

Abstract
Paradigm shifting studies in the mouse have identified tissue macrophage heterogeneity as a critical determinant of immune responses. In contrast, surprisingly little is known regarding macrophage heterogeneity in humans. Macrophages within the mouse heart are partitioned into CCR2- and CCR2+ subsets with divergent origins, repopulation mechanisms, and functions. Here we demonstrate that the human myocardium also contains distinct subsets of CCR2- and CCR2+ macrophages. Analysis of sex mismatched heart transplant recipients revealed that CCR2- macrophages are a tissue-resident population exclusively replenished through local proliferation, whereas CCR2+ macrophages are maintained through monocyte recruitment and proliferation. Moreover, CCR2- and CCR2+ macrophages have distinct functional properties, analogous to reparative CCR2- and inflammatory CCR2+ macrophages in the mouse heart. Clinically, CCR2+ macrophage abundance is associated with LV remodeling and systolic function in heart failure patients. Collectively, these observations provide initial evidence for the functional importance of macrophage heterogeneity in the human heart.
Keywords: Heart Failure, Inflammation, Macrophages
Introduction
Over the past 40 years the prevailing view has been that tissue macrophages originate from circulating blood monocytes. Recently, a growing body of literature has challenged this dogma and revised our understanding of macrophage origins. Studies performed in mouse models have provided evidence that tissue macrophages represent a heterogeneous population of cells derived from a variety of lineages1–3. In the mouse, many tissue resident macrophages in the brain, skin, liver, kidney, lung, and heart are established during embryonic development and persist into adulthood independent of blood monocyte input4–12. Embryonic-derived macrophages are long-lived and replenished locally independent of peripheral monocyte input through cell proliferation4, 13. In contrast, classically described macrophages originate from definitive hematopoietic progenitors located within the bone marrow and spleen and are replenished under steady state and inflammatory conditions through monocyte recruitment in a CCR2 (receptor for CCL2/MCP1 and CCL7/MCP3) dependent manner6, 7. These advancements in knowledge relied on the establishment of sophisticated genetic lineage tracing, parabiosis, and monocyte tracking strategies only available in rodent systems14–16.
While tissue resident macrophage populations and their exact embryonic origins continue to be defined across a variety of organs and tissues, it is immediately apparent that macrophage origin is a critical determinant of cell behavior. This is particularly important as macrophages of distinct origin often coexist within tissues11. For example, the heart contains several macrophage populations that can be distinguished based on lineage tracing and cell surface expression of CCR2. By employing flow cytometry, genetic lineage tracing, and parabiosis strategies, we previously demonstrated that the mouse heart contains distinct populations of CCR2- and CCR2+ macrophages. CCR2- macrophages are derived from primitive yolk sac and fetal monocyte progenitors. CCR2- macrophages establish residency within the heart during embryonic development, and in the absence of disease, are maintained throughout life independent of blood monocyte input. In contrast, CCR2+ macrophages originate from definitive hematopoietic progenitors, enter the heart after the first few weeks of life, and are maintained through a combination of mechanisms including gradual monocyte recruitment and proliferation8, 17, 18.
Importantly, CCR2- and CCR2+ macrophages have distinct functions in the heart. CCR2- macrophages are involved in various forms of tissue remodeling such as coronary development, vascular expansion, and cardiac tissue repair17, 18. For example, following neonatal cardiomyocyte injury, CCR2- macrophages orchestrate cardiac tissue regeneration and functional recovery of the heart through expansion of the coronary vasculature, cardiomyocyte proliferation, and physiological cardiomyocyte hypertrophy. In the absence of CCR2- macrophages, the pediatric mouse heart demonstrates little regenerative capacity. While the exact functions of CCR2+ macrophages within the adult heart under steady state conditions are not completely defined, it is likely that these cells participate in the initiation of inflammation. Following myocardial ischemia reperfusion injury, CCR2+ macrophages are activated in a TLR9 dependent manner and orchestrate neutrophil extravasation into the injured myocardium through production of the neutrophil chemokines, CXCL2 and CXCL519.
Collectively, these studies have established that in mice the heart contains a heterogeneous population of functionally distinct macrophages with remarkable effects on cardiovascular disease pathogenesis. However, macrophage heterogeneity in human tissues remains largely unexplored. In this study, we tested the hypothesis that the human heart contains distinct macrophage populations that are functionally analogous to mouse cardiac CCR2- and CCR2+ macrophages.
Results
The human heart contains distinct subsets of CCR2- and CCR2+ macrophages
To define appropriate cell surface markers for human cardiac macrophages, we performed immunostaining on LV myocardial specimens obtained from patients with dilated and ischemic cardiomyopathies. Transmural LV specimens were collected at the time of left ventricular assist device (LVAD) implantation or heart transplantation. All specimens were obtained from either the apical or lateral LV walls. We first tested whether cardiac macrophages express CD14, a GPI anchored TLR4 co-receptor preferentially expressed on human monocytes and macrophages20. Dual immunostaining for CD68 (pan-macrophage marker) and CD14 revealed that human cardiac macrophages uniformly express CD14 (Fig. 1a). Quantification of CD14+CD68+ cells in specimens obtained from patients with dilated cardiomyopathy (DCM) and ischemic cardiomyopathy (ICM) revealed that the vast majority of cardiac macrophages are CD14+ (94.1 and 94.4%, respectively) (Fig. 1b). Based on the finding that human cardiac macrophages uniformly express CD14, we devised a flow cytometry gating scheme to identify and characterize human cardiac macrophage populations.
Figure 1. The human heart contains distinct populations of CCR2- and CCR2+ macrophages.
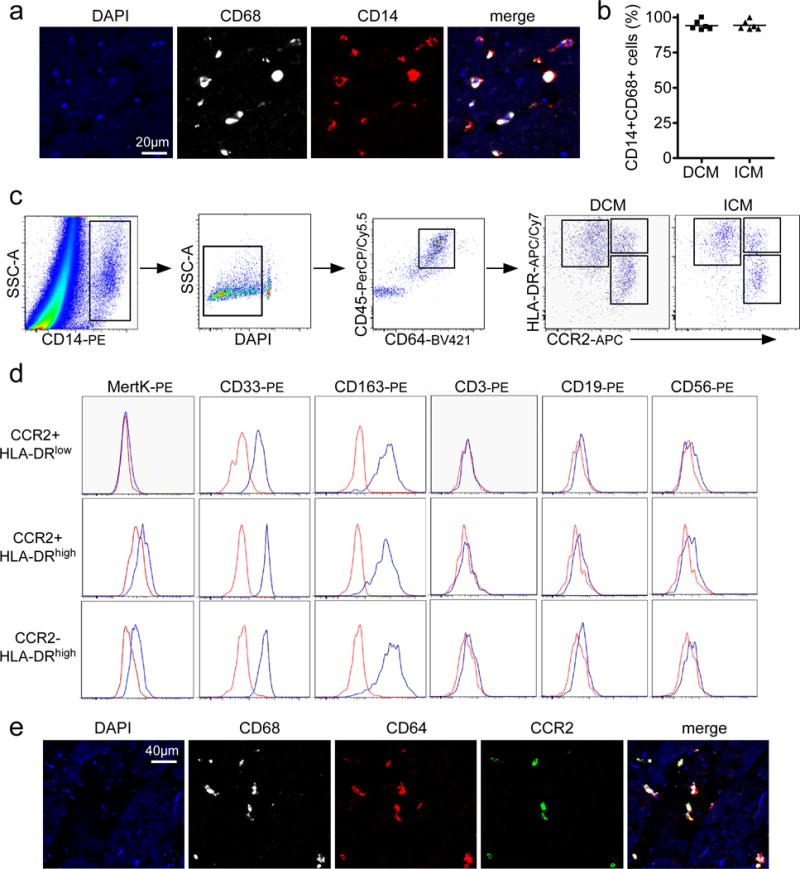
a, Immunostaining of human cardiac macrophages (CD68, white) for CD14 (red). Blue: DAPI. b, Percentage of CD14+CD68+ cells in specimens obtained from patients with dilated (DCM) and ischemic (ICM) cardiomyopathy. Each data point (n=6) represents a biologically independent heart failure sample. The line indicates the mean value. c, Flow cytometry gating scheme utilized to identify and characterize cardiac macrophage populations. d, Flow cytometry plots showing expression of MertK (macrophage marker), CD33 and CD163 (monocyte/macrophage markers), CD3 (T-cell marker), CD19 (B-cell marker), and CD56 (NK cell marker) in CCR2+HLA-DRlow, CCR2-HLA-DRhigh, and CCR2+HLA-DRhigh cells. Red: isotype control, Blue: indicated antibody. e, Immunostaining for CD68 (white), CD64 (red), and CCR2 (green) indicates that both CCR2- and CCR2+ macrophages are present within the left ventricular myocardium. These experiments were independently repeated 3 times with similar results. Blue: DAPI. a: 400X magnification, e: 200X magnification.
