

Abstract
Extracellular matrix biomaterials have been shown to promote constructive remodeling in many preclinical and clinical applications. This response has been associated with the promotion of a timely switch from pro-inflammatory (M1) to anti-inflammatory (M2) macrophages. A previous study has shown that this beneficial response is lost when these biomaterials are derived from aged animals. This study examined the impact of small intestine submucosa (SIS) derived from 12, 26 and 52 week old pigs on the phenotype and function of bone marrow macrophages derived either from 2 or 18 month old mice. Results showed that 52 week old SIS promoted less iNOS in 2 month macrophages and Fizz1 expression in 2 and 18 month compared to 12 week SIS. Pro-inflammatory cytokine exposure to 52 week SIS-treated macrophages resulted in higher iNOS in 18 month macrophages and reduced MHC-II expression in 2 month macrophages, as well as reduced nitric oxide production in comparison to 12 week SIS. These results indicate that ECM derived from aged animals promotes an altered macrophage phenotype compared to young controls. This suggests that sourcing of ECM from young donors is important to preserve constructive remodeling outcomes of ECM biomaterials. Alteration of macrophage phenotype by aged ECM also raises the hypothesis that alterations in aged ECM may play a role in immune dysfunction in aged individuals.
Keywords: aging, macrophage polarization, extracellular matrix, small intestine submucosa
Graphical abstract
1. Introduction
Extracellular matrix (ECM)-based biomaterials have been used successfully in a wide variety of pre-clinical and clinical soft tissue reconstruction applications [1–5]. Positive outcomes of ECM implantation have been associated with “constructive remodeling” of the biomaterials, which involves the formation of new site-appropriate host tissue [6]. ECM-based materials are most commonly derived from mammalian tissues using decellularization processes [7]. These protocols, when successful, remove a significant portion of the immunogenic components of tissues including cellular proteins and DNA, while preserving the components and microarchitecture of the native extracellular matrix [8–10]. This results in a scaffold that is acellular and possesses inherent bioactivity due to the many ligands present on source tissue ECM macromolecules [11]. Upon implantation, these materials serve not only as a structural scaffold for cellular adhesion and a substrate for constructive remodeling [12], but also rapidly degrade, releasing bioactive molecules that have been shown to stimulate signaling in the tissue remodeling pathways [13–16]. Recent work has shown these bioactive constituents to include matrix-bound vesicles (MBVs) which carry unique protein and nucleic acid cargo that can affect the macrophage response [17]. Constructive remodeling responses from ECM scaffolds have been associated with a timely shift in macrophage polarization from pro-inflammatory (M1) to anti-inflammatory (M2) phenotypes [18–20]. The degradation of ECM scaffolds, which promotes the release of matricryptic peptides, has been shown to be a necessary event for this promotion of constructive remodeling [18, 21].
While ECM and other natural scaffolds possess advantages of inherent bioactivity, they are often criticized for their variability. One potential source of variability is the age of the donor animal or cadaver from which the tissues are harvested. The extracellular matrix has been shown to vary in composition with the age of an organism [22]. For example, muscle ECM shows increased deposition of collagen I and decreased collagen III with age [23–25]. Collagen IV is increased whereas laminin decreases with age in the basal lamina [26], a key component of many ECM-based scaffold materials [27]. Increased deposition and reduced collagen turnover leads to accumulation of glycation end-products (AGEs) and associated crosslinks which can interfere with normal cell-tissue interactions [28, 29]. ECM changes may also lead to stem cell senescence or induced differentiation through stiffer matrix [23, 30]. These aging-related changes have been suggested to affect the ability to regenerate tissue following injury.
It is logical, therefore, that differences observed in native tissues would persist in ECM scaffolds following decellularization and may have implications for their effectiveness following implantation. A recent study showed that small intestinal submucosa (SIS) derived from 52 week old pigs promoted less constructive remodeling and increased collagen deposition when implanted in to a rat abdominal wall defect model compared to SIS derived from 12 and 26 week old pigs [31]. This response was also associated with increased pro-inflammatory (M1) macrophage polarization with increased source animal age, suggesting these poorer outcomes were due to alteration in macrophage phenotype.
The immune system also experiences age-related changes as well, becoming dysfunctional in activation and polarization of macrophages as well as changes in other cell types [32]. This phenomenon, termed “inflamm-aging,” is associated with increased levels of cytokines and reactive oxygen species (ROS) production which can lead to cellular and tissue damage [33, 34]. The aging immune system is also susceptible to “immunosenescence,” which results in alteration of normal immune cell circulation, migration and cell signaling [35]. While the exact mechanisms underlying these observations are not clear, recent studies have demonstrated that the local tissue microenvironment (i.e. ECM) may play an important role [36, 37].
The current study sought to examine the impact of changes in ECM scaffolds with different source animal age upon macrophage polarization responses. In order to accomplish this an in vitro primary cell model of macrophage polarization to ECM scaffolds was utilized to understand these age-related changes in ECM. Murine bone marrow-derived macrophages were chosen as a model system to reduce variability in the host immune responses due to genetic and/or environmental factors. As aging is a multifactorial condition, genetically similar mice with similar environments and diets were necessary to eliminate comorbidities associated with aging such as obesity, diabetes, and cardiovascular disorders that could be present in blood drawn from young and aged humans [38–41]. Additional studies can take advantage of murine genetic models in order to understand pathways involved in the macrophage response to biomaterials. Nevertheless, future studies involving human monocyte-derived macrophages will be necessary to assess the response to such materials in humans.
To model the responses to extracellular matrix biomaterials, we used ECM degradation products to treat macrophages. Degradation of biomaterials has been shown to be important to the host response, with non-degrading scaffolds eliciting increased pro-inflammatory responses [18]. Extracellular matrix degradation products have been shown to be bioactive and elicit macrophage phenotypes similar to those observed to whole scaffolds in vivo [16, 42, 43]. Macrophage response to biomaterials at acute time-points of 7-14 days, time when biomaterials are already being degraded and remodeled, has been correlated to functional downstream outcomes [16, 18, 44]. Therefore, the response to degradation products is relevant to the key timeframe of phenotypic shift necessary to alter downstream remodeling outcomes.
2. Materials & Methods
2.1 Scaffold Preparation
Jejunum were harvested from 12 week, 26 week, and 52 week-old Landrace pigs immediately following euthanasia (Tissue Source, Lafayette, IN). The animals were of similar genetic heritage, diet and immunization history. Small intestine submucosa (SIS) extracellular matrix biomaterials were prepared from these tissues as previously described [45]. Briefly, the tissues were rinsed with water and the mesenteric tissues were removed. Tissues were cut longitudinally then mechanically delaminated to remove the tunica serosa, tunica muscularis externa, and the luminal tunica mucosa, including most of the lamina propria. After delamination, the tunica submucosa and the basilar layer of the tunica mucosa including the muscularis mucosa and the stratum compactum of the lamina propria remained. The material was further decellularized using mechanical agitation with an orbital shaker at 300 rpm in a solution of 0.1% (v/v) peracetic acid (Rochester Midland Corporation, Rochester, NY). Tissues were then rinsed multiple times with saline and deionized water. Decellularized scaffolds were frozen at −80°C and then lyophilized. The resultant scaffolds were considered “intact ECM scaffolds.” Scaffolds were digested at a concentration of 10 mg/mL in an acid-pepsin solution (pH 2) under constant magnetic stir for 48 hours. These digested scaffolds were considered “ECM degradation products.”
2.2 Scaffold Characterization
Hydrated native tissue and decellularized scaffolds were fixed in 10% neutral buffered formalin (NBF) and then embedded in paraffin. Sections of these scaffolds were stained separately with hematoxylin & eosin or DAPI to confirm removal of nuclei. Proteinase K digests of native and decellularized scaffolds underwent phenol: chloroform: isoamyl alcohol (25:24:1) extraction for DNA and were resuspended in 1X TE buffer. DNA extracts were separated using electrophoresis on a 2% agarose gel in 0.5X TBE buffer to confirm reduction of DNA content and fragmentation of remnant DNA in decellularized scaffolds compared to native controls. DNA extracts were also quantified for double-stranded DNA content using a PicoGreen assay (Thermo) according to manufacturer’s instructions.
