

Summary
The circadian clock coordinates behavioral and circadian cues with availability and utilization of nutrients. Proteasomal degradation of clock repressors, e.g., cryptochrome (CRY)1 maintains periodicity. Whether macroautophagy, a quality control pathway, degrades circadian proteins remains unknown. Here we show that circadian proteins BMAL1, CLOCK, REV-ERBα, and CRY1 are lysosomal targets, and that macroautophagy impacts the circadian clock by selectively degrading CRY1. Autophagic degradation of CRY1, an inhibitor of gluconeogenesis, occurs in a diurnal window when rodents rely on gluconeogenesis, suggesting that CRY1 degradation is time-imprinted to maintenance of blood glucose. High fat feeding accelerates autophagic CRY1 degradation and contributes to obesity-associated hyperglycemia. CRY1 contains several light chain 3 (LC3)-interacting region (LIR) motifs, which facilitate the interaction of cargo proteins with the autophagosome marker LC3. Using mutational analyses, we identified two distinct LIRs on CRY1 that exert circadian glycemic control by regulating CRY1 degradation, revealing LIRs as potential targets for controlling hyperglycemia.
eTOC blurb
Toledo et al. show that autophagy controls the liver clock by timely degradation of a circadian protein cryptochrome 1 (CRY1). CRY1 lowers glucose production in liver and its timely removal by autophagy allows glucose production. Obesity accentuates CRY1 degradation by autophagy, increasing glucose production and blood sugar levels.
Introduction
The circadian clock synchronizes behavioral and circadian cues with nutrient availability and energy metabolism (Bass and Takahashi, 2010; Perelis et al., 2015). Disrupting circadian rhythm contributes to development of type II diabetes (Marcheva et al., 2010). The mammalian clock consists of complex feedback loops wherein heterodimeric complexes of transcription factors CLOCK (circadian locomotor output cycles kaput) and BMAL1 (brain and muscle Arnt-like protein 1) initiate expression of period (PER)1/2 and cryptochrome (CRY)1/2, which repress their transcriptional activity (Darlington et al., 1998; Gekakis et al., 1998; Griffin et al., 1999; Kume et al., 1999; Sangoram et al., 1998). Expression of Bmal1 is controlled by a second loop consisting of nuclear hormone receptor REV-ERBα, which represses Bmal1 expression, and retinoid-related orphan receptors (ROR) that drive Bmal1 expression (Guillaumond et al., 2005; Preitner et al., 2002; Sato et al., 2004). CRY1 is a core clock repressor that along with PER determines circadian periodicity (Griffin et al., 1999; Kume et al., 1999), and degradation of PER and CRY impacts period length. While mutations in casein kinase 1ε tau lead to PER hyperphosphorylation and its proteasomal degradation, which shortens period (Meng et al., 2008); mutations in F-box protein FBXL3 stabilize CRY1 by blocking its proteasomal degradation that lengthens period (Busino et al., 2007; Godinho et al., 2007; Siepka et al., 2007). Ubiquitination of CRY1 by SCF (Skp1-Cul1-FBXL3) complex (Xing et al., 2013) and its proteasomal degradation regulate CRY1 stability. Recent studies indicate that the F-box protein, FBXL21, opposes the effect of FBXL3 on CRY1 degradation (Hirano et al., 2013; Yoo et al., 2013) suggesting that multiple mechanisms regulate turnover of CRY proteins. Whether macroautophagy (hereafter autophagy) degrades circadian proteins remains unknown.
In addition to serving as a clock repressor, CRY1 suppresses hepatic gluconeogenesis by regulating CREB/cAMP signaling, by rhythmic repression of glucocorticoid receptor, and by decreasing nuclear FoxO1 levels that downregulates gluconeogenic gene expression (Jang et al., 2016; Lamia et al., 2011; Zhang et al., 2010). Since the liver clock is regulated by nutrients, insulin, and glucagon, and since glucagon activates autophagy (Deter and De Duve, 1967), we considered the possibility that autophagy, regulation of liver clock, and glucose metabolism are interlinked. Autophagy degrades cytoplasmic contents in lysosomes (He and Klionsky, 2009). Induction of autophagy occurs by activation of class III PI3K complex and generation of PI3Ps, which recruit autophagy (ATG) proteins to cellular sites of autophagosome (APh) formation (He and Klionsky, 2009). Activation of ATG7 initiates distinct conjugation cascades that lipidate cytosolic LC3-I into APh-bound LC3-II (He and Klionsky, 2009). APhs sequester cytoplasmic contents for degradation in lysosomes. Cargo sequestration requires interaction of their LC3-interacting region (LIR) motifs, which are W/F/Y-X-X-I/L/V sequences with LC3 (Birgisdottir et al., 2013). Analyses of CRY1 protein revealed several LIR motifs that formed the mechanistic basis to explore whether CRY1 interacts with LC3. Studies in Atg7 knockout livers have revealed roles for autophagy in glycogen (Kern et al., 2016), glucose (Karsli-Uzunbas et al., 2014), and lipid metabolism (Singh et al., 2009); however, the mechanisms by which autophagy regulates glucose metabolism remain unclear. Since loss of hepatic Atg7 via tail vein injections of adeno-associated viruses (AAVs) (Atg7KOAAV-Cre) led to hypoglycemia, we hypothesized that autophagy maintains gluconeogenesis and blood glucose levels by degrading CRY1. Here, we show that circadian proteins are lysosomal substrates, and that CRY1 is degraded by autophagy in a temporal manner. We show that autophagic degradation of CRY1 releases its repression on gluconeogenesis and increases blood glucose levels. We unravel the mechanism of autophagic CRY1 degradation by identifying the specific LIR motifs on CRY1 that determine its interaction with LC3. Finally, we show that high fat feeding decreases CRY1 protein levels in an autophagy-dependent manner, and that restoring hepatic CRY1 levels reverses obesity-associated hyperglycemia. Our studies reveal CRY1 LIR motifs as potential targets to control obesity-associated hyperglycemia.
Results
Temporal degradation of core circadian proteins in lysosomes
To determine if, and when during the circadian period, core circadian proteins are degraded by autophagy, we administered the lysosomal inhibitor leupeptin i.p. in mice 2 hr prior to collection of livers at 7am, 11am, 3pm, 7pm, 11pm and 3am (Fig. 1A). Livers were homogenized and immunoblotted for LC3, clock inducers BMAL1, CLOCK, and repressors PER1, PER2, CRY1, and REV-ERBα (Fig. 1B, and Fig. S1A). Accumulation of a protein at a given timepoint in presence of leupeptin indicates its flux through lysosomes indicating degradation. Quantification of LC3-II flux (Fig. 1C) revealed that induction of autophagy in livers from ad libitum-fed mice occurred between 3pm and 11pm with maximal activation at 7pm. Strikingly, lysosomal degradation of CRY1 (Fig. 1D) also occurred between 3pm–11pm and peaked at 7pm—coinciding with induction of autophagy. Immunoblotting of lysosome fractions from livers at each timepoint (as in Figure 1A) confirmed that CRY1 is indeed a lysosomal substrate that undergoes maximal degradation at 7pm (Fig. S1B). By contrast, analyses of PER1/2 flux excluded these proteins as lysosomal targets (Fig. 1E, 1F). Interestingly, BMAL1, CLOCK, and REV-ERBα (Fig. 1G, 1H, and 1I) each displayed greater net flux/degradation in lysosomes when compared to CRY1 (comparisons of individual flux rates in Fig. 1J); however, the temporal pattern of degradation of each of these proteins was distinct from that of LC3 and CRY1 (Fig. 1C, 1D). For instance, lysosomal degradation of BMAL1 occurred at high rates between 7am–3pm and at 3am that were separated by decreased lysosomal degradation at 11pm (Fig. 1G). The lysosomal degradation of CLOCK occurred maximally at 11am and 3am with reduced turnover at 7pm (Fig. 1H). By contrast, lysosomal degradation of REV-ERBα increased gradually from 7am–3pm following by a rapid decline in its degradation for the rest of the period (Fig. 1I). Increases in circadian protein levels in leupeptin-treated samples were due to their accumulation in lysosomes and not from increased gene expression since equivalent expression of Bmal1, Clock, and Cry1 genes was observed in livers from control and leupeptin-treated mice (Fig. S1C–1E). These results demonstrate that BMAL1, CLOCK, REV-ERBα, and CRY1 proteins are lysosomal substrates and exhibit distinct temporal patterns of lysosomal degradation (Fig. 1J).
