

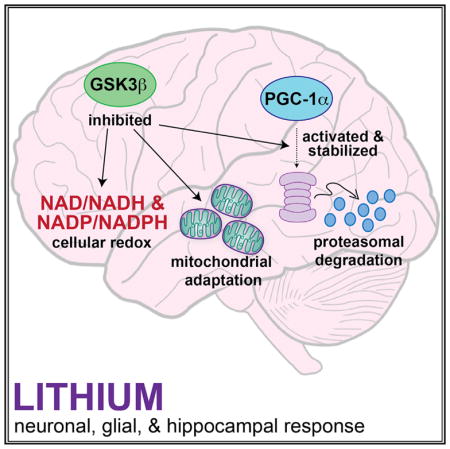
SUMMARY
GSK3β is a serine threonine kinase implicated in the progression of Alzheimer’s disease. Although the role of GSK3β in growth and pathology has been extensively studied, little is known about the metabolic consequences of GSK3β manipulation, particularly in the brain. Here, we show that GSK3β regulates mitochondrial energy metabolism in human H4 neuroglioma cells and rat PC12-derived neuronal cells and that inhibition of GSK3β in mice in vivo alters metabolism in the hippocampus in a region-specific manner. We demonstrate that GSK3β inhibition increases mitochondrial respiration and membrane potential and alters NAD(P)H metabolism. These metabolic effects are associated with increased PGC-1α protein stabilization, enhanced nuclear localization, and increased transcriptional co-activation. In mice treated with the GSK3β inhibitor lithium carbonate, changes in hippocampal energy metabolism are linked to increased PGC-1α. These data highlight a metabolic role for brain GSK3β and suggest that the GSK3β/PGC-1α axis may be important in neuronal metabolic integrity.
In Brief
Martin et al. demonstrate that GSK3β is a regulator of energy metabolism in the brain. They show that GSK3β inhibition stimulates mitochondrial regulator PGC-1α and leads to activation of mitochondrial and redox pathways in glia, in neurons in culture, and in the hippocampus in mice in vivo.
INTRODUCTION
Many of the most common neurodegenerative disorders share a phenotype of protein aggregation and proteostatic crisis that ultimately lead to neuronal loss (Hetz and Mollereau, 2014); however, these disorders also exhibit a common phenotype of mitochondrial dysfunction (Schon and Przedborski, 2011). Mitochondrial efficiency is critical in maintaining neuronal function and plasticity (Yin et al., 2014), and mitochondrial integrity is an essential component in learning/memory (Pei et al., 2015). GSK3β (glycogen synthase kinase 3 beta) is a growth-signaling-sensitive kinase negatively regulated by inhibitory phosphorylation downstream of the insulin receptor, Wnt, and mTOR growth signaling pathways (Patel and Woodgett, 2017). Genetic studies demonstrate a mechanistic role for GSK3β in memory, behavior, and neuronal fate determination (Beurel et al., 2015; Kaidanovich-Beilin and Woodgett, 2011). GSK3β has also been implicated in Alzheimer’s disease (AD), where activation of GSK3β can promote tau hyperphosphorylation, neurofibrillary tangles, and amyloid plaques (DaRocha-Souto et al., 2012; Serenó et al., 2009). In contrast, levels of GSK3β are lower in the brains of monkeys that are protected from age-related brain atrophy by the dietary intervention of caloric restriction (CR) (Colman et al., 2009; Martin et al., 2016).
GSK3 was originally identified as an insulin sensitive kinase involved in the activation of glycogen synthesis (Parker et al., 1983). Two isoforms of GSK3, GSK3α and GSK3β, have been identified (Woodgett, 1990) that have distinct non-redundant functions: global knockout of GSK3α is phenotypically silent (Patel et al., 2011), whereas global knockout of GSK3β is embryonic lethal (Hoeflich et al., 2000). We and others have identified a role for GSK3β in regulating the stability and activity of PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), a key regulator of mitochondrial function (Anderson et al., 2008; Olson et al., 2008); however, the involvement of GSK3β specifically in energy metabolism is not well defined. In this study, we utilize lithium, a robust GSK3β inhibitor (Klein and Melton, 1996; Stambolic et al., 1996) and common psychiatric drug used in the treatment of schizophrenia and bipolar disorder (Geddes et al., 2004), to determine the role of GSK3β brain cell energy metabolism.
RESULTS
GSK3β Inhibition Stimulates Glial Mitochondrial Energy Metabolism
Human H4 neuroglioma cells were treated with lithium chloride (15 mM), a direct inhibitor of GSK3β (Zhang et al., 2003). Consistent with a growth regulatory role of GSK3, GSK3β inhibition by lithium negatively affected cellular growth in the absence of a difference in cell viability (Figures S1A and S1B). An increase in mitochondrial membrane potential was induced by 24 hr of lithium treatment (Figure 1A) in a dose-dependent manner (Figure S1C). Inhibitor VIII (15 μM), a selective non-competitive inhibitor of GSK3β, also induced an increase in mitochondrial membrane potential in H4 glioma (Figure 1B). To determine if the metabolic effects of lithium were GSK3β dependent, we used small interfering RNA (siRNA) to knockdown GSK3β at the RNA and protein levels (Figure S1D). GSK3β knockdown significantly increased mitochondrial membrane potential, phenocopying the effect of lithium, but abrogated the increase in membrane potential induced by lithium treatment confirming the requirement for GSK3β in lithium’s metabolic effects (Figure 1C). The metabolic impact of inhibitor VIII was similarly disrupted by knockdown of GSK3β (Figure S1E). Furthermore, expression of constitutively active GSK3β with the phosphorylation site serine 9 mutated to alanine (Figure S1F), significantly decreased mitochondrial membrane potential in H4 glioma, and abrogated lithium’s ability to increase mitochondrial membrane potential (Figure 1D). Lithium induced an increase in mitochondrial respiration (Figure 1E), with basal respiration, maximal respiration, and spare capacity significantly higher in treated cells (Table S1). Despite subtle differences in the impact of lithium and inhibitor VIII on membrane potential, the effect of GSK3β inhibition by either modality on respiration was equivalent and non-additive (Figure S1G) and was phenocopied by siRNA against GSK3β (Figure S1H). In terms of the broader metabolic effects, lithium induced an increase in free intracellular levels of co-factor NAD+ as detected by biochemical assay, although redox ratios were not significantly different (Figures 1F and S1I). These metabolic changes occurred in the absence of overt differences in mitochondrial abundance: mitochondrial content, as assessed by staining for the outer membrane protein Tomm20 (translocase of outer mitochondrial membrane 20) was not different (Figures 1G and S1J), and levels of proteins involved in the electron transport system (ETS) were unchanged (Figure S1K). These data indicate that mitochondria were activated but biogenesis was not induced. At the protein level, lithium-induced inhibitory phosphorylation of GSK3β at serine 9, confirming efficacy of lithium-induced GSK3β inhibition (Figures 1H and S1L). Coincident with this, a 1.3-fold significant increase in PGC-1α protein levels was detected in lithium-treated cells, and although the impact of inhibitor VIII on PGC-1α levels was more modest, treatment with both GSK3β inhibitors was not additive (Figure S1M). A difference in PGC-1α cellular distribution was evident upon knockdown of GSK3β with an apparent increase in nuclear PGC-1α (Figure S1N).
