

Abstract
Inflammatory Bowel Disease (IBD) has been associated with the dysregulation of T cells specific to antigens derived from the intestinal microbiota. How microbiota-specific T cells are regulated is not completely clear but is believed to be mediated by a combination of Immunglobulin A (IgA), regulatory T cells (Tregs) and Type 3 Innate Lymphoid cells (ILC3s). To test the role of these regulatory components on microbiota-specific T cells, we bred CBir1 TCR transgenic mice (CBir1Tg; specific to flagellin from common intestinal bacteria) onto a lymphopenic Rag1−/− background (CBirRag). Surprisingly, CBirRag T cells could not induce colitis and did not differentiate to become effectors under lymphopenic conditions, despite deficits in immunoregulatory factors such as IgA, Tregs and ILC3s. In fact, upon transfer of conventional CBir1Tg T cells into lymphopenic mice, the vast majority of proliferating T cells responded to antigens other than CBir1 flagellin, including those found on other bacteria, such as Helicobacter spp. Thus, we discovered a caveat in the CBir1 TCR Tg model within our animal facility that illustrates the limitations of using TCR transgenics at mucosal surfaces, where multiple TCR specificities can respond to the plethora of foreign antigens. Our findings also indicate that T cell specificity to the microbiota alone is not sufficient to induce T cell activation and colitis. Instead, other interrelated factors, such as the composition and ecology of the intestinal microbiota and host access to antigen, are paramount in controlling the activation of microbiota-specific T cell clones.
Introduction
The barrier surfaces of mammals are covered in a vast consortium of microorganisms that together comprises the microbiome. Mammals also possess millions of B and T lymphocytes with variable specificities that include responsiveness to antigens present in the microbial flora. Microbiota-specific T cells have been described as part of the healthy T cell repertoire in both mice and humans but are believed to contribute to the development of Inflammatory Bowel Disease (IBD) under certain contexts (1, 2). Many of the genetic polymorphisms associated with IBD have been shown to directly affect T cells or intestinal barrier function and therefore access of intestinal antigens to the host immune system (3). Elucidating the mechanisms that maintain homeostasis among microbiota-specific T cells is critical to our understanding of mucosal immunology and the development of IBD (4, 5).
In the T cell mediated model of colitis, transfer of naïve CD4+ T cells into lymphopenic mice leads to colitis associated with large numbers of activated helper T cells (IL-17A+; IFNγ+) and a relative absence of regulatory T (Treg) cells. This model requires the intestinal microbiota, and many of the colitogenic T cells in these mice are specific to various intestinal bacteria, including Helicobacter spp. and CBir1 flagellin-expressing Clostridria (6–13). Presumably, presentation of antigens in the lymphopenic lymph node to transferred naïve CD4+ T cells in the absence of immunoregulatory factors leads to the development of colitogenic T cell clones. Regulation of microbiota-specific T cells is believed to be controlled by a variety of interrelated factors, including Interleukin 10 (IL-10), regulatory T cells (Tregs), Immunoglobulin A (IgA) and Type 3 Innate Lyphoid Cells (ILC3s), though how these factors work together is not clearly understood (14–17). On a lymphoreplete ‘wild-type’ background, CBir1-specific transgenic (CBirWt) mice do not spontaneously develop colitis, and physical breakdown of the intestine by infection (Toxoplasma gondii) or chemical irritation (Dextran Sodium sulfate) is necessary to activate CBir1Tg T cells in such hosts (14, 18). This is in contrast to TCR transgenic cells specific to either segmented filamentous bacteria or Helicobacter spp., where colonization is sufficient to induce spontaneous T cell responses, due to the residence of these organisms directly on the epithelial surface (19–21). However, it is not known whether T cell responses in lymphopenic animals are strictly limited to bacteria that live close to the intestinal surface, as it appears to be in lymphoreplete animals, or whether such responses are broadened to other members of the microbiota.
To test the contributions of these various regulatory factors on a genetically lymphopenic background, we bred CBir1Tg mice to a Rag1−/− background (CBirRag). These mice also allowed us to study a population of CBir1-specific T cells that is not contaminated by alternate specificities produced by rearrangements of the endogenous TCR locus. Surprisingly, CBirRag T cells did not become spontaneously activated in vivo nor were they capable of inducing colitis upon transfer to either Rag1−/− or Rag2−/−γc−/− mice, despite the lack of Tregs, IgA and ILC3s. The majority of CBirWt T cells that accumulated in Rag1−/− mice post-transfer did not bind a tetramer bearing the relevant peptide from CBir1 flagellin and were responsive to other intestinal bacteria, such as Helicobacter spp., due to the expression of endogenously rearranged non-transgenic TCRα chains. Taken together, our data uncovers a complication of the CBir1 Tg model of T cell mediated colitis and show that antigen specificity alone is not sufficient for the activation of T cells against the microbiota.
Materials and Methods
Mice
C57BL/6 mice were purchased from Taconic. Rag1−/− and Rag2−/−γc−/− mice were obtained from Jackson Laboratories. CBir1 Tg mice were produced by Dr. Charles Elson (Univ. Alabama-Birmingham) as described, obtained under an MTA and back-crossed to CD45.1 expressing mice, TCRα−/− mice, or Rag1−/− mice for at least three generations (14). Smarta TCR transgenic mice were obtained from Dr. Ronald Germain (NIH/NIAID). In some experiments, mice were given autoclaved drinking water supplemented with either vancomycin (0.5mg/mL, Sigma) or a cocktail of metronidazole (1mg/mL, Sigma), ampicillin (1mg/mL, Sigma), neomycin (1mg/mL, Sigma), and vancomycin (0.5 mg/mL, Sigma). Sucralose (Splenda™) (0.8mg/mL) was added to make the antiobiotic-containing water more palatable. Antibiotic treatment was started 1 week prior to adoptive T cell transfer or other uses of the mice. Gender-matched and age-matched mice were used and co-housed whenever possible. We used males and females equally in all experiments with the provision that cell transfers were always carried out within one sex group. Donor and recipient mice for cell transfers and analyses were used at 5–10 weeks of age. All mice were maintained at and all experiments were performed in an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the University of Pittsburgh and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Mice were housed in specific pathogen-free (SPF) conditions.