Previously, we and others have performed detailed lineage tracing, flow cytometry, and transcriptomic analyses to define cell surface markers for cardiac macrophages in the mouse including CD45, CD64, MHC-II, and CCR28, 17, 18, 21, 22. To test the hypothesis that human cardiac macrophages can be identified using an evolutionarily conserved set of cell surface markers, we performed the following experiments. Flow cytometry analysis revealed that CD14+ cells present in the human heart co-express both CD45 (common leukocyte antigen) and CD64 (Fc gamma receptor 1A) (Fig. 1c). CD64 is an evolutionarily conserved receptor that is exclusively expressed on mouse and human monocytes and macrophages. CD64 is not expressed on other myeloid cells including neutrophils, eosinophils, or dendritic cells20, 23. Using a gating strategy similar to that employed in our previous studies focused on mouse cardiac macrophages8, we demonstrated that CD14+CD45+CD64+ cells can be divided into 3 distinct subsets based on the expression of HLA-DR (human homologue of MHC-II) and CCR2: CCR2+HLA-DRlow, CCR2+HLA-DRhigh, and CCR2-HLA-DRhigh cells. The distribution of these cell types did not differ between ICM and DCM (Supplemental Fig. 1a). To more precisely define the identity of these cell populations, we performed further flow cytometry assays examining the expression of established cell markers. Previous studies have demonstrated that MertK (MER proto-oncogene tyrosine kinase) is an evolutionarily conserved marker specific for mouse and human macrophages20, 23. Compared to isotype control, MertK staining could only be detected on CCR2+HLA-DRhigh and CCR2-HLA-DRhigh cells suggesting that CCR2+HLA-DRhigh and CCR2-HLA-DRhigh cells represent macrophages and CCR2+HLA-DRlow cells are monocytes. Consistent with monocyte and macrophage identity CCR2+HLA-DRlow, CCR2+HLA-DRhigh, and CCR2-HLA-DRhigh cells expressed both CD33 (Siglec-3, myeloid marker) and CD163 (monocyte/macrophage marker) and lacked expression of CD3 (T-cell marker), CD19 (B-cell marker), and CD56 (NK-cell marker) (Fig. 1d).
To provide confirmatory evidence that human cardiac macrophages can be divided into CCR2- and CCR2+ subsets using a separate technique, we developed an immunostaining assay to identify CCR2- and CCR2+ macrophages in formalin fixed paraffin-embedded human heart tissue. For immunostaining experiments we identified monocytes and macrophages using CD68, a marker routinely used in clinical practice. Intracellular flow cytometry confirmed that CD45+CD14+CD64+ cells also expressed CD68 and that monocytes, CCR2- macrophages, and CCR2+ macrophages were found within the CD45+CD14+CD64+CD68+ gate (Supplemental Fig. 1b). Immunostaining with antibodies specific for CD64, CD68, and CCR2 revealed the presence of CD64+CD68+CCR2- and CD64+CD68+CCR2+ cells within the LV myocardium (Fig. 1e, Supplemental Fig. 1c). Together, these data demonstrate that the human heart contains a heterogeneous population of monocytes and macrophages that can be divided into distinct subsets based on the expression of CCR2 and HLA-DR.
Tissue localization of CCR2- and CCR2+ macrophages
To determine whether CCR2- macrophages and CCR2+ monocytes/macrophages occupy distant locations within the LV myocardium, we performed CD68 and CCR2 immunostaining on specimens obtained from DCM and ICM patients. Tissues were perfused with saline prior to fixation to remove intravascular immune cells including monocytes. Flow cytometry and immunostaining analyses indicated that tissue perfusion substantially reduced monocyte numbers and that the majority of CD68+ cells were CCR2-HLA-DRpos and CCR2+HLA-DRpos macrophages (Supplemental Fig. 2a-c). Rare monocytes (CCR2+HLA-DRneg) were only found adjacent to blood vessels located within areas of dense fibrosis (Supplemental Fig. 2d). Within viable appearing areas of myocardium (defined by the absence of scar tissue) the majority of CD68+ cells represented CCR2- macrophages. Co-staining with either CD34 or eNOS antibodies (vascular markers) revealed that CCR2- macrophages were closely associated with coronary endothelial cells (Supplemental Fig. 3a, d). In contrast, CCR2+ macrophages preferentially occupied areas containing scar or fibrotic tissue where they were found embedded in areas infiltrated with type I collagen (Supplemental Fig. 3b, d). We have previously demonstrated that mouse CCR2- and CCR2+ macrophages are activated in response to cardiomyocyte cell death17, 19. To determine whether human CCR2- and CCR2+ macrophages might similarly recognize dying cardiomyocytes, we performed CD68, CCR2, and TUNEL staining. CCR2- and CCR2+ macrophages were both present adjacent to TUNEL+ cardiomyocytes at equivalent ratios. However, areas of cell death were rare and the majority of CCR2- and CCR2+ macrophages were not located adjacent to TUNEL+ cardiomyocytes (Supplemental Fig. 3c-d).
CCR2- and CCR2+ macrophages are maintained through distinct mechanisms
To delineate whether human cardiac CCR2- and CCR2+ macrophages are maintained through similar or distinct mechanisms we measured contributions from peripheral monocyte recruitment and cell proliferation. We chose to focus on peripheral recruitment and cell proliferation as previous studies in the mouse have demonstrated that these activities represent the primary mechanisms responsible for maintenance and repopulation of mouse tissue macrophages11.
To measure the contribution of peripheral monocyte recruitment to the maintenance of human cardiac CCR2- and CCR2+ macrophage subsets, we examined endomyocardial biopsy specimens obtained from sex mismatched heart transplant recipients. All included endomyocardial biopsy specimens were obtained from male patients who received a heart from a female donor. The mean time from transplant was 8.8 years and none of the included biopsy specimens showed evidence of rejection or allograft dysfunction (Supplemental Table 1). Using a combination of immunostaining for CD68 and CCR2 and in situ hybridization for Y chromosomes, we quantified the percentage of CCR2- and CCR2+ macrophages that were derived from the recipient avoiding intravascular CD68+ cells (Fig. 2a-b). Recipient derived (Y chromosome+) macrophages were interpreted as originating from recruited monocytes. Consistent with being a tissue resident population, only a small percentage of CCR2- macrophages (0.70±1.4%) contained a Y chromosome. In contrast, 30.6±16.8% of CCR2+ macrophages contained a Y chromosome, suggesting that peripheral monocyte recruitment represents an important mechanism by which CCR2+ macrophages are maintained in the human heart (Fig. 2c).
Figure 2. CCR2- and CCR2+ cardiac macrophage populations are maintained through distinct mechanisms.
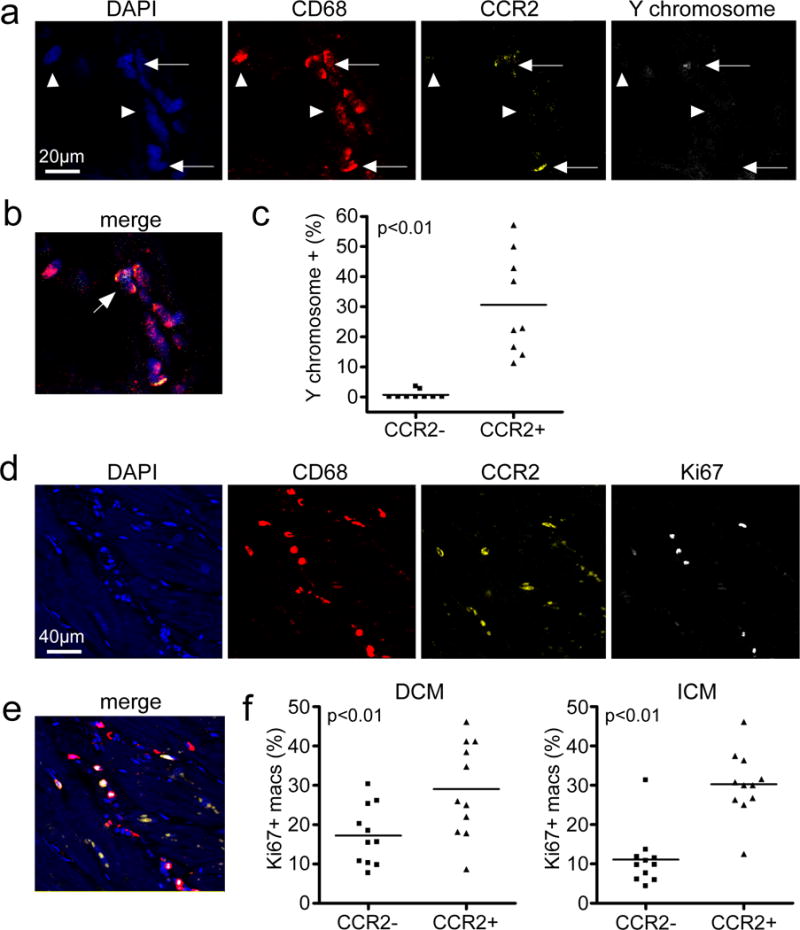
a, In situ hybridization and immunostaining of endomyocardial biopsy specimens obtained from recipients of sex mismatch heart transplants (n=9). All specimens were obtained from male patients who had received a heart from a female donor >1 year prior to biopsy. DAPI (blue), CD68 (red), CCR2 (yellow), and Y chromosome (white). Arrows: CCR2+ macrophages, arrowheads: CCR2- macrophages. b, Merged image from a. 400X magnification. Arrow denotes CCR2+ macrophage containing a Y chromosome. c, Percentages of CCR2- and CCR2+ macrophages that contain a Y chromosome (n=9). Each data point represents a biologically independent biopsy specimen and the line refers to the mean value. Mann Whitney test (two-sided), p<0.0001. d, Cell proliferation of CCR2- and CCR2+ macrophages, as assessed by immunostaining for CD68 (red), CCR2 (yellow), and Ki67 (white). Each data point represents a biologically independent heart failure specimen and the line refers to the mean value. Mann Whitney test (two-sided): DCM, p=0.0036 and ICM, p=0.006. e, Merged image from d. 200X magnification. f, Percentage of CCR2- and CCR2+ macrophages (macs) staining for Ki67 in hearts from DCM (n=11) and ICM (n=11) patients.