Fig. 1. BMAL1, CLOCK, REV-ERBα, and CRY1 are degraded by lysosomes.

(A) Scheme depicting use of lysosomal inhibitors (Lys Inh) intraperitoneally (i.p.) in mice at indicated timepoints.
(B–I) Immunoblots (IB) and quantifications for net lysosomal flux of each indicated protein in livers at indicated timepoints in ad libitum-fed male mice injected with vehicle or Lys Inh as in panel A, n=4–8.
(J) Graph comparing relative rates of lysosomal degradation of each indicated circadian protein, n=4–8.
(K) IB for indicated proteins in homogenate (Hom) and autophagosome (APh) fractions from livers at 7pm. Two independent experiments are shown (n=3 livers per APh sample).
(L, M) Co-immunoprecipitation (co-IP) of LC3 and IB for indicated proteins from livers at 7pm (n=7) and 3pm (n=3). Two independent co-IP experiments are shown for 7pm.
Values are mean ± s.e.m. In panels 1C and 1D, *P<0.05, **P<0.01, ***P<0.001 compare indicated values to the corresponding 7pm value. One-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S1.
To validate that CRY1 is degraded in lysosomes in a temporal manner, we subjected livers harvested at 7am and 7pm from ad libitum-fed mice to immunostaining for CRY1 and lysosomal marker LAMP1. Consistent with results from lysosomal flux analyses (Fig. 1B), immunofluorescence (IF) revealed an increase in colocalization of CRY1 with LAMP1 at 7pm but not at 7am (Fig. S1F). We also noted a remarkable increase in LAMP1 signal in livers at 7pm (Fig. S1F) indicating expansion of lysosome-related acidic organelles at 7pm when compared to livers at 7am, which we confirmed by demonstrating ~50% increase in Lamp1 gene expression at 7pm when compared to 7am (Fig. S1G). Lysosomal expansion likely contributed to induction of LC3-II/autophagy flux and CRY1 degradation at 7pm (Fig. 1C, 1D).
Since LC3 and CRY1 were degraded over a similar time course, we next explored whether degradation of CRY1 is LC3-dependent. Consequently, we first tested whether CRY1 is sequestered by APhs. APhs were isolated from liver by density gradient centrifugation (Marzella et al., 1982) and fractions were validated by their enrichment of APh marker LC3-II by immunoblotting (Fig. 1K). Consistent with increased CRY1 flux at 7pm (Fig. 1D), APhs from 7pm livers were enriched in CRY1 but not BMAL1/CLOCK (Fig. 1G, 1H, and 1K) indicating that CRY1 is sequestered by APhs and thereby degraded by autophagy. Although lack of PER2 accumulation in presence of leupeptin excludes it as an autophagy substrate (Fig. 1F), PER2 co-purified with CRY1 in APhs at 7pm (Fig. 1K). Co-immunoprecipitation (co-IP) analyses from livers at 7pm confirmed that LC3 interacts with CRY1 and PER2 but not PER1, BMAL1 or CLOCK (Fig. 1L). Co-IP analyses at 3pm revealed the interaction of CRY1 with LC3 but not PER2 (Fig. 1M) supporting that CRY1 is degraded by autophagy.
Autophagy degrades CRY1 in a time-dependent manner
Three distinct mechanisms deliver cargo to lysosomes – macroautophagy (or autophagy) (He and Klionsky, 2009), chaperone-mediated autophagy (CMA) (Cuervo, 2010), and endosomal microautophagy (eMI) (Sahu et al., 2011). Loss of Atg7 blocks APh formation and autophagy but has no effect on CMA (Park et al., 2015; Rodriguez-Muela et al., 2013) and eMI (Sahu et al., 2011). Since CRY1 was detected in APhs and interacted with LC3, we sought to confirm whether CRY1 is an autophagy substrate. Consequently, we depleted the autophagy gene Atg7 in livers of Atg7 floxed mice by tail vein injections of AAVs expressing Cre recombinase (hereafter Atg7KO) for 7 and 13 days. We intended to establish a model of short-term autophagy inhibition in vivo such that adverse effects of prolonged autophagy suppression are eliminated. Depleting Atg7 in livers for 7 and 13 days each blocked autophagy indicated by decreased ATG7 and LC3-II levels, although complete loss of ATG7 and accumulation of autophagy cargo p62 were observed after 13 days of Cre injections (Fig. 2A). Importantly, loss of Atg7 for 7 or 13 days each led to accumulation of CRY1 but not CRY2 (Fig. 2A). In addition, accumulation of CRY1 in Atg7-deficient livers was detected in tissue homogenates and cytosol, and in nuclear fractions (validated by enrichment of nuclear marker Lamin A/C) where CRY1 translocates to repress BMAL1/CLOCK transcriptional activity (Fig. 2B). Since the SCF complex ubiquitinates CRY1 for proteasomal degradation (Xing et al., 2013), and since autophagy degrades cargo ‘in bulk’, we explored whether SCF components that associate with CRY1 might also be degraded by autophagy at 7pm. Interestingly, APhs from livers of wildtype mice at 7pm were enriched in SCF components Skp1 and FBXL3 (Fig. S2A). Furthermore, Atg7-null livers displayed ~2-fold increase in CRY1, ~4-fold increase in Skp1, and ~30% increase in FBXL3 levels (Fig. S2B) suggesting that autophagy degrades components of the SCF complex in addition to CRY1. Although lysosomal flux analyses showed decreased turnover of BMAL1 and CLOCK in Atg7KO livers (Fig. S2C), BMAL1 and CLOCK did not interact with LC3 or enrich in APhs (Fig. 1K–1M) or accumulate in Atg7-deficient livers (Fig. 2C) excluding them as macroautophagy targets. Dampened BMAL1/CLOCK flux in Atg7KO livers likely resulted from modest decreases in BMAL1/CLOCK protein levels (Fig. 2C) secondary to decreased Bmal1 and Clock gene expression (Fig 2J–2M).
Fig. 2. Autophagy specifically degrades CRY1 and regulates the liver clock.

(A, B) IB for indicated proteins in livers from Atg7flox/flox mice subjected to tail vein injections of control (Con) and Cre-expressing AAVs for 7 or 13 days, and liver homogenates (Hom), cytosol (Cyt), and nuclear (Nuc) fractions from mice injected with Cre-expressing AAVs for 13 days, n=6. Quantification for panel B is shown.
(C–E, G, I, J, L, N) IB and quantification for indicated proteins in blots in panel C, and (F, H, K, M, O) RT-PCR for corresponding circadian genes from livers harvested at six indicated timepoints across 24 hr from ad libitum-fed male mice injected with Con or Cre-expressing AAVs (Atg7KO) for 13 days, n=5–7.
Values are mean ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compare values in Atg7KO mice to corresponding values in Con mice. Student’s T-test or Two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S2.
Loss of autophagy disrupts the circadian clock in liver
Since CRY1 is degraded by autophagy, we investigated the effect of loss of Atg7 on oscillations of the liver clock across period. Deletion of Atg7 resulted in loss of LC3-II, and accumulation of LC3-I at each timepoint indicating loss of autophagy (Fig. 2C, 2D). As anticipated, loss of autophagy resulted in accumulation of CRY1 at each timepoint; however, no increases in CRY2 protein levels were noted (Fig. 2C, 2E, Fig. S2D). Increased CRY1 protein levels blocks BMAL1/CLOCK transcriptional activity leading to repression of Cry1 gene, hence abundance of CRY1 in Atg7-deficient livers should, in principle, dampen Cry1 expression. Indeed, Atg7-deficient livers showed significantly decreased Cry1 expression across period (Fig. 2F) suggesting a functional increase in CRY1-driven repression of BMAL1/CLOCK activity. Atg7-deficient livers displayed a reduction in PER1 protein levels that could not be explained by changes in Per1 expression while PER2 protein levels remained largely unaffected (Fig. 2G–2I). Atg7-deficient livers also displayed decreased BMAL1 protein levels and Bmal1 gene expression (Fig. 2J, 2K), and modestly reduced CLOCK levels at 3pm that associated with decreased Clock expression (Fig. 2L, 2M). Levels of REV-ERBα protein (Fig. 2N) and Rev-erbα and Rev-erbβ gene expression were also suppressed in Atg7-deficient livers (Fig. 2O, Fig. S2E) reflecting an overall trend towards dampening of the oscillations of the liver clock in absence of autophagy. Reduction of Bmal1 expression in Atg7KO livers likely occurred due to decreased expression of Rorα (Fig. S2F), which encodes for RORα that drives Bmal1 gene expression (Guillaumond et al., 2005; Sato et al., 2004). In sum, these results indicate that loss of autophagy disrupts the liver clock.