Cell isolation, stimulation, and culture
Cells from spleens, mesenteric lymph nodes (mLN), and colonic lamina propria (cLP) were isolated and stimulated for cytokine production with PMA/Ionomycin (Sigma) in the presence of Brefeldin A (ThermoFisher) as previously described (22).
For in vitro culture assays, splenic and LN CD4+ T cells were isolated by magnetic purification (StemCell) and stained with 1μM CFSE (ThermoFisher) as described previously (23). Fecal bacterial antigens were obtained by boiling fecal pellets for 1 min at 100°C and centrifuging at 12,000 × g for 5 min to collect the supernatant. CD4+ T cells were co-cultured for 96 hours with magnetically purified CD11c+ splenic dendritic cells from a CD45.2+ congenic C57BL/6NTac mouse and either the cleared fecal lysates or 10 ng/mL CBir1 peptide. Proliferation and activation of CD4+ T cells were then assessed via flow cytometry.
Antibodies, tetramers, and flow cytometry
All antibodies used for flow cytometry were purchased from either ThermoFisher, BD Biosciences, or BioLegend. The following antibodies were used to discriminate cell surface or intracellular phenotype: TCRβ (H57-597), CD3 (500A2), CD90.2 (53-2.1), CD4 (RM4-5), CD8b (H35-17.2), CD45.1 (A20), CD45.2 (104) CD44 (IM7), IFN-γ (XMG1.2), TNFα (MP6-XT22), IL-17α (eBio17B7), IL-2Rα/CD25 (PC61), Vα2 (B20.1) and Foxp3 (FJK-16S). Dead cells were discriminated in all experiments using LIVE/DEAD fixable dead stain (ThermoFisher). All stains were carried out in media containing anti-CD16/32 blocking antibody (clone 93, ThermoFisher). For intracellular cytokine staining, cells were fixed in BD Cytofix buffer (BD Biosciences) and stained in BD PermWash buffer (BD Biosciences). For Foxp3 staining, cells were fixed and permabilized using the FoxP3/Transcription factor staining buffer set according to the manufacturer’s directions (ThermoFisher). APC-conjugated CBir1464-72 tetramers (YSNANILSQ) and PE-conjugated HH1713172-86 and HH1713230-44 tetramers (QESPRIAAAYTIKGA and GNAYISVLAHYGKNG, respectively) were provided by the NIH Tetramer Core Facility (Atlanta, GA). All tetramer stains were performed at room temperature for 45–60 minutes. All flow cytometry was acquired on an LSRFortessa FACS analyzer, and cell sorting was carried out on a FACS Aria (BD Biosciences).
T cell transfer colitis and Histopathologic assessment
Cells from host spleen and lymph nodes were isolated, and CD4+ T cells were enriched via magnetic separation (StemCell) and then sort purified via FACs for naïve CD44loCD45RBhiCD25− CD4+ T cells (>99% purity). Of these, 0.5–1.5 × 106 cells were injected i.v. into groups of Rag1−/− or Rag2−/−γc−/− mice. In some experiments, transferred cells were labelled with CFSE or Cell Trace Violet (ThermoFisher), and recipient mice were sacrificed to assess T cell responses in the spleen, mLN, and cLP at 9–13 days post-transfer. In other experiments, unlabeled naïve CD4+ T cells were transferred, recipient mice were weighed weekly to assess colitis and sacrificed after 7–10 weeks post-transfer to assess transferred T cell responses in the spleen, mLN, and cLP. Mice were euthanized if their weights drop below 70% of baseline. Sections of colons were fixed in 10% formalin and paraffin embedded. Slides were stained with hematoxylin and eosin by the University of Pittsburgh Pathology Department, and lymphocyte infiltration and villous structural integrity were evaluated via microscopy.
Statistical analyses
Statistical tests used are indicated in the figure legends. All scatter plots show the mean, and all other graphs show the mean ± SD. All statistical analysis was calculated using Prism software (GraphPad). In experiments where cells were transferred to Rag1−/− mice, the resulting number of isolated T cells varied considerably due to the complex nature of the experiments and therefore a Kruskal-Wallis test was used because the data were non-parametric.
Results
To investigate microbiota-specific T cell development without the influence of IgA, B cells, and secondary TCRs, we generated CBirRag mice. We hypothesized that these mice would develop spontaneous colitis because their T cells would express only a single TCR specific to a common intestinal antigen and develop within a lymphopenic environment. In further support of our hypothesis, recent studies show that CBir1 flagellin may be transported by goblet cell-associated antigen passages early in the pre-weaning period of development (24). In light of these findings, we were surprised to discover that CBirRag mice in our facility develop colitis at a rate that is not significantly greater than littermate non-transgenic Rag1−/− mice (CBirRag 1/49; Rag1−/− 2/48). To help explain the lack of disease, we compared CD4+ T cells from spleens, mesenteric lymph nodes (mLN), and colonic lamina propria (cLP) of CBirWt, CBirRag, and Smarta TCR Tg (specific to an antigen-derived from Lymphochoriomeningitis virus, which is absent from the standard murine microbiota (25)) mice. In accord with the lack of disease, these studies revealed almost no T cell activation in CBirRag mice, as measured by the expression of CD44 and the production of the cytokines IFNγ, TNFα and IL-17A (Fig. 1A–D and G).