To examine whether cell proliferation also contributes to human cardiac CCR2- and CCR2+ macrophage maintenance, we performed immunostaining for CD68, CCR2, and Ki67 (Fig. 2d-e). Both CCR2- and CCR2+ macrophages populations displayed significant numbers of cells that were Ki67+, indicating that cell proliferation is an important mechanism of cell maintenance for each macrophage subset. However, CCR2+ macrophages displayed higher frequencies of Ki67+ cells compared to CCR2- macrophages (DCM: 29.0±11.4% vs. 17.2±7.2%, p<0.01 and ICM: 30.3±8.0% vs. 11.1±6.9%, p<0.01) (Fig. 2f). Together, these data suggest that CCR2- macrophages represent a tissue resident population that is maintained through cell proliferation, while CCR2+ macrophages are maintained through a combination of monocyte recruitment and cell proliferation. These data are consistent with previous work suggesting that monocyte recruitment and local proliferation are important mechanisms contributing to macrophage expansion in the chronically failing mouse heart22 and suggest that human cardiac CCR2+ macrophages may have higher turnover rates compared to human cardiac CCR2- macrophages.
Gene expression profiling of CCR2- macrophages, CCR2+ macrophages, and CCR2+ monocytes suggests differential cell origins and functions
To provide further evidence that human cardiac CCR2- and CCR2+ macrophages comprise functionally distinct macrophage populations, we performed transcriptomic profiling of RNA isolated from purified CCR2- macrophages (n=19 patients), CCR2+ macrophages (n=19 patients), and CCR2+ monocytes (n=10 patients) using microarray technology. Macrophages and monocyte populations were isolated from patients with DCM (n=8) and ICM (n=11) using flow cytometry based cell sorting. Prior to performing our transcriptomic profiling studies, we examined the morphology of flow cytometry sorted CCR2+HLA-DRlow monocytes, CCR2+HLA-DRhigh macrophages, and CCR2-HLA-DRhigh macrophages using cytospin preparations. Compared to CCR2+HLA-DRlow monocytes, CCR2+HLA-DRhigh and CCR2-HLA-DRhigh macrophage subsets displayed increased granularity consistent with known distinctions between monocyte and macrophage morphology. In addition, the morphology of CCR2+HLA-DRhigh and CCR2-HLA-DRhigh macrophages differed with CCR2+HLA-DRhigh macrophages being larger in size compared to CCR2-HLA-DRhigh macrophages (Fig. 3a).
Figure 3. Microarray gene expression profiling of CCR2+ monocytes, CCR2- macrophages, and CCR2+ macrophages in the failing human heart.
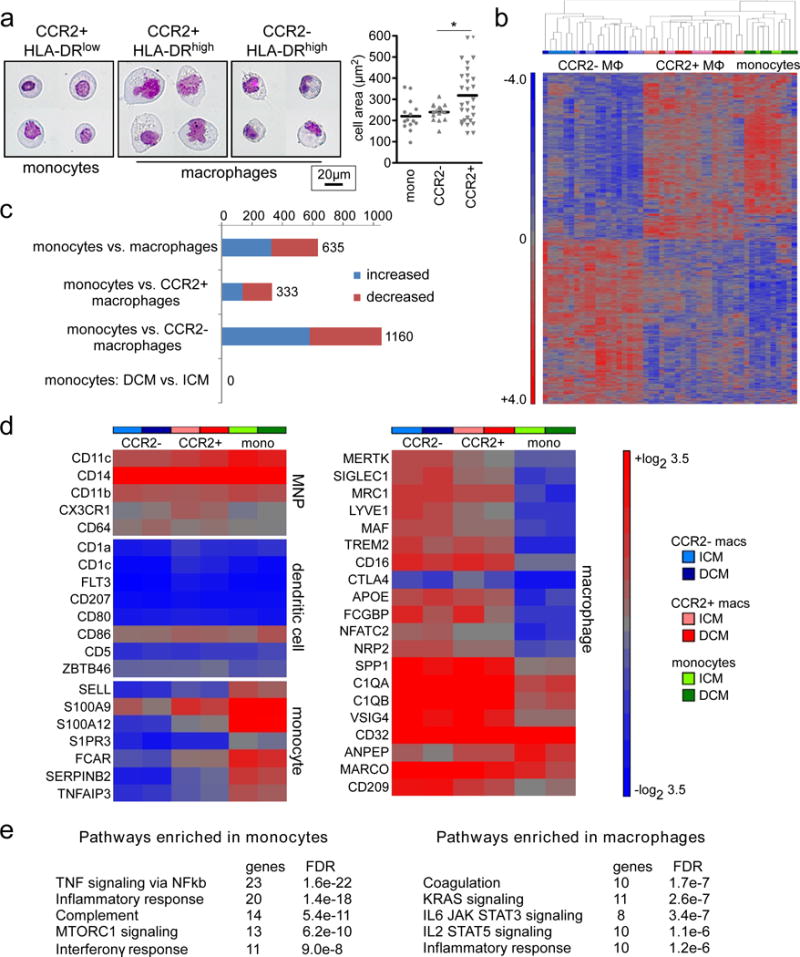
a, Left, representative images of CCR2+HLA-DRlow monocytes (n=14), CCR2+HLA-DRhigh macrophages (n=16), and CCR2-HLA-DRhigh macrophages (n=29) isolated from 4 biologically independent failing hearts (ICM and DCM) using FACS. Wright staining, 800X magnification. Right, quantification of cell area. Asterisks denotes p<0.05. Each data point represents an individual cell and the line represents the median value. Mann Whitney test (two-sided) p=0.025. b, Hierarchical clustering highlighting the relationships among CCR2+ monocytes (n=10), CCR2- macrophages (n=19) and CCR2+ macrophages (n=19) in the failing heart (DCM, n=8 and ICM, n=11). Sample color scheme is identical to the legend in d. MΦ: macrophages. c, Bar graph displaying the number of differentially regulated genes, using a threshold of 2X fold change and FDR<0.05. Comparisons include both DCM and ICM samples except when otherwise indicated. Blue: increased expression, Red: decreased expression. d, Heat maps showing the absolute expression values of genes that are associated with human mononuclear phagocytes (MNPs), dendritic cells, monocytes, and macrophages. Data are shown for CCR2+ monocytes, CCR2- macrophages and CCR2+ macrophages obtained from specimens of patients with ICM or DCM and the results are displayed as average expression values. e, GSEA pathway analysis revealing pathways enriched in cardiac monocytes versus macrophages. Analysis combines ICM and DCM specimens. Statistical significance was evaluated using false discovery rate (FDR).
Consistent with the concept that CCR2- macrophages, CCR2+ macrophages, and CCR2+ monocytes represent distinct cell types, hierarchical clustering demonstrated that each cell population clustered tightly together. Furthermore, CCR2+ macrophages preferentially clustered with CCR2+ monocytes suggesting that these populations are closely related (Fig. 3b). These data are consistent with our finding that monocytes exclusively contribute to maintenance of CCR2+ macrophages. Differential gene expression analysis revealed 635 genes that were differentially expressed between cardiac monocytes and macrophages (CCR2- and CCR2+) using a threshold of 2-fold change and FDR<0.05. Consistent with our hierarchical cluster analysis, CCR2- macrophages had a greater number of genes (n=1160) that were differentially expressed compared to monocytes than did CCR2+ macrophages (n=333). Of note, no differentially expressed genes were identified in monocytes isolated from patients with DCM versus ICM (Fig. 3c).
To place human cardiac monocytes and macrophages within the broader context of what is known regarding human myeloid populations, we examined the expression of previously described mononuclear phagocyte, dendritic cell, monocyte, and macrophage cell markers20, 24. Consistent with previous reports describing human mononuclear phagocytes, human cardiac CCR2+ monocytes, CCR2+ macrophages, and CCR2- macrophages uniformly expressed CD11c/ITGAX, CD14, CD11b/ITGAM, CX3CR1, and CD64/FCGR1. In contrast, human cardiac CCR2+ monocytes, CCR2+ macrophages, and CCR2- macrophages lacked the expression of numerous dendritic cell markers including CD1a, CD1c, FLT3, CD207/Langerin, CD80/B7, CD5, and ZBTB46. CCR2+ monocytes, CCR2+ macrophages, and CCR2- macrophages did express CD86, which is found on both macrophages and dendritic cells25. The previously reported monocyte cell markers SELL/L-selectin, S100A9, and S100A8 were differentially expressed on CCR2+ monocytes compared to CCR2- and CCR2+ macrophage subsets. Other identified genes differentially expressed on CCR2+ monocytes included S1PR3. FCAR, SERPINB2, and TNFAIP3. Consistent with macrophage cell identity, human cardiac CCR2- and CCR2+ macrophages displayed robust expression of MERTK, SIGLEC1, MRC1, LYVE1, MAF, TREM2, CD16, CD32, SPP1/Osteopontin, and MARCO (Fig. 3d). GSEA pathway analysis demonstrated that genes upregulated in monocytes displayed enrichment for pathways involved in TNF/NFκb signaling, inflammatory response, complement, MTORC1, and interferonγ signaling. In contrast, genes upregulated in macrophages displayed enrichment for pathways involved in coagulation, K-RAS, IL6/STAT3, IL2/STAT5, and inflammatory signaling (Fig. 3e).
To evaluate whether human cardiac CCR2- and CCR2+ macrophages represent functionally distinct subsets, we further examined our microarray data. Both hierarchical clustering (Fig. 3b) and principal component analysis (Fig. 4a) demonstrated that CCR2- and CCR2+ macrophages display distinct gene expression profiles. Differential gene expression analysis revealed 1194 genes that were differentially expressed between CCR2- and CCR2+ macrophages using a threshold of 1.5 fold change and FDR<0.05. Stratification by cardiomyopathy etiology (DCM vs ICM) revealed only 6 genes differentially regulated in CCR2+ macrophages and 4 genes differentially regulated in CCR2- macrophages, all of which were upregulated in ICM specimens (Fig. 4b). Genes upregulated in CCR2- macrophages included OR2A9P (pseudogene), SUV39H2 (Histone-lysine N-methyltransferase), G6PC3 (glucose-6-phosphatase catalytic subunit 3), and ST7-OT4 (non-coding RNA). Genes upregulated in CCR2+ macrophages included TEX37 (Testis Expressed 37), GNPDA (Glucosamine-6-Phosphate Deaminase 1), PLEKHA7 (Pleckstrin homology domain-containing family A member 7), L3MBTL4-AS1 (antisense RNA), THC2493232 (not characterized), and LNC-TWSG1-1 (non-coding RNA).
Figure 4. CCR2- and CCR2+ macrophages display distinct gene expression profiles.
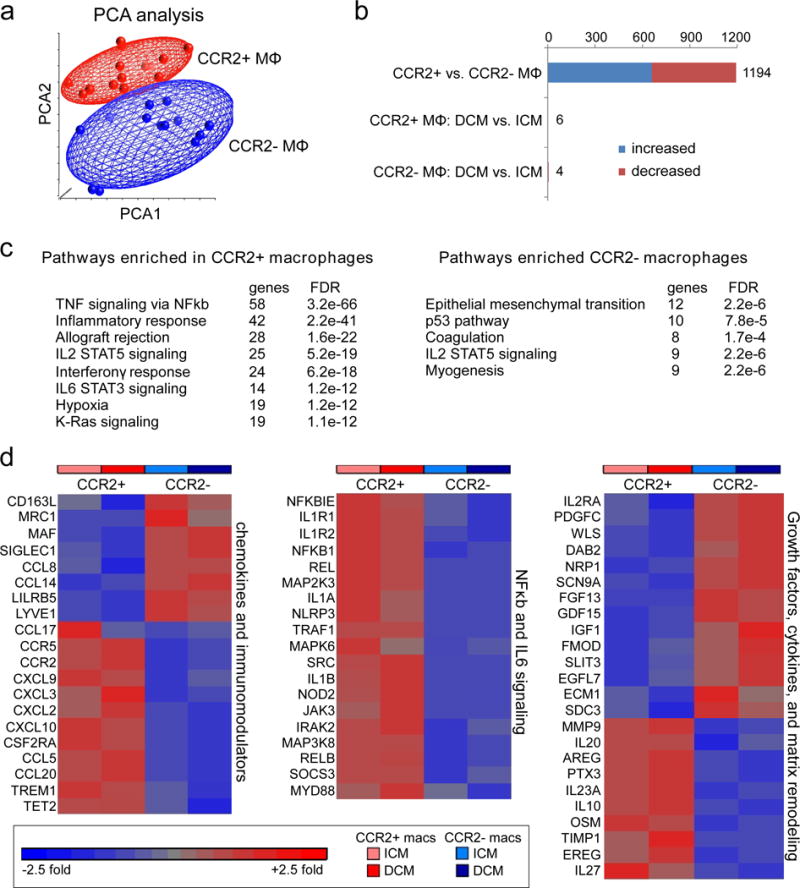
a, Principal component analysis of CCR2- (n=19) and CCR2+ (n=19) cardiac macrophages (ICM and DCM). Red: CCR2+ macrophages, Blue: CCR2- macrophages. b, Bar graph displaying the number of genes that were differentially expressed between all CCR2- and CCR2+ macrophages (DCM and ICM) and the number of genes that were differentially expressed in CCR2- and CCR2+ macrophages stratified by DCM and ICM designation using a threshold of 1.4X fold change and FDR<0.05. Blue: increased expression, Red: decreased expression. c, GSEA pathway analysis revealing pathways enriched in cardiac CCR2+ versus CCR2- macrophages (ICM and DCM). Statistical significance was evaluated using false discovery rate (FDR). d, Heat maps showing relative fold changes in genes associated with chemokine and immunomodulatory signaling, NFκb and IL6 signaling, as well as selected growth factors, cytokines, and extracellular matrix remodeling factors. Data are shown for CCR2+ and CCR2- macrophages obtained from specimens of patients with ICM or DCM and the results are displayed as average expression values. All genes displayed on the heat maps were differentially expressed (FDR<0.05) between CCR2+ and CCR2- macrophages.
GSEA pathway analysis highlighted that genes upregulated in CCR2+ macrophages were associated with inflammatory pathways including TNF/NFκb signaling, inflammatory response, allograft rejection, IL2/STAT5, IL6/STAT3, interferonγ, hypoxia and K-RAS signaling. In contrast, genes upregulated in CCR2- macrophages were associated with epithelial mesenchymal transition, coagulation, myogenesis, p53, and IL2/STAT5 signaling (Fig. 4c). To more precisely gauge the inflammatory potential of CCR2- and CCR2+ macrophage subsets, we examined known chemokines, immunomodulators, cytokines and associated signaling pathways. Compared to CCR2- macrophages, which differentially expressed negative immunomodulators and tissue macrophage markers such as LILRB5, CD163, MRC1, MAF, SIGLEC1, and LYVE1, CCR2+ macrophages expressed large numbers of chemokines, chemokine receptors, and mediators of IL1, NFκb, and IL6 signaling. In contrast, CCR2- macrophages expressed numerous growth factors, extracellular matrix components, and conduction genes such as IGF1, PDGFC, EGFL7, GDF15, NRP1, SLIT3, ECM1, SDC3, SCN9A, and FGF13. CCR2+ macrophages expressed growth factors known to promote fibrosis and hypertrophy including AREG, EREG, OSM, and PTX326–28 as well as genes associated with extracellular matrix degradation such as MMP9 and TIMP1 (Fig. 4d). Only some classic markers of M1 and M2 macrophage phenotypes were differentially expressed between CCR2- and CCR2+ macrophages, highlighting the limitations of this approach (Supplemental Fig. 4). Collectively, these data support the conclusion that CCR2+ monocytes, CCR2+ macrophages, and CCR2- macrophages represent distinct cell types and suggest that CCR2+ monocytes and CCR2+ macrophages likely represent inflammatory populations, while CCR2- macrophages are enriched with genes with the potential to orchestrate tissue repair.
CCR2+ macrophages represent an inflammatory population
To test the hypothesis that CCR2+ macrophages represent an inflammatory population, we purified CCR2- and CCR2+ macrophages from human LV specimens using flow cytometry based cell sorting and cultured cells in vitro. Macrophages were then treated with either vehicle control or the TLR4 agonist LPS. Using quantitative RT-PCR, we then measured mRNA expression of the pro-inflammatory mediators, IL1β and CCL7. Following stimulation with either vehicle or LPS, CCR2+ macrophages expressed substantially higher levels of IL1β and CCL7 mRNA compared to CCR2- macrophages. While CCR2- macrophages did display increased IL1β and CCL7 mRNA expression following LPS treatment (compared to vehicle) the overall magnitude of IL1β and CCL7 mRNA expression was substantially lower than that of CCR2+ macrophages (Fig. 5a-b). Measurement of IL1β protein concentration in the cell culture supernatant further demonstrated that CCR2+ macrophages produce more IL1β than CCR2- macrophages (Fig. 5c).
Figure 5. CCR2+ cardiac macrophages represent an inflammatory population.