Loss of autophagy impacts nuclear and cytoplasmic levels of core circadian proteins
Nucleocytoplasmic shuttling of circadian proteins and degradation of PER/CRY each modulate period length. Because blocking autophagy resulted in accumulation of CRY1 and decreases in levels of BMAL1, CLOCK and REV-ERBα, we characterized in greater detail the effect of loss of autophagy on period-wide oscillations of these proteins in nuclear and cytoplasmic compartments. To that purpose, livers were harvested at 7am, 11am, 3pm, 7pm, 11pm and 3am from control and Atg7-deficient livers (13 days after a single AAV-Cre injection) following which nuclear and cytosolic fractions were generated. Autophagy deficiency was verified by accumulation of LC3-I in both nuclear and cytosolic fractions from Atg7KO mice (Fig. 3A–3C), while nuclear enrichment was confirmed by abundance of nuclear marker Lamin A/C and its absence in cytosolic fractions (Fig. S3A). As expected, biochemical fractionation and IF revealed ~2–3-fold increase in CRY1 accumulation in nucleus in livers from Atg7KO mice (Fig 3A, 3D, 3E). Although both nuclear and cytosolic fractions from Atg7KO mice showed increased CRY1 accumulation (Fig. 3A–3E), greater retention of CRY1 was observed in the nucleus than cytoplasm (Fig. 3D). These findings are consistent with the fact that CRY1 exerts its repressive function by nuclear translocation. We next explored the effect of loss of autophagy on oscillations of additional core circadian proteins in nuclear and cytoplasmic compartments. Consistent with decreased BMAL1/CLOCK protein levels in homogenates (Fig. 2J, 2L), BMAL1 and CLOCK levels were also decreased in nuclear fractions from Atg7KO livers when compared to controls (Fig. 3F, 3G). Modest reduction in BMAL1 protein was also detected in cytoplasm from Atg7KO livers at 7am (Fig. 3F) although the significance of this finding remains unclear. Consistent with our observations in total homogenates, nuclear and cytoplasmic levels of PER1 and REV-ERBα were also significantly reduced across period in Atg7KO livers, while no changes in PER2 levels were detected (Fig. S3B–S3D). Taken together, these findings reflect a remarkable dampening in the oscillations of core circadian genes - a hallmark of age-associated changes in rhythmicity (Cornelissen and Otsuka, 2017; Zwighaft et al., 2015). Given that autophagy decreases with age (Rubinsztein et al., 2011), our data suggest that defects in autophagy may contribute to the recently elucidated effects of aging on the molecular clock.
Fig. 3. Loss of autophagy impacts nuclear levels of core circadian proteins.

(A–D, F–H) IB for nuclear and cytosolic distribution of indicated circadian proteins, autophagy-related protein LC3, and FBXL3, Skp1, and FBXL21 in livers across 24 hr from ad libitum-fed male mice injected with Con or Cre-expressing AAVs (Atg7KO) for 13 days, n=4–8. Quantifications for nuclear and cytosolic distribution of LC3, CRY1, BMAL1, CLOCK, and FBXL3 are depicted. Quantifications for nuclear and cytosolic distribution of PER1, PER2, REVERBα, Skp1, and FBXL21 are shown in Fig. S3. Scale bar: 10 μm.
(E) Indirect immunofluorescence for CRY1 (green) in livers and quantification for nuclear and total cellular CRY1 content at 7pm in livers from ad libitum-fed male mice injected with Con or Cre-expressing AAVs (Atg7KO) for 13 days, n=4. Values are mean ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compare values in Atg7KO mice to corresponding values in Con mice. Student’s T-test or Two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S3.
Because loss of Atg7 led to accumulation of CRY1 and SCF complex (FBXL3 and Skp1) (Fig. S2B) that facilitate ubiquitination/degradation of CRY1 (Xing et al., 2013), we next characterized the effect of loss of autophagy on period-wide changes in nuclear and cytoplasmic levels of FBXL3 and Skp1. Interestingly, loss of autophagy led to accumulation of FBXL3 and Skp1 in the nucleus across the period although increases in both molecules occurred most significantly at 3am (Fig. 3H and S3E). FBXL3 did not accumulate in cytosolic fractions; however, a significant increase in cytosolic Skp1 accumulation was noted in Atg7KO livers at 3am (Fig. S3E). While the mechanism of sequestration of nuclear FBXL3 by autophagy is unclear, given the enrichment of CRY1, FBXL3, and Skp1 in APhs, our results suggest that CRY1 and SCF are likely degraded by autophagy as a complex. Since cytoplasmic FBXL21, another member of the F-box-type E3 ubiquitin ligase family (Jin et al., 2004) opposes the function of FBXL3 and promotes CRY1 stability (Hirano et al., 2013; Yoo et al., 2013), we tested whether loss of autophagy led to accumulation of FBXL21 that, in turn, increased CRY1 stability/levels in Atg7KO livers. Loss of autophagy did not affect FBXL21 protein levels (Fig. S3F) excluding the possibility that accumulation of CRY1 in Atg7KO livers occurred from an FBXL21-dependent increase in CRY1 stability.
Autophagic degradation of CRY1 sustains gluconeogenesis
Since depleting ATG7 in livers increases CRY1 levels, and since CRY1 suppresses gluconeogenesis (Jang et al., 2016; Lamia et al., 2011; Zhang et al., 2010), we hypothesized that autophagic degradation of CRY1 maintains blood glucose levels by driving gluconeogenesis. Consistent with previous work showing a role for autophagy in preventing hypoglycemia (Karsli-Uzunbas et al., 2014), Atg7 floxed mice displayed significant reduction in blood glucose levels when tested at 7pm as early 3 days after Cre injections (Fig. 4A). After a week of Cre injections, Atg7KO mice showed lower blood glucose at each timepoint across the circadian period, demonstrating that hepatic autophagy is required to maintain blood glucose levels (Fig. 4B). Reduced blood glucose levels did not occur from decreased glycogen breakdown by autophagy since acute loss of Atg7 in livers associated with diminished hepatic glycogen stores across the period (Fig. S4A). In fact, livers from Atg7KO mice were largely depleted of glycogen when compared to controls indicating significant deficiency in glucose production/storage in liver. Although Atg7KO mice consumed less food in the nocturnal cycle, both control and Atg7KO mice consumed equal amounts of food in the diurnal cycle excluding differences in diurnal feeding as the reason for reduced blood glucose levels (Fig. S4B).
Fig. 4. Autophagic degradation of CRY1 in liver controls gluconeogenesis.

(A) Blood glucose levels at 7pm on indicated days after Atg7flox/flox male mice were injected with control (Con) or Cre-expressing AAVs (Atg7KO), n=4.
(B–F) Blood glucose levels (n=6–12) and expression of gluconeogenic genes, G6p, Pck1, and Pc in liver, and i.p. PTT from male mice at indicated timepoints across 24 hr on day 7 after injections of Con or AAV-Cre, n=4.
(G–J) IB for indicated proteins in the nucleus in livers at indicated timepoints across 24 hr from Con and Atg7KO male mice. Quantifications for nuclear levels of indicated proteins are shown, n=4–5.
(K, L) Experimental plan showing assessment of blood glucose levels at indicated timepoints on day 7 after Con and AAV-Cre injections, i.e., 1 day after tail vein injections of AAVs expressing Con or shCRY1 constructs on day 6, n=3–10. Graph depicts blood glucose values at 7pm, n=10.
Values are mean ± s.e.m. *P<0.05, **P<0.01, ***P<0.001 compare values in Atg7KO mice to corresponding values in Con mice. ###P<0.001 compares indicated values in Con mice to 7am values in Con mice. Student’s T-test or one-way or two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S4.