Figure 1. GSK3β Regulates Mitochondrial Metabolism and PGC-1α Stability, Localization, and Activity in H4 Glioma.

(A–D) JC-1 measurement of mitochondrial membrane potential following (A) LiCl (15 mM) or (B) inhibitor VIII (15 μM) treatment and following LiCl treatment (15 mM) in cells transfected with GSK3β siRNA (C) or GSK3β-S9A (D).
(E–J) Basal and maximal cellular respiration (E); NAD+, NADH, and NAD+/NADH ratio (F); immunodetection of mitochondrial Tomm20 (G); PGC-1α, GSK3β, GSK3β, and actin protein levels following 2- or 24-hr LiCl treatment (H); PGC-1α, cyclin D, and actin protein detection in cells treated with cyclohexamide (100 μM) in the absence or presence of LiCl (15 mM) (I); or inhibitor VIII (15 μM) (J).
(K–N) Protein levels of PGC-1α, pGSK3β, GSK3β, tubulin, and PARP protein in cytoplasmic and nuclear fractions following 24-hr LiCl treatment (K); immunodetection of tubulin, PGC-1α, and GSK3β (L); RT-PCR detection of PGC-1α (M); and the indicated transcripts following LiCl treatment (N).
n = 3–6 biological replicates per assay; data are shown as an average ± SEM; *p < 0.05 ANOVA.
To investigate PGC-1α protein turnover, we exposed H4 glioma to the ribosomal inhibitor cycloheximide (100 μM) resulting in significant depletion of PGC-1α after 24 hr. Cycloheximide-induced depletion of PGC-1α levels was blocked by concomitant treatment with lithium (Figure 1I) or GSK3β inhibitor VIII (Figure 1J), confirming the role of GSK3β in regulating PGC-1α protein stability. Next, we investigated PGC-1α subcellular distribution and detected increased nuclear accumulation of PGC-1α in lithium-treated cells by subcellular fractionation (Figure 1K) and confirmed by immunofluorescent detection (Figure 1L). GSK3β was also enriched in the nuclei of lithium-treated cells consistent with the increase in the phosphorylated form, which is almost exclusively nuclear in H4 glioma (Figure S1O) and increased upon treatment with lithium. Next, we investigated the regulation of PGC-1α at the transcript level. Lithium resulted in increased expression of PGC-1α isoforms α1 and α4 and a subset of brain-specific isoforms (Martinez-Redondo et al., 2016) (Figure 1M). Gene targets of PGC-1α were also differentially expressed, including the glycolytic regulator PDK4 (pyruvate dehydrogenase kinase 4), BDNF (brain-derived neurotrophic factor), SCD1 (stearoyl coenzyme A [CoA] desaturase 1), and COX5b (cytochrome c oxidase subunit 5b) (Figure 1N). As before, siRNA knockdown of GSK3β partially mimicked the effect of lithium (Figure 1SP). These data demonstrate that GSK3β regulation of mitochondrial activity in H4 glioma is associated with changes in PGC-1α activity, stability, and subcellular localization.
GSK3β Inhibition Stimulates Neuronal Mitochondrial Energy Metabolism
The rat PC12 neuroblastic cell line can be induced to differentiate into electrically excitable neuron-like cells upon treatment with nerve growth factor (NGF) (Fujita et al., 1989). Treatment of PC12-derived neurons with lithium chloride (15 mM) for 24 hr resulted in a 1.5-fold increase in PGC-1α protein levels and increased phosphorylation of GSK3β, recapitulating the outcomes observed in H4 glioma (Figures 2A and S2A). At the transcript level, lithium induced expression of the PGC-1α4 isoform, but a significant impact on other isoforms, including a novel rat-specific isoform (Figures 2B and S2B), was not detected. Lithium affected the expression of PGC-1α targets, with induction of both BDNF and PDK4 (Figure 2C); however, the impact of lithium on expression of genes encoding subunits of complex IV of ETS was not equivalent to that observed in H4 glioma, suggesting that there are differences in PGC-1α gene target specificity between these cell types. Similar to H4 glioma, lithium induced an increase in intracellular levels of NAD+ (Figure 2D). These effects were not explained by a difference in mitochondrial content; Tomm20 staining intensity was equivalent in untreated and lithium-treated PC12-derived neurons (Figure 2E), and protein levels of components of ETS complexes I–V were equivalent (Figure S2C). Unlike the case for H4 glioma, PGC-1α was almost entirely nuclear in PC12-derived neurons and subcellular localization was not changed in response to lithium. GSK3β was predominantly cytosolic in untreated PC12-derived neurons and a clear shift to nuclear localization was detected following lithium treatment (Figure 2F). Phospho-GSK3β was predominantly cytosolic and treatment with lithium resulted in the formation of discrete puncta of phospho-GSK3β in the perinuclear region (Figure S2D); however, the significance of this change in localization is currently unclear. These data show that the effects of GSK3β inhibition on cellular NAD metabolism and PGC-1α protein levels are common to H4 glioma and PC12-derived neurons.
Figure 2. GSK3β Regulation of PGC-1α Activity in PC12-Derived Neuron-like Cells.