2.3 Growth Factor ELISA Quantification
Urea heparin extracts were performed on lyophilized native and decellularized SIS samples as previously described [46]. Briefly, 100 mg of lyophilized tissue was extracted using 3 mL urea-heparin extraction buffer (2M urea, 50mM Tris Base, 5 mg/mL heparin, 10mM N-Ethylmaleimide, 5mM benzamidine, 1mM PMSF in DI H2O). Samples were agitated overnight at 4°C. Samples were centrifuged at 10,000 xg for 10 min at 4°C. Supernatants were collected, volumes recorded, and stored at −80°C until time of assay. Extraction procedure was repeated once more to ensure complete extraction. Growth factor ELISAs were purchased from R&D Biosystems for basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), and heparin-binding EGF-like growth factor (HB-EGF). Urea-heparin extracts were assayed for growth factor content using the ELISA assays above according to manufacturing instructions.
2.4 Cell Isolation
Bone marrow-derived macrophages were harvested from 2 month or 18-20 month-old C57/BL6 mice as previously described [16]. Briefly, femurs and tibiae were harvested and separated from muscle and connective tissue. Bones were cut at either end to expose bone marrow. Sterile syringe and needles were used to flush out bone marrow using macrophage differentiation media (DMEM/10% FBS/10% L-929 Supernatant/1% PenStrep/2% MEM non-essential amino acids/1% HEPES/0.1% 55μM β-2 mercaptoethanol). Bone marrow lysate was reconstituted in media and filtered through a sterile cell filter. Cells were cultured for 7 days in media to differentiate them into macrophages, changing differentiation media every 2 days.
2.5 Cell Treatment
Following 7 days of differentiation culture as described above, macrophages were treated with acute polarizing regimens to distinguish phenotypes over 24 hours. Naïve macrophage (M0) controls were treated with basal media for 24 hours. M1 (20 ng/mL IFN-γ and 100 ng/mL LPS) and M2 (20 ng/mL IL-4) polarizing cytokines were used to create positive controls for classical pro- and anti-inflammatory macrophages. ECM degradation products were neutralized and diluted to 250 μg/mL in macrophage media to isolate biochemical effects of degradation products and prevent structural moieties from forming. Pepsin buffer (1 mg/mL pepsin in 0.01 M HCl) diluted in macrophage media was used as a control. Another set of treatment groups involved 24-hour exposure of ECM degradation products followed by 24-hour treatment with either the M1 or M2 treatment regimen.
2.6 Indirect Immunofluorescent Antibody Staining
Cells were fixed with 2% paraformaldehyde (PFA) for 30 minutes then washed in 1XPBS. Cells were incubated in blocking buffer (2% donkey serum, 1% bovine serum albumin (BSA), 0.1% Triton X-100, 0.1% Tween-20) for 1 hour at room temperature. Primary antibodies were diluted in this blocking buffer as follows and incubated overnight at 4 °C: F4/80 (1:200, Abcam 6640), iNOS (1:100, Abcam 3523), Arginase-1 (1:200, Abcam 91279), Fizz1 (1:200, Peprotech 500-P214), CCR2 (1:200, Abcam 21667), CX3CR1 (1:200, Abcam 8021), MHC-II (1:100, Abcam). F4/80 is a marker of macrophage differentiation. iNOS and MHC-II are classical M1 macrophage markers whereas Arginase-1 and Fizz1 are classical M2 macrophage markers. CCR2 is a chemokine receptor associated with macrophage migration whereas CX3CR1 is a fractalkine receptor associated with tissue-resident macrophages. Cells were washed in 1XPBS then incubated in the appropriate fluorescently-labeled secondary antibody solution in blocking buffer for 1 hour at room temperature (donkey anti-rat Alexa Fluor 488, Invitrogen, 1:200; donkey anti-rabbit Alexa Fluor 488, Invitrogen, 1:300). Cell nuclei were counterstained with DAPI. Cells from five young (2 month) and aged (18 month) mice were imaged nine times in the center of each well at 10× magnification using automated position capture function to remove bias from subjective image location acquisition. This set of 9 images per well was counted as one biological replicate and was repeated in cells from n=5 mice for a total of 5 sets of 9 images per group. All imaging was performed on an Axio observer T1 microscope. Mean fluorescence intensity of cells was analyzed using Cell Profiler software (Broad Institute). Briefly, DAPI images were used by the program to identify cell nuclei then FITC images were used to identify cell borders around the identified nuclei. The mean fluorescent intensity was calculated by averaging the pixel intensities (scale of 0 to 1) across the entire cell area. Mean fluorescence intensity values were averaged for all imaged cells in each well.
2.7 Phagocytosis Assay
Following treatments, cells were assayed for phagocytic ability using Vybrant Phagocytosis Assay Kit (Invitrogen). Cells were incubated in FITC-labeled dead E. Coli particles for 2 hours in the cell culture incubator. Following washing, the cells were fixed with 2% PFA for 30 minutes then washed with 1X PBS. Cells were counterstained with DAPI then imaged and analyzed as described above.
2.8 Taqman Gene Expression Analysis
Following treatments, macrophages (n=5 biological replicates from 5 young (2 month) and 5 aged (18 month) mice) were harvested for RNA using Qiagen RNEasy MiniPrep RNA Isolation Columns following standard protocol. RNA was quantified using a NanoDrop Lite Spectrophotometer (Thermo). cDNA templates were created from 1 μg of RNA using Invitrogen High Capacity RNA-to-cDNA kits (Thermo). Taqman Gene Expression Assays (Thermo) were performed for the following commonly reported M1 and M2 macrophage genes: Nos2 (Mm00440502_m1), IL1b (Mm00434228_m1), IL12b (Mm01288989_m1), TNFa (Mm00443258_m1), MHC-II (Mm01181326_m1), Arg (Mm00475988_m1), Retlna (Fizz1) (Mm00445109_m1), Mrc1 (Mm01329362_m1), IL10 (Mm01288386_m1), and PPARg (Mm00440940_m1).
2.9 Nitric Oxide Assay
Following treatments, 50 μL of supernatant from macrophages were transferred into a fresh plate. Nitric oxide content was assayed using a Greiss Reagent system. Briefly, 50 μL of sulfanilamide (1% in 5% phosphoric acid) was added to supernatants for 10 minutes. Then, 50 μL of 0.1% N-1-napthylethylenediamine in water was added to the mixture for an additional 10 minutes. The absorbance at 540 nm was measured using a BioTek SYNERGY HTX spectrophotometer.
2.10 Statistical Analysis
Quantitative results were analyzed using a two-way ANOVA (treatment, age) with Tukey post-hoc analysis using GraphPad PRISM 7 software. Significance was determined at a p-value less than 0.05.
3. Results
3.1 Confirmation of Decellularization
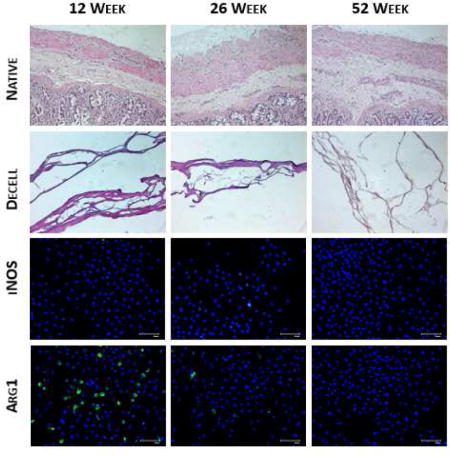
Hematoyxlin & eosin staining of native and decellularized small intestinal submucosa samples confirmed decellularization. Outer intestinal musculature and inner luminal mucosa were removed in decellularized samples as previously described [47]. Nuclear removal was confirmed through visual observation in H&E (Figure 1A) and DAPI staining (Figure 1B). DNA fragmentation was confirmed through DNA agarose gel electrophoresis (Figure 1C). DNA extracts from native samples show large continuous bands indicative of large DNA fragments. Extracts from decellularized samples show significant removal of DNA material, with remnant DNA existing as lower molecular weight (Figure 1D). This is indicative of effective removal and fragmentation of any remnant DNA. Results from PicoGreen DNA quantification (Figure 1D) showed that scaffolds were effectively decellularized [48]. Effective and similar levels of decellularization confirm that DNA content should not cause differences in immune responses between different source age samples.