Mice rely on gluconeogenesis to maintain diurnal blood glucose levels since they consume a significant proportion of their total calories in the early hours of the dark cycle. To begin to test whether lower blood glucose levels in Atg7KO mice occurred from decreased gluconeogenesis, we first characterized the temporal changes in expression of gluconeogenic genes in normal mice across the circadian period. While livers from ad libitum-fed control mice showed progressive increases (~2-fold) in expression of gluconeogenic genes G6p, Pck1, and Pc between 3pm–7pm, Atg7-deficient livers failed to induce these genes at similar timepoints (Fig. 4C–4E). Atg7KO mice were also unable to increase their blood glucose levels when subjected to pyruvate tolerance tests (PTT) at 6pm indicating impaired gluconeogenesis (Fig. 4F). The transcription factor FoxO1 translocates to the nucleus and drives gluconeogenesis during reduced feeding when insulin levels are low (Puigserver et al., 2003), and because CRY1 participates in the degradation of nuclear FoxO1 (Jang et al., 2016), we tested whether decreased gluconeogenesis in Atg7KO mice associates with reduced nuclear FoxO1 content. Indeed, Atg7KO livers displayed a significant reduction in nuclear FoxO1 levels when compared to controls (Fig. 4G, 4H), while cytoplasmic FoxO1 levels remained largely unaffected (data not shown). Reduction in nuclear FoxO1 levels did not occur from its retention in the cytoplasm since circulating levels of insulin, a factor that drives FoxO1 phosphorylation and cytoplasmic retention, was largely reduced in Atg7KO mice (Fig. S4C). Given the accumulation of CRY1 in Atg7KO livers (Fig. 3A, 3D, 3E), it is likely that reduction in nuclear FoxO1 results from CRY1-mediated degradation of FoxO1 as shown recently (Jang et al., 2016). Decreased gluconeogenesis in Atg7KO mice could not be explained by changes in nuclear levels of phosphorylated-CREB, which is known to activate gluconeogenesis (Koo et al., 2005). In fact, nuclear levels of phosphorylated-CREB were elevated from 7pm–11pm in Atg7KO livers (Fig. 4I) and circulating glucagon (Fig. S4D) that phosphorylates CREB and drives gluconeogenesis (Koo et al., 2005) was also significantly elevated in Atg7KO livers (Fig. 4G, 4I, 4J). In sum, decreased gluconeogenesis in Atg7KO livers correlated with reduced nuclear FoxO1 levels, which suggest that FoxO1 plays a key role in aberrant glucose homeostasis in these animals, while induction of glucagon/CREB signaling in Atg7KO mice is a compensatory attempt to unsuccessfully restore blood glucose levels.
More importantly, to determine whether suppression of gluconeogenesis in Atg7KO mice occurs from increased CRY1 levels, we tested whether dampening CRY1 levels in Atg7-deficient livers (by injecting shRNAs against CRY1) (Fig. 4K) prevents the reduction in blood glucose levels. While reducing Cry1 expression by ~80% (Fig. S4E) and CRY1 protein levels by ~50% (Fig. S4F) failed to affect blood glucose levels in control mice (Fig. 4L), dampening Cry1 expression in Atg7KO livers increased blood glucose levels from 75±2 to 102±4 mg/dl at 7pm (P<0.001) (Fig. 4L). Increases in blood glucose levels in Atg7KO mice after depletion of CRY1 occurred specifically between 3pm–9pm, which coincides with the circadian interval when autophagy maximally degrades CRY1 (Fig. 1C, 1D). These data show that degradation of CRY1 by autophagy occurs during the day when mice consume less food, and that autophagic degradation of CRY1 sustains blood glucose levels.
Identifying CRY1 LIR motifs that regulate its degradation by autophagy
To determine the mechanism of degradation of CRY1 by autophagy, we first examined for presence of canonical LIR motif W/F/Y-X-X-I/L/V sequences in CRY1 (Birgisdottir et al., 2013). We identified thirteen putative LIRs on CRY1 (Fig. 5A), which were prioritized for mutagenesis based on their accessibility determined by visualization of crystal structure of mouse CRY1 (PDB: 5T5X) (Michael et al., 2017) using the Swiss-PdbViewer software (http://www.expasy.org/spdbv/) (Guex and Peitsch, 1997), and on presence of negatively charged residues in the vicinity of LIR motifs that facilitate binding to LC3 (Birgisdottir et al., 2013). This revealed four LIR motifs on CRY1 as candidates for interaction with LC3, which were individually inactivated in FLAG-tagged CRY1 plasmid by mutating their tyrosine, valine or leucine residues to alanine (Fig. 5B). We then determined the role of each LIR motif in degradation of CRY1 by expressing FLAG-tagged WT-CRY1 plasmid and each CRY1 mutant LIR plasmids (mLIR1, mLIR2, mLIR3, mLIR4 as in Fig. 5B) in liver via tail vein injections using jet-PEI in vivo transfection system (Chang et al., 2014) (Fig. S5A). We also explored whether blocking autophagic degradation of CRY1 by inactivating its LIRs impacts gluconeogenesis. Prior to injecting mice with mLIR CRY1 plasmids, we tested the robustness of the experimental system by determining the degree to which expression of FLAG-tagged WT-CRY1 suppresses hepatic gluconeogenesis when compared to empty vector. As reported in cultured hepatocytes (Jang et al., 2016), expressing WT-CRY1 in livers (Fig. S5B) decreased the expression of gluconeogenic genes Pck1 and G6p by ~80–90% (Fig. S5C) and lowered blood glucose levels by 50% (~50mg/dl) at 7pm (Fig. S5D). We then titrated the dose of injected WT-CRY1 plasmid such that blood glucose levels were consistently maintained at ~80–90mg/dl between 1pm–7pm. Consistent with the idea that inactivating LIR motifs on CRY1 prevents its degradation, expressing each of the four FLAG-tagged mLIR CRY1 in liver led to varying degrees of accumulation of FLAG when compared to WT-CRY1 (Fig. 5C). Strikingly, mLIR1-CRY1 and mLIR4-CRY1 accumulated significantly greater than mLIR2-CRY1 and mLIR3-CRY1 or WT-CRY1. Levels of mLIR2 and mLIR3 CRY1 were moderately higher than WT-CRY1 but differences between these groups remained statistically insignificant (Fig. 5C). These results indicate that LIR1 (271DLYKKV276) and LIR4 (492SRYRGL497) are required for degradation of CRY1 by autophagy. Accumulation of mLIR1-CRY1 and mLIR4-CRY1 did not occur from reduced autophagy per se indicated by equivalent LC3-II flux in livers injected with WT or mLIR-CRY1 plasmids (Fig. 5D) or from changes in transfection determined by qPCR showing equivalent Cry1 expression in livers injected with WT and mLIR-CRY1 plasmids (Fig. S5E). Co-IP in 293T cells confirmed that interactions of mLIR1-CRY1 and mLIR4-CRY1 with GFP-tagged LC3 were remarkably reduced when compared to WT-CRY1, mLIR2-CRY1 or mLIR3-CRY1 (Fig. S5F). These results are consistent with significant accumulation of mLIR1-CRY1 and mLIR4-CRY1 and moderate accumulation of mLIR2 and mLIR3 CRY1 in liver when compared to WT-CRY1 (Fig. 5C). Consistent with our observation that accumulation of CRY1 in Atg7KO mice associates with decreased BMAL1 levels (Fig. 2J), accumulation of mLIR1-CRY1 or mLIR4-CRY1 each associated with reduced BMAL1 levels (Fig. 5E) indicating that expressing autophagy-resistant forms of CRY1 is sufficient to disrupt the liver clock.
Fig. 5. LIR motifs determine CRY1 degradation and regulation of gluconeogenesis.

(A) Murine CRY1 has 13 LIR motifs (green).
(B) Inactivation of selected LIR motifs (mLIR1-4) on CRY1 via mutagenesis of tyrosine (Y), valine (V) or leucine (L) residues (green) to alanine (A) (red).
(C) IB for FLAG in livers from male mice at 7pm expressing FLAG-tagged wildtype (WT)-CRY1 plasmid or each FLAG-tagged CRY1 LIR mutant plasmid (mLIR1-4), n=12.
