

SUMMARY
The inhibitory effects of cancer on T cell metabolism have been well established, but the metabolic impact of immunotherapy on tumor cells is poorly understood. Here, we developed a CD4+ T cell-based adoptive immunotherapy protocol that was curative for mice with implanted colorectal tumors. By conducting metabolic profiling on tumors, we show that adoptive immunotherapy profoundly altered tumor metabolism, resulting in glutathione depletion and accumulation of reactive oxygen species (ROS) in tumor cells. We further demonstrate that T cell-derived tumor necrosis factor alpha (TNF-α) can synergize with chemotherapy to intensify oxidative stress and tumor cell death in an NADPH (nicotinamide adenine dinucleotide phosphate hydrogen) oxidase-dependent manner. Reduction of oxidative stress, by preventing TNF-α-signaling in tumor cells or scavenging ROS, antagonized the therapeutic effects of adoptive immunotherapy. Conversely, provision of pro-oxidants after chemotherapy can partially recapitulate the antitumor effects of T cell transfer. These findings imply that reinforcing tumor oxidative stress represents an important mechanism underlying the efficacy of adoptive immunotherapy.
In Brief
Using a preclinical model of colorectal tumors treated with CD4+ T cell-based adoptive immunotherapy, Habtetsion et al. show that profound metabolic changes occur in tumors before tumor regression. T cells shape tumor metabolism through TNF-α, which can synergize with chemotherapy, to increase tumor cell oxidative stress through an NOX-dependent mechanism.
INTRODUCTION
Cancer cells can alter their metabolism to meet the increased energy needs and biosynthetic requirements of uncontrolled cell growth (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Targeting the metabolic pathways pivotal for cancer cell survival and growth represents an attractive cancer treatment strategy (Martinez-Outschoorn et al., 2017; Vander Heiden, 2011). A class of chemotherapeutic agents termed antimetabolites has been developed based on this principle (Kaye, 1998). However, antimetabolite drugs face the challenge of development of drug resistance, which largely accounts for the poor long-term patient outcomes in most solid tumors.
T cell adoptive immunotherapy (ACT) has increasingly become a viable treatment option for patients with cancer (Rosenberg and Restifo, 2015; Vonderheide and June, 2014). T cells used for adoptive immunotherapy can come from ex vivo expanded tumor-infiltrating lymphocytes, or T cells engineered to express a tumor antigen-specific T cell receptor (TCR) or a chimeric antigen receptor (CAR). It has been shown that pre-conditioning hosts with a lymphodepletive chemotherapy regimen, which often contains the alkylating agent cyclophosphamide (CTX), can promote the expansion and persistence of the infused T cells (Dudley et al., 2008; Klebanoff et al., 2005). Adoptive immunotherapy has manifested significant, sometimes curative, therapeutic effects in treating certain types of cancer. Recent studies have shown that T cell metabolic attributes largely shape donor T cell persistence and memory development, which are key determinants of therapy efficacy (Kawalekar et al., 2016; Kishton et al., 2017; Sukumar et al., 2013).
Mounting evidence has revealed a dynamic metabolic crosstalk between cancer cells and T cells (Herbel et al., 2016; Kouidhi et al., 2017). In the tumor microenvironment (TME), activated T cells have to compete against cancer cells for energy and nutrients in order to expand and acquire effector function. Cancer cells appear to outcompete T cells in exploiting the nutrient-deficient milieu, making T cells metabolically stressed (Beckermann et al., 2017; Delgoffe, 2016). It is evident that the metabolic constraints imposed by cancer cells compromise T cell metabolic fitness and render T cells dysfunctional even in the face of antigenic stimulation (Chang et al., 2015; Scharping et al., 2016; Siska et al., 2017; Zhao et al., 2016). There is increasing interest in developing strategies to modulate T cell metabolism so as to strengthen T cell metabolic fitness and improve antitumor T cell responses (Chang and Pearce, 2016; O’Sullivan and Pearce, 2015; Sukumar et al., 2017). So far, much attention has focused on unraveling the metabolic impact of tumor cells on T cells; however, little is known about the reciprocal impact of T cells on tumor cells. A better understanding of the metabolic changes in tumor cells during the course of an effective immunotherapy, such as adoptive T cell therapy, may identify key metabolic pathways that can be therapeutically targeted.