Figure 1.
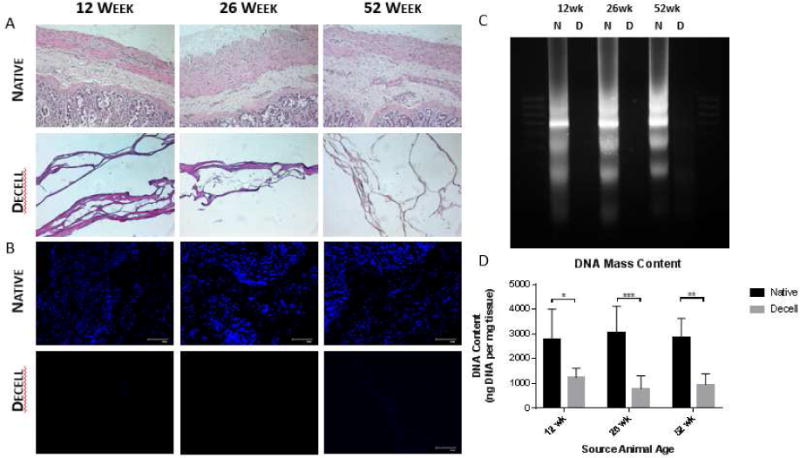
Confirmation of decellularization using hematoxylin & eosin (A) and DAPI (B) staining, as well as DNA agarose gel electrophoresis between native (N) and decell (D) samples (C) and DNA quantification (D). *p<0.05
3.2 Analysis of ECM Composition
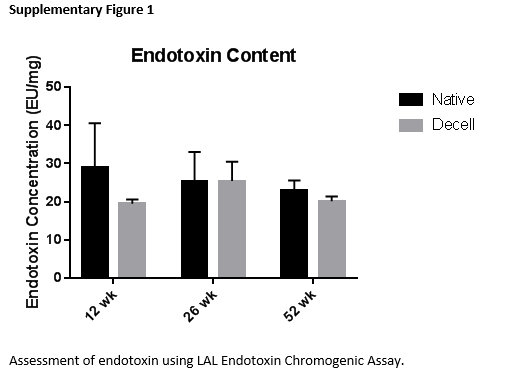
SIS scaffolds of different source animal age were analyzed for content of major extracellular matrix structural proteins, specifically collagens and glycosaminoglycans. Hydroxyproline content was assessed to approximate collagen composition in SIS scaffolds (Figure 2A). In native small intestine submucosa, hydroxyproline content significantly increased with source animal age. However, in decellularized scaffolds, there was no significant difference in hydroxyproline content. Glycosaminoglycan content was assessed using dimethylmethylene blue dye (Figure 2B). Native small intestine submucosa showed a significant increase in glycosaminoglycan content with increasing source animal age. However, no trend appeared with glycosaminoglycan content in decellularized scaffolds of different source animal age. Endotoxin content was also assessed using an LAL Endotoxin Assay. However, our results show that endotoxin content did not differ in ECM degradation products (Supplementary Figure 1).
Figure 2.
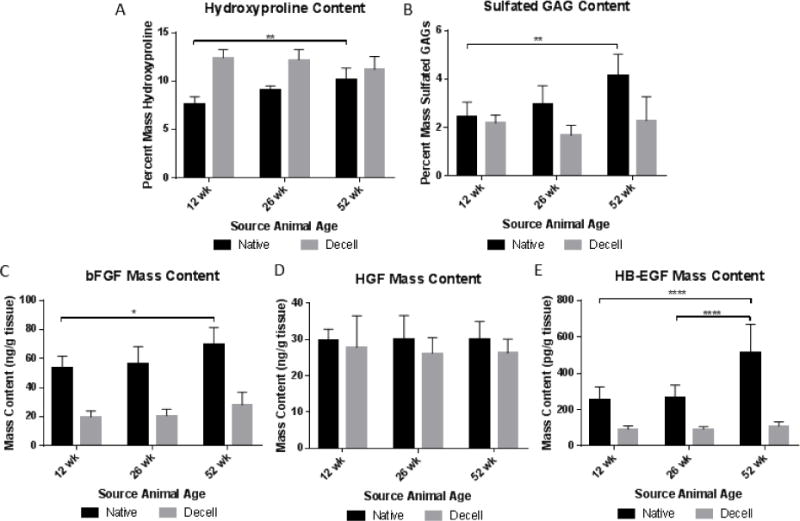
Compositional analysis of ECM scaffolds from different source animal ages for hydroxyproline (A) and sulfated glycosaminoglycan (B) content. Growth factor content assessed by ELISA for basic fibroblast growth factor (bFGF) (C), hepatocyte growth factor (HGF) (D), and heparin-binding EGF-like growth factor (HB-EGF) (E). *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Growth factors laden within extracellular matrix scaffolds can contribute to the overall biochemical microenvironment created by the scaffold. Analysis of growth factor content change with source animal age is important to the overall characterization of ECM’s effect on macrophage phenotype. Due to previous work showing ECM scaffolds of different animal source age affected both skeletal muscle regeneration and macrophage phenotype, growth factors were selected that are known to affect both of these phenomena and have also been found in whole small intestine tissue [49–51]. Native small intestine tissue showed significant increases in bFGF and HB-EGF. However, no significant changes in content were observed following decellularization (Figure 2C–E).
3.3 Immunofluorescent Antibody Labeling
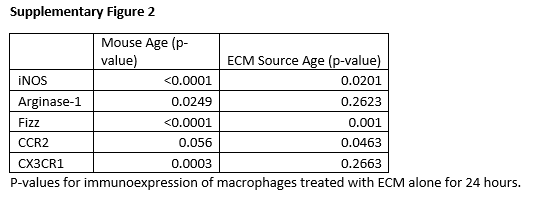
Differentiation into a macrophage phenotype was confirmed with antibody immunolabeling for F4/80, a classical macrophage marker. All treatment groups displayed staining for F4/80 with M1 treatment resulting in significant increases in F4/80 labeling (Figure 3A). Macrophages treated with ECM degradation products were stained for classical differentiation markers of macrophage phenotype. Staining for iNOS, a classical M1, pro-inflammatory marker, showed upregulation in M1 controls and downregulation in M2 controls (Figure 3B). Macrophages treated with ECM degradation products showed slight iNOS activation. Increasing source animal age resulted in significantly decreased iNOS surface marker expression with increasing age in macrophages from 2 month mice (Figure 3B). Macrophages derived from 18 month mice did not have a significant decrease in iNOS expression between 12 and 52 week ECM but ECM groups in 18 month macrophages had significantly higher iNOS expression over 2 month macrophages (p<0.0001) (Supplementary Figure 2). Staining for Arginase-1, a marker for M2, alternative activation, showed large upregulation in M2 controls and slight downregulation in M1 controls (Figure 3C). ECM-treated macrophages showed minor Arginase-1 surface marker expression. There were no significant changes in Arginase-1 immunoexpression with source animal age (Figure 3C).
Figure 3.
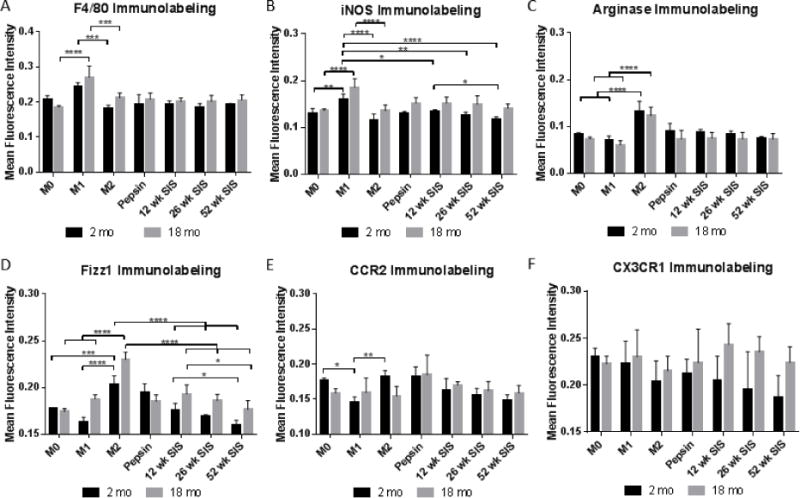
Mean intensity of indirect immunofluorescent antibody staining of macrophages treated with ECM degradation products for F4/80 (A), iNOS (B), Arginase-1 (C), Fizz1 (D), CCR2 (E), and CX3CR1 (F). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Fizz1, a marker for alternative macrophage activation, showed upregulation in M2 controls and downregulation in M1 controls (Figure 3D). Fizz1 expression of ECM-treated macrophages was intermediate, neither pro- nor anti-inflammatory. Source animal age significantly decreased Fizz1 expression from 12 week to 52 week SIS. Fizz1 expression was significantly higher in macrophages derived from 18 month mice compared to 2 month old mice (p<0.0001) (Supplementary Figure 2).