(D) IB for LC3 in livers from male mice at 7pm expressing FLAG-tagged WT-CRY1 or each CRY1 LIR mutant (mLIR1-4) and cultured in presence or absence of Lys Inh for 2 hr, n=7–10.
(E, F) IB for BMAL1 and G6P in livers from male mice at 7pm expressing FLAG-tagged WT-CRY1 plasmid or each CRY1 LIR mutant plasmid (mLIR1-4), n=8–12.
(G–K) Blood glucose levels at indicated timepoints, n=9–16, and i.p. PTT at 6pm from male mice expressing FLAG-tagged WT-CRY1 plasmid or each CRY1 LIR mutant plasmid in liver, n=5.
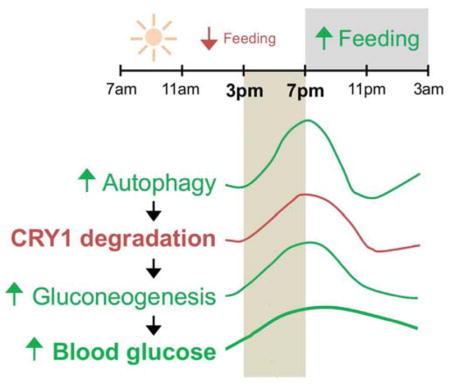
(L) Cartoon summarizing temporal autophagic degradation of CRY1 and its effects on gluconeogenesis and blood glucose levels.
Values are mean ± s.e.m. *P<0.05, **P<0.01, ***P<0.001. Student’s T-test, One-way or Two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S5.
LIR motifs link CRY1 degradation to regulation of gluconeogenesis
Since increases in hepatocellular CRY1 protein inhibit gluconeogenesis, we next tested whether accumulation of mLIR1-CRY1 or mLIR4-CRY1 was sufficient to suppress hepatic glucose production/blood glucose levels. Indeed, mice injected with mLIR1-CRY1 or mLIR4-CRY1, but not mLIR2-CRY1 and mLIR3-CRY1 constructs, displayed reduced G6P protein levels (Fig. 5F) and decreased blood glucose levels at 7pm (Fig. 5G–5J). Furthermore, mLIR1-CRY1 or mLIR4-CRY1-injected mice, and not the mLIR2-CRY1 and mLIR3-CRY1-injected mice, exhibited less glucose production when subjected to PTTs (Fig. 5K) – indicating that LIR motifs 271DLYKKV276 and 492SRYRGL497 link CRY1 turnover by autophagy to regulation of gluconeogenesis. Most surprisingly, CRY1 LIR2 (285SLYGQL290) and LIR4 (492SRYRGL497) are also required for degradation of SCF components Skp1 and FBXL3 by autophagy, since livers injected with mLIR2-CRY1, mLIR3-CRY1, and mLIR4-CRY1 each showed accumulation of these proteins when compared to WT-CRY1 or mLIR1-CRY1 (Fig. S5G). These data suggest that SCF components Skp1 and Fbxl3 are likely targeted for autophagic degradation via their association with CRY1 (Xing et al., 2013) that, in turn, binds to LC3 via its LIR motifs.
HFD feeding decreases CRY1 protein levels in liver
Because obesity dysregulates gluconeogenesis and given our finding that autophagic turnover of CRY1 derepresses hepatic gluconeogenesis under physiological states, we hypothesized that obesity accelerates the degradation of CRY1 by autophagy, which increases glucose production. To test this hypothesis, C57BL/6J male mice were fed a regular chow diet (RD) or high fat diet (HFD; 60% calories in fat) for 8 weeks following which, livers were collected at 7am, 11am, 3pm, 7pm, 11pm and 3am and assessed for period-wide changes in: (i) expression of gluconeogenic genes, and (ii) levels of core circadian proteins and the autophagy protein LC3. As expected, HFD feeding for 8 weeks significantly increased expression of gluconeogenic genes, G6p, Pck1, Fbp1, and Pc in liver (Fig. S6A–S6D). Although we noted qualitative differences in the patterns of their expression across period, a unifying feature was maximal induction of gluconeogenic genes at 7pm. Consequently, we tested whether increased gluconeogenic gene expression at 7pm correlated with decreased CRY1 protein levels in nuclear and cytoplasmic compartments from livers of HFD-fed mice. Indeed, period-wide assessments in livers revealed that HFD feeding progressively decreased nuclear and cytosolic CRY1 levels (Fig. 6A, 6B, and 6D) that associated with increased levels of the autophagosome marker LC3-II in both nuclear and cytosolic fractions (Fig. 6A, 6B, and 6C). Reduction in CRY1 levels did not occur from changes in expression of repressor genes Cry1 or Per1 (Fig. S6E, S6F). Indeed, while HFD feeding moderately decreased Cry1 gene expression at 3am and 7am, no differences were observed between RD and HFD-fed mice from 11am–11pm—the broad interval when CRY1 regulates gluconeogenesis (Fig. S6E). These data suggest that early obesity, as observed with 8 weeks of HFD feeding, is sufficient to promote CRY1 protein instability.
Fig. 6. High fat feeding decreases CRY1 protein levels in liver.

(A–K) Immunoblots and quantifications for indicated proteins in nuclear and cytosolic fractions from livers at indicated timepoints from male mice fed regular chow (RD) or high fat diet (HFD; 60% calories in fat) for 8 weeks. n=4–8.
Values are mean ± s.e.m. *P<0.05. **P<0.01, ***P<0.001. Two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S6.
HFD feeding did not have a remarkable effect on levels of the other core circadian proteins (Fig. 6), although we did note an interesting trend towards slight increases in nuclear levels of PER1, PER2, and REV-ERBα and reduction in their corresponding levels in the cytosol (Fig. 6E, 6F, and 6I). Nuclear and cytosolic levels of BMAL1 were modestly decreased in the early hours of the light cycle that correlated with decreased Bmal1 gene expression in livers from HFD-fed mice (Fig. 6G, and S6G) as shown previously (Eckel-Mahan et al., 2013). Conversely, modest increases in cytosolic CLOCK protein after 3pm correlated with increased Clock gene expression (Fig. 6H, and S6H). HFD feeding modestly increased cytoplasmic FBXL3 and nuclear FBXL21 levels and did not affect levels of Skp1 (not shown); however, these changes do not explain the instability of CRY1 in HFD-fed mice (Fig. 6J, 6K). Taken together, these data show that moderate duration of HFD feeding is sufficient to decrease CRY1 protein levels in liver—forming the basis to explore whether decreases in CRY1 through autophagic degradation promote gluconeogenesis during early obesity.
Obesity promotes degradation of CRY1 in an autophagy-dependent manner
Given these results, we asked whether HFD feeding-induced CRY1 degradation is autophagy-dependent. To that purpose, we subjected liver lysates from RD and HFD-fed control and Atg7KO mice to immunoblotting for CRY1, while CLOCK served as a control. While Atg7 loss in RD or HFD-fed mice (Fig. S7A) did not affect CLOCK protein levels at 7pm (Fig. 7A, and 2L), loss of Atg7 (as early as 7 days after AAV-Cre injection) blocked the degradation of CRY1 in livers from HFD-fed mice by ~50% (Fig. 7A) demonstrating that autophagy contributes to CRY1 instability in HFD-fed mice (Fig. 6D).
Fig. 7. Restoring CRY1 prevents obesity-associated dysregulation of gluconeogenesis.

(A) IB for CRY1 and CLOCK in livers from male mice fed RD or HFD for 8 weeks and then subsets of mice were injected with control (Con) or Cre-expressing AAVs (Atg7KO), as in 7D, n=5.
(B, C) Blood glucose and PTT at indicated timepoints in male mice fed HFD for indicated duration of time (plan in Fig. S7B) following which a subset of mice was injected with WT-CRY1 for 1 day, n=5–8.
(D, E) Experimental plan and PTT at indicated timepoints (before HFD feeding; 7 weeks after HFD feeding; and 1 week after AAV-Cre injection) in male mice fed RD or HFD for 7 weeks and then subsets of mice were injected with Con or Cre-expressing AAVs (Atg7KO), n=5–10.
(F–J) RT-PCR for indicated genes in livers at 7pm as per plan in panel 7D, n=4–8.