(A) Detection of PGC-1α, pGSK3β, GSK3β, and actin proteins following LiCl treatment (15 mM).
(B and C) RT-PCR detection of PGC-1α (B) and indicated transcripts following 24-hr LiCl treatment (C).
(D) Total NAD (NADt), NAD+, NADH, and NAD+/NADH ratio following 24-hr LiCl treatment.
(E) Immunodetection of mitochondrial Tomm20.
(F) Immunodetection of PGC-1α, GSK3β, and tubulin.
(G) Protein levels of PGC-1α, pGSK3β, GSK3β, actin, and tubulin in H4 glioma and PC12-derived neuronal cells.
(H) RT-PCR detection PGC-1α isoforms in H4 glioma and PC12.
(I) Protein levels of subunits of complexes I–V of the electron transport system.
(J) Total NAD (NADt), NAD+, NADH, and NAD+/NADH ratio in H4 glioma and PC12-derived neuronal cells.
(K) Protein levels of NAMPT, Sirt1, and PARP proteins in H4 glioma and PC12-derived neuronal cells.
n = 3–6 biological replicates per assay; data are shown as an average ± SEM; *p < 0.05 Student’s t test.
The above experiments suggested that metabolic status might differ at baseline between H4 glioma and PC12-derived neurons. Although levels of GSK3β were not significantly different between cell types, protein levels of PGC-1α were significantly higher in PC12-derived neurons than in H4 glioma (Figures 2G and S2E). PGC-1α transcript isoform distribution was also different between the two cell types. The predominant isoform in glia was PGC-1α1 that was expressed at twice the level of PGC-1α4, with a lesser contribution from the brain-specific isoform (Figure 2H). PGC-1α2 and PGC-1α3 were not detected in H4 glioma. In PC12-derived neurons, PGC-1αx1 was the predominant isoform expressed, with PGC-1α4 expressed at ~30% of that level and PGC-1αx2 lower again. The αx1 and αx2 isoforms are previously unreported but predicted variants of rat PGC-1α closest in equivalence to murine PGC-1α1. Next, we looked at metabolic parameters and identified significantly higher levels of proteins from mitochondrial ETS complexes II through V in PC12-derived neurons (Figure 2I), and significant differences in basal redox state between the cell types (Figure 2J). Abundance of NAMPT (nicotinamide phosphoribosyltransferase), the rate-limiting enzyme of the NAD salvage pathway, was not significantly different between cell types; however, NAD-dependent enzymes, including the deacetylase SIRT1 and the DNA repair enzyme PARP1 (poly-ADP-ribose polymerase 1), were significantly lower in PC12-derived neurons, pointing to potential differences in NAD consumption between these cell types (Figure 2K).
NAD Metabolism Is Responsive to GSK3β Inhibition
Multi-photon laser-scanning microscopy (MPLSM) is a high-resolution imaging-based technique that allows for quantitation of differences in cellular metabolism among cells or as a function of treatment. This technology takes advantage of the autofluorescence of reduced forms of NAD and NADP (Denk et al., 1990). Kinetic properties of photon release can be quantified by fluorescence lifetime imaging microscopy (FLIM), informing of the cellular environment of the fluorophores (Lakowicz et al., 1992). Mean fluorescence lifetime (τm) summarizes a first order decay curve including a fast component (τ1) and a slow component (τ2) that correspond to free and protein-bound pools of NAD(P)H, respectively. The relative contribution of τ1 to τm is indexed by the a1 coefficient where τm = a1•τ1 + a2•τ2. Decay curves were generated over multiple pulses, repeated for each pixel in the image capture field, and were quantified on a by-pixel basis and color-coded by picoseconds of decay (Figures 3A and 3B). Remarkably, the mean fluorescence lifetime was completely different in nuclear and cytosolic compartments for both cell types (Figures 3C, 3D, and S3), with significantly shorter τm detected for nuclear pools. This difference is primarily explained by the significantly higher values of a1 in the nuclei, indicating that most of the nuclear co-factor pool is in the unbound state.
Figure 3. GSK3β Affects Cellular NAD(P)H Metabolism.

(A and B) Representative image showing mean fluorescence lifetime (τm) in picoseconds (exλ780) in the absence or presence of LiCl (15 mM) for H4 glioma (A) and PC12-derived neurons (B).
(C and D) Distributions of mean fluorescence lifetime τm (top rows), short component τ1 (upper middle rows), long component τ2 (lower middle rows), a1, the relative contribution of τ1 to τm (bottom row) before and after 24 hr LiCl treatment for H4 glioma (C) and PC12-derived neurons (D).
(E and F) NAD(P)H fluorescent intensity within the nucleus and cytoplasm following 24-hr LiCl treatment (15 mM) for H4 glioma (E) and PC12-derived neurons (F).
n = 6–8 biological replicates per measure; data are shown as a distribution or as an average ± SEM; *p < 0.05, linear mixed model.
In general, factors influencing the decay values (τ1 and τ2) include the immediate local environment, including hypoxia, pH, redox, and, in the case of τ2, the proteins to which the fluorophores are bound. Changes in a1 values are indicative of a shift in the balance of free and bound NAD(P)H, where lower a1 values are associated with greater reliance on oxidative metabolism (Bird et al., 2005). Lithium treatment increased the τm in H4 glioma (Figure 3C), with main effects of treatment, cellular compartment, and a treatment by compartment interaction (Table S2). These differences extended to all parameters of the decay curve including higher τ1 (free pool), a more modest increase in τ2 (bound pool), and a substantial decrease in a1 that was most evident in the cytosolic pool. These outcomes are consistent with the respiratory measures described earlier.
Larger magnitude changes in FLIM were detected in lithium-treated PC12-derived neurons, with an increase in τm and main effects of treatment, cellular compartment, and a treatment by compartment interaction detected (Table S2). The impact of lithium extended to main effects and interaction for all parameters of the decay curve with significant increases detected for τ1 and τ2, and a substantial decrease in a1, again suggesting a shift toward increased proportion of bound co-factors in cellular NAD(P)H pools (Figure 3D).