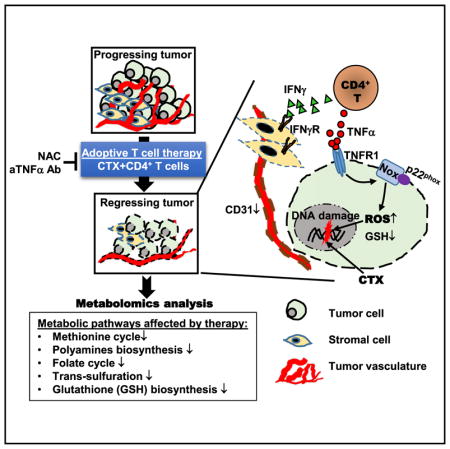
In the present study, we set out to address this issue in a preclinical model in which mice with large implanted colorectal tumors were treated by CD4+ T cell-based adoptive immunotherapy. We showed that adoptive transfer (AT) of tumor-specific CD4+ T cells following CTX pre-conditioning gave rise to polyfunctional CD4+ effector cells capable of concomitantly producing multiple inflammatory cytokines, including tumor necrosis factor alpha (TNF-α) and interferon gamma (IFNγ). These CD4+ effector cells drove complete regression of well-vascularized tumors. By conducting comprehensive metabolomics analysis on resected tumors, we found that the combination of CTX and CD4 AT induced profound metabolic changes in tumors before tumor regression was evident. Disruptions in multiple metabolic pathways converged to cause defective synthesis of the major cellular antioxidant glutathione (GSH), resulting in severe GSH deficiency, heightened reactive oxygen species (ROS) accumulation, and oxidative DNA damage in tumor cells. We demonstrated that tumor cell-intrinsic TNF-α signaling was required to synergize with chemotherapy to intensify ROS production in tumor via a mechanism involving nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidases. Furthermore, administration of antioxidant N-acetylcysteine (NAC) to mice abrogated the curative effect of CTX + CD4 AT, highlighting the importance of ROS in tumor rejection. Our data provide new insight into the impact of an effective immunotherapy on tumor metabolism, and identify tilting tumor redox toward excessive oxidative stress as an important mechanism underlying the efficacy of adoptive immunotherapy.
RESULTS
Chemotherapy Conditioning Followed by CD4 AT Leads to Eradication of Established Colorectal Tumors in Mice
To investigate how a successful adoptive immunotherapy would impact tumor metabolism, first we sought to establish a curative adoptive T cell therapy model. We previously characterized a CD4+ T cell-based AT model system in which mice with tumors expressing the model tumor antigen HA receive CTX pre-conditioning followed by infusion of HA-specific CD4+T cells (Ding et al., 2010). We used this AT protocol to treat mice with established HA-expressing colorectal tumor CT26 (Figure 1 schema). As shown in Figure 1A, the combination of CTX and CD4+ T cells (CTX + CD4 AT) led to tumor shrinkage, starting around day 10, followed by progressive regression that resulted in complete elimination of tumors in 14–21 days in most of the mice. In contrast, CD4+ T cell transfer without CTX pre-conditioning had minimal impact on tumor growth. Although CTX alone retarded tumor growth initially, tumors subsequently regrew in all mice. In line with our published studies using the same AT protocol (Ding et al., 2010, 2012), the donor CD4+ T cells in CTX-conditioned mice acquired a polyfunctional phenotype, characterized by their ability to simultaneously produce inflammatory cytokines such as IFNγ and TNF-α (Figure 1B). We examined the cytokine milieu in the tumor tissues resected from mice under different treatment conditions. As shown in Figure 1C, CTX treatment alone led to increased transcripts of a number of inflammatory cytokines and chemokines, including Ifnγ, Tsp1, Il-6, Ccl3, Ccl5, and Tnf-α. Of note, CTX + CD4 AT further boosted the levels of Ifnγ, Tsp1, Il-6, Ccl3, and Ccl5. Moreover, increases of Il-1β, Ifnβ, and Gmcsf transcripts were detected only after CTX + CD4 AT treatment. Tumors from mice treated with CD4 AT alone exhibited a similar gene expression profile as tumors from untreated mice (data not shown). The results revealed a heightened inflammatory immune milieu in the TME after the treatment of CTX + CD4 AT.
Figure 1. CTX Pre-conditioning Followed by CD4 AT Leads to Eradication of Established Colorectal Tumors in Mice.

(A) The schema outlines the timeline of experimental procedures. Tumor-bearing mice were untreated (No Tx) or subject to one of three treatment conditions: CD4 T cell transfer only (CD4 AT), CTX only (CTX), or CTX followed by CD4 AT (CTX + CD4 AT). Tumor growth curves of each mouse are shown.
(B) Polyfunctional status of the transferred CD4+ T cells. Tumor-bearing mice received CD4 AT or CTX + CD4 AT. Seven days later, tumor-infiltrating CD4+ T cells were isolated and stimulated with phorbol myristate acetate (PMA)/ionomycin in the presence of GolgiPlug for 4 hr before intracellular cytokine staining. The cytokine profiles of the donor CD4+ T cells are shown by representative dot plots.
(C) Cytokine/chemokine profile in tumor mass. Total RNA was extracted from resected tumor tissues and subject to quantitative real-time PCR for the indicated genes. Each sample was measured in triplicate for each gene. The amount of target mRNA was first normalized to β-actin, mRNA fold changes relative to the no-treatment sample (No Tx) are shown in bar graphs.