Figure 1. CBir1 flagellin-specific T cells do not become activated during development in lymphopenic hosts (A–D).

Lymphocytes from spleen, mesenteric lymph nodes (mLN) and colonic lamina propria (cLP) of various mouse strains were analyzed by flow cytometry. (A) CD44 expression on splenic CD4+TCRβ+ cells; numbers represent mean percentage ± SEM. (B–D) Cells were stimulated with PMA/ionomycin and analyzed for (B and C) TNFα/IFNγ co-expression or (D) IL-17A expression. Numbers on flow cytometry plots (C) represent mean positive events in cLP ± SEM. (E and F) Percent of cells from various tissues (E) expressing Foxp3; the flow cytometry plots (F) represent cells from the cLP. Numbers on flow cytometry plots show the mean percent of Foxp3+ cells in the cLP ± SEM. (G) Percent of CD4+ T cells from the cLP expressing IL-10 and IL-17A. Numbers on flow cytometry plots represent mean positive events in cLP ± SEM. Flow cytometry plots were gated on Live CD90.2+TCRb+CD4+CD8− cells (and additional Foxp3− for A–D). Data are representative of ≥3 independent experiments, n=8–10 mice/group. Statistical test: one way ANOVA with Tukey’s test for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
One explanation for the relative quiescence of CBirRag T cells could be that they develop into Tregs. Previous studies have shown that TCR transgenic T cells specific to foreign antigens, such as chicken ovalbumin, do not develop into Tregs (26). In contrast, microbiota-specific T cells, including CBir1, can differentiate into peripheral Tregs (pTregs) (14, 24, 27). Foxp3+CD4+ Tregs were almost undetectable in the secondary lymphoid tissues of CBirRag mice and present at significantly reduced frequencies in the cLP (Fig. 1E and F). Another possible explanation for the lack of T cell activation in CBirRag mice was T regulatory 1 cells that lack FoxP3 expression and produce IL-10. Analysis of colonic T cells from CBirWt, CBirRag and Smarta mice revealed that fewer CBirRag T cells produce IL-10 than CBirWt or Smarta T cells, indicating that this is unlikely to be preventing T cell activation and colitis (Fig. 1G). Thus, Tregs, IgA and T cell-derived IL-10 are not necessary to prevent the spontaneous activation and colitogenic potential of CBir1Tg T cells developing on a lymphopenic Rag−/− background.
It remained possible that CD4+ T cells in CBirRag are being prevented from spontaneous activation by either the small population of Tregs found in CBirRag mice or other immunoregulatory cells, such as CD8+ regulatory T cells (Supplemental Fig. 1A) (28). Thus, we sought to determine if the transfer of purified CD4+ CBirRag T cells to lymphopenic hosts will induce disease, as shown previously for CBirWt T cells (8, 13). We transferred FACs-purified naïve CBirWt, CBirRag or Smarta T cells to Rag1−/− mice and monitored colitis development via weight loss. In accordance with previous studies, Rag1−/− mice containing CBirWt T cells started losing weight 3 weeks post-transfer, and histological analysis of the recipient colons showed significant hyperplasia and pathology (Fig. 2A and B). In contrast, Rag1−/− recipients of CBirRag T cells gained weight and showed no obvious pathology (Fig. 2A and B). CBirWt T cells accumulated in >10-fold larger quantities in Rag1−/− recipients and displayed significantly higher expression of IFNγ, TNFα and IL-17A in the cLP compared to CBirRag T cells (Fig. 2C–E). The lack of accumulation among CBirRag T cells was not due to increased conversion into Tregs since they were almost undetectable among transferred CBirRag T cells (Fig. 2F). Therefore, despite the fact that both CBirRag and CBirWt T cells express the same TCR, only CBirWt T cells are capable of inducing colitis, implying that responsiveness to CBir1 flagellin alone is not sufficient for colitogenic T cell activation in lymphopenic hosts.
Figure 2. Transfer of CBir1 flagellin-specific T cells on a Rag1−/− background to lymphopenic hosts does not result in the development of colitis (A–F).

5 × 105 FACs-sorted (CD4+CD44loCD45RBhiCD25−) CD4+ T cells were transferred to gender-matched Rag1−/− recipients (n=3). (A) Weight change was tracked, and (B) colons were isolated for H&E histological staining (scale bars = 250μm). (C–F) Lymphocytes were isolated from Rag1−/− recipient mice in (A), stimulated with PMA and ionomycin, and quantified via flow cytometry for (C) total donor CD4+ T cells, (D) IFNγ+TNFα+ CD4+ T cells and (E) IL-17a+ CD4+ T cells. (F)Number Foxp3+ Treg cells isolated from different tissues, as indicated. Statistical test: Kruskal-Wallis for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
It was possible that CBir1 specific T cells are not becoming spontaneously activated because CBir1 flagellin-bearing bacteria were lost from the microbiota of our mouse colony. To confirm the presence of CBir1 flagellin in our mice, we co-cultured the supernatants from boiled fecal samples from various mouse strains (CBirWt, CBirRag, CBir1Tg x TCRα−/− (CBirTCR), Rag1−/−) with CBirWt T cells and dendritic cells (DCs). All samples tested were capable of inducing T cell proliferation, confirming the presence of CBir1-bearing bacteria in our colony (Fig. 3A). Furthermore, CBirWt T cells did not proliferate in response to fecal antigens mice treated with vancomycin, which depletes Gram-positive anaerobes (including CBir1-bearing Clostridia) (Fig. 3B). A previous study had shown that TCR transgenic T cells on a Rag1−/− background do not proliferate as well as those on a conventional background (29). However, CBirRag T cells proliferate just as well as CBirWt cells and CBirTCR cells in response to CBir1 peptide in vitro, indicating that these cells do not have any cell-intrinsic proliferative defects and do not require a second TCR specificity to respond to CBir1 antigen (Fig. 3C). Therefore, neither the loss of CBir1 flagellin nor proliferative potential amongst CBirRag CD4+ T cells could explain their inability to cause colitis.
Figure 3. CBirRag T cells proliferate normally and respond specifically to CBir1 flagellin, which is found in all mouse strains used in this study (A and B).
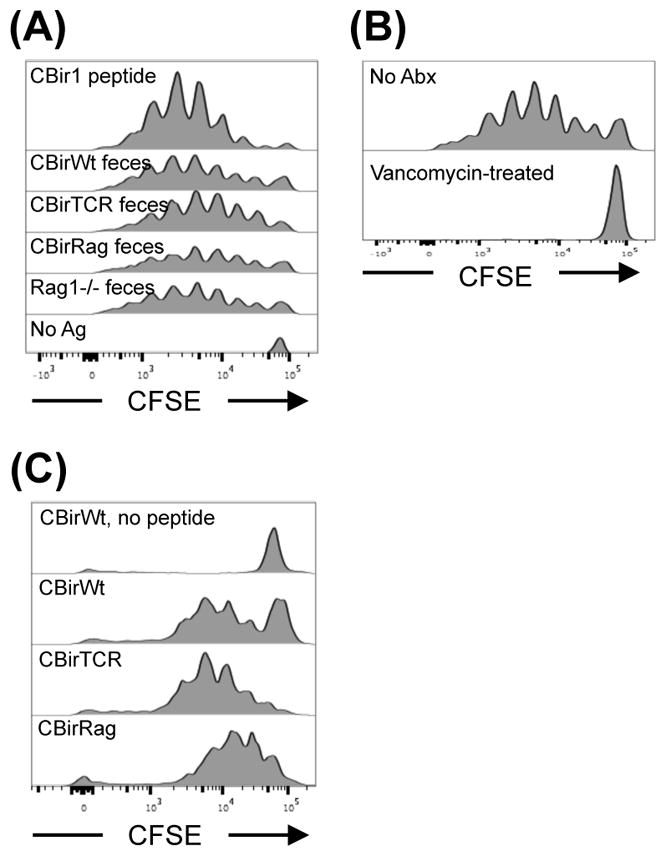
CFSE-labeled CBirWt splenocytes were cultured with splenic CD11c+ dendritic cells for 4 days and stimulated with either (A) 10ng/mL CBir1 peptide or supernatant from boiled fecal pellets of indicated mouse strains or (B) fecal pellets from Rag1−/− mice orally treated with vancomycin. (C) Splenocytes from CBirWt, CBirTCR and CBirRag mice were labeled with CFSE and cultured for 4 days with splenic CD11c+ dendritic cells and 10ng/mL CBir1 peptide. Shown is one representative experiment of three independent experiments
Another possibility that could explain the selective inability of CBirRag T cells to cause colitis was interactions with Innate Lymphoid cells type 3 (ILC3s). ILC3s have been shown to control the accumulation of CBirWt T cells and are present in Rag1−/− hosts. The lack of a possible secondary TCRα chain may also render CBirRag T cells more susceptible to deletion by ILC3s (15, 16). To test the importance of ILC3s, we transferred CBirRag or CBirWt T cells to ILC3-deficient Rag2−/−γc−/− mice. Activated CBirRag T cells did not accumulate in significant numbers in the GI tract of Rag2−/−γc−/− mice nor did they cause colitis as measured by weight loss and histology (Fig. 4A–C). Thus, while ILC3s may delete activated microbiota-specific T cells, they are not the primary regulatory element that prevents CBir1-specific T cell activation in lymphopenic settings.
Figure 4. ILCs are not the primary regulatory element that prevents CBir1-specific T cell activation in lymphopenic settings (A–C).

5 × 105 FACs-sorted naïve CD4+ T cells were transferred to Rag2−/−γc−/− mice (n=6). (A) Weight change was measured. (B) Post-transfer cLP lymphocytes were stimulated with PMA/ionomycin and analyzed for TNFα/IFNγ co-expression. (C) Colons were isolated for H&E histological staining (scale bars = 250μm). Numbers on flow cytometry plots represent mean positive events in cLP. Flow cytometry plots were gated on Live CD45.1+CD90.2+TCRβ+CD4+CD8− cells. Graph shows mean ± SD. Data are representative of 2 independent experiments, n=6 mice/group.
It remained unclear why CBirWt, but not CBirRag, T cells became activated and induced colitis upon transfer to Rag1−/− mice. One explanation is that re-arrangement of endogenous TCRα chains in CBirWt mice can lead to TCR transgenic T cells that express multiple functional TCRs and that these secondary specificities confer additional functionality. Analysis of CBirWt T cells showed that while all cells express Vβ8, associated with the CBir1 TCR, some cells (~4%) co-expressed a secondary Vα2 chain (the CBir1 TCR transgene expresses Vα6), indicating that they exhibit multiple specificities (Supplemental Fig. 1B) (14). Therefore, we sought to measure the TCR expression of CBirWt and CBirRag T cells (with non-transgenic C57BL/6 as controls) using MHC class II tetramers bearing the relevant peptide from CBir1 flagellin. These experiments confirmed that nearly all CBirRag T cells bind the CBir1 tetramer, as re-arrangement of endogenous TCRα loci is not possible in CBirRag mice (Fig. 5A). However, surprisingly, a significant fraction of T cells in CBirWt mice do not express sufficient levels of transgenic CBir1 TCR to bind the tetramer (Fig. 5A and B). Strikingly, splenic CBir1 tetramer-positive cells in CBirWt and CBirRag mice were phenotypically indistinguishable in that they almost completely lacked the expression of Vα2 (as a surrogate for the ability to re-arrange endogenous TCRα chains), high level expression of CD44 and differentiation to FoxP3+ Tregs (Fig. 5A, C and D). Conversely, the expression of CD44, Vα2 and IL-2Rα/FoxP3 was almost solely attributable to CBir1 tetramer negative T cells from CBirWt mice (Fig. 5C and D). Thus, perhaps the difference in the ability to induce colitis was not due to phenotypic differences in CBir1-specific T cells from CBirWt and CBirRag mice but rather due to CBirWt T cells that have re-arranged a second TCRα chain, leading to the potential for novel specificities.
Figure 5. Endogenous rearrangement of the TCRα chain in CBir1Tg mice allows for multiple TCR specifities and consequent T cell activation and colitis induction (A–D).

Lymphocytes from spleen and cLP from various mouse strains analyzed by flow cytometry. (A and B) CD44 expression and binding to CBir1 tetramer by CD4+ CD3+ cells; numbers represent percent of cells in each gate. (C) Vα2 expression and (D) Foxp3 and CD25 co-expression in CD4+CD3+ T cells. (E–G) 106 FACs-sorted (CD44loCD45RBhiCD25− CD4+), CFSE-labelled naïve CD4+ T cells were transferred to gender-matched Rag1−/− recipients. Ten days post-transfer, lymphocytes were analyzed via flow cytometry to determine (E) percent of CFSE− cells and tetramer expression, Numbers on flow cytometry plots represent mean positive events ± SEM. (F) CBir1 MHCII tetramer specificity among CFSE− CD4+ T cells, and (G) Vβ expression among transferred CBirWt CD4+ T cells from the spleen of Rag1−/− recipients (mean ± SD). Flow cytometry plots were gated on Live CD45.1+CD90.2+CD4+CD8− cells. Data are representative of 2–3 independent experiments, n=3–8 mice/group. Statistical test: one way ANOVA with Tukey’s test for multiple comparisons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To test the hypothesis that the ability of CBirWt T cells to induce colitis was associated with non-transgenic specificities, we measured the TCR expression of proliferating CBir1Tg T cells in lymphopenic hosts. We transferred CFSE-labeled naïve CBirWt or CBirRag CD4+ T cells to Rag1−/− recipients and analyzed the transferred CBir1Tg cells ten days later. Analysis of mLN and spleens (cLPs of CBirRag transferred mice contained no transferred cells) indicated that most CBirWt T cells showed rapid proliferation (complete loss of CFSE), and the majority (~60%) of rapidly proliferating T cells lacked the ability to bind CBir1 tetramer (Fig. 5E and F). This is in contrast to transferred CBirRag T cells, which maintained CBir1 tetramer binding and exhibited antigen-independent ‘slow’ proliferation associated with the increased availability of homeostatic cytokines such as IL-7 (Fig. 5E and F) (30). It is possible that the lack of the CBir TCR surface expression is due to cognate T cell activation (with CBir flagellin) and receptor endocytosis. However, overnight culture of CFSE− CBirWt T cells (sorted from Rag1−/− recipient mice) without antigen did not result in restoration of the ability to bind CBir1 tetramer, making cognate antigen-driven TCR down-regulation an unlikely explanation (Supplemental Fig. 2). We tested for downregulation of the CBir1-specific TCR directly by transferring either FACs-sorted tetramer positive or negative CBirWt naïve CD4+ T cells to Rag1−/− recipients (Supplemental Fig. 3A). In accord with our in vitro experiments, analysis of transferred cells 13 days later revealed minimal re-expression of the CBir1-specific TCRs on cells sorted as tetramer negative (Supplemental Fig. 3B). Further, sorted CBir1 tetramer negative T cells proliferated and accumulated in much greater numbers than their tetramer positive counterparts adding further evidence that these cells dominate the response in lymphopenic hosts. (Supplemental Fig. 3C). Interestingly, in contrast to CBirRag T cells, recipients of CBir1 tetramer positive CBirWt T cells came to be dominated by tetramer negative T cells, indicating that lymphopenic conditions may select for the outgrowth of cells expressing alternative TCR specificities and that such expression may exclude the transgenic Vα chain (Supplemental Fig. 3B and C). It should be noted that those CBirWt T cells that maintained expression of the transgenic TCR are much more likely to exhibit ‘slow’ proliferation further indicating that the expression of a second TCR specificity may favor rapid proliferation (Supplemental Fig. 3C). Surface staining of CFSE− CBir1Wt T cells from Rag1−/− mice revealed no significant expression of TCRβ variable regions, aside from the transgenic Vβ8.3 (expressed on all transferred cells), negating the possibility that tetramer negative CBirWt T cells are escaping β exclusion to produce complete non-transgenic TCRs or downregulating the TCR altogether (Fig. 5G). Taken together our results support the hypothesis that the dominant proliferating population of CBirWt T cells transferred to Rag1−/− hosts do not respond to CBir1 antigen and instead carry endogenously re-arranged TCRα chains that provide alternate specificities.
To further test if CBir1 flagellin-bearing bacteria were required for the activation of the tetramer negative CBirWt cells, we transferred CBirWt T cells to Rag1−/− mice that had been treated with either vancomycin (previously shown in Figure 3B to deplete CBir1 flagellin+ bacteria) or a broad-spectrum collection of antibiotics (metronidazole, ampicillin, neomycin and vancomycin; MANV), which has been shown to reduce intestinal bacteria >100-fold and largely prevent ‘rapid’ microbiota-driven T cell proliferation and the development of T cell-induced transfer colitis (8, 30, 31). CBirWt T cells isolated from untreated and vancomycin-treated Rag1−/− mice were dominated by rapidly proliferating (CellTrace−) T cells in their mLN and colon (Fig. 6A). In contrast, CBirWt T cells transferred to Rag1−/− mice treated with MANV showed significantly fewer rapidly proliferating T cells in their mLN, indicating that the rapid response requires an intact microbiota but not CBir1 flagellin (Fig. 6A) (8, 30). CBirWt T cells that accumulated in the cLP of MANV-treated mice had mostly diluted their CellTrace dye, indicating rapid proliferation, but the reduced numbers (~8–20X) of transferred cells in the cLP compared to both untreated and vancomycin-treated controls indicates the importance of the microbiota for the accumulation of CD4 T cells (Supplemental Fig. 4). Furthermore, our data show that CBirWt T cell rapid proliferation and accumulation in the colon post-transfer into lymphopenic hosts is driven by the microbiota but does not require the presence of the CBir1 flagellin. In fact, tetramer-positive (CBir1-responsive) CBirWt T cells and CBirRag T cells that can only express the CBir1-specific TCR were at a significant disadvantage compared to tetramer-negative cells with regard to proliferation and accumulation in the colon, as is evident from the frequency of CBir tetramer binding cells in the spleen of CBirWt mice compared to those cells only 10 days post-transfer to a Rag1−/− host (Fig. 5A and E). Thus, while specificity against the microbiota may be necessary for colitogenic T cells to induce disease, it is not a sufficient property.
Figure 6. CBir1 flagellin is not the main antigenic driver of proliferation of CBir1-specific T cells in lymphopenic hosts (A and B).
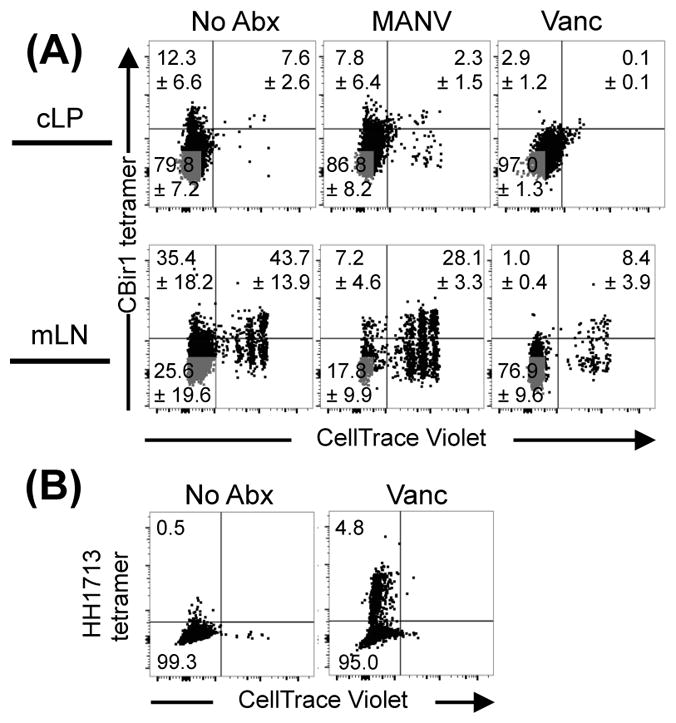
Rag1−/− mice were treated with antibiotics as indicated for 1 week and then injected with 106 FACs-sorted, CellTrace Violet-stained naïve CD4+ T cells. Ten days later, transferred cells were analyzed for dilution of CellTrace dye and binding to either (A) CBir1 tetramer or (B) Helicobacter hepaticus MHCII tetramer. Numbers on flow cytometry plots show mean percentage of gated cells ± SEM. Flow cytometry plots were gated on Live CD45.1+CD90.2+CD4+CD8− cells. Data are representative of 2 independent experiments; n=3 mice/group.
It was still not clear whether the actively proliferating CBirWt T cells were specific to microbiota-derived antigens or responding to microbiota-dependent inflammation, as previous studies have indicated the importance of both (8, 30). Therefore, we looked into whether CBir1 tetramer-negative T cells were specific to antigens derived from other members of the bacterial microbiota. Multiple publications suggest T cells specific to Helicobacter spp. may dominate the repertoire of colonic resident microbiota-specific T cells, and Helicobacter-specific T cells are sufficient to induce disease in multiple models of colitis (10, 17, 20). Therefore, we stained CBirWt T cells transferred to Rag1−/− mice with a recently described MHC II tetramer specific to Helicobacter spp (32). Despite the limited repertoire of CBirWt T cells, a modest proportion of cLP CBirWt T cells were specific to peptides derived from Helicobacter spp. (Fig. 6B). This is similar to observations made with OT-II (ovalbumin-specific) Tg T cells that respond to Segmented Filamentous bacteria (SFB) in the small intestine (33). Depletion of Gram-positive anaerobes with vancomycin increased Helicobacter-specific responses, indicating that these tetramers are not cross-reactive to CBir1 flagellin and also that removal of Gram positive anaerobes may increase the availability of antigens derived from Helicobacter (Fig. 6B). These results are consistent with a growing body of studies indicating that the most immunogenic members of the microbiota are those capable of making direct contact with the host intestinal epithelium, such as SFB in the ileum and Helicobacter in the colon (19, 20). Further, they underscore the finding that specificity for CBir1 flagellin is not the driving force behind the proliferation and accumulation of CBir1Wt T cells and that instead these are provided via other TCR specificities associated with re-arrangment of the endogenous TCRα locus.