Next, we investigated the impact of lithium on fluorescence intensity of NAD(P)H. Fluorescence intensity reflects total fluorophore pools and can be influenced by changes in abundance of the total NAD(P)H pool and also by changes in the bound proteome within that pool. Here, the outcomes differed between H4 glioma and PC12-derived neurons where lithium had no impact on fluorescence intensity in H4 glioma (Figure 3E) but significantly increased fluorescence intensity in PC12-derived neurons (Figure 3F). Taken together, these data demonstrate that GSK3β inhibition affects cellular NAD metabolism and the metabolic environment in both PC12-derived neurons and H4 glioma.
Inhibition of GSK3β Affects Brain Energy Metabolism
To determine the impact of GSK3β inhibition on PGC-1α processing in vivo, male C3B6-F1 hybrid mice were treated with lithium carbonate (0.6–2.4 mg/g in the diet) for 4 months from 2 months of age. At higher doses lithium had an impact on body weight even though food intake was identical among all animals of the cohort (Table S3). PGC-1α4 isoform was detected in whole-brain extracts by RT-PCR, with PGC-1α1 expression at about two-thirds that level and PGC-1α2 lower again (Figure 4A). Investigation of PGC-1α expression in ex-vivo-isolated cells indicated that PGC-1α1 isoform expression was significantly higher in glia than in neurons while PGC-1α4 isoform was the major isoform detected in neurons (Figure 4B). Lithium-induced increases in PGC-1α1 and PGC-1α4 transcripts in neurons, and PGC-1α4, but not PGC-1α1 in glia (Figure 4C), although the effect was dose specific. Histochemical measures of mitochondrial cytochrome c oxidase activity (complex IV of the ETS) revealed cell type and regional differences within the hippocampus, consistent with prior reports (Martin et al., 2016) (Figure 4D). Activity stain intensity was quantified by region, including the dentate gyrus granular layer (GL), the neurons of the polymorphic layer (PL), the molecular layer (ML), and the outer hippocampal CA1 and CA3 regions, where cell bodies and neuropil were separately quantified (Figure S4A). Main effects of region and treatment as well as an interaction of region and treatment were detected (Table S4). The impact of lithium was region and dose dependent: the GL and cell bodies of the CA1 and CA3 were refractory to treatment; activity stain intensity was increased in the PL and ML, but the response was non-linear; and the CA1 neuropil exhibited a linear dose-dependent impact of lithium to increase cytochrome c oxidase activity. These data show a hippocampal region and cell-type-specific mitochondrial response to lithium treatment in vivo.
Figure 4. Inhibition of GSK3β Regulates Hippocampal Energy Metabolism in Mice.

(A and B) RT-PCR detection of PGC-1α isoforms in the whole-mouse brain (A) or in isolated neurons and glia (B).
(C) RT-PCR detection of PGC-1α isoforms in neurons and glia isolated from the whole brain of mice fed the indicated doses of dietary lithium carbonate (Li2CO3) for 4 months.
(D–F) Representative images and quantification of cytochrome C oxidase mitochondrial activity stain (D), GSK3β immunodetection (E), and PGC-1α immunodetection in the indicated hippocampal regions from Li2CO3 fed mice (F).
(G and H) Representative images of mean fluorescence lifetime (τm) in picoseconds (exλ780) in the dentate gyrus from Li2CO3 fed mice (G) and τm distributions separated by region (top panel) and by dose (bottom panel) (H).
n = 4–6 mice per Li2CO3 dosage; data shown as an average ± SEM or distributions; *p < 0.05, linear mixed models. WH, whole hippocampus; DG, dentate gyrus; GL, granular layer; PL, polymorphic layer; ML, molecular layer; CB, cell bodies; and NP, neuropil.
Hippocampal protein levels of GSK3β (Figure 4E) and PGC-1α (Figure 4F) were highly region and cell type specific (Figures S4B and S4C). A main effect of region was detected for both proteins (Table S4), and a region by dose interaction was detected. Dose-dependent effects of lithium were non-linear within responsive regions, where PGC-1α levels were increased in neurons of the PL, GL, and cell bodies of the CA1 and CA3 at lower doses (1.2 and 1.8 mg/kg). The impact of lithium on phospho-GSK3β showed a similar dose response (Figure S4D), where increased levels detected at lower doses in neurons of the GL, PL, and the cell bodies of CA1 were not observed at the higher dose (2.4 mg/kg). Multi-photon imaging of hippocampal sections from lithium-treated mice revealed no difference in fluorescence intensity, but main effects of region, treatment, and an interaction of region by treatment were detected by FLIM in the GL, PL, and ML (Figures 4G and S5; Table S5). Lithium increased τm at 0.6 mg/kg for all three regions, similar to what was observed in lithium-treated cultured cells (Figure 4H). Unexpectedly, values returned to those of untreated at 1.2 mg/kg; however, this dynamic was explained by differences in components contributing to τm, where τ1, τ1, and a1 values each differed between lithium treatment and no treatment. These data demonstrate a clear NAD-associated metabolic response to hippocampal GSK3β inhibition in vivo.
DISCUSSION
Several lines of evidence presented here are indicative of a role for GSK3β in the regulation brain energy metabolism. In cultured cells, inhibition of GSK3β by treatment with either lithium or Inhibitor VIII stimulated mitochondrial energy metabolism, including increases in the mitochondrial proton motive force and concomitant increases in mitochondrial respiration. Genetic approaches confirm the role of GSK3β in cellular metabolic regulation, where lithium’s effects were diminished when levels of GSK3β were lowered by RNA interference and were abrogated by expression of a GSK3β mutant (GSK3β-S9A) that is resistant to the effects of inhibitory phosphorylation. This GSK3β-associated change in mitochondrial function was accompanied by a lengthening of NAD(P)H fluorescence lifetime, an adaptation that is also consistent with a shift toward an oxidative phenotype. The metabolic response to GSK3β inhibition was largely conserved between cell culture models of glia and neurons despite critical differences in the underlying biology of these cell types including differences in innate oxidative metabolic capacity and differences in levels of proteins associated with the GSK3β/PGC-1α axis. Lithium’s effects on metabolism in mice in vivo were consistent with those observed in cultured cells. GSK3β inhibition increased mitochondrial activity the hippocampus and lengthened NAD(P)H fluorescent lifetime. A role for PGC-1a is implied in the metabolic response to GSK3b inhibition, where stability of PGC-1α protein was increased by either lithium or inhibitor VIII treatment in cultured cells, and expression of PGC-1α target genes was altered in both H4 glioma and PC12-derived neurons. In mice treated in vivo, hippocampal levels of PGC-1α protein were increased in response to lithium and PGC-1α transcripts were increased in isolated glia and neurons. Minor differences between the cell culture and in vivo responses to lithium might be explained by crosstalk among cell types in the brain (Bélanger et al., 2011). Co-culturing and 3D growth experiments with neurons and glia together may shed light on secondary signaling, metabolic coupling, and adaptive mechanisms involved in the brain metabolic response to GSK3β inhibition that are not captured in homogeneous culture models.