CCR2, a chemokine receptor for CCL2, is classically a marker for invading macrophages. CCR2 showed decreased surface marker expression in M1 controls (Figure 3E). There were no significant differences in CCR2 immunolabeling based on source animal age of ECM. However, 18 month macrophages responded to ECM with increased CCR2 immunoexpression compared to 2 month macrophages (p = 0.0463) (Supplementary Figure 2). CX3CR1, a fractalkine receptor, is a marker for tissue-resident macrophages. There were no significant changes in fractalkine receptor expression in cytokine controls or ECM treatment (Figure 3F).
In order to determine whether ECM degradation product exposure altered the ability of macrophages to polarize, macrophages were treated with ECM degradation products for 24 hours then treated with M1 or M2 cytokine regimes for the subsequent 24 hours. 52 week SIS significantly increased iNOS immunoexpression in ECM→M1 groups over IFN-γ/LPS treatments in 2 month old macrophages and 12 week SIS treatments in 18 month old macrophages (Figure 4A). Arginase-1 immunoexpression was significantly upregulated with 12 week pre-treatment with subsequent IL-4 treatment compared to M2 controls (Figure 4B). In 18 month macrophages, 52 week SIS caused a significant increase in Arginase-1 immunoexpression over 12 week SIS with IL-4 post-stimulation. The only significant change in Fizz1 immunoexpression was a significant increase in Fizz1 in all ECM pre-treatments with IFN-γ/LPS post-stimulation over M1 controls in the 2 month macrophage group (Figure 4C). MHC-II immunoexpression was significantly decreased with 26 or 52 week SIS pre-stimulation over M1 and 12 week SIS pretreatment samples in 2 month old macrophages (Figure 4F). SIS from 26 and 52 week SIS only decreased MHC-II expression compared to M1 stimulation in 18 month old macrophages.
Figure 4.

Mean intensity of indirect immunofluorescent antibody staining of macrophages treated with ECM degradation productions for 24 hours followed by 24 hours of M1 or M2 stimulus for iNOS (A), Arginase-1 (B), Fizz1 (C), CCR2 (D), CX3CR1 (E), and MHC-II (F). *p<0.05, **p<0.01, ***p<0.001, ****P<0.0001.
3.4 Taqman Gene Expression Assays
Transcriptional levels of classical pro-inflammatory and anti-inflammatory markers and cytokines were assayed in 24 hour macrophage RNA extracts using Taqman gene expression assays (Figures 5). Pro-inflammatory markers NOS2, IL1b, IL12b, and Tnfa showed large increases in fold expression change in M1 treatment over naïve M0 macrophages (Supplementary Figure 3). The anti-inflammatory gene targets Arg1, Fizz1, IL10, and Mrc1 showed significant increases in IL-4-stimulated M2 macrophages (Supplementary Figure 4). Fifty two week SIS treatment resulted in significant increases of IL-1ra expression over 12 and 26 week SIS treatment in 2 month old macrophages (Supplementary Figure 3E). Eighteen month old macrophages treated with 52 week SIS resulted in significant increases in Fizz1 expression over 12 week SIS treatment (Supplementary Figure 4E).
Figure 5.

Heat map of Taqman gene expression analysis on bone marrow macrophages compared to M0 GAPDH. Gene expression analysis performed on 2 month (Young) and 18 month (Old) macrophages. Color for heat map normalized to highest values within all samples for a gene target and color based on a logarithmic scale for fold expression change values. Green indicates increased fold expression change and red indicates reduced fold expression change.
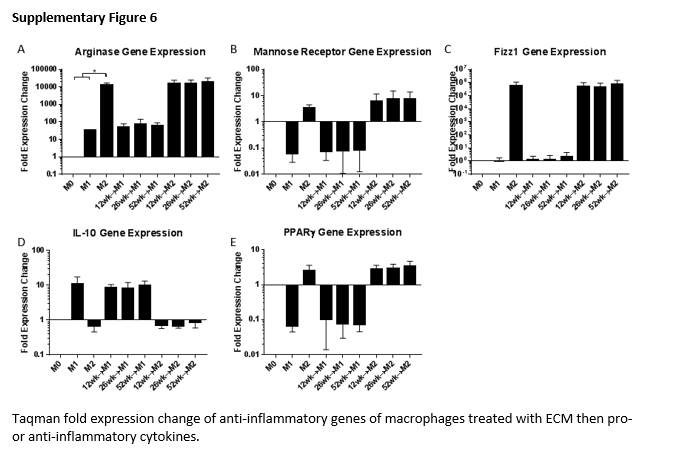
Gene expression assays were also conducted in challenge experiments where macrophages were exposed to ECM degradation products for 24 hours then challenged with M1 or M2 polarizing cytokines for an additional 24 hours (Figure 6). Pro-inflammatory (M1) controls showed large fold expression increase in iNOS, IL-12β, IL-1β, MHC-II, TNF-α, and IL-10 (Supplementary Figure 3). Anti-inflammatory (M2) controls showed large fold expression increase in Arginase-1 and Fizz1 and smaller increases in Mrc1, PPARγ, MHC-II and IL-1β with decreases in TNFα and IL-10 (Supplementary Figure 4). ECM pretreatment did not alter expression of any genes following M1 or M2 treatment (Supplementary Figure 5 & 6).
Figure 6.

Taqman gene expression analysis on young (2 month) bone marrow macrophages compared to M0 GAPDH. Color for heat map normalized to highest values within all samples for a gene target and color based on a logarithmic scale for fold expression change values. Green indicates increased fold expression change and red indicates reduced fold expression change.
3.5 Nitric oxide production
Nitric oxide was assayed in the supernatants of macrophages treated with ECM degradation products (Figure 7A). The stable form of nitric oxide, nitrite, was identified using the Greiss reagent system. Results showed an increase in nitric oxide production in M1 controls but no increase in M2 controls. ECM treatment showed limited levels of nitric oxide production. There were no significant differences in nitric oxide production from macrophages treated with ECM alone. ECM pre-treatment followed by M1 stimulus increased nitric oxide production in 12 week SIS samples compared to M1 controls and 52 week SIS pre-treatment.
Figure 7.
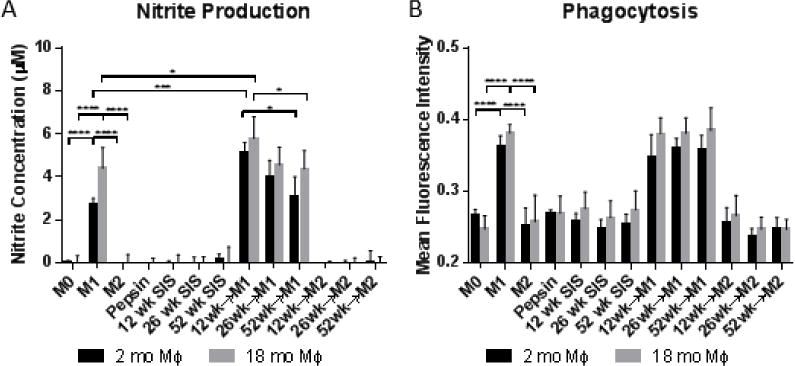
(A) Nitrite production from macrophages exposed to different source animal age SIS. (B) Mean intensity of macrophages exposed to FITC-labeled E. Coli particles for phagocytic analysis.