Discussion
In aggregate, our data show that specificity towards a common intestinal antigen, CBir1 flagellin, is not sufficient for the spontaneous activation of T cells nor for the development of colitis. This is in line with our previous study, which showed that activation of CBir1 Tg T cells required the breakdown of the intestinal lining, which presumably allowed greater access of the antigen to antigen presenting cells and T cells (18). In these experiments, despite the activation of microbiota-specific T cells after gastrointestinal infection, spontaneous colitis never developed, which was presumably due to immunoregulatory mechanisms (18). In contrast, CBir1 Tg T cells have been used commonly by multiple laboratories to look at immune responses against the microbiota under lymphopenic conditions where the barrier integrity is not obviously compromised (8, 13, 15, 16, 34, 35). Here we show under both genetically lymphopenic conditions (CBirRag) and adoptive transfer into Rag1−/− mice that CBir1-specific T cells do not become activated and differentiate to either effector or regulatory states nor do they traffic or accumulate significantly in the colon. Instead, CBir1-specific T cells appear to mostly undergo ‘lymphopenia-induced’ proliferation, which is antigen independent and driven by the increased availability of IL-7 in lymphopenic mice (30). We also show that upon transfer to lymphopenic mice, proliferating T cells are dominated by clones that lack the surface expression of the CBir1 TCR, and at least some of the proliferating T cells are specific to other bacteria, such as Helicobacter spp.
Interestingly, all T cells in CBirWt mice express the Vβ8.3 chain associated with the transgenic TCR, indicating that those cells lacking high avidity binding to the CBir1 tetramer have not lost the ability to express the transgene. This implies that the endogenously rearranged TCRα chain is favored and potentially provides the cell with a dominant second specificity. The expression of secondary TCRs responsive to environmental antigens presumably is a possibility for all TCR transgenic mice on conventional backgrounds. However, it is perhaps most critical when trying to use them to interrogate responses at mucosal sites populated with various environmental antigens and particularly microbiota-specific TCRs, which respond to organisms that typically do not invade and proliferate in the host and thus provide low concentrations of their antigens. In that respect, it is somewhat surprising that Smarta transgenic mice on a wild-type background do not induce colitis over the time frame examined. There are a number of possible explanations for this discrepancy, but tetramer staining (I-Ab:LCMVgp66-77) of Smarta transgenic T cells reveals a significantly higher frequency of tetramer positive cells (85+%) compared to CBir1Wt (35–80%), indicating that the repertoire of ‘alternative’ TCR specificities may be smaller. Our work also underscores the importance of confirming the results of experiments using TCR transgenics with tetramer-based measurements of the response of the endogenous repertoire to the same antigen (18, 21, 36).
The differences between what we have observed for CBir1 specific T cells and what has been widely observed for SFB and Helicobacter spp. highlight the importance of understanding the ecology of intestinal bacteria (20, 21, 32). Recent studies suggest that spontaneous microbiota-specific T cell activation is largely made against organisms that live in close apposition to the intestinal epithelium, as this allows for a steady traffic of antigens from mucosa spanning antigen-presenting cells without the need for intestinal breach (37, 38). Indeed, we cannot exclude the possibility that in other mouse colonies, CBir1 flagellin may be expressed by intestinal bacteria that live in or on the mucosa, allowing for spontaneous T cell activation and CBir1-specific T cell colitis. However, our results using CBirWt T cells are consistent with previous studies in regard to the incidence and timing of disease, so it is reasonable to presume that our findings are not unique to the microbiota within our facility and are broadly applicable (8, 13, 16). The observation that not all microbiota-derived antigens induce spontaneous T cell activation is seemingly at odds with studies indicating broad specificity to microbiota-derived antigens (including CBir1 flagellin) among activated T and B cells in human subjects (1, 2). However, the ecological behavior of most bacteria within the intestine remains unknown, and human lifestyles and infectious histories may lead to bacterial translocation events that are not modeled in murine systems within carefully controlled environments. We hypothesize that responses to the dominant immunogenic organisms of the GI tract are required to initiate immune responses that later spread to include other specificities, such as CBir1, as damage to the intestine intensifies. Therefore, the frequency of a given T cell clone in clinical IBD may not correlate to their etiological importance. Thus, we posit that the organisms most critical to the initiation of IBD either secrete products that directly interact with the host (such as toxins) or live within the mucosal barrier itself and therefore access to intestinal antigens precedes the necessity for more active forms of T cell regulation (Tregs, IgA, ILCs). For example, Adherent-Invasive E. coli are commonly associated with IBD, and mouse experiments indicate that their association with the intestinal epithelium is required for the induction of inflammation (39, 40). We assert that future studies on the role of the microbiota in IBD should focus on the interaction between the immune system and those organisms whose ecological behavior initiates spontaneous T cell responses and that particular attention should be addressed to those organisms living on the surface of the intestine.
Supplementary Material
Acknowledgments
The authors would like to thank J. Michel, A. Styche, M. Woolford and C. Coogan for assistance with cell sorting, the Children’s Hospital of Pittsburgh Histology core for the preparation of tissue slides, and the University of Pittsburgh Division of Laboratory Animal Research. We would like to thank A. Poholek, Y. Belkaid, M. McGeachy, S. Canna and the members of the Hand lab for discussion and critical reading of the manuscript. The authors declare no competing financial interests.
Funding: TWH: NIH (K22AI108719) and Richard King Mellon Institute for Pediatric Research
Abbreviations used in this article
- CBirWt
CBir1 TCR transgenic
- CBirTCR
CBir1 x TCRα−/−
- CBirRag
CBir1 x Rag1−/−
- cLP
colonic lamina propria
- DC
dendritic cell
- IBD
inflammatory bowel disease
- IgA
immunoglobulin A
- ILC3
innate lymphoid cell type 3
- MANV
metronidazole, ampicillin, neomycin, vancomycin
- mLN
mesenteric lymph node
- SFB
segmented filamentous bacteria
- Tg
transgenic
- Treg
regulatory T cell
References
- 1.Christmann BS, Abrahamsson TR, Bernstein CN, Duck LW, Mannon PJ, Berg G, Bjorksten B, Jenmalm MC, Elson CO. Human seroreactivity to gut microbiota antigens. J Allergy Clin Immunol. 2015;136:1378–1386. e1371–1375. doi: 10.1016/j.jaci.2015.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, This S, Danne C, Campion S, Duncan SH, Owens BMJ, Uhlig HH, McMichael A, Oxford IBDCI, Bergthaler A, Teichmann SA, Keshav S, Powrie F. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology. 2017;153:1320–1337. e1316. doi: 10.1053/j.gastro.2017.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belkaid Y, Bouladoux N, Hand TW. Effector and memory T cell responses to commensal bacteria. Trends Immunol. 2013;34:299–306. doi: 10.1016/j.it.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, Chai C, Kim M, Diehl GE, Ajami NJ, Petrosino JF, Zhou XK, Schwartzman S, Mandl LA, Abramowitz M, Jacob V, Bosworth B, Steinlauf A, Scherl EJ, Wu HJ, Simpson KW, Longman RS. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65:3126–3131. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Kullberg MC, Andersen JF, Gorelick PL, Caspar P, Suerbaum S, Fox JG, Cheever AW, Jankovic D, Sher A. Induction of colitis by a CD4+ T cell clone specific for a bacterial epitope. Proc Natl Acad Sci U S A. 2003;100:15830–15835. doi: 10.1073/pnas.2534546100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossini V, Zhurina D, Radulovic K, Manta C, Walther P, Riedel CU, Niess JH. CX3CR1(+) cells facilitate the activation of CD4 T cells in the colonic lamina propria during antigen-driven colitis. Mucosal Immunol. 2014;7:533–548. doi: 10.1038/mi.2013.70. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, Marriott CL, Brucklacher-Waldert V, Veldhoen M, Kelsen J, Baldassano RN, Sonnenberg GF. Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med. 2016;22:319–323. doi: 10.1038/nm.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A. 2009;106:19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W, Eberl G, Baldassano RN, Laufer TM, Elson CO, Sonnenberg GF. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science. 2015;348:1031–1035. doi: 10.1126/science.aaa4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, Wherry EJ, Koni PA, Bushman FD, Elson CO, Eberl G, Artis D, Sonnenberg GF. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117. doi: 10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kullberg MC, Rothfuchs AG, Jankovic D, Caspar P, Wynn TA, Gorelick PL, Cheever AW, Sher A. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infection and immunity. 2001;69:4232–4241. doi: 10.1128/IAI.69.7.4232-4241.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO, 3rd, Belkaid Y. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337:1553–1556. doi: 10.1126/science.1220961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov, Sugiyama T, Nunez G, Camp JG, Hattori M, Umesaki Y, Honda K. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell. 2015;163:367–380. doi: 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z, Perry JSA, Knoop KA, Tanoue T, Narushima S, Honda K, Elson CO, Newberry RD, Stappenbeck TS, Kau AL, Peterson DA, Fox JG, Hsieh CS. Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci Immunol. 2017:2. doi: 10.1126/sciimmunol.aal5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, Sczesnak A, Liao JJ, Torres VJ, Jenkins MK, Lafaille JJ, Littman DR. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert AE, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, Belkaid Y. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity. 2011;34:435–447. doi: 10.1016/j.immuni.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci U S A. 2007;104:11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, McCrate S, Kim D, Hsieh CS, Hogan SP, Elson CO, Tarr PI, Newberry RD. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci Immunol. 2017:2. doi: 10.1126/sciimmunol.aao1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 26.DiPaolo RJ, Shevach EM. CD4+ T-cell development in a mouse expressing a transgenic TCR derived from a Treg. Eur J Immunol. 2009;39:234–240. doi: 10.1002/eji.200838772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akane K, Kojima S, Mak TW, Shiku H, Suzuki H. CD8+CD122+CD49dlow regulatory T cells maintain T-cell homeostasis by killing activated T cells via Fas/FasL-mediated cytotoxicity. Proc Natl Acad Sci U S A. 2016;113:2460–2465. doi: 10.1073/pnas.1525098113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94–107. doi: 10.1016/j.cell.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang HQ, Dummer W, Shen H, Cebra JJ, Surh CD. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 31.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2009 doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, Littman DR. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature. 2018 doi: 10.1038/nature25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, Huang Y, Gerner MY, Belkaid Y, Germain RN. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. 2018 doi: 10.1038/nature25437. [DOI] [PubMed] [Google Scholar]
- 35.Liu HP, Cao AT, Feng T, Li Q, Zhang W, Yao S, Dann SM, Elson CO, Cong Y. TGF-beta converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol. 2015;45:1010–1018. doi: 10.1002/eji.201444726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, Furtado GC, Lira SA, Shakhar G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity. 2013;38:581–595. doi: 10.1016/j.immuni.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 39.Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009;206:2179–2189. doi: 10.1084/jem.20090741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rolhion N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1277–1283. doi: 10.1002/ibd.20176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.