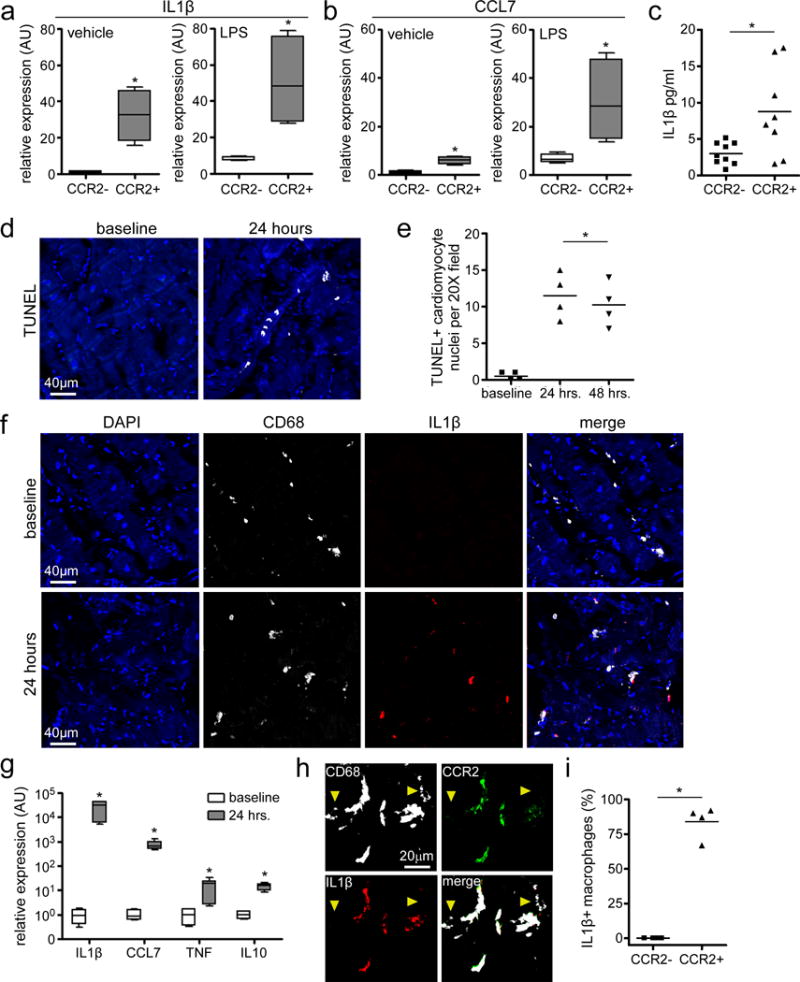
a-b, IL1β (a) and CCL7/MCP3 (b) expression in CCR2- and CCR2+ macrophages treated with vehicle or LPS, as assessed by quantitative RT-PCR. Asterisks denote p<0.05 (Mann Whitney test, 2-sided) compared to CCR2- macrophages. n=3 independent experiments from 4 biologically independent heart failure specimens (DCM and ICM). Data displayed as box and whisker plots. The box denotes the 25th and 75th percentiles, the line indicates the median value, and the whiskers reflect the minimum and maximum values. c, IL1β secretion by cultured CCR2- and CCR2+ macrophages, as assessed by ELISA. Each data point represents a biologically independent replicate derived from 4 individual heart failure specimens (DCM and ICM). Line indicates the mean values. Asterisks denote p<0.05 (Mann Whitney test, 2-sided). d-e, Cardiomyocyte cell death in the human myocardial slice culture system. d, Representative images of TUNEL staining showing evidence of cardiomyocyte cell death after 24 hours of slice culture. e, Quantification of TUNEL staining at 24 and 48 hours of slice culture. Baseline refers to examination of myocardial tissue immediately after slice preparation. Asterisks denote p<0.05 (ANOVA) compared to baseline. Each data point (n=4) is derived from a biologically independent heart failure specimen (DCM and ICM) and lines denote mean values. f, Immunostaining for CD68 (white) and IL1β (red) showing induction of IL1β expression in macrophages after 24 hours of slice culture. g, IL1β, CCL7/MCP3, TNF, and IL10 mRNA expression after 24 hours of slice culture. Data displayed as box and whisker plots. The box denotes the 25th and 75th percentiles, the line indicates the median value, and the whiskers reflect the minimum and maximum values. Asterisks denote p<0.05 (Mann Whitney test) compared to baseline. n=3 independent experiments. h, Immunostaining for CD68 (white), CCR2 (green), and IL1b (red) indicates that IL1β is preferentially expressed in CCR2+ macrophages. Yellow arrowheads, CCR2- macrophages. i, Percentages of CCR2- and CCR2+ macrophages with detectable IL1β antibody staining. Each symbol refers to data derived from a biologically independent heart failure specimen and lines indicate mean values. Asterisks denotes p<0.05 (Mann Whitney test) d, f: 200X magnification. h: 400X magnification. Blue: DAPI.
To provide further evidence that human cardiac CCR2+ macrophages are pro-inflammatory, we developed a human organotypic slice culture system based on previously described reports29, 30. Briefly, human heart explants were obtained from patients undergoing cardiac transplantation and the LV lateral wall trimmed into transmural rectangular specimens. Using a Krumdieck Tissue Slicer, 300 μM tissue slices were generated and cultured on semi-porous tissue culture inserts. TUNEL staining performed 2 hours (baseline), 24 hours, and 48 hours after slice culture revealed that 0.5±0.6, 11.5±3.1, and 10.25±3.0 cardiomyocytes per 20X field underwent cell death after 2 hours, 24 hours, and 48 hours of slice culture, respectively (Fig. 5d-e). These data indicate that while the majority of cardiomyocytes remain viable after 48 hours slice culture, foci of cardiomyocyte cell death reproducibly emerge within 24 hours of slice culture. As such, we took advantage of this system to model how cardiac macrophage populations might respond to cardiomyocyte cell death ex vivo. While it is possible that macrophages may respond to other stimuli, this system allowed us to interrogate macrophage behavior in their native environment.
Consistent with previous studies in mouse models demonstrating that cardiomyocyte cell death results in cardiac macrophage activation and expression of pro-inflammatory mediators, immunostaining of human cardiac tissue slices cultured for 24 hours revealed marked induction of IL1β expression in CD68+ macrophages compared to baseline (Fig. 5f, Supplemental Fig. 5). Quantitative RT-PCR further demonstrated robust increases in IL1β, CCL7, TNF, and IL10 mRNA expression in human cardiac tissue slices cultured for 24 hours (Fig. 5g). Consistent with the conclusion that CCR2+ macrophages represent an inflammatory subset, IL1β expression specifically co-localized with CCR2+CD68+ cells (Fig. 5h-i).
CCR2+ macrophage abundance is associated with persistent LV systolic dysfunction following mechanical unloading
Given that human cardiac CCR2- and CCR2+ macrophages represent distinct macrophage subsets and likely have divergent functions, we hypothesized that these populations may differentially impact on cardiac function and LV remodeling. To test this hypothesis, we examined whether human cardiac macrophage subset composition was associated with LV systolic function in a well described cohort of patients who underwent LVAD implantation31, 32. Based on echocardiographic analysis, 34% of patients within this cohort displayed sustained improvements in LV ejection fraction (>50% relative increase) and reduced LV volumes 6 months following LVAD implantation. Using immunostaining, we measured macrophage composition in LV specimens obtained at the time of LVAD implantation (n=36) and at the time of transplantation (n=26). Patients were stratified into 2 groups based on changes in LV systolic function at 6 months as originally described: 1) persistent LV dysfunction (<50% relative improvement in LV EF, n=18) and 2) improved LV systolic function (>50% relative increase in LV EF or absolute EF>40%, n=18) (Fig. 6a, Supplemental Fig. 6). Analysis of clinical and demographic data revealed balanced covariates between these groups and further showed that patients who experienced improved LV systolic function displayed concomitant reductions in LV chamber dimensions (Supplemental Table 2).
Figure 6. Macrophage subpopulations are associated with outcome following mechanical unloading.
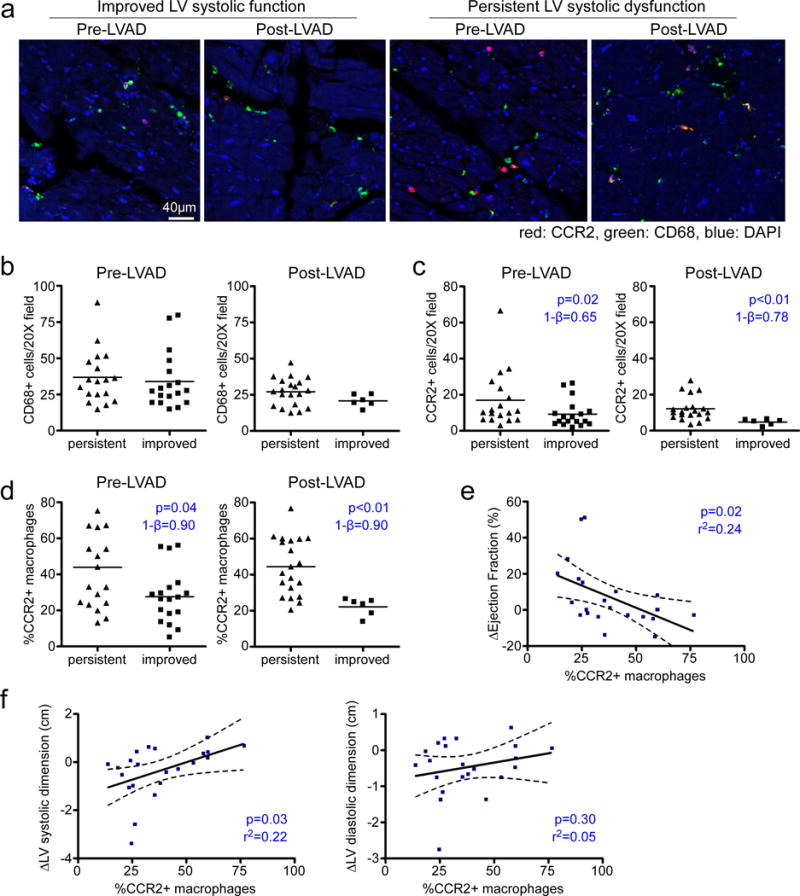
a, Immunostaining for CD68 (green) and CCR2 (red) in myocardial tissue specimens obtained from heart failure patients at the time of left ventricular assist device placement (pre-LVAD) and at the time of transplant (post-LVAD). Patients were stratified into those who displayed persistent LV systolic dysfunction (n=17) and those who displayed improved LV systolic function (n=18). Blue: DAPI, 200X magnification. b-c, Numbers of total (CD68+) (b) and CCR2+ (c) macrophages in pre-LVAD and post-LVAD specimens. Mann Whitney test (two-sided): CD68+ pre-LVAD, p=0.40 and CD68+ post-LVAD, p=0.16. d, Percentage of CCR2+ macrophages in pre-LVAD and post-LVAD. All data points represent biologically independent specimens and the lines indicate mean values. Asterisks indicate statistically significant p-values using Matt Whitney test (two-sided). e-f, Linear regression analysis for the association of the percentage of CCR2 macrophages and absolute changes in EF (e) and LV systolic dimension (f) over time. Dashed lines indicate 95% confidence intervals. Asterisks denotes p<0.05. 1-β denotes statistical power. Each data point (n=22) represents a biologically independent sample.
Quantification of macrophage composition demonstrated that CD68+ macrophage abundance was not associated with improvements in LV systolic function either at the time of LVAD implantation or transplantation (Fig. 6b). In contrast, both the abundance and percentage of CCR2+ macrophages correlated with LV systolic function following LVAD implantation. Specifically, patients who displayed improvement in LV systolic function 6 months after LVAD implantation had lower absolute numbers and percentage of CCR2+ macrophages both at the time of LVAD implantation and at the time of explant (Fig. 6c-d). The percent of CCR2+ macrophages at the time of explant was associated with absolute changes in ejection fraction and LV systolic dimension (Fig. 6e-f). Collectively, these findings suggest that cardiac macrophage composition is associated with LV systolic function and cardiac remodeling following mechanical unloading and support the concept that the human heart contains functionally distinct subsets of macrophages that may have clinically important effects on heart failure outcomes.
Discussion
The goal of this study was to determine whether emerging concepts of tissue macrophage heterogeneity are translatable to humans. By examining LV myocardial specimens obtained from patients with heart failure, we tested the hypothesis that the human heart contains a heterogeneous population of macrophages with divergent origins and functions. We demonstrated that the human myocardium is populated by distinct subsets of CCR2- macrophages, CCR2+ macrophages, and CCR2+ monocytes. CCR2- macrophages represent a tissue resident population that is maintained outside of monocyte input through local proliferation, while CCR2+ macrophages are likely derived from monocytes and expand locally through cell proliferation. Gene expression profiling, cell culture, and organotypic slice culture substantiated that CCR2- macrophages and CCR2+ macrophages represent distinct cell types with divergent reparative and inflammatory functions, respectively. Consistent with a pathological role for CCR2+ macrophages, the abundance of CCR2+ macrophages was associated with persistent LV systolic dysfunction and adverse LV remodeling following mechanical unloading in heart failure patients. Collectively, these data demonstrate that the human heart contains distinct macrophage subsets that are functionally analogous to mouse CCR2- and CCR2+ macrophages and provides initial evidence that human macrophage heterogeneity is functionally important.
Paradigm shifting studies have revealed that mice contain a complex and heterogeneous array of tissue macrophages with distinct origins, life cycles, and functions. We have previously demonstrated that mouse cardiac macrophage populations can be divided into CCR2-MHCIIlow, CCR2-MHCIIhigh, and CCR2+MHCIIhigh subsets. Single cell RNA sequencing of mouse cardiac macrophages confirmed the presence of these subsets and suggested that CCR2 and MHCII expression is sufficient to resolve macrophage subset heterogeneity under homeostatic conditions21. CCR2- (MHCIIlow and MHCIIhigh) macrophages are derived from embryonic progenitors (yolk sac and fetal monocytes), seed the heart during development, are maintained independent of monocyte input through local proliferation, possess minimal inflammatory potential, and display robust pro-angiogenic activity. In contrast, CCR2+ macrophages are derived from adult hematopoietic progenitors, maintained through CCR2+MHCIIlowLy6Chigh monocyte recruitment and subsequent proliferation, and dramatically increase in number following cardiac tissue injury or in models of heart failure8, 17, 22, 33, 34. Recruited CCR2+MHCIIlowLy6Chigh monocytes and CCR2+MHCIIhigh macrophages express a broad array of inflammatory mediators and contribute to heart failure progression through exaggerated neutrophil and monocyte recruitment, oxidative injury, and collateral tissue damage. Intriguingly, in the context of aging, monocyte-derived macrophages progressively replace embryonic-derived macrophages in some tissues including the heart35.
Similar to mouse models, myocardial specimens obtained from patients with DCM and ICM contained CCR2- macrophages, CCR2+ macrophages, and CCR2+ monocytes. These data are in line with previous reports that human monocytes and monocyte-derived macrophages express CCR236, 37. Consistent with mouse subsets, CCR2+ macrophages express high levels of the MHC class II homologue, HLA-DR, while CCR2+ monocytes express low levels of HLA-DR. One distinction between mouse and human macrophages is that mouse CCR2- macrophages are divided into MHCIIlow and MHCIIhigh subsets, while human CCR2- macrophages are predominately HLA-DRhigh. To date, beyond antigen presentation, no functional differences have been described between mouse CCR2-MHCIIlow and CCR2-MHCIIhigh macrophages8.
Human CCR2+ monocytes, CCR2+ macrophages, and CCR2- macrophages uniformly expressed common markers of monocytes and macrophages (CD14, CD64/FCGR1A, CD32/FCGR2A) and lacked the expression of known dendritic cell markers (ZBTB46, CD1a, CD1c, CD80, CD5, Flt3)20, 23, 24. CCR2+ monocytes within the heart express high levels of CD14 and low levels of CD16 suggesting that they may be most related to human blood CD14+CD16- monocytes, the functional equivalent of mouse blood Ly6ChighCCR2+ monocytes24. The presence of CCR2+ monocytes within myocardial tissue is consistent with prior reports describing the ability of monocytes to retain their identity and survey antigens within tissue10. Consistent with the paradigm that macrophages can be distinguished from monocytes and dendritic cells based on the expression of MerK20, 23, human cardiac CCR2- macrophages and CCR2+ macrophages expressed MertK on the mRNA and protein level, while CCR2+ monocytes lacked MertK expression. Gene expression profiling identified additional markers that distinguished tissue macrophages from monocytes. CCR2- and CCR2+ macrophages differentially expressed SIGLEC1, MRC1, LYVE1, MAF, TREM2, CD16, APOE, FCGBP, NFATC2, and NRP2. CCR2+ monocytes differentially expressed SELL/CD62L, S100A12, FCAR, SERPINB2, and TNFAIP3. Whether these markers differentiate monocytes and tissue macrophages in other organs remains to be clarified.
To decipher the contribution of monocyte recruitment to the maintenance of human CCR2- and CCR2+ macrophages, we examined patients who underwent sex mismatched heart transplantation (male patients who received a female heart). Subjects had normal allograft function, were free from rejection, and underwent transplantation >1 year prior to routine surveillance endomyocardial biopsy. While transplant studies are influenced by exposure to immunosuppressive medications, this analysis revealed that similar to mouse CCR2- macrophages, human CCR2- macrophages exist independent of monocyte input. In contrast, monocyte recruitment contributed to maintenance of at least a subset of human CCR2+ macrophages. Cell proliferation appeared to be an important mechanism for both CCR2- and CCR2+ macrophages. These data are consistent with described repopulation dynamics of mouse CCR2- and CCR2+ macrophage subsets. Of note, these findings do not provide meaningful information regarding the rate of CCR2+ macrophage turnover and thus do not exclude the possibilities that CCR2+ macrophages may represent a long lived monocyte-derived population or that CCR2+ macrophages may represent a mixed population of newly recruited and long-lived monocyte-derived macrophages.
Gene expression profiling revealed several features that were shared between mouse and human cardiac macrophages. Both mouse and human CCR2- macrophage subsets expressed higher levels of tissue resident macrophage markers (MRC1, CD163, SIGLEC1, LYVE1) and growth factors (IGF1, PDGF-C) compared to human and mouse CCR2+ macrophages. Human CCR2- macrophages differentially expressed several other growth factors and extracellular matrix genes implicated in tissue morphogenesis and remodeling. Consistent with a recently described role in electrical conduction21, human CCR2- macrophages expressed the sodium channel SCN9A and sodium channel modulator FGF13. Conversely, human and mouse CCR2+ macrophages selectively expressed inflammatory mediators including monocyte and neutrophil chemokines, the inflammatory cytokine IL1β, and associated components of the inflammasome8, 17, 19, 21. Human CCR2+ macrophages also differentially expressed several genes implicated in adverse cardiac remodeling including MMP9, TIMP1, PTX3, EREG, and OSM26–28, 38.
Consistent with a role for CCR2+ macrophages in inflammation, adverse remodeling and heart failure pathogenesis, isolated CCR2+ macrophages produced robust quantities of IL1β following either LPS stimulation or exposure to necrotic cardiomyocytes. In contrast, mouse and human CCR2- macrophage displayed markedly less inflammatory activity17. Importantly, human CCR2+ macrophage abundance was associated with worsened LV systolic dysfunction and adverse remodeling in heart failure patients. Together, these observations suggest that interventions that target CCR2+ macrophages, may represent a favorable approach to suppress inflammation and adverse remodeling in the context of heart failure. In addition, the finding that mouse and human CCR2+ macrophages are functionally analogous implies that dissecting mechanisms by which mouse CCR2+ macrophages are activated and exert their inflammatory effects is translationally relevant and will likely lead to critical insights into the development of effective strategies to intervene on the inflammatory functions of human CCR2+ macrophages.
Prior studies have provided clues to suggest that macrophage heterogeneity may be applicable to other human tissues. Examination of transplant recipients has suggested that admixtures of resident and recruited macrophage populations exist in human skin and lung39–42. Further evidence supporting the existence of tissue resident populations in the skin is provided by examination of patients with deficiencies in bone marrow myelopoiesis. Subjects carrying either GATA2 or biallelic IRF8 mutations demonstrate preservation of epidermal Langerhans cells and dermal macrophages despite marked impairments in peripheral monocyte and dendritic cell differentiation43–45. Outside of these early studies, little is known regarding human tissue mononuclear phagocyte diversity and function. Intriguingly, a recent study exploring human lung mononuclear phagocytes obtained from explanted lung specimens identified immense diversity among lung monocyte, macrophage, and dendritic cell subsets46. Future studies will undoubtedly delineate whether other human tissues harbor heterogeneous mononuclear phagocyte populations with unique or differing recruitment dynamics and functions.
We acknowledge that there are important limitations to our study. In contrast to mouse models, it is not possible to perform lineage tracing or detailed cell tracking studies in humans. As a result, we are not able to make any meaningful conclusions regarding macrophage ontogeny as it relates to embryonic or adult hematopoietic origins. However, by examining sex mismatch transplant recipients, we are able to gain valuable insights into resident versus recruited populations. Even though in situ hybridization may underestimate the number of recipient derived macrophages, we believe that the robust differences observed between populations indicate that CCR2- macrophages are a tissue resident population and CCR2+ macrophages are replenished through monocyte recruitment. We also recognize that inferences regarding macrophage function are limited to gene expression, in vitro assays, and clinical associations in a relatively small sized cohort of patients.
In conclusion, we have demonstrated that the human heart contains distinct macrophage subsets with differing repopulation dynamics and gene expression profiles that are functionally analogous to tissue resident CCR2- and inflammatory monocyte-derived CCR2+ macrophages found in the mouse heart. Our findings provide evidence that macrophage heterogeneity is functionally important in the human heart and suggest that therapeutics targeting inflammatory functions of CCR2+ macrophages may represent a novel therapeutic target for patients with heart failure.
Materials and Methods
Study Approval
This study was approved by the Washington University in St. Louis Institutional Review Board (#201305086). All subjects provided informed consent prior to sample collection and the experiments were performed in accordance with the approved study protocol.
Pathologic Specimens used for immunostaining and flow cytomertry
Cardiac tissue specimens were obtained from adult patients with DCM (idiopathic and familial) and ICM undergoing LVAD implantation or cardiac transplantation. Patients with secondary causes of DCM including cardiac amyloidosis, cardiac sarcoidosis, viral myocarditis, giant cell myocarditis, peripartum cardiomyopathy, chemotherapy associated cardiomyopathy, and complex congenital heart disease were excluded from this study. In addition, patients with established autoimmune disease, active infections, HIV, and hepatitis C were excluded. Tissues consisted of transmural specimens obtained from the apical or lateral wall of the LV. Explanted hearts were flushed by cannulating the left and right coronary artery ostia and perfusing 200ml of cold saline. LVAD apical cores were flushed by cannulating an epicardial vessel and perfusing 50ml of cold saline. Specimens were then immersed in cold saline and were either immediately flash frozen or fixed in 10% formalin upon collection to preserve tissue integrity.
Flow cytometry
To generate single cell suspensions, saline perfused cardiac tissue specimens were finely minced, and digested in DMEM with Collagenase type 1 (450 U/ml) Hyaluronidase (60 U/ml) and DNase I (60 U/ml) for 1 hour at 37°C. All enzymes were sourced from Sigma. Digested samples were then filtered through 40 μM cell strainers and washed with cold HBSS that was supplemented with 2% FBS and 0.2% BSA. Red blood cell lysis was performed with ACK lysis buffer (Thermo Fisher Scientific). Cells were washed with HBSS and resuspended in 100 μL of FACS buffer (DPBS containing 2% FBS and 2 mM EDTA). For monocyte and macrophage sorting, cells were then stained with CD45-PercpCy5.5 (2D1), CD14-PE (M5E2), CD64-FITC (10.1), CCR2-APC (K036C2), and HLA-DR APC/Cy7 (L243) at 4°C for 30 minutes in the dark. Stained single cell suspensions were washed twice with FACS buffer and resuspended in a 0.35 ml volume. DAPI was used to exclude dead cells. For Intracellular flow cytometry, myocardial tissue was processed as outlined above to generate a single cell suspension. Following labeled with appropriate cell surface antibodies, cells were fixed (PFA, Biolegend 420801) and permeablized (Permeabilization Wash Buffer) Biolegend 421002) and stained for CD68. FACS analysis and sorting was performed on BD LSR II and BD FACSAria™III platforms. A complete list of antibodies is shown in Supplemental Table 3.
Immunohistochemistry
Paraffin embedded sections were dewaxed in xylene, rehydrated, endogenous peroxide activity quenched in 10% methanol and 3% hydrogen peroxide, processed fro antigen retrieval by boiling in citrate buffer pH 6.0 containing 0.1% Tween-20, blocked in 1% BSA, and stained with the following primary antibodies overnight at 4 degrees C: CD68 (KP1 eBiosceince 1:2000), CCR2 (7A7 Abcam 1:2000), CD34 (Q/bend1 Abcam 1:2000), Collagen 1 (COL-1 Abcam 1:2000), IL-1β (NB600-633 NOVUS 1:2000), Ki67 (ab15580 Abcam 1:1000), CD14 (ab183322 Abcam 1:2000), CD64 (ab119843 Abcam 1:4000), iNOS (ab76198 Abcam 1:1000), HLA-DR (clone L243 Biolegend 1:1000). The primary antibody was detected using a biotin conjugated anti-mouse or anti-rabbit secondary antibodies (Vector Labs) in conjunction with streptavidin HRP (ABC Elite, Vector Labs). The PerkinElmer Opal Multicolor IHC system was utilized to visualize antibody staining per manufacturer protocol. TUNEL staining (Roche) was performed per manufacturer’s protocol. Immunofluorescence was visualized on a Zeiss confocal microscopy system. Macrophages were quantified by examining at least 4 similarly oriented sections from 4 independent samples in blinded fashion.
Microarray
To isolated RNA, macrophages were directly sorted into QLT buffer containing 2-mercaptoethanol and RNA isolated using the RNeay micro kit (Qiagen) per manufacturer’s instructions. Gene expression profiling was performed using microarray analysis in collaboration with the Genome Technology Access Core at Washington University. RNA was amplified using the WTA (Sigma) system and hybridized to Agilent 8×60 gene chips. Data analysis was performed using Partek genome suite software.
Transplant specimens and in situ hybridization
To identify sex mismatched patients who underwent cardiac transplantation, we performed a retrospective analysis of all patients who either received a cardiac transplant at Barnes Jewish Hospital between 1994 and 2008 or received a heart transplant elsewhere and were followed in our post-transplant program between 1994 and 2008. Male patients who received a heart from a female donor were included. Exclusion criteria included any episode of >2R/3A rejection, prior or ongoing antibody mediated rejection, established cardiac allograft vasculopathy, or LV ejection fraction <60%. Standard demographic information was obtained from the medical record including age, sex, reason for transplantation, cardiovascular comorbidities, transplant and heart failure medicines, rejection episodes, CMV status, blood type, echocardiographic data, stress testing, LV end diastolic pressure, heart rate and blood pressure as derived from diagnostic catheterization data.
Myocardial biopsy specimens that were previously collected for routine clinical care were obtained from the department of pathology. Collected tissues were previously fixed in 10% formalin and stored in paraffin blocks. For each patient, the most recent biopsy specimen was obtained. Myocardial biopsy specimens were cut into 4 micron sections using standard techniques and mounted on glass slides. Each paraffin block typically contained 3-4 myocardial specimens. Immunohistochemistry was performed for CCR2 and CD68 as outlined above using the PerkinElmer Opal Multicolor IHC kit. In situ hybridization for Y-chromosomes (STARFISH) was performed after IHC using a biotinylated probe per manufacturer’s instructions. The biotin conjugated probe was detected using streptavidin-FITC and visualized on a Zeiss confocal microscopy system. Macrophages were quantified by examining at least 4 sections from each independent sample in blinded fashion.
Organotypic Slice Culture
To prepare cardiac slices, Krumdieck Tissue Slicer (Alabama Research and Development) was used. Tissue was first placed in tissue embedding unit containing 4% low melting agarose dissolved in HBSS and allowed to set at 4°C. The embedding unit was then placed on to the sample holder of the microtome assembly. The reservoir was filled with ice cold HBSS (without calcium and magnesium). The arm and blade speed were set to medium speed, and thickness of the slices were set to 300 μM. Following slice generation and collection, slices were then cultured at a liquid–air interface using semi-porous tissue culture inserts (Millipore). Inserts containing cardiac tissue slices were placed in a six well tissue culture plate with 1 mL of IMDM supplemented with 20% FBS and 1% penicillin/streptomysin. Slices were cultured for 24-48 hours at 37°C in humidified air with 5% CO2.
Macrophage cell culture
CCR2+ and CCR2- macrophages were purified from myocardial tissue using flow cytometry as described above. Cells were sorted on a BD FACSAria™III platform with 85 μM nozzle and flow rate set to 1 μL/min. The pre- and post- sort collection tube holders were maintained at 4°C to preserve cell viability. Cells were sorted directly into culture medium (DMEM Supplemented with 10% FBS and 1% penicillin/streptomysin) and immediately plated into 96 well tissue culture plates and allowed to adhere overnight. The following day, fresh media was added and cells were stimulated with vehicle control or LPS (10 ng/ml) for 6 hours. IL1β concentration in the tissue culture supernatant was measured using the human IL1β Quantikine HS ELISA kite (R&D systems).
cDNA amplification and RT-PCR
RNA was extracted from the cardiac slices or cultured cells using the RNeasy RNA micro kit (Qiagen). RNA concentration was measured using a nanodrop spectrophotometer (ThermoFisher Scientific). For cardiac slices, cDNA synthesis was performed using the High Capacity RNA to cDNA synthesis kit (Applied Biosystems). For cultured macrophages, cDNA was synthesized using the iScript™ Reverse Transcription Supermix (Bio-Rad) and pre-amplified using the Sso Advanced PreAmp Supermix kit (Bio-Rad). Quantitative real time PCR reactions were prepared with sequence-specific primers (IDT) with PowerUP™ Syber Green Master mix (ThermoFisher Scientific) in a 20 μL volume. Real time PCR was performed using QuantStudio 3 (ThermoFisher Scientific). mRNA expression was normalized to β2 Microglobulin (B2M). IL-1β: Forward ATG CAC CTG TAC GAT CAC TG, Reverse ACA AAG GAC ATG GAG AAC ACC; CCL7: Forward AGA CCA AAC CAG AAA CCT CC, Reverse AGT ATT AAT CCC AAC TGG CTG AG; IL-10: Forward CGC ATG TGA ACT CCC TGG, Reverse TAG ATG CCT TTC TCT TGG AGC; TNF: Forward ACT TTG GAG TGA TCG GCC, Reverse GCT TGA GGG TTT GCT ACA AC; β2M: Forward TGC TGT CTC CAT GTT TGA TGT ATC T, Reverse TCT CTG CTC CCC ACC TCT AAG.
Statistical Analysis
Fisher’s exact and Mann Whitney tests were used to identify statistically significant differences between groups. Data are presented as dot plots, box whisker plots, or linear regression plots generated in PRISM. The exact sample size used to calculate statistical significance is stated in the appropriate figure legend. Replicates were defined as individual human specimens or experiments and described in the figure legends.
Data Availability
Source Data for all experiments have been provided. All other data are available from the corresponding author on reasonable request. Microarray data was deposited in GEO (GSE112630). Additional details can be found in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We acknowledge the Translational Cardiovascular Biorepository at Washington University for providing cardiac tissue specimens. This project was made possible by funding provided from the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CH-II-2015-462, CH-II-2017-628), Foundation of Barnes-Jewish Hospital (8038-88), and the NHLBI (R01 HL138466, R01 HL139714). K.J.L. is supported by NIH K08 HL123519 and Burroughs Welcome Fund (1014782). Histology was performed in the DDRCC advanced imaging and tissue analysis core supported by Grant #P30 DK52574. The Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. S.G.D. is supported by NHLBI R01 HL135121-01, AHA HF 16SFRN29020000-Project 1 and the Nora Eccles Treadwell Foundation. M.N. is supported by NIH HL139598 and the MGH Research Scholar Program. Y.L. is supported by NIH R01HL131908 and R01HL125655. D.K. is supported by NIH P01AI116501 and R01 HL094601, Veterans Administration Merit Review grant 1I01BX002730 and The Foundation for Barnes-Jewish Hospital. M.H. was supported by an MGH ECOR Tosteson and Fund for Medical Discovery Fellowship (2017A052660).
Footnotes
Author contributions
G.B. performed the flow cytometry, gene expression profiling, and macrophage in vitro assays. C.S. performed and analyzed the immunostaining experiments. A.I. provided cardiac specimens. N.W. Assisted with quantitative data analyses. S.G.D., C.H.S. and T.S.S. provided cardiac specimens and clinical data for the LVAD patient cohort. G.B., D.K., M.H., M.N., S.E., Y.L., and A.B. assisted with study design, data interpretation, and manuscript production. K.J.L. was responsible for all aspects of this study including study design, experimental execution, data analysis, data interpretation, and manuscript production.
Competing financial interests
The authors have no competing financial interests to disclose.
Reference List
- 1.Van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128:415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volkman A, Chang NC, Strausbauch PH, Morahan PS. Differential effects of chronic monocyte depletion on macrophage populations. Lab Invest. 1983;49:291–298. [PubMed] [Google Scholar]
- 3.Sawyer RT, Strausbauch PH, Volkman A. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab Invest. 1982;46:165–170. [PubMed] [Google Scholar]
- 4.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 6.Yona S, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashimoto D, et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epelman S, et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakubzick C, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017;15:53. doi: 10.1186/s12915-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell. 2011;9:64–73. doi: 10.1016/j.stem.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoeffel G, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209:1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342:1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 15.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavine KJ, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111:16029–16034. doi: 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leid J, et al. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res. 2016;118:1498–1511. doi: 10.1161/CIRCRESAHA.115.308270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, et al. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 2016;1 doi: 10.1172/jci.insight.87315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hulsmans M. Macrophages Facilitate Electrical Conduction in the Heart. Cell. 2017;169:510–522. doi: 10.1016/j.cell.2017.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sager HB, et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res. 2016;119:853–864. doi: 10.1161/CIRCRESAHA.116.309001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds G. Human and Mouse Mononuclear Phagocyte Networks: A Tale of Two Species? Front Immunol. 2015;6:330. doi: 10.3389/fimmu.2015.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zarif JC. A phased strategy to differentiate human CD14+monocytes into classically and alternatively activated macrophages and dendritic cells. Biotechniques. 2016;61:33–41. doi: 10.2144/000114435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garlanda C, Bottazzi B, Bastone A, Mantovani A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol. 2005;23:337–366. doi: 10.1146/annurev.immunol.23.021704.115756. [DOI] [PubMed] [Google Scholar]
- 27.Fujiu K, et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med. 2017;23:611–622. doi: 10.1038/nm.4326. [DOI] [PubMed] [Google Scholar]
- 28.Kubin T, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9:420–432. doi: 10.1016/j.stem.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 29.Vickers AE, et al. Organ slice viability extended for pathway characterization: an in vitro model to investigate fibrosis. Toxicol Sci. 2004;82:534–544. doi: 10.1093/toxsci/kfh285. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburger M, et al. Organotypic slice culture from human adult ventricular myocardium. Cardiovasc Res. 2012;93:50–59. doi: 10.1093/cvr/cvr259. [DOI] [PubMed] [Google Scholar]
- 31.Diakos NA, et al. Myocardial atrophy and chronic mechanical unloading of the failing human heart: implications for cardiac assist device-induced myocardial recovery. J Am Coll Cardiol. 2014;64:1602–1612. doi: 10.1016/j.jacc.2014.05.073. [DOI] [PubMed] [Google Scholar]
- 32.Drakos SG, et al. Magnitude and time course of changes induced by continuous-flow left ventricular assist device unloading in chronic heart failure: insights into cardiac recovery. J Am Coll Cardiol. 2013;61:1985–1994. doi: 10.1016/j.jacc.2013.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heidt T, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. 2014;115:284–295. doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molawi K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211:2151–2158. doi: 10.1084/jem.20140639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufmann A, Salentin R, Gemsa D, Sprenger H. Increase of CCR1 and CCR5 expression and enhanced functional response to MIP-1 alpha during differentiation of human monocytes to macrophages. J Leukoc Biol. 2001;69:248–252. [PubMed] [Google Scholar]
- 37.Weber C, et al. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 38.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanitakis J, Petruzzo P, Dubernard JM. Turnover of epidermal Langerhans’ cells. N Engl J Med. 2004;351:2661–2662. doi: 10.1056/NEJM200412163512523. [DOI] [PubMed] [Google Scholar]
- 40.Eguiluz-Gracia I, et al. Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax. 2016;71:1006–1011. doi: 10.1136/thoraxjnl-2016-208292. [DOI] [PubMed] [Google Scholar]
- 41.Bittmann I, et al. Cellular chimerism of the lung after transplantation. An interphase cytogenetic study. Am J Clin Pathol. 2001;115:525–533. doi: 10.1309/GAFN-5MPA-LY8E-DTPQ. [DOI] [PubMed] [Google Scholar]
- 42.Haniffa M. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med. 2009;206:371–385. doi: 10.1084/jem.20081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dickinson RE, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–2658. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hambleton S. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bigley V, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208:227–234. doi: 10.1084/jem.20101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desch AN. Flow Cytometric Analysis of Mononuclear Phagocytes in Nondiseased Human Lung and Lung-Draining Lymph Nodes. Am J Respir Crit Care Med. 2016;193:614–626. doi: 10.1164/rccm.201507-1376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source Data for all experiments have been provided. All other data are available from the corresponding author on reasonable request. Microarray data was deposited in GEO (GSE112630). Additional details can be found in the Life Sciences Reporting Summary.