3.6 Phagocytosis
To assess phagocytic function, Vybrant Phagocytosis Beads produced from FITC-labeled E. coli bacteria particles were used. Phagocytic activity increased in pro-inflammatory controls but not in anti-inflammatory controls (Figure 7B). ECM degradation products did not significantly change baseline phagocytosis or phagocytosis following cytokine regimens.
4. Discussion
The present study investigated the response of macrophages to extracellular matrix (ECM) degradation products within an aging context. Specifically, this study investigates porcine small intestine submucosa (SIS) scaffolds that differ in only the variable of source animal age (the age of the animal from which it was derived). Previous work has shown that these scaffolds, when implanted into a rat abdominal wall reconstruction model, elicited different potentials for muscle healing [31]. This previous study showed that increasing source animal age resulted in increased fibrosis and decreased skeletal muscle formation, which was related to increased pro-inflammatory macrophage phenotype at early time-points. In order to identify macrophages as being able to respond to these age-related changes, the present study utilized an in vitro model of macrophage response to ECM biomaterials. Results showed that ECM biomaterials caused the greatest shifts in macrophage phenotype when examined in combination with inflammatory cytokine polarization. The lack of robust macrophage response to ECM scaffolds alone is logical as the ECM is naturally occurring and involved in the homeostasis of tissues.
There are currently 69 extracellular matrix-based products registered with the FDA, many of which are derived from animal sources [70]. Due to known differences in composition of biologic materials due to individual organism variabilities, it is important to understand what characteristics of donor animals have effects on resultant biomaterial properties and their response upon implantation. Quality compliance of source animal facilities (SAFs) is regulated under FDA guidance “Source Animal, Product, Preclinical, and Clinical Issues Concerning the Use of Xenotransplantation Products in Humans” which ensures that animals used for clinical products are infection and disease free. However, factors such as age are not included in this guidance and could potentially have an effect on the performance of medical devices derived from aged animals. Understanding how changes in the extracellular matrix changes with source animal age could lead to reduced variability in xenogeneic biomaterials and improved outcomes.
It is well documented that there are changes with age in extracellular matrix composition of many tissues. Aged colon exhibits increases in collagen content and decreases in strength [71]. Macrophages have been shown to respond differently to different collagen subtypes with collagen I increasing CCL18, IL-1ra, CCL2, CD204 and p-Akt expression whereas collagen III only increased CCL18 and IL-1ra [72]. Therefore, changes in extracellular matrix composition with age could alter the macrophage response to biomaterials derived from different aged source animals, as shown in this study. This could compromise normally beneficial host responses and remodeling outcomes downstream of implantation. Furthermore, the influence of ECM on responses to inflammatory cytokines suggest the ECM environment may play a role in the nature and progression of the host inflammatory responses to injury and infection. Biochemical analyses for hydroxyproline and glycosaminoglycan content were unable to detect changes in decellularized ECM scaffolds. Therefore, more rigorous analysis including mass spectrometry is needed to understand changes in composition of specific components within aged ECM.
The importance of changes in animal tissue product composition is most important with regard to how these changes effect the host response following implantation. A recent study has shown that aged animals elicited an altered macrophage response to polypropylene mesh implanted subcutaneously compared to young controls [73]. Results in this study show that bone marrow-derived macrophages from young and aged mice have similar abilities to polarize to M1 and M2 profiles in vitro. This suggests that changes in the macrophage response to implantable biomaterials cannot be explained by macrophage-intrinsic changes with age alone. It is logical to infer that the aged local microenvironment, consisting of the extracellular matrix and cellular signals, could be responsible for these changes in responses to biomaterials. These in vivo results further findings in this paper that aged ECM microenvironments affect macrophage phenotypes which could lead to altered immune responses.
There are documented changes in macrophage populations and phenotypes in aged organisms. Peritoneal macrophage populations increased in the percentage of monocyte-derived and mature macrophages. Aged peritoneal macrophages also exhibited increased pro-inflammatory cytokine IL-1b and IL-6 expression when stimulated with GM-CSF or IL-4. Aged macrophages also exhibited a decreased nitric oxide/urea ratio [74]. Aged splenic macrophages exhibited decreased expression of IL-6, iNOS, IL-1b and TNFa when stimulated with LPS. Aged splenocytes also had reduced M2 markers when stimulated with IL-4 compared with young controls [75]. Aged macrophages have also been shown to have decreased MHC-II expression following IFN-y treatment, suggesting onset of immunosenescence [76]. Our results showed 52 week ECM reduced MHC-II immunoexpression following IFN-y/LPS suggesting aged ECM may contribute to this immunosenescent phenotype. Further elucidation of these mechanisms is necessary to determine how aged ECM causes changes in macrophage phenotype.
Treatment of macrophages with aged ECM in vitro results in similar alterations in phenotype compared to young ECM, reduced iNOS and Fizz1 immunoexpression. Aged ECM treatment with IFN-y/LPS stimulus increased iNOS immunoexpression and reduced nitric oxide production at 24 hours. This could suggest that aged ECM incites a delayed iNOS response as nitric oxide production is a result of the cumulative production in media over 24 hours whereas immunolabeling assays protein level at 24 hours only. This suggests that age-related changes in the ECM may contribute to the altered immune responses observed in aged wound healing. Increased numbers of M2a macrophages have been reported in aged muscle, which may be due to the influence of aged microenvironments [77]. These changes in aged muscle are corrected by transfection of nNOS, suggesting that reduced nitric oxide in aged muscle could be a factor in this macrophage phenotype shift. Our findings that aged ECM reduces the acute production of nitric oxide could be a cause of the increased fibrosis in aged muscle.
Previous studies have noted that the aging microenvironment is responsible for alterations in macrophage populations. Aging muscle exhibits an increase in M2a macrophage populations that could be the cause of increased fibrosis [77]. Transplantation of young bone marrow cells into aged muscle reduced the number of M2a macrophages, which could be caused by altered phenotypes in aged macrophages or be a result of the shift toward myeloid lineages in aged bone marrow [77]. Blau et al. also suggest that the aging microenvironment impacts the function of muscle stem cells [36]. Some changes were attributed to intrinsic changes in MuSCs with age but others have been attributed to the local microenvironment. Age-related changes in the ECM microenvironment thus represents a cell-extrinsic mechanism of altered wound healing and regeneration in aged individuals.
Aging has an effect not only on macrophage phenotypes and populations, but also macrophage function. Linehan et al. showed that the aged peritoneal microenvironment significantly impaired phagocytic function of macrophages independent of intrinsic changes in bone marrow macrophage populations [82]. Some of these changes were attributed to changes in T and B cell populations and levels of IL-10. Reduced phagocytic function could lead to impaired responses to pathogens or delayed debris removal in wound environments. However, our results did not show any change in phagocytic function due to aged ECM exposure. Aged alveolar macrophages were also shown to have reduced production of nitric oxide [83]. This matches our findings that macrophages exposed to aged ECM produced less nitric oxide upon IFN-y/LPS exposure. This reduction in macrophage function could impair pathogen immunity and alter angiogenic responses.
The effects of aging have also been well documented in the area of wound healing. Wound healing in aged individuals is associated with delayed monocyte infiltration, increases in mature macrophages, and impaired macrophage function including reduced phagocytic capacity and increased secretion of pro-inflammatory mediators [37, 78, 79]. There are also delays in angiogenesis, collagen production and re-epithelialization, all of which are functions that are orchestrated by macrophages and their interaction with other cells of the injury response [80, 81]. In spinal cord injury, macrophages have been shown to have reduced IL-10 expression in aged individuals [84]. Macrophages in aged vascular injury exhibited increased IL-18 expression which was associated with higher fibrinogen deposition [85]. All these examples show the impact of shifts in aged macrophage populations and phenotypes upon normal wound healing. Aged microenvironments may be responsible for these shifts in maintained or recruited macrophage populations or alterations from expected phenotypic responses.