The role of metabolism in age-related disease has become a major focus in aging research, and NAD specifically, due to its central role in intermediate metabolism and as a regulator of sirtuin activity, has moved to the fore in this arena (Chini et al., 2017; Verdin, 2015; Yoshino et al., 2017). In peripheral tissues, PGC1α activity is stimulated by the actions of the NAD-dependent deacetylase SIRT1 (Nemoto et al., 2005; Rodgers et al., 2005) and studies in cultured neurons have implicated SIRT1 in neuroprotection (Qin et al., 2006). Genetic studies have independently revealed parallel roles for GSK3β and PGC-1α in vital brain processes. GSK3β has a long established role in cytoskeletal regulation, including dendritic spine stability (Ochs et al., 2015), as well as regulation of neurogenesis and memory consolidation, while PGC-1α has been shown regulate dendritic arborization (Cheng et al., 2012). An imbalance in GSK3β impedes neuroregenerative processes in mice (Kondratiuk et al., 2013) and disruptions in PGC-1α produce a neurodegenerative phenotype (St-Pierre et al., 2006). GSK3β and PGC-1α are both influenced by aging within the hippocampus. Aged rats exhibit significantly elevated levels of GSK3β in the dentate gyrus and CA regions of the hippocampus, while aged mice exhibit decreased hippocampal PGC-1α protein (Lee et al., 2006; Martin et al., 2016). In the context of Alzheimer’s disease, GSK3β inhibition has been previously demonstrated to ameliorate disease pathology and improve cognitive function (Forlenza et al., 2012; Ly et al., 2013), while hippocampal PGC-1α gene delivery protects against neurodegeneration and cognitive impairment (Katsouri et al., 2016). A number of studies place mitochondrial dysfunction downstream of aging-related neurodegeneration and Alzheimer’s pathology (Reddy, 2013; Rhein et al., 2009), but it is possible that in normal disease progression metabolic dysfunction and pathology arise concomitantly. Indeed, recent studies suggest that mitochondrial dysfunction may be a driver of cognitive decline (Grimm and Eckert, 2017). The ability of GSK3β to regulate brain mitochondrial function and NAD metabolism brings a new aspect to its role in aging and neurodegenerative disease, with implications for neurotransmitters and neurotrophic signaling pathways that impinge on GSK3β as part of normal inter- and intracellular communication.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-PGC-1α | Santa Cruz | sc-13067 |
| Rabbit anti-GSK3β | Cell Signaling Tech. | 9315S |
| Rabbit anti-pGSK3β | Cell Signaling Tech. | 9336 |
| Mouse anti-β–actin | Sigma Aldrich | A1978 |
| Mouse anti-α–tubulin | Sigma Aldrich | T6199 |
| Mouse anti-OxPhos Cocktail | AbCam | ab110413 |
| Mouse anti-Sirt1 | Santa Cruz | sc-74504 |
| Rabbit anti-NAMPT | Sigma Aldrich | V9139 |
| Rabbit anti-PARP | Cell Signaling Tech. | 9542 |
| Rabbit anti-cyclin d1 | AbCam | ab7958 |
| Biotinylated anti-mouse Ig | Vector Labs | BA-9200 |
| Biotinylated anti-mouse Ig | Vector Labs | BA-1000 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lithium Chloride (8M) | Sigma Aldrich | L7026 |
| Cycloheximide | Sigma Aldrich | C7698 |
| GSK3β Inhibitor VIII | Millipore | 361549-5mg |
| ABC Solution | Vector Labs | PK-6200 |
| NovaRed Reagent | Vector Labs | SK-4850 |
| Critical Commercial Assays | ||
| JC-1 Reagent | Invitrogen | T3168 |
| Cell Mito Stress Test Kit | Agilent | 103015-100 |
| NAD+/NADH Quantification Kit | Biovision | K337-100 |
| Oxoplate | Presens | OP96U |
| Nuclear/Cytosol Fractionation Kit | BioVision | K266-100 |
| Neuronal Isolation Kit | Miltenyi Biotec | 130-098-754 |
| LIVE/DEAD Viability/Cytotoxicity Kit | Molecular Probes | L3224 |
| Experimental Models: Cell Lines | ||
| H4 | ATCC | HTB-148 |
| PC12 | ATCC | CRL-1721 |
| Experimental Models: Organisms/Strains | ||
| B6C3F1 | Harlan | N/A |
| Oligonucleotides | ||
| See Table S6 | N/A | N/A |
| Other | ||
| Custom Mouse Diet w/Lithium Carbonate | Bio-Serv | AIN-93M |
| GSK3b siRNA | ThermoFisher | S6239 |
| Negative Control siRNA | ThermoFisher | AM4615 |
| HA-GSK3b S9A pcDNA3 | Addgene | 14754 |
| Lipofectamine 2000 | ThermoFisher | 11668019 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Rozalyn Anderson (rozalyn.anderson@wisc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Six-week-old male B6C3F1 hybrid mice were obtained from Harlan Laboratories (Madison, WI, USA) and housed under controlled pathogen-free conditions in accordance with the recommendations of the University of Wisconsin Institutional Animal Care and Use Committee. Mice were fed 87 kcal week−1 of control diet (F05312; Bio-Serv, Flemington, NJ, USA) and were individually housed with ad libitum access to water. This level of food intake is ~95% ad libitum for the B6C3F1 strain so all food was consumed. Following two weeks of facility acclimation, mice were randomized into five treatment groups fed the control diet supplemented with increasing concentrations of dietary lithium carbonate (2 months old; n = 10/group): Group 1) 0.0 g/kg/day Li2CO3; Group 2) 0.6 g/kg/day Li2CO3; Group 3) 1.2 g/kg/day Li2CO3; Group 4) 1.8 g/kg/day Li2CO3; Group 5) 2.4 g/kg/day Li2CO3. Li2CO3 supplemented mice were administered an additional drinking bottle containing saline (0.45% NaCl) to offset polyuria, a common side effect of lithium treatment. Mice consumed dietary lithium for 4 months, and were euthanized at 6 months of age. Brains were isolated, bisected, embedded in OCT, frozen in liquid nitrogen, and stored at −80°C until further processing.
Cell Culture
H4 cells were obtained from ATCC (HTB-148; Manassas, VA, USA) and cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. PC-12 cells were obtained from ATCC (CRL-1721) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% horse serum, 5% fetal bovine serum, and 1% penicillin/streptomycin.
METHOD DETAILS
PC-12 cell differentiation
All PC-12 experiments were conducted on fully differentiated PC-12 cells (7 days of differentiation). For differentiation, PC-12 cells were plated on collagen coated plates and cultured for 7 days in DMEM containing 0.1% horse serum, 100 ng/ml 2.5S nerve growth factor (N-100; Alomone Labs, Jerusalem, Israel), and 1% penicillin/streptomycin. Media was changed every 2 days.
Lithium Treatment
Lithium time-course experiments were carried out using cell culture media supplemented with 15mM lithium chloride (LiCl). Media change was carried out 2 or 24 hours prior to collection as indicated. For PGC-1α protein stability experiments, cells were pretreated for 1 hour with media containing 15mM LiCl or control media, followed by the direct addition of cycloheximide (100nM) or eqivolume DMSO to the plates for the indicated time.
Lipofectamine Transfection
H4 cells were seeded in 6-well plates 24 hours prior to lipofectamine transfection (6ul/well). HA-GSK3b[S9A] and pcDNA3.1 control vectors were transfected at 3ug/well and media change was carried out 4 hours after transfection and prior to lithium treatment. GSK3b siRNA was transfected at 30nM for 24h and media was changed prior to lithium treatment.
Metabolic Assays
Mitochondrial membrane potential
Mitochondrial membrane potential was determined using the JC-1 assay (T-3168; Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Cells were incubated in 1 μg/mL JC-1 dye for 15-minutes prior to counting and re-suspension in D-PBS for assay. Fluorescent emission was measured at 590nm and 530nm with excitation at 535nm and 485nm respectively.
Mitochondrial Respiration (Seahorse Assay)
Basal and maximal respiration of H4 cells was determined using a Seahorse extracellular flux (XF) Cell Mito Stress Test Kit (103015-100; Agilent Technologies, Santa Clara, CA, USA). Cells were plated at 4.0×104/well in a 96-well microplate using growth media in the presence or absence of 15mM LiCl and incubated overnight. One hour prior to assay, cells were switched into assay media in accordance with the manufacturer’s instructions and incubated at 37 C in a non-CO2 incubator. At the time of assay, cells were loaded into the XF analyzer along with a loaded sensor cartridge containing the following ETS inhibitors: Oligomycin (100 uM), FCCP (100uM), and Rotenone/antimycin A (50 uM). Inhibitors were sequentially injected into the assay media and basal respiration, ATP production, maximal respiration, and non-mitochondrial respiration were measured as a function of cellular oxygen consumption.
Cellular Respiration (OxoPlate Assay)
Oxygen consumption of H4 cells was determined using an Oxoplate (OP96U; Presens) oxygen monitoring system. Cells were suspended in respiration buffer (pH 7.4) containing D-Mannitol, Potassium Chloride (KCL), Magnesium Chloride (MgCl2), and Monopotassium Phosphate (KH2PO4) at 4.0×105/well in quadruplicate. Fluorescent emission was measured at 650nm and 590nm with excitation at 540nm at 10-minute intervals at 37 C.
Biochemical NAD(H) Assays
NAD/NADH quantification was determined using the Biovision NAD/NADH quantification colorimetric assay kit (K337-100; Biovision) per manufacturer’s instructions.
Cell Proliferation Analysis
Cellular proliferation was quantified using a CyQUANT direct cell proliferation assay (C35011; Thermo Fisher Scientific) per manufacturer’s instructions.
Live/Dead Assay
Cellular viability in response to lithium and GSK3b inhibitor VIII treatment was determined using a Molecular Probes LIVE/DEAD Viability/Cytotoxicity Kit (L3224; Thermo Fisher Scientific).
Immunoblotting and qRT-PCR
Western Blotting and Antibodies
Cells were lysed and protein was extracted in modified RIPA buffer containing protease and phosphatase inhibitors (P8340 and 524624, respectively; Sigma Aldrich, St. Louis, MO, USA). Proteins were detected by immunoblotting using standard techniques. Antibodies used were PGC-1α (sc-13067; Santa Cruz Biotechnology, Santa Cruz, CA, USA) GSK3β (9315; Cell Signaling Technologies, Boston, MA), serine 9 phospho-GSK3β (9336; Cell Signaling Technology), Total OXPHOS (ab110413; Abcam, Cambridge, MA, USA), Cyclin-d1 (ab7958; Abcam) beta-actin (A1978; Sigma Aldrich) and Sirt1 ((sc-74504, Santa Cruz), PARP (9542S; Cell Signaling). Subcellular fractionation was performed using nuclear/cytoplasmic fractionation kit (K266-100; Biovision, Milpitas, CA, USA) per manufacturer’s instructions. Equivalent protein amounts were loaded for both the nucleus and cytoplasm.
RNA Analysis
Cells were lysed in Trizol (15596018; Thermo Fisher Scientific, Waltham, MA, USA) and RNA was isolated using Direct-zol RNA Miniprep kit (R2072; Zymo Research, Irvine, CA, USA) in accordance with manufacturers instructions. cDNA was synthesized using a High Capacity reverse transcription cDNA kit (4368813; Thermo Fisher Scientific). Quantitive real-time reverse transcription PCR was performed on an Applied Biosystems Prism 7900 using TaqMan and SYBR Green gene expression assays (Thermo Fisher Scientific)
Microscopy
Immunofluorescence Microscopy
Cellular localization was analyzed by immunofluorescence using standard techniques. H4 cells were cultured on glass coverslips and PC-12 cells were cultured on Nunc Lab-Tek CC2 chamber slides (154917; Thermo Fisher Scientific). Following 24h LiCl treatment, cells were fixed in 3.7% formaldehyde for 10 minutes. Cells were incubated overnight in primary antibodies (sc-13067; Santa Cruz Biotechnology, Santa Cruz, CA, USA) GSK3β (9315; Cell Signaling Technologies, Boston, MA), serine 9 phospho-GSK3β (9336; Cell Signaling Technology), alpha-tubulin (T6199; Sigma Aldrich)), and Tomm20 (ab 56783; Abcam). Cellular distribution of proteins was visualized using fluorescent-tagged secondary antibodies (Fl-2000, Fl-1000; Vector Laboratories) USA. F-actin was visualized using Rhodamine Phalloidin (PDHR1; Cytoskeleton Inc, Denver, CO, USA). Nuclei were visualized using Hoechst Solution (62249; Thermo Fisher Scientific). All images were captured using uniform exposure settings on a Leica DM4000B microscope (Leica Microsystems, Wetzlar, Germany) and photographed with a Retiga 4000R digital camera (QImaging Systems, Surrey, BC, Canada)
Multiphoton laser scanning Microscopy
Immediately prior to multiphoton imaging, cells were fixed for 10 minutes with formalin and mounted onto glass coverslips using Vectashield (Vector Labs) hard-mount mounting solution. Cryostat sections were dried onto glass coverslips and mounted using Clear-mount mounting solution (Thermo Fisher Scientific). The instrument response function of the optical system was calibrated before each imaging session. A Nikon CFI Plan Apo 60x lens (Melville, NY, USA) was used for all imaging. Data were collected using an excitation wavelength of 780 nm, and emission was filtered at 457 ± 50 nm, the spectral peak for NADH/NADPH. The data collection time was 120 s using a pixel frame size of 256 × 256. The system has multiple detectors including a 16 channel combined spectral lifetime detector (utilizes a Hamamatsu PML-16 PMT), detection range 350 to 720 nm, and a H7422P GaAsP photon counting PMT (Hamamatsu) for intensity and lifetime imaging. Acquisition was performed with WiscScan, a LOCI developed acquisition package software. Autofluorescence intensity and fluorescence lifetime data were analyzed in SPCImage (Becker & Hickl, v.3.9.7, Berlin, Germany) where a Levenberg–Marquardt routine for nonlinear fitting was used to fit the fluorescence decay curve collected for each pixel in the 256 × 256 frame to a model multi-exponential decay function. Data were assessed by the minimized chi-square value generated during the fit so that analysis was unbiased. To eliminate background fluorescence a threshold for analysis was applied based on photon counts. Additionally, pixels were assigned a bin of 2 for optimal fitting of the data. For NAD(P)H autofluorescent intensity, data were analyzed in ImageJ (NIH, Wayne Rasband; https://imagej.nih.gov/ij/) and regions were defined by cellular compartment or hippocampal region. For fluorescence lifetime, regions of interest were defined by the same criteria using the inclusion tool in SPC image.
Histochemistry and Immunodetection
Serial cryostat sections 10 μm in thickness were cut at −14°C with a Leica Cryostat, defrosted and air-dried, and stained for cytochrome c oxidase enzymatic activity as previously described (Martin et al., 2016). For each experiment, tissues were sliced, batches were processed, and data were captured start to finish within 24 h. Immunodetection of PGC-1α and GSK3β was conducted as previously described (Martin et al., 2016) following antigen retrieval on 10-μm cryosections (mouse) tissues. Antibodies and reagents used are as follows: biotinylated anti-mouse Ig (BA-9200; Vector Labs, Burlingame, CA, USA) or biotinylated anti-rabbit IgG (BA-1000; Vector Labs), peroxidase-labeled avidin biotin complex (ABC) solution (PK-6200; Vector Labs), ImmPACT NovaRED reagent (SK-4805; Vector Labs), PGC-1α (sc-13067; Santa Cruz Biotechnology), total GSK3b (9315; Cell Signaling Technology), serine 9 phospho-GSK3β (9336; Cell Signaling Technology). With the exception of the multiphoton imaging, stained slides were imaged with a Leica Microsystems DM4000B microscope and photographed with a Retiga 4000R digital camera (QImaging Systems, Surrey, BC, Canada). Camera settings were optimized for each stain; for uniformity, all images for a given stain were taken on the same day with identical settings, fixed light levels, and fixed shutter speed optimized at each magnification. Digital images were converted from color to monochrome and inverted, so that greater stain intensity is shown as brighter pixels. All image analysis was performed using Adobe Photoshop (Adobe Systems, San Jose, CA, USA). Stain intensity was measured using either the rectangular marquee tool or the lasso outline tool in the hippocampal region of interest. Within each region for each stain, the size of the capture box was uniform with an average inclusion of ~30K pixels.
Neuron Isolation
Mouse neurons and non-neuronal cells were isolated from whole brain suspensions using the Neuron Isolation Kit (130-098-754; Miltenyi Biotec, San Diego, CA, USA) per manufacturer’s instructions. Briefly, brain tissue was enzymatically dissociated and debris was removed through centrifugation at 4°C. Non-neuronal cells were labeled using the Non-Neuronal Cells Biotin-Antibody Cocktail and depleted using magnetic separation. Isolated cell populations were lysed in Trizol and RNA analysis was conducted as previously described.
QUANTIFICATION AND STATISTICAL ANALYSES
Independent Student’s t test and ANOVA with post hoc analyses were used to evaluate statistical significance in all cell culture studies. Statistical analyses for the hippocampal immunohistochemistry and MPLSM-FLIM was conducted as previously described (Martin et al., 2016). Briefly, to account for the dependence among observations due to multiple measurements per animal, we performed linear mixed models (LMM) assuming a compound symmetric covariance structure using SAS PROC MIXED (Littell et al., 2006). The LMMs included full factorial with Type 3 tests of the main effects and interactions. To explore the lithium carbonate dosage-by-region interaction, simple main effects were investigated to determine whether there were dosage effects within each region. We employed no formal multiple testing correction. Instead, consistent with published guidelines for statistical reporting (Saville, 1990), exact p values are reported. All data are reported as mean ± SEM.
Supplementary Material
Highlights.
GSK3β regulates mitochondrial energy metabolism in neurons and in glia
Metabolic regulation by GSK3β extends to cellular NAD metabolism
Inhibition of GSK3β alters PGC-1α protein stability, localization, and activity
GSK3β inhibition alters hippocampal energy metabolism
Acknowledgments
This work was supported by The Glenn Foundation for Medical Research, the American Federation for Aging Research, and the UW-ADRC: NIH/NIA (AG033514). S.A.M., D.C.S., and J.P.C. were supported by fellowships from NIH/NIA (T32 AG000213). K.N.M. was supported by a fellowship from NIH/NIDDK (T32 DK007665). This work was conducted with the use of resources and facilities at the William S. Middleton Memorial Veterans Hospital (Madison, WI).
Footnotes
Supplemental Information includes five figures and six tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.045.
AUTHOR CONTRIBUTIONS
S.A.M., D.C.S., and R.M.A. designed the study; S.A.M., D.C.S., K.N.M., and J.P.C. conducted the experiments; A.K.S. and K.W.E. consulted on the multi-photon imaging; L.P. provided reagents and expertise; S.A.M. and T.M.B. conducted the statistical analyses; and S.A.M., D.C.S., and R.M.A. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, White JG, Ramanujam N. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res. 2005;65:8766–8773. doi: 10.1158/0008-5472.CAN-04-3922. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini CCS, Tarragó MG, Chini EN. NAD and the aging process: role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRocha-Souto B, Coma M, Pérez-Nievas BG, Scotton TC, Siao M, Sánchez-Ferrer P, Hashimoto T, Fan Z, Hudry E, Barroeta I, et al. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol Dis. 2012;45:425–437. doi: 10.1016/j.nbd.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Forlenza OV, de Paula VJ, Machado-Vieira R, Diniz BS, Gattaz WF. Does lithium prevent Alzheimer’s disease? Drugs Aging. 2012;29:335–342. doi: 10.2165/11599180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Fujita K, Lazarovici P, Guroff G. Regulation of the differentiation of PC12 pheochromocytoma cells. Environ Health Perspect. 1989;80:127–142. doi: 10.1289/ehp.8980127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM. Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry. 2004;161:217–222. doi: 10.1176/appi.ajp.161.2.217. [DOI] [PubMed] [Google Scholar]
- Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143:418–431. doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouri L, Lim YM, Blondrath K, Eleftheriadou I, Lombardero L, Birch AM, Mirzaei N, Irvine EE, Mazarakis ND, Sastre M. PPARγ-coactivator-1α gene transfer reduces neuronal loss and amyloid-β generation by reducing β-secretase in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2016;113:12292–12297. doi: 10.1073/pnas.1606171113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratiuk I, Devijver H, Lechat B, Van Leuven F, Kaczmarek L, Filipkowski RK. Glycogen synthase kinase-3beta affects size of dentate gyrus and species-typical behavioral tasks in transgenic and knockout mice. Behav Brain Res. 2013;248:46–50. doi: 10.1016/j.bbr.2013.03.045. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, Szmacinski H, Nowaczyk K, Johnson ML. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci USA. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Chung YH, Joo KM, Lim HC, Jeon GS, Kim D, Lee WB, Kim YS, Cha CI. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci Lett. 2006;409:134–139. doi: 10.1016/j.neulet.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Littell R, Milliken G, Stroup W, Wolfinger R, Schabenberger O. SAS System for Mixed Models. SAS Institute; 2006. [Google Scholar]
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SA, DeMuth TM, Miller KN, Pugh TD, Polewski MA, Colman RJ, Eliceiri KW, Beasley TM, Johnson SC, Anderson RM. Regional metabolic heterogeneity of the hippocampus is nonuniformly impacted by age and caloric restriction. Aging Cell. 2016;15:100–110. doi: 10.1111/acel.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Redondo V, Jannig PR, Correia JC, Ferreira DM, Cervenka I, Lindvall JM, Sinha I, Izadi M, Pettersson-Klein AT, Agudelo LZ, et al. Peroxisome proliferator-activated receptor γ coactivator-1 α isoforms selectively regulate multiple splicing events on target genes. J Biol Chem. 2016;291:15169–15184. doi: 10.1074/jbc.M115.705822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Ochs SM, Dorostkar MM, Aramuni G, Schön C, Filser S, Pöschl J, Kremer A, Van Leuven F, Ovsepian SV, Herms J. Loss of neuronal GSK3β reduces dendritic spine stability and attenuates excitatory synaptic transmission via β-catenin. Mol Psychiatry. 2015;20:482–489. doi: 10.1038/mp.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, Reed SI. SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22:252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur J Biochem. 1983;130:227–234. doi: 10.1111/j.1432-1033.1983.tb07140.x. [DOI] [PubMed] [Google Scholar]
- Patel P, Woodgett JR. Glycogen synthase kinase 3: a kinase for all pathways? Curr Top Dev Biol. 2017;123:277–302. doi: 10.1016/bs.ctdb.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Patel S, Macaulay K, Woodgett JR. Tissue-specific analysis of glycogen synthase kinase-3α (GSK-3α) in glucose metabolism: effect of strain variation. PLoS ONE. 2011;6:e15845. doi: 10.1371/journal.pone.0015845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Mu Y, Leblanc M, Alaynick W, Barish GD, Pankratz M, Tseng TW, Kaufman S, Liddle C, Yu RT, et al. Dependence of hippocampal function on ERRγ-regulated mitochondrial metabolism. Cell Metab. 2015;21:628–636. doi: 10.1016/j.cmet.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Reddy PH. Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer’s disease: implications for synaptic dysfunction and neuronal damage. Biochim Biophys Acta. 2013;1832:1913–1921. doi: 10.1016/j.bbadis.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci USA. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Saville DJ. Multiple comparison procedures–the practical solution. Am Stat. 1990;44:174–180. [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20:353–371. doi: 10.1089/ars.2012.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Baur JA, Imai SI. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2017;27:513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.