Values are mean ± s.e.m. *P<0.05, **P<0.01, ***P<0.001. Student’s T-test or Two-way ANOVA and Bonferroni correction. Ponceau is the loading control. See also Figure S7.
Restoring CRY1 levels prevents obesity-associated dysregulation of gluconeogenesis
To mechanistically link autophagic degradation of CRY1 to increased hepatic gluconeogenesis in obese mice, we tested whether two distinct approaches to reconstitute hepatic CRY1 levels in obese mice will normalize glucose metabolism. Consequently, RD-fed C57BL/6J mice were subjected to PTT on day 1 prior to initiating them on HFD (60% calories in fat) for 8 weeks (plan in Fig. S7B). After 7 weeks of HFD feeding, mice were subjected to a second PTT and then rested for 7 days following which mice were tail vein-injected with cDNA expressing WT-CRY1 and administered a PTT the next day (Fig. S7B). Restoring CRY1 levels (~5-fold increase in Cry1 expression in liver) (Fig. S7C) was sufficient to lower blood glucose levels by ~50% in HFD-fed mice (Fig. 7B), which associated with remarkable inhibition in expression of Pck1, G6p, and Fbp1 (Fig. S7D–S7F) and normalization of glucose production in a PTT when compared to HFD-fed controls (Fig. 7C). In addition, knocking out Atg7 in 8-week HFD-fed mice (Fig. 7D) increased hepatic CRY1 levels by ~50% (Fig. 7A), reduced glucose production in a PTT (Fig. 7E) and decreased the expression of Fbp1, G6p, Pc and Pck1 when compared to HFD-fed controls (Fig. 7F–7I). Normalization of glucose homeostasis in HFD-fed Atg7KO mice occurred due to accumulation of CRY1 (Fig. 7A) and not due to differences in Cry1 gene expression (Fig. 7J). These results demonstrate that inhibiting autophagy in liver or targeting specific LIR motifs on CRY1 to prevent their autophagic degradation is sufficient to normalize obesity-associated dysregulation of gluconeogenesis—revealing LIRs on CRY1 as therapeutic targets against hyperglycemia.
Discussion
In this study, we show that autophagy in the liver degrades CRY1 during a temporal window when rodents typically eat less and subsist on gluconeogenesis. The enrichment of CRY1 in APhs, its interaction with LC3, and accumulation of CRY1 in autophagy-deficient livers demonstrate that CRY1 is an autophagy substrate. Site-directed mutagenesis to inactivate distinct LIR motifs on CRY1 reveal two LIRs that regulate CRY1 degradation by autophagy. Degradation of CRY1 allows induction of gluconeogenesis and maintenance of blood glucose levels. Consistent with this notion, mice with Atg7-deficient livers display increased CRY1 levels and reduced blood glucose levels, which is rescued in part by decreasing their hepatic CRY1 content. Rescue of blood glucose occurs selectively between 3pm–7pm, suggesting that autophagic CRY1 degradation is time-imprinted to a specific physiological function—maintenance of blood glucose levels during reduced feeding. A well-defined signaling module that induces autophagy during nutrient depletion is AMPK, and phosphorylation by AMPK promotes CRY1 degradation (Lamia et al., 2009). Although AMPK primes CRY1 for proteasomal degradation, whether phosphorylation by AMPK facilitates CRY1 sequestration by autophagy remains unknown. It is thought that phosphorylation of residues in proximity to LIR motifs facilitates LIR-LC3 interaction (Birgisdottir et al., 2013). Examination of CRY1 protein revealed that Serine 280, a residue phosphorylated by AMPK, lies distal to LIR 271DLYKKV276 inactivating which led to CRY1 accumulation. It is thus possible that introducing a negative charge in the vicinity of CRY1 LIR could increase its degradation via autophagy, and from that perspective, Ser280 phosphorylation could drive CRY1 degradation by autophagy. Whether such cooperativity determines the fate of CRY1 protein remains to be seen. We were intrigued by the finding that PER2 and CRY1 each interact with LC3 at 7pm when CRY1 is maximally degraded by autophagy yet PER2 does not accumulate in autophagy-deficient livers. We speculate that PER2, a known interacting partner of CRY1, is a cargo adapter that facilitates interaction of CRY1 with LC3, although future studies will be required to test this notion. We were also intrigued by the finding that SCF components Skp1 and Fbxl3, which determine CRY1 degradation by the proteasome are also autophagy substrates. In context of this study, it would seem that reduced feeding at specific diurnal intervals directs hepatocytes to prioritize autophagic degradation of CRY1 and possibly CRY1-associated proteins, e.g., Skp1 and Fbxl3, such that suppressive effects of CRY1 on gluconeogenesis are effectively neutralized. Loss of autophagy resulted in pronounced accumulation of FBXL3 and Skp1 in the nucleus, which begs further investigation on mechanisms of sequestration of these nuclear-localized proteins by autophagy; however, these extensive cell biological excursions are beyond the scope of the present work. Taken together, we show that temporal degradation of CRY1 by autophagy derepresses hepatic gluconeogenesis, and that LIR motifs mechanistically link degradation of CRY1 to regulation of gluconeogenesis revealing LIRs as potential targets for glycemic control.
Limitations of study
We find that HFD feeding accelerates the degradation of CRY1 by autophagy, which is surprising given that chronic lipid stimulus is thought to suppress autophagy (Singh et al., 2009). This is an exciting finding that begs the question—how is CRY1 degradation accelerated when autophagy is suppressed? These results allow us to consider the possibility that despite the inhibitory effect of obesity on autophagy, chronic lipid availability may trigger post-translational modifications on autophagic cargo, for instance, phosphorylations in the vicinity of LIR motifs, that could make a subset of proteins more efficiently recognized by the residual autophagic machinery. The second limitation of our study is the lack of experiments that reveal the relative contribution of the proteasome and autophagy to the degradation of CRY1. It could be possible that autophagy and the proteasome each regulate CRY1 levels at distinct intervals across period and likely respond to different environmental cues. Future studies will be required to address these limitations.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ATG7 | Cell Signaling Technology | Cat# 2631; RRID: AB_2227783 |
| Rabbit monoclonal anti-BMAL1 | Cell Signaling Technology | Cat# 14020 |
| Rabbit monoclonal anti-CLOCK | Cell Signaling Technology | Cat# 5157; RRID:AB_10695411 |
| Rabbit polyclonal anti-CRY1 | Origene | Cat# TA342728 |
| Rabbit polyclonal anti-CRY2 | Abcam | Cat# ab38872, RRID:AB_731755 |
| Mouse monoclonal anti-DYKDDDDK Tag | Cell Signaling Technology | Cat# 8146S; RRID:AB_10950495 |
| Rabbit polyclonal anti-Fbxl3 | Abcam | Cat# ab96645, RRID:AB_10679381 |
| Rabbit polyclonal anti-G6Pase | Abcam | Cat# ab83690, RRID:AB_1860503 |
| Rat monoclonal anti-LAMP1 | DSHB | Cat# 1d4b, RRID:AB_2134500 |
| Rabbit polyclonal anti-LC3B | Cell Signaling Technology | Cat# 2775; RRID: AB_915950 |
| Human polyclonal anti-p62 | Enzo Life Sciences | Cat# BML-PW9860, RRID:AB_2196009 |
| Rabbit polyclonal anti-Pyruvate Carboxylase | Novus | Cat# NBP1-49536, RRID:AB_10011589 |
| Rabbit polyclonal anti-PER1 | Novus | Cat# NBP2-24589 |
| Rabbit polyclonal anti-PER2 | Origene | Cat# TA337016 |
| Rabbit monoclonal anti-Rev-erb α | Cell Signaling Technology | Cat# 13418, RRID:AB_2630359 |
| Rabbit polyclonal anti-Skp1 | Cell Signaling Technology | Cat# 2156S; RRID:AB_10692507 |
| Rabbit polyclonal anti-Fbxl3 | Abcam | Cat# ab96645, RRID:AB_10679381 |