There were several limitations to the present study. This study only analyzed the effects of aging on porcine small intestine submucosa over the ages of 12 to 52 weeks. Not only does the extracellular matrix from different organs have different compositions but there are also organ-specific changes in this ECM makeup with age. Therefore, these results may not apply to ECM biomaterials derived from different sources. Furthermore, an in-depth assessment of what changed in these SIS scaffolds was not performed. More work would need to be done to understand the specific compositional changes and the signaling pathways which dictate changes in ECM-treated macrophage phenotype, which are currently unknown. Assessment of the presence and change in composition of matrix bound nanovesicles was also not completed. Changes in MBV content with age could be a potential mechanism for altered responses to these biomaterials. This study also focused on the effects of extracellular matrix biomaterials on the phenotypes of macrophages alone. Many other cells participate during, before and after the involvement of macrophages during wound healing and biomaterial remodeling, including neutrophils, fibroblasts, adaptive immune cells, endothelial cells and stem cells. Little investigation has been performed on the synergistic roles of these cells during regenerative processes and further work is warranted in order to understand how these cells function together.
5. Conclusion
Extracellular matrix scaffolds vary significantly due to their biological origin, and age effects their properties significantly. These changes in ECM structure are recognized by macrophages which change both their expression and function in response. Aged ECM degradation products alter macrophage phenotype and function both alone and in the presence of polarizing cytokines. These findings suggest that age-related changes in the ECM microenvironment have direct effects on the immune system. This suggests that age-related changes in wound healing and regeneration could be a result, in part, due to alterations in the extracellular matrix. This suggests further investigation into the ECM from aged individuals and its direct effect on immune and host response cells could help to elucidate mechanisms of altered healing responses in elderly populations.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Age-related changes in ECM directly impacts macrophage phenotype and function
52 week old SIS causes decreases in iNOS immunoexpression in 2 month macrophages and Fizz immunoexpression in 2 and 18 month macrophages
52 week old SIS with IFN-y/LPS exposure causes increased iNOS in 18 month macrophages and decreased MHC-II in 2 month macrophages
12 week old SIS causes enhanced nitric oxide production with IFN-y/LPS exposure
12 week old SIS causes increased arginase immunoexpression with IL-4 exposure
Acknowledgments
This work was supported by National Institutes of Health awards K12HD043441, R03AG043606, R01AG055564 and T32EB001026. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Sameer Patil, Sterling Bowers, Tomisin Ojo-Aromokudu and Siddhartha Dash are acknowledged for contributions to experimental completion, data collection and analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors have no financial or personal interests to disclose that interfere with the objectivity of the published work.
References
- 1.Keane TJ, Dziki J, Sobieski E, Smoulder A, Castleton A, Turner N, White LJ, Badylak SF. Restoring Mucosal Barrier Function and Modifying Macrophage Phenotype with an Extracellular Matrix Hydrogel: Potential Therapy for Ulcerative Colitis. Journal of Crohn’s & colitis. 2016 doi: 10.1093/ecco-jcc/jjw149. [DOI] [PubMed] [Google Scholar]
- 2.Wu Y, Wang J, Shi Y, Pu H, Leak RK, Liou AK, Badylak SF, Liu Z, Zhang J, Chen J, Chen L. Implantation of Brain-derived Extracellular Matrix Enhances Neurological Recovery after Traumatic Brain Injury. Cell transplantation. 2016 doi: 10.1177/0963689717714090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faulk DM, Londono R, Wolf MT, Ranallo CA, Carruthers CA, Wildemann JD, Dearth CL, Badylak SF. ECM hydrogel coating mitigates the chronic inflammatory response to polypropylene mesh. Biomaterials. 2014;35(30):8585–95. doi: 10.1016/j.biomaterials.2014.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sicari BM, Rubin JP, Dearth CL, Wolf MT, Ambrosio F, Boninger M, Turner NJ, Weber DJ, Simpson TW, Wyse A, Brown EH, Dziki JL, Fisher LE, Brown S, Badylak SF. An acellular biologic scaffold promotes skeletal muscle formation in mice and humans with volumetric muscle loss. Science translational medicine. 2014;6(234):234ra58. doi: 10.1126/scitranslmed.3008085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badylak SF, Hoppo T, Nieponice A, Gilbert TW, Davison JM, Jobe BA. Esophageal preservation in five male patients after endoscopic inner-layer circumferential resection in the setting of superficial cancer: a regenerative medicine approach with a biologic scaffold. Tissue engineering Part A. 2011;17(11–12):1643–50. doi: 10.1089/ten.tea.2010.0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown BN, Badylak SF. Extracellular matrix as an inductive scaffold for functional tissue reconstruction. Translational research : the journal of laboratory and clinical medicine. 2014;163(4):268–85. doi: 10.1016/j.trsl.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crapo PM, Gilbert TW, Badylak SF. An overview of tissue and whole organ decellularization processes. Biomaterials. 2011;32(12):3233–43. doi: 10.1016/j.biomaterials.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keane TJ, Dziki J, Castelton A, Faulk DM, Messerschmidt V, Londono R, Reing JE, Velankar SS, Badylak SF. Preparation and characterization of a biologic scaffold and hydrogel derived from colonic mucosa. Journal of biomedical materials research Part B, Applied biomaterials. 2015 doi: 10.1002/jbm.b.33556. [DOI] [PubMed] [Google Scholar]
- 9.Keane TJ, Londono R, Carey RM, Carruthers CA, Reing JE, Dearth CL, D’Amore A, Medberry CJ, Badylak SF. Preparation and characterization of a biologic scaffold from esophageal mucosa. Biomaterials. 2013;34(28):6729–37. doi: 10.1016/j.biomaterials.2013.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Hu ZQ, Turner NJ, Teng SF, Cheng WY, Zhou HY, Zhang L, Hu HW, Wang Q, Badylak SF. Perfusion-decellularized skeletal muscle as a three-dimensional scaffold with a vascular network template. Biomaterials. 2016;89:114–26. doi: 10.1016/j.biomaterials.2016.02.040. [DOI] [PubMed] [Google Scholar]
- 11.Schlie-Wolter S, Ngezahayo A, Chichkov BN. The selective role of ECM components on cell adhesion, morphology, proliferation and communication in vitro. Experimental cell research. 2013;319(10):1553–61. doi: 10.1016/j.yexcr.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Hodde J, Record R, Tullius R, Badylak S. Fibronectin peptides mediate HMEC adhesion to porcine-derived extracellular matrix. Biomaterials. 2002;23(8):1841–8. doi: 10.1016/s0142-9612(01)00310-6. [DOI] [PubMed] [Google Scholar]
- 13.Agrawal V, Kelly J, Tottey S, Daly KA, Johnson SA, Siu BF, Reing J, Badylak SF. An isolated cryptic peptide influences osteogenesis and bone remodeling in an adult mammalian model of digit amputation. Tissue engineering Part A. 2011;17(23–24):3033–44. doi: 10.1089/ten.tea.2011.0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal V, Tottey S, Johnson SA, Freund JM, Siu BF, Badylak SF. Recruitment of progenitor cells by an extracellular matrix cryptic peptide in a mouse model of digit amputation. Tissue engineering Part A. 2011;17(19–20):2435–43. doi: 10.1089/ten.tea.2011.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dziki JL, Wang DS, Pineda C, Sicari BM, Rausch T, Badylak SF. Solubilized extracellular matrix bioscaffolds derived from diverse source tissues differentially influence macrophage phenotype. Journal of biomedical materials research Part A. 2017;105(1):138–147. doi: 10.1002/jbm.a.35894. [DOI] [PubMed] [Google Scholar]
- 16.Sicari BM, Dziki JL, Siu BF, Medberry CJ, Dearth CL, Badylak SF. The promotion of a constructive macrophage phenotype by solubilized extracellular matrix. Biomaterials. 2014;35(30):8605–12. doi: 10.1016/j.biomaterials.2014.06.060. [DOI] [PubMed] [Google Scholar]
- 17.Huleihel L, Hussey GS, Naranjo JD, Zhang L, Dziki JL, Turner NJ, Stolz DB, Badylak SF. Matrix-bound nanovesicles within ECM bioscaffolds. Science advances. 2016;2(6):e1600502. doi: 10.1126/sciadv.1600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown BN, Londono R, Tottey S, Zhang L, Kukla KA, Wolf MT, Daly KA, Reing JE, Badylak SF. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta biomaterialia. 2012;8(3):978–87. doi: 10.1016/j.actbio.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration, American journal of physiology. Regulatory, integrative and comparative physiology. 2010;298(5):R1173–87. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials. 2009;30(8):1482–91. doi: 10.1016/j.biomaterials.2008.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valentin JE, Stewart-Akers AM, Gilbert TW, Badylak SF. Macrophage participation in the degradation and remodeling of extracellular matrix scaffolds. Tissue engineering Part A. 2009;15(7):1687–94. doi: 10.1089/ten.tea.2008.0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tottey S, Johnson SA, Crapo PM, Reing JE, Zhang L, Jiang H, Medberry CJ, Reines B, Badylak SF. The effect of source animal age upon extracellular matrix scaffold properties. Biomaterials. 2011;32(1):128–36. doi: 10.1016/j.biomaterials.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacraz G, Rouleau AJ, Couture V, Sollrald T, Drouin G, Veillette N, Grandbois M, Grenier G. Increased Stiffness in Aged Skeletal Muscle Impairs Muscle Progenitor Cell Proliferative Activity. PloS one. 2015;10(8):e0136217. doi: 10.1371/journal.pone.0136217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramaswamy KS, Palmer ML, van der Meulen JH, Renoux A, Kostrominova TY, Michele DE, Faulkner JA. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. The Journal of physiology. 2011;589(Pt 5):1195–208. doi: 10.1113/jphysiol.2010.201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hindle AG, Horning M, Mellish JA, Lawler JM. Diving into old age: muscular senescence in a large-bodied, long-lived mammal, the Weddell seal (Leptonychotes weddellii) The Journal of experimental biology. 2009;212(Pt 6):790–6. doi: 10.1242/jeb.025387. [DOI] [PubMed] [Google Scholar]
- 26.Kovanen V, Suominen H, Risteli J, Risteli L. Type IV collagen and laminin in slow and fast skeletal muscle in rats–effects of age and life-time endurance training. Collagen and related research. 1988;8(2):145–53. doi: 10.1016/s0174-173x(88)80026-8. [DOI] [PubMed] [Google Scholar]
- 27.Brown B, Lindberg K, Reing J, Stolz DB, Badylak SF. The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue engineering. 2006;12(3):519–26. doi: 10.1089/ten.2006.12.519. [DOI] [PubMed] [Google Scholar]
- 28.Labat-Robert J. Age-dependent remodeling of connective tissue: role of fibronectin and laminin. Pathologie-biologie. 2003;51(10):563–8. doi: 10.1016/j.patbio.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Snow MH. The effects of aging on satellite cells in skeletal muscles of mice and rats. Cell and tissue research. 1977;185(3):399–408. doi: 10.1007/BF00220299. [DOI] [PubMed] [Google Scholar]
- 30.Brack AS, Muñoz-Cánoves P. The ins and outs of muscle stem cell aging. Skeletal Muscle. 2015;6 doi: 10.1186/s13395-016-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sicari BM, Johnson SA, Siu BF, Crapo PM, Daly KA, Jiang H, Medberry CJ, Tottey S, Turner NJ, Badylak SF. The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials. 2012;33(22):5524–33. doi: 10.1016/j.biomaterials.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holt D, Grainger D. Senescence and quiescence induced compromised function in cultured macrophages. Biomaterials. 2012;33(30):7497–507. doi: 10.1016/j.biomaterials.2012.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases, The journals of gerontology. Series A. Biological sciences and medical sciences. 2014;69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 34.Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mechanisms of ageing and development. 2007;128(1):92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 35.Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007;120(4):435–46. doi: 10.1111/j.1365-2567.2007.02555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nature medicine. 2015;21(8):854–62. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashcroft GS, Horan MA, Ferguson MW. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Laboratory investigation; a journal of technical methods and pathology. 1998;78(1):47–58. [PubMed] [Google Scholar]
- 38.Labat-Robert J, Robert L. Longevity and aging. Mechanisms and perspectives. Pathologie-biologie. 2015;63(6):272–6. doi: 10.1016/j.patbio.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Bucala R. Diabetes, aging, and their tissue complications. The Journal of clinical investigation. 2014;124(5):1887–8. doi: 10.1172/JCI75224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corica F, Bianchi G, Corsonello A, Mazzella N, Lattanzio F, Marchesini G. Obesity in the Context of Aging: Quality of Life Considerations. PharmacoEconomics. 2015;33(7):655–72. doi: 10.1007/s40273-014-0237-8. [DOI] [PubMed] [Google Scholar]
- 41.North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circulation research. 2012;110(8):1097–108. doi: 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennan EP, Tang XH, Stewart-Akers AM, Gudas LJ, Badylak SF. Chemoattractant activity of degradation products of fetal and adult skin extracellular matrix for keratinocyte progenitor cells. Journal of tissue engineering and regenerative medicine. 2008;2(8):491–8. doi: 10.1002/term.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vorotnikova E, McIntosh D, Dewilde A, Zhang J, Reing JE, Zhang L, Cordero K, Bedelbaeva K, Gourevitch D, Heber-Katz E, Badylak SF, Braunhut SJ. Extracellular matrix-derived products modulate endothelial and progenitor cell migration and proliferation in vitro and stimulate regenerative healing in vivo. Matrix biology : journal of the International Society for Matrix Biology. 2010;29(8):690–700. doi: 10.1016/j.matbio.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Huleihel L, Dziki JL, Bartolacci JG, Rausch T, Scarritt ME, Cramer MC, Vorobyov T, LoPresti ST, Swineheart IT, White LJ, Brown BN, Badylak SF. Macrophage phenotype in response to ECM bioscaffolds. Seminars in immunology. 2017;29:2–13. doi: 10.1016/j.smim.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Badylak S, Kokini K, Tullius B, Simmons-Byrd A, Morff R. Morphologic study of small intestinal submucosa as a body wall repair device. The Journal of surgical research. 2002;103(2):190–202. doi: 10.1006/jsre.2001.6349. [DOI] [PubMed] [Google Scholar]
- 46.Reing JE, Brown BN, Daly KA, Freund JM, Gilbert TW, Hsiong SX, Huber A, Kullas KE, Tottey S, Wolf MT, Badylak SF. The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials. 2010;31(33):8626–33. doi: 10.1016/j.biomaterials.2010.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilbert TW, Stewart-Akers AM, Sydeski J, Nguyen TD, Badylak SF, Woo SL. Gene expression by fibroblasts seeded on small intestinal submucosa and subjected to cyclic stretching. Tissue engineering. 2007;13(6):1313–23. doi: 10.1089/ten.2006.0318. [DOI] [PubMed] [Google Scholar]
- 48.Keane TJ, Londono R, Turner NJ, Badylak SF. Consequences of ineffective decellularization of biologic scaffolds on the host response. Biomaterials. 2012;33(6):1771–81. doi: 10.1016/j.biomaterials.2011.10.054. [DOI] [PubMed] [Google Scholar]
- 49.Guthridge M, Wilson M, Cowling J, Bertolini J, Hearn MT. The role of basic fibroblast growth factor in skeletal muscle regeneration. Growth factors (Chur, Switzerland) 1992;6(1):53–63. doi: 10.3109/08977199209008871. [DOI] [PubMed] [Google Scholar]
- 50.Leroy MC, Perroud J, Darbellay B, Bernheim L, Konig S. Epidermal Growth Factor Receptor Down-Regulation Triggers Human Myoblast Differentiation. PloS one. 2013;8(8):e71770. doi: 10.1371/journal.pone.0071770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller KJ, Thaloor D, Matteson S, Pavlath GK. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle, American journal of physiology. Cell physiology. 2000;278(1):C174–81. doi: 10.1152/ajpcell.2000.278.1.C174. [DOI] [PubMed] [Google Scholar]