(D) Representative images of tumors in mice before treatment and at different times after receiving CTX + CD4 AT treatment.
(E and F) (E) Immunofluorescence (IF) stain of cleaved caspase 3 in tumor tissues. Tumors were resected 7 days after treatment, and a fraction of tumor tissues were embedded in optimal cutting temperature compound (OCT) for cryosections and IF stain. Scale bar, 20 μm. Fractions of tumor tissues were formalin fixed and paraffin embedded for H&E histology (F). Scale bar, 100 μm.
(G) The presence of red blood cell patches (marked by arrows) in tumors resected from mice receiving CTX + CD4 AT treatment. (H) IF stain of CD31 in tumor tissues. Scale bar, 100 μm.
*p < 0.05, **p < 0.01, ***p < 0.001, error bars indicate SEM. See also Figure S1.
A marked change in tumors after CTX + CD4 AT therapy was the appearance of necrotic lesions in the central area of tumor, which expanded progressively to the whole tumor over time, and eventually fell off as mice became cured (Figure 1D). Immunofluorescence (IF) staining revealed the extensive presence of cleaved caspase 3 (cCasp3) in the tumor tissues of CTX + CD4 AT-treated mice (Figure 1E), indicating that tumor cells were constantly undergoing apoptosis. Furthermore, hematoxylin and eosin (H&E) staining revealed that necrosis was prevalent in tumors from CTX + CD4 AT-treated mice (Figure 1F). Notably, red blood cell patches were frequently found in tumors under the CTX + CD4 AT treatment condition (Figure 1G, arrowheads) but were absent under other conditions. These features are reminiscent of hemorrhagic tumor necrosis induced by endotoxin or TNF-α (Carswell et al., 1975). Hemorrhagic tumor necrosis is often associated with tumor vasculature destruction. Indeed, we found that CTX + CD4 AT treatment led to marked reduction of CD31+ cells in tumor tissues (Figure 1H).
It should be noted that the observed curative effect was antigen driven because CTX followed by AT of HA-specific CD4+ T cells eliminated HA-expressing tumors, but not wild-type tumors (Figure S1A). Moreover, we found that host response to IFNγ was critical for the curative effects of CTX + CD4 AT because tumor rejection failed to occur in IFNγ receptor knockout (IFNγRKO) mice (Figure S1B). It has been reported that activated macrophages and inflammatory myeloid cells can also mediate tumor rejection (Corthay et al., 2005; Haabeth et al., 2011; Iida et al., 2013). However, we found that these innate immune cells were not involved in tumor rejection in our model because depletion of either population did not have any impact on the curative effect of CTX + CD4 AT (Figure S1C). Furthermore, mice cured by CTX + CD4 AT were resistant to tumor re-challenge (Figure S1D), indicating the development of immune memory in cured mice.
CTX + CD4 AT Reprograms Tumor Metabolism Prior to Overt Tumor Regression
We employed the above-described model to gain insight into the impact of adoptive immunotherapy on tumor metabolic profiling. To this end, mice with established CT26HA tumors were divided into three groups and received no treatment, CTX alone, or CTX + CD4 AT. Tumor tissues were collected on day 5 when tumor regression was absent, and day 10 when tumor regression just started in the combination treatment group (Figure 2). Since CD4 AT alone was virtually identical to the no-treatment group in terms of tumor growth (Figure 1A), we omitted this treatment condition in the following metabolomic analyses. Global metabolic profiling analyses were determined using Metabolon ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS)-based metabolomics platform.
Figure 2. Global Metabolomic Analysis Reveals Profound Changes in Tumor Metabolism after CTX + CD4 AT Treatment.

(A) Top: the schema depicts the timeline of tumor sample collection. Global metabolic profiling analyses were conducted using Metabolon UPLC-MS/MS-based metabolomics platform. Bottom: the principal component analysis (PCA).
(B) Heatmap visualization of the metabolomics data. Samples are arranged by treatment group, and compounds are arranged by major biochemical class (super pathway).
(C) Summary of the numbers of biochemicals that achieved statistical significance (p ≤ 0.05). ANOVA contrasts were used to identify biochemicals that differed significantly between experimental groups.
(D–G) (D) Simplified schematic presentation of the inter-relationship of some affected metabolic pathways. Metabolic data were scaled to the median value for each compound, then missing values were imputed with the minimum detected value for that compound. Line plot graphics shown as mean ± SEM are provided for metabolites in the methionine and folate cycles (one-carbon metabolism) (E), the trans-sulfuration pathway (F), and glutathione biosynthesis (G).
(H) CTX + CD4 AT leads to decrease in GSH/GSSG ratio in tumor. The scaled imputed GSH and GSSG data were used to calculate the ratio and presented as mean ± SEM.
(I) Metabolic changes in polyamine biosynthesis.
SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; MTA, 5-methