room temperature for 1 hour. Membranes were washed two times with TBS-T and once in TBS and the expressed proteins were visualized using the ECL Western Blotting Substrate (Pierce). Image acquisition was done using a Chemidoc UVP imaging system (UltraViolet Products Ltd). Images were analyzed using ImageJ software, and plots correspond to one representative example of at least three experiments.
iPSC-derived neurons: generation, culture and caspase assay
iPSC-derived neuron-like cells from HD patients and controls (non-HD sibling) were cultured in NIM (1% N2 in DMEM:F12) for 48 hrs after transfection with pools from the HTRF hit siRNAs (including HTT3 and Scramble controls), before BDNF removal. Phenotype was detected by the caspase activity assay from NucView 488 caspase-3 dye (Biotium, cat. no. 30029) used as indicated on manufacturer’s protocols. We measured caspase activation as a function of time.
Quantification and statistical analysis
Statistical tests and chart representations were performed using jmp8 software. For gene expression analysis we used the indicated R suites (Key Resources Table).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HTT | Novartis | 2B7 |
| Mouse monoclonal anti-polyglutamine | Hybridoma Bank | MW1 |
| Rabbit anti-LC3 | Novus Biologicals | Cat#NB100-2220 |
| Mouse anti-LC3 | Biolegend (Covance) | Cat#848801 |
| Mouse anti-actin | Sigma | A5441 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| MEM cell culture media | Life Technologies | Cat#10370 |
| Fetal Bovine Serum | Life Technologies | Cat#10082-147 |
| GlutaMax | Life Technologies | Cat#35050079 |
| BDNF | Protech | Cat#450-02 |
| GDNF | Protech | Cat#450-10 |
| IGF1 | Protech | Cat#100-11 |
| Vitamin C | Sigma | Cat#D-0260 |
| EDTA-free protease inhibitor | Calbiochem | Cat#535140 |
| Critical Commercial Assays | ||
| Pierce ECL Western Blotting Substrate | Thermofisher scientific | Cat#32132 |
| Caspase activity assay NucView | Biotium | Cat#30029 |
| Deposited Data | ||
| GSE3790 - Postmortem human HD microarray data | Hodges et al., 2006 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3790 |
| GSE10202 - CHL2Q150/Q150 microarray data | Kuhn et al., 2007 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10202 |
| GSE9038 - HdhQ111/HdhQ111 microarray data | Fossale et al., 2011 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9038 |
| Microarray data from 9 month old R6/2 mice | Strand et al., 2007 | http://www.ncbi.nlm.nih.gov/geo |
| GSE65776 – Rnaseq from 6- and 10-month-old HdhQ92, HdhQ111, HdhQ140 and HdhQ175 mice | Langfelder et al., 2016 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65776 |
| Experimental Models: Cell Lines | ||
| Wild-type female fibroblast line | Coriell cell repositories | GM02153 |
| HTT[Q45] female fibroblast line | Coriell cell repositories | GM03868 |
| HTT[Q68] male fibroblast line | Coriell cell repositories | GM21757 |
| iPS-derived neurons | Boxun Lu laboratory | Yao et al., 2015 |
| Experimental Models: Organisms/Strains | ||
| P{GawB}elav[C155] | Bloomington Drosophila stock center | 458 |
| UAS-HTTN231Q128 | Botas Lab | Branco et al., 2008 |
| Classical loss of function and over expression Drosophila mutants | Bloomington Drosophila stock center | summarized in Tables-S2, S3, S5 |
| Drosophila shRNAs | VDRC | summarized in Tables-S2, S3, S5 |
| Oligonucleotides | ||
| siRNAs | Dharmacon | indicated in Table-S6 |
| siRNAs | Qiagen | indicated in Table-S6 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| DESeq2 R package | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| jmp8 | jmp Statistical Discovery. From SAS. | https://www.jmp.com/en_us/software.html |
| Other | ||
Gene expression analysis (human and murine)
Gene expression profiles for postmortem human tissue were obtained from GSE3790 (Hodges et al., 2006). We employed a cutoff of Benjamini & Hochberg (BH) adjusted p-value less than 0.1 (Student t-test) and fold-change greater than 1.5 to select differentially expressed genes in the caudate nucleus of the human brain samples from control (N=32) and Vonsattel grades 0-2 HD brains (N=32). HD Vonsattel grades were extracted from the annotation file GSE3790. Spearman correlation coefficients between gene expression and HD Vonsattel grades were computed for each of the probe sets. A hierarchical clustering on the correlated genes was computed using Euclidian distance and complete link functions.
Gene expression profiles for the striatum from mice bearing full-length HTT were obtained from GSE10202 (CHL2Q150/Q150) (Kuhn et al., 2007) and GSE9038 (HdhQ111/HdhQ111) (Fossale et al., 2011) at http://www.ncbi.nlm.nih.gov/geo. Gene expression profiles for the striatum of R6/2 mice at 9 weeks of age (Strand et al., 2007) were obtained from hdbase.org. There are 22,690 probe sets on the affymetrix MOE430A 2.0 arrays. A Student t-test with BH adjusted p-value threshold at 0.2 and fold-change greater than 1.2 were used to extract genes that are differentially expressed between the wild-type controls and the mouse samples.
For re-analyses of the RNAseq data published in (Langfelder et al., 2016), gene expression fold-changes from 6- and 10-month-old HdhQ92, HdhQ111, HdhQ140 and HdhQ175 mice (Table S1) were calculated using the DESeq2 R package (Love et al., 2014), with the raw counts as input and a false discovery rate of 0.1. Table S1 lists the genes whose expression is altered early in human HD progression and in at least one HD mouse model; the direction of the alteration in each species is also indicated. Note that for certain genes, the direction of changes is not the same between models.
Drosophila motor performance test
We plotted for each day the average % of animals climbing past 9cm. Significant differences were assessed performing ANOVA followed by Tukey’s post hoc test for each time point. N=15 individuals and 10 trials per day represented.
Network and pathway analysis
The InWeb network (version 1.0) was downloaded from http://www.broadinstitute.org/mpg/dapple/. The network contains 12,348 nodes and 158,377 edges (network contained 31 of 38 compensatory genes and 36 of 44 pathogenic genes). The STRING network (version 9.1) was downloaded from http://string-db.org/ and only gene interactions with a confidence score greater than 0.5 were used. The network contains 16,203 nodes and 312,491 edges (network contained 36 of 38 compensatory genes and 41 of 44 pathogenic genes).
Node degree is computed as the number of edges connected to the node in a network. Average shortest path length is computed as the average number of steps along the shortest paths for all possible pairs of a set of nodes. Betweenness centrality is computed as the number of shortest paths from all nodes to all others that pass through that node (Newman, 2010). For comparison of degree and betweenness with whole genome background, we used 12,275 genes in the InWeb network or 16,120 genes in the STRING network that are not among the compensatory or pathogenic genes. We evaluated significance using the ‘Wilcoxon rank sum test,’ and the value is indicated in each figure.
To measure whether the compensatory and pathogenic genes have smaller shortest path length than would be expected by chance, we randomly sampled 39 genes from the Inweb and STRING networks and calculated their pairwise shortest path length and average shortest path length. We repeated this sampling 100,000 times, and obtained the random distributions of the pairwise average shortest path length shown in Figure 2E, F, I and J, then we plotted the value for compensatory or pathogenic network and calculated the p-value.
To calculate the network properties of the striatally-expressed genes we followed the same procedures as above. The genes expressed in the human striatum where identified by downloading RNAseq data from human samples from the Allen Brain Atlas (http://www.brain-map.org/) site (donor10021 and donor9861). We averaged expression in the striatum across the different samples (n=16) and eliminated all those genes whose expression was 0. To be on the safe side and ensure that every gene picked was truly expressed, we also eliminated the genes in the bottom 20% quantile. In the Striatum gene expression dataset, we mapped 10,023 genes to Inweb and 12,316 to STRING.
Pathway enrichment was calculated using the tools in DAVID/KEGG database (http://david.abcc.ncifcrf.gov/). The EASE score (a modified Fisher Exact Test, https://david.ncifcrf.gov/helps/functional_annotation.html) was used for hypergeometric testing. This was followed by the Benjamini correction for multiple hypothesis test adjustment. Data for direct genetic and functional interactions among nodes, used to build Figure 4A and C and Figure S5E–F and to perform the pathway expansion to identify additional genetic modifiers, were obtained from the DAVID/KEGG and IPA (Ingenuity Pathway Analysis) databases. Network figures were built using Cytoscape (Shannon et al., 2003).
Table S4 lists the pathways enriched among genes presenting direct protein-protein interactions in the compensatory network; Table S4 also lists the pathways enriched among the pathogenic network.
HTRF in fibroblasts
The blank-corrected signals were normalized to Z scores based on the negative siRNA control samples: Z = (sample signal – median signal of negative siRNA samples on the same plate) / s.d. of negative siRNA samples in the library). Z > 2 or Z < 2 (typically more than 15 % changes of the blanked corrected HTRF signals) were used as the threshold. siRNAs that led to consistently above-threshold changes were selected.
Data and Software Availability
Raw data from Drosophila, HD patient cells and bioinformatic analysis are available on request.
Supplementary Material
For the majority of these genes (67%), the direction of the transcriptional alteration (up or down) is the same between HD patients and at least one of the mouse HD models, i.e., not all models show transcriptional change in the same direction.
Highlights.
Neurons implement changes at the transcriptional level to counteract excitotoxicity
Elevated NFKB/actin cytoskeletal signaling increases mHTT levels, driving disease
Downregulating inflammatory pathways reduces mHTT protein and neurotoxicity
Antagonizing HD-related expression changes in NFKB/ RAC genes enhances autophagy
Acknowledgments
We thank Vicky Brandt for critical input on the manuscript and Huda Y. Zoghbi, Mirjana Maletic-Savatic, Joshua Shulman and Alejandro Botas for thoughtful discussions. This work was supported by grants to JB from CHDI, the Robert A. and Renée E. Belfer Family Foundation and NIH grant NS42179. IA was supported by the Hereditary Disease Foundation, the Darrell K Royal Research Fund for Alzheimer’s Disease, NRSA-NS043124 (NIH) and R21NS096395 (NIH). ZL is supported by NSF-DMS1263932. BL is sponsored by the Chinese Ministry of Science and Technology (2014AA02502) and National Natural Science Foundation of China (31371421, 31422024). The project was supported in part by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. Cores: Human Cell Lines.
Footnotes
Author Contributions
I.A. and J.B. conceived the study. I.A., B.L., J.P., Z.L., D.L.M. and J.B. contributed with experimental design. I.A., B.L., S.D., K.P., I.P., T.G, N.T.M., K.E., G.F, L.E.R., J.D., M.d.H., B.A.B, M.V., M.H, A.M.L. and Z.L. performed the experiments. I.A., and J.B. wrote and edited the paper with contributions of B.L. and D.L.M.
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Ament SA, Pearl JR, Grindeland A, St Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, et al. High resolution time-course mapping of early transcriptom-ic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet. 2017;26:913–922. doi: 10.1093/hmg/ddx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre R, Carty L, Tabrizi SJ. Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr Opin Pharmacol. 2016;26:33–38. doi: 10.1016/j.coph.2015.09.008. [DOI] [PubMed] [Google Scholar]
- Bando SY, Silva FN, da Costa LF, Silva AV, Pimentel-Silva LR, Castro LH, Wen HT, Ama-ro E, Jr, Moreira-Filho CA. Complex network analysis of CA3 transcriptome reveals pathogenic and compensatory pathways in refractory temporal lobe epilepsy. PLoS One. 2014;8:e79913. doi: 10.1371/journal.pone.0079913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco J, Al-Ramahi I, Ukani L, Perez AM, Fernandez-Funez P, Rincon-Limas D, Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- Choi AJ, Ryter SW. Autophagy in inflammatory diseases. Int J Cell Biol. 2011;2011:732798. doi: 10.1155/2011/732798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19:963–971. doi: 10.1016/j.drudis.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, Glass CK. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015;36(6):364–73. doi: 10.1016/j.it.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Zhang S. Selective autophagy and Huntingtin: learning from disease. Cell Cycle. 2015;14:1617–1618. doi: 10.1080/15384101.2015.1039365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Toro D, Alberch J, Lazaro-Dieguez F, Martin-Ibanez R, Xifro X, Egea G, Canals JM. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineu-rin/Rab8 complex from the Golgi apparatus. Mol Biol Cell. 2009;20:1478–1492. doi: 10.1091/mbc.E08-07-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossale E, Seong IS, Coser KR, Shioda T, Kohane IS, Wheeler VC, Gusella JF, MacDonald ME, Lee JM. Differential effects of the Huntington’s disease CAG mutation in stri-atum and cerebellum are quantitative not qualitative. Hum Mol Genet. 2011;20:4258–4267. doi: 10.1093/hmg/ddr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19(1):1–49. doi: 10.1101/gad.1256405. Review. [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28(8):425–33. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Go-lenbock DT, Tschopp J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, O’Callaghan B, Zoghbi HY, Henzler C, Orr HT. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron. 2016;89:1194–1207. doi: 10.1016/j.neuron.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb Perspect Med. 2017;7 doi: 10.1101/cshperspect.a024240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol. 2013;91:250–258. doi: 10.1038/icb.2012.82. [DOI] [PubMed] [Google Scholar]
- Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Hun-tingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31:18492–18505. doi: 10.1523/JNEUROSCI.3219-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pou-ladi MA, Sathasivam K, Cha JH, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016;19:623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz GP, Weiss A. Immuno-based detection assays to quantify distinct mutant huntingtin conformations in biological samples. Methods Mol Biol. 2013;1017:163–171. doi: 10.1007/978-1-62703-438-8_12. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Al-Ramahi I, Valencia A, Wang Q, Berenshteyn F, Yang H, Gallego-Flores T, Ichcho S, Lacoste A, Hild M, et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci. 2013;16:562–570. doi: 10.1038/nn.3367. [DOI] [PubMed] [Google Scholar]
- Lu XH, Mattis VB, Wang N, Al-Ramahi I, van den Berg N, Fratantoni SA, Waldvogel H, Greiner E, Osmand A, Elzein K, et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. 2014;6:268ra178. doi: 10.1126/scitranslmed.3010523. [DOI] [PubMed] [Google Scholar]
- Macdonald NJ, Perez-Polo JR, Bennett AD, Taglialatela G. NGF-resistant PC12 cell death induced by arachidonic acid is accompanied by a decrease of active PKC zeta and nuclear factor kappa B. J Neurosci Res. 1999;57(2):219–26. doi: 10.1002/(SICI)1097-4547(19990715)57:2<219::AID-JNR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Martin DD, Heit RJ, Yap MC, Davidson MW, Hayden MR, Berthiaume LG. Identification of a post-translationally myristoylated autophagy-inducing domain released by caspase cleavage of huntingtin. Hum Mol Genet. 2014a;23:3166–3179. doi: 10.1093/hmg/ddu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2014b;38:26–35. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autoph-agy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Rennert P. NF-kappaB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003;4(1):31–6. doi: 10.1038/sj.embor.embor704. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Im-munol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M. Networks: An Introduction. 1. Oxford: University Press, Inc; 2010. [Google Scholar]
- Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L, Mao K, Lau AL, Yeung SY, Humbert S, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014;111:16889–16894. doi: 10.1073/pnas.1420103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang K, Wan YW, Choi WT, Donehower LA, Sun J, Pant D, Liu Z. Combinatorial therapy discovery using mixed integer linear programming. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 2017 doi: 10.1016/j.ceca.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1243–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Raymond LA. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem Biophys Res Commun. 2017;483:1051–1062. doi: 10.1016/j.bbrc.2016.07.058. [DOI] [PubMed] [Google Scholar]
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron. 2008;57:27–40. doi: 10.1016/j.neuron.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EY, Wexler EM, Versano R, Coppola G, Gao F, Winden KD, Oldham MC, Martens LH, Zhou P, Farese RV, Jr, et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, Daly MJ. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by au-tophagy. FEBS J. 2008;275:4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara H, Yasuda T, Aiba Y, Sanjo H, Hamadate M, Watarai H, Sakurai H, Kurosaki T. PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J Exp Med. 2005;202(10):1423–31. doi: 10.1084/jem.20051591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Cop-pola G, Horvath S, Loo JA, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Koo-perberg C, Olson JM, et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroedicke M, Bounab Y, Strempel N, Klockmeier K, Yigit S, Friedrich RP, Chaurasia G, Li S, Hesse F, Riechers SP, Russ J, Nicoletti C, Boeddrich A, Wiglenda T, Haenig C, Schnoegl S, Fournier D, Graham RK, Hayden MR, Sigrist S, Bates GP, Priller J, Andrade-Navarro MA, Futschik ME, Wanker EE. Systematic interaction network filtering identifies CRMP1 as a novel suppressor of huntingtin misfolding and neurotoxicity. Genome Res. 2015;25(5):701–13. doi: 10.1101/gr.182444.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, Teitell M, Leitges M, Kawakami T, Rawlings DJ. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3(8):780–6. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013;9:586–592. doi: 10.1038/nchembio.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A, Abramowski D, Bibel M, Bodner R, Chopra V, DiFiglia M, Fox J, Kegel K, Klein C, Grueninger S, et al. Single-step detection of mutant huntingtin in animal and human tissues: a bioassay for Huntington’s disease. Anal Biochem. 2009;395:8–15. doi: 10.1016/j.ab.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Weiss A, Trager U, Wild EJ, Grueninger S, Farmer R, Landles C, Scahill RI, Lahiri N, Haider S, Macdonald D, et al. Mutant huntingtin fragmentation in immune cells tracks Hun-tington’s disease progression. J Clin Invest. 2012;122:3731–3736. doi: 10.1172/JCI64565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayán J, et al. Venezuelan kindreds reveal that genetic and envronmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. 2004;101(10):3498–503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi SJ, Bjorkqvist M. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS Curr. 2011;3:RRN1231. doi: 10.1371/currents.RRN1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yao Y, Cui X, Al-Ramahi I, Sun X, Li B, Hou J, Difiglia M, Palacino J, Wu ZY, Ma L, et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. Elife. 2015;4 doi: 10.7554/eLife.05449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflam-masome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For the majority of these genes (67%), the direction of the transcriptional alteration (up or down) is the same between HD patients and at least one of the mouse HD models, i.e., not all models show transcriptional change in the same direction.