- 52.Huleihel L, Bartolacci JG, Dziki JL, Vorobyov T, Arnold B, Scarritt ME, Pineda Molina C, LoPresti ST, Brown BN, Naranjo JD, Badylak SF. Matrix-Bound Nanovesicles Recapitulate Extracellular Matrix Effects on Macrophage Phenotype. Tissue engineering Part A. 2017;23(21–22):1283–1294. doi: 10.1089/ten.tea.2017.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reing JE, Zhang L, Myers-Irvin J, Cordero KE, Freytes DO, Heber-Katz E, Bedelbaeva K, McIntosh D, Dewilde A, Braunhut SJ, Badylak SF. Degradation products of extracellular matrix affect cell migration and proliferation. Tissue engineering Part A. 2009;15(3):605–14. doi: 10.1089/ten.tea.2007.0425. [DOI] [PubMed] [Google Scholar]
- 54.Daly KA, Liu S, Agrawal V, Brown BN, Huber A, Johnson SA, Reing J, Sicari B, Wolf M, Zhang X, Badylak SF. The host response to endotoxin-contaminated dermal matrix. Tissue engineering Part A. 2012;18(11–12):1293–303. doi: 10.1089/ten.TEA.2011.0597. [DOI] [PubMed] [Google Scholar]
- 55.Gryder RM, Lamon M, Adams E. Sequence position of 3-hydroxyproline in basement membrane collagen. Isolation of glycyl-3-hydroxyprolyl-4-hydroxyproline from swine kidney. The Journal of biological chemistry. 1975;250(7):2470–4. [PubMed] [Google Scholar]
- 56.Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavsky I. A heparin-binding angiogenic protein–basic fibroblast growth factor–is stored within basement membrane. The American journal of pathology. 1988;130(2):393–400. [PMC free article] [PubMed] [Google Scholar]
- 57.Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17(1):109–18. doi: 10.1007/s10456-013-9381-6. [DOI] [PubMed] [Google Scholar]
- 58.Galimi F, Cottone E, Vigna E, Arena N, Boccaccio C, Giordano S, Naldini L, Comoglio PM. Hepatocyte growth factor is a regulator of monocyte-macrophage function. Journal of immunology (Baltimore, Md : 1950) 2001;166(2):1241–7. doi: 10.4049/jimmunol.166.2.1241. [DOI] [PubMed] [Google Scholar]
- 59.Matsumoto S, Kishida K, Shimomura I, Maeda N, Nagaretani H, Matsuda M, Nishizawa H, Kihara S, Funahashi T, Matsuzawa Y, Yamada A, Yamashita S, Tamura S, Kawata S. Increased plasma HB-EGF associated with obesity and coronary artery disease. Biochemical and biophysical research communications. 2002;292(3):781–6. doi: 10.1006/bbrc.2002.6720. [DOI] [PubMed] [Google Scholar]
- 60.Edwards JP, Zhang X, Mosser DM. The expression of heparin-binding epidermal growth factor-like growth factor by regulatory macrophages. Journal of immunology (Baltimore, Md : 1950) 2009;182(4):1929–39. doi: 10.4049/jimmunol.0802703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. Journal of immunology (Baltimore, Md : 1950) 2004;172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 62.Yamaji-Kegan K, Su Q, Angelini DJ, Myers AC, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. Journal of immunology (Baltimore, Md : 1950) 2010;185(9):5539–48. doi: 10.4049/jimmunol.0904021. [DOI] [PubMed] [Google Scholar]
- 63.Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, Pasparakis M, Eming SA. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120(3):613–25. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- 64.Vestergaard C, Just H, Baumgartner Nielsen J, Thestrup-Pedersen K, Deleuran M. Expression of CCR2 on monocytes and macrophages in chronically inflamed skin in atopic dermatitis and psoriasis. Acta dermato-venereologica. 2004;84(5):353–8. doi: 10.1080/00015550410034444. [DOI] [PubMed] [Google Scholar]
- 65.Ostuni MA, Guellec J, Hermand P, Durand P, Combadiere C, Pincet F, Deterre P. CX3CL1, a chemokine finely tuned to adhesion: critical roles of the stalk glycosylation and the membrane domain. Biology open. 2014;3(12):1173–82. doi: 10.1242/bio.20149845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng J, Yang M, Shao J, Miao Y, Han J, Du J. Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Molecular cancer. 2013;12(1):141. doi: 10.1186/1476-4598-12-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xaus J, Comalada M, Barrachina M, Herrero C, Gonalons E, Soler C, Lloberas J, Celada A. The expression of MHC class II genes in macrophages is cell cycle dependent. Journal of immunology (Baltimore, Md : 1950) 2000;165(11):6364–71. doi: 10.4049/jimmunol.165.11.6364. [DOI] [PubMed] [Google Scholar]
- 68.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annual review of immunology. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 69.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. Journal of immunology (Baltimore, Md : 1950) 1998;160(12):5729–34. [PubMed] [Google Scholar]
- 70.Costa A, Naranjo JD, Londono R, Badylak SF. Biologic Scaffolds. Cold Spring Harbor perspectives in medicine. 2017;7(9) doi: 10.1101/cshperspect.a025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christensen H, Andreassen TT, Oxlund H. Age-related alterations in the strength and collagen content of left colon in rats. International journal of colorectal disease. 1992;7(2):85–8. doi: 10.1007/BF00341292. [DOI] [PubMed] [Google Scholar]
- 72.Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, Prasse A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PloS one. 2013;8(11):e81382. doi: 10.1371/journal.pone.0081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hachim D, Wang N, Lopresti ST, Stahl EC, Umeda YU, Rege RD, Carey ST, Mani D, Brown BN. Effects of Aging upon the Host Response to Implants. Journal of Biomedical Materials Research Part A. 2017 doi: 10.1002/jbm.a.36013. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dimitrijevic M, Stanojevic S, Blagojevic V, Curuvija I, Vujnovic I, Petrovic R, Arsenovic-Ranin N, Vujic V, Leposavic G. Aging affects the responsiveness of rat peritoneal macrophages to GM-CSF and IL-4. Biogerontology. 2016;17(2):359–71. doi: 10.1007/s10522-015-9620-x. [DOI] [PubMed] [Google Scholar]
- 75.Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2012;32(1):18–26. doi: 10.1089/jir.2011.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herrero C, Sebastian C, Marques L, Comalada M, Xaus J, Valledor AF, Lloberas J, Celada A. Immunosenescence of macrophages: reduced MHC class II gene expression. Experimental gerontology. 2002;37(2–3):389–94. doi: 10.1016/s0531-5565(01)00205-4. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Wehling-Henricks M, Samengo G, Tidball JG. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell. 2015;14(4):678–88. doi: 10.1111/acel.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. The Journal of investigative dermatology. 2001;117(5):1027–35. doi: 10.1046/j.0022-202x.2001.01539.x. [DOI] [PubMed] [Google Scholar]
- 79.Ashcroft GS, Mills SJ, Ashworth JJ. Ageing and wound healing. Biogerontology. 2002;3(6):337–45. doi: 10.1023/a:1021399228395. [DOI] [PubMed] [Google Scholar]
- 80.Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Laboratory investigation; a journal of technical methods and pathology. 1999;79(12):1479–87. [PubMed] [Google Scholar]
- 81.Reed MJ, Karres N, Eyman D, Vernon RB, Edelberg JM. Age-related differences in repair of dermal wounds and myocardial infarcts attenuate during the later stages of healing. In vivo (Athens, Greece) 2006;20(6b):801–6. [PubMed] [Google Scholar]
- 82.Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. 2014;13(4):699–708. doi: 10.1111/acel.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koike E, Kobayashi T, Mochitate K, Murakami M. Effect of aging on nitric oxide production by rat alveolar macrophages. Experimental gerontology. 1999;34(7):889–94. doi: 10.1016/s0531-5565(99)00061-3. [DOI] [PubMed] [Google Scholar]
- 84.Zhang B, Bailey WM, Braun KJ, Gensel JC. Age decreases macrophage IL-10 expression: Implications for functional recovery and tissue repair in spinal cord injury. Experimental neurology. 2015;273:83–91. doi: 10.1016/j.expneurol.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez-Menocal L, Faridi MH, Martinez L, Shehadeh LA, Duque JC, Wei Y, Mesa A, Pena A, Gupta V, Pham SM, Vazquez-Padron RI. Macrophage-derived IL-18 and increased fibrinogen deposition are age-related inflammatory signatures of vascular remodeling, American journal of physiology. Heart and circulatory physiology. 2014;306(5):H641–53. doi: 10.1152/ajpheart.00641.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.