| Rabbit polyclonal anti-Fbxl21 | Abcam | Cat# ab222894 |
| Secondary HRP Antibody Rabbit anti-Mouse IgG | Invitrogen | Cat# 61-6520 RRID: AB_2533933 |
| Secondary HRP Antibody Goat anti-Rabbit IgG | KPL | 074-1506 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | Cat# R37116, RRID:AB_2556544 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21247, RRID:AB_141778 |
| Bacterial and Virus Strains | ||
| pMC1SG5_mmCry1 | Addgene | 31282 |
| mLIR1-CRY1 (271DLAKKA276) | GenScript | N/A |
| mLIR2-CRY1 (285SLAGQA290) | GenScript | N/A |
| mLIR3-CRY1 (486QIAQQA491) | GenScript | N/A |
| mLIR4-CRY1 (492SRARGA497) | GenScript | N/A |
| AAV-TBG-GFP | Vector Biolabs | VB1743 |
| AAV-TBG-iCre | Vector Biolabs | VB1724 |
| Ad-CMV-Null | Vector Biolabs | 1300 |
| Ad-m-CRY1-shRNA | Vector Biolabs | shADV-256159 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| High Fat Diet (60% of calories in fat) | Research Diets | D12492 |
| Leupeptin hemisulfate | Fisher Scientific | BP2662100 |
| Metrizamide | Sigma-Aldrich | 860506 |
| Trizol Reagent | Invitrogen | 15596018 |
| Triton X-100 | Sigma-Aldrich | X100-500ml |
| Superscript II Reverse Transcriptase | Invitrogen | 18064014 |
| Sodium Pyruvate | Sigma-Aldrich | P2256 |
| Power SYBR Green PCR Master Mix | Invitrogen | 4368708 |
| RNeasy Plus Mini kit | Qiagen | 74136 |
| 30% Acrylamide/ Bis Solution 37-5-1 | BioRad | 161-0158 |
| Paraformaldehyde | Electron Microscopy Sciences | 15714-S |
| Sodium Pyrophosphate | Sigma-Aldrich | AB02014 |
| Sodium Chloride | AmericanBio | AB01915 |
| β-Glycerophosphate | Sigma-Aldrich | G9891 |
| Sodium Orthovanadate | Sigma-Aldrich | S6508 |
| PMSF | Sigma-Aldrich | 10837091001 |
| Sucrose | AmericanBio | AB01900-01000 |
| Super Signal West Femto Maximum Sensitivity Substrate, ECL | Pierce | 34096 |
| DAPI Fluoromount-G | SouthernBiotech | 0100-20 |
| Critical Commercial Assays | ||
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermo Fisher Scientific | 78835 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 wildtype | The Jackson Laboratory | JAX: 000664 |
| Mouse Atg7flox/flox | Komatsu et al., (2006), Nature 441, 880–884. | N/A |
| Oligonucleotides | ||
| RT-PCR primers Please see Table S1 | Sigma-Aldrich | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/index.html; RRID: SCR_003070 |
| Prism | Graph Pad | https://www.graphpad.com/scientificsoftware/prism/; RRID:SCR_002798 |
| Other | ||
| StepOne Plus Real-Time PCR System | Thermo Fisher Scientific | 4376600 |
| CLAMS open-circuit indirect calorimetry | Columbus Instruments | N/A |
| Leica SP5 AOBS Inverted DMI6000 confocal microscope | Leica Microsystems | N/A |
| Ascensia Contour Glucometer | Bayer | 7151H |
| Ascensia Contour strips | Bayer | 7080G |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Rajat Singh (rajat.singh@einstein.yu.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Studies were performed in 4–6-month-old male C57BL/6 mice, and Atg7flox/flox mice (Komatsu et al., 2006) that was a kind gift from Drs. M. Komatsu and K. Tanaka, Tokyo Metropolitan Institute of Medical Science, Japan). Atg7flox/flox mice were on the C57BL/6 background. Mice were used under a protocol approved by the Institutional Animal Care and Use Committee. Mice were fed a regular chow (5058; Lab Diet, St Louis, MO) or 8 weeks of HFD providing 60% calories in fat D12492; Research Diets, New Brunswick, NJ, USA. No inclusion/exclusion criteria were used in the selection of mice for the study.
Housing
Mice were maintained at 22–23°C on 12 hr light/dark cycles (7am–7pm) in the institutional barrier facility along with sentinel cages and are specific pathogen-free. Mice in sentinel cages are routinely tested by the Institute for Animal Studies for specific pathogens, and health reports are evaluated at regular intervals to determine whether rodents are pathogen-free or whether a specific treatment is required.
METHOD DETAILS
Autophagy flux assay
Autophagy/LC3-II flux was performed by intraperitoneal (i.p.) injection of lysosomal inhibitor (Lys Inh), leupeptin, at 40mg/kg mouse weight injected 2 hr prior to liver collection at 7am, 11am, 3pm, 7pm, 11pm and 3am. Livers were homogenized in a buffer containing protease and phosphatase inhibitors and immunoblotted for LC3. Autophagy flux was calculated by subtracting the densitometry values of LC3-II or p62 in Lys Inh-untreated from Lys Inh-treated samples (Martinez-Lopez et al., 2016).
Isolation of autophagic structures and nuclear fractions
Autophagosomes (APh) and lysosomes (Lys) were isolated from livers by differential centrifugation using discontinuous density gradients of metrizamide. Briefly, liver homogenates were centrifuged at 2,000 g for 5 min followed by centrifugation of supernatants at 17,000 g for 12 min. We resuspended the pellets in 1.9 ml 0.25 M sucrose and then 2.8 ml of metrizamide (85.6%) was added. We centrifuged samples on a 26–24–20–15% metrizamide gradient at 75,334 g for 3 hr, following which the APh fraction was collected from 15–20% interface, and Lys fraction from the 24–26% interface. Fractions were centrifuged at 24,000 g and pellets were resuspended in 0.25 M sucrose and analyzed. Cytosolic and nuclear fractions were isolated using ProteoExtract subcellular proteome extraction kit (EMD Millipore, Billerica, MA) as per manufacturer’s instructions. Equivalent protein loading was verified using Ponceau and antibodies for Lamin A/C (nucleus), LC3-II (APh), and LAMP1 (Lys).
RNA isolation and qPCR analyses
Total RNA was isolated using the Trizol Reagent (Invitrogen). The aqueous phase containing the RNA was loaded onto a gDNA Eliminator Spin Column (Qiagen, USA) to eliminate genomic DNA, and RNA was isolated using the RNeasy Plus kit (Qiagen) according to manufacturer’s instructions. Total RNA (1μg) was reverse transcribed into cDNA using Superscript II (Invitrogen), and quantitative RT-PCR analyses was performed using Power SYBR® Green PCR Master Mix (Applied Biosystems, UK) on a StepOne Plus Real-Time PCR System (Applied Biosystems, UK). Values were normalized to expression of the housekeeping gene TATA-binding protein (TBP). The mRNA expression in control samples was considered as 1 and mRNA expression in experimental samples was represented as fold-change compared to expression in controls. Comparisons were only made for expression levels between the same gene in control or KO samples. All reactions were performed in triplicate. Values were expressed in arbitrary units (a.u).
Plasmids and adeno-associated virus (AAV) injections in vivo
Wildtype (WT) CRY1 plasmid (pMC1SG5_mmCry1) was a gift from Dr. Aziz Sancar (Addgene plasmid #31282) (5). CRY1 mLIR1-4 mutant plasmids were generated by site-directed mutagenesis (GenScript USA, Inc. Piscataway, NJ) and individual LIR mutations were validated by sequencing. We injected 50 μg of WT FLAG-tagged CRY1 or mLIR CRY1 plasmids via tail vein injections using the in vivo jet polyethylenimine (PEI) transfection reagent (Polyplus-transfection Inc., NY) as per manufacturer’s protocols (Chang et al., 2014; Wahlquist et al., 2014). Mice were injected at 8am and sacrificed at 7pm. Expression of the plasmid in liver was determined by immunoblotting or RT-PCR analyses. Deletion of Atg7 in liver was accomplished by tail vein injections of 109 PFU of AAVs expressing Cre recombinase and mice were humanely killed 7–13 days after injection. Knockdown of CRY1 in liver was accomplished by 1010 PFU of adenoviruses expressing CRY1-shRNA purchased from Vector Biolabs (Malvern, PA, USA) and injected 2 days prior to tissue collection.
Plasmid transfections in vitro
Cells (293T) were co-transfected with 0.5 μg of GFP-tagged LC3 and 0.5 μg FLAG-tagged WT-CRY1 or mutant CRY1 by lipofectamine 3000. After 36 hr of transfection, cells were lysed and precipitated with FLAG M2 gel (Sigma-Aldrich) and eluted with FLAG peptide (Sigma-Aldrich). Protein samples (10 μg) were resolved in 4–10% gradient gels and then transferred and membranes were probed with anti-FLAG antibody (Sigma-Aldrich) and GFP antibody (Cell Signaling technology). Plasmid only control samples were transfected with pcDNA3 plasmid.
Pyruvate tolerance test (PTT)
Mice were fasted at noon and PTT was performed at 6pm, after i.p. injections of sodium pyruvate (1.5 g/kg body weight). Blood glucose levels were measured immediately prior to injection and at indicated timepoints after pyruvate injection using an Ascensia Contour glucometer (Bayer).
Western Blotting
Total protein from liver was isolated in buffer containing 20mM Tris, pH 7.5, 1% Triton X-100, 1mM EDTA, 1mM EGTA, and protease/phosphatase inhibitors. Lysates were centrifuged, and supernatants were subjected to immunoblotting by denaturing 20–50μg of protein at 100°C for 5 min in Laemmli sample buffer containing 62.5mM Tris, 2% SDS, 25% glycerol, 0.01% bromophenol blue, and 5% β-mercaptoethanol. Samples were resolved on SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare, USA) in transfer buffer containing 25mM Tris, 192mM glycine, 0.01% SDS, and 15% methanol using a Bio-Rad semidry transfer cell at 150mA for 90 minutes. Membranes were blocked in 5% nonfat dry milk, 20mM Tris, 500mM sodium chloride, and 0.5% Tween-20 for 1 hr and probed with primary antibodies.
Indirect Immunofluorescence
Livers were fixed with 4% paraformaldehyde, blocked and incubated with primary and corresponding secondary antibodies (Alexa Fluor 488 and/or Alexa Fluor 647 conjugated) (Invitrogen). Mounting medium contained DAPI (4′,6-diamidino-2-phenylindole) to visualize the nucleus (Invitrogen). Images were acquired on a Leica SP5 AOBS Inverted DMI6000 confocal microscope (Leica Microsystems, Germany) using X63/1.4 oil objective. Images in each experiment were acquired at same exposure times within the same imaging session. All images were prepared using Adobe Photoshop and subjected to identical post-acquisition manipulations. Representative native images are shown. Quantification was performed after appropriate thresholding using the ImageJ software (NIH) in a minimum of 30 cells from three or more experiments. Cellular fluorescence intensity was expressed as mean integrated density as a function of individual cell size. Percentage colocalization was calculated using the JACoP plugin in single Z-stack sections of native images. Acquisition and quantification of images were performed in a blinded manner.
Assessments of Circadian Feeding Patterns
Circadian feeding patterns were assessed in CLAMS/metabolic cages (Columbus Instruments, USA).
Biochemical Analyses
Serum Insulin and Glucagon (ALPCO, NH, USA) and liver glycogen levels (Sigma Aldrich, USA) were assessed using commercial kits according to manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mean and standard error were calculated for each studied variable. Statistical significance was determined using One-way or Two-way ANOVA followed by Bonferroni multiple comparison test or by two-tailed unpaired Student’s t-test. *P<0.05, **P<0.01, ***P<0.001 and ###P<0.001. We performed the Shapiro-Wilk test to determine the normal distribution of the variables being tested. All statistical analyses were performed using Prism Graph Pad (La Jolla, CA). Statistical details for each experiment including n value are provided in the Figure legend.
Supplementary Material
Highlights.
Core circadian proteins are temporally degraded by lysosomes
Loss of autophagy promotes CRY1 accumulation and disrupts the circadian clock
Autophagy drives gluconeogenesis by degrading CRY1
LIR motifs link CRY1 degradation to regulation of glucose homeostasis
Acknowledgments
We thank Drs. M. Komatsu and K. Tanaka (Tokyo Metropolitan Institute of Medical Science, Japan) for Atg7F/F mice (Komatsu et al., 2006). This work was supported by R01 AG043517 (R.S.), P30 DK020541 (Einstein Diabetes Research Center), P01 AG031782 (Project leader, R.S.), American Diabetes Association (ADA) postdoctoral fellowship #1-17-PMF-011 (M.T.), and ADA Grant 1-18-IBS-062 (R.S.). E.M. is supported by R01 GM120358, and A.B.-G. is supported by T32 AG023475. We thank Dr. J. Bass (Northwestern University Feinberg School of Medicine) for his thought-provoking suggestions.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Author contributions: Conceptualization, R.S..; Methodology, M.T.; Investigation, M.T., A.B.-G., E.M., M.L.A., E.T., D.F., J.S.; Writing – Original Draft, R.S.; Writing – Review & Editing, M.T. and R.S.; Funding Acquisition, R.S., M.T., and J.E.P.; Resources, P.M., F.B., G.J.S, and J.E.P.; Supervision, R.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126:3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- Chang RM, Yang H, Fang F, Xu JF, Yang LY. MicroRNA-331-3p promotes proliferation and metastasis of hepatocellular carcinoma by targeting PH domain and leucine-rich repeat protein phosphatase. Hepatology. 2014;60:1251–1263. doi: 10.1002/hep.27221. [DOI] [PubMed] [Google Scholar]
- Cornelissen G, Otsuka K. Chronobiology of Aging: A Mini-Review. Gerontology. 2017;63:118–128. doi: 10.1159/000450945. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21:142–150. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington TK, Wager-Smith K, Ceriani MF, Staknis D, Gekakis N, Steeves TD, Weitz CJ, Takahashi JS, Kay SA. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science. 1998;280:1599–1603. doi: 10.1126/science.280.5369.1599. [DOI] [PubMed] [Google Scholar]
- Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–449. doi: 10.1083/jcb.33.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, Sassone-Corsi P. Reprogramming of the circadian clock by nutritional challenge. Cell. 2013;155:1464–1478. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- Griffin EA, Jr, Staknis D, Weitz CJ. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;286:768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythms. 2005;20:391–403. doi: 10.1177/0748730405277232. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152:1106–1118. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- Jang H, Lee GY, Selby CP, Lee G, Jeon YG, Lee JH, Cheng KK, Titchenell P, Birnbaum MJ, Xu A, et al. SREBP1c-CRY1 signalling represses hepatic glucose production by promoting FOXO1 degradation during refeeding. Nat Commun. 2016;7:12180. doi: 10.1038/ncomms12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern L, Spreckels J, Nist A, Stiewe T, Skevaki C, Greene B, Mernberger M, Elsasser HP. Altered glycogen metabolism causes hepatomegaly following an Atg7 deletion. Cell Tissue Res. 2016;366:651–665. doi: 10.1007/s00441-016-2477-8. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, Downes M, Evans RM. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ, Singh R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzella L, Ahlberg J, Glaumann H. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. J Cell Biol. 1982;93:144–154. doi: 10.1083/jcb.93.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng QJ, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sladek M, Semikhodskii AS, Glossop NR, Piggins HD, et al. Setting clock speed in mammals: the CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron. 2008;58:78–88. doi: 10.1016/j.neuron.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AK, Fribourgh JL, Chelliah Y, Sandate CR, Hura GL, Schneidman-Duhovny D, Tripathi SM, Takahashi JS, Partch CL. Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proc Natl Acad Sci U S A. 2017;114:1560–1565. doi: 10.1073/pnas.1615310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015;6:6823. doi: 10.1038/ncomms7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science. 2015;350:aac4250. doi: 10.1126/science.aac4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell. 2013;12:478–488. doi: 10.1111/acel.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, Fruechte EM, Vitaterna MH, Shimomura K, King DP, et al. Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron. 1998;21:1101–1113. doi: 10.1016/s0896-6273(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA, Hogenesch JB. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JS. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508:531–535. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, Zheng N. SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496:64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–1105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, Nusinow DA, Sun X, Landais S, Kodama Y, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwighaft Z, Aviram R, Shalev M, Rousso-Noori L, Kraut-Cohen J, Golik M, Brandis A, Reinke H, Aharoni A, Kahana C, et al. Circadian Clock Control by Polyamine Levels through a Mechanism that Declines with Age. Cell Metab. 2015;22:874–885. doi: 10.1016/j.cmet.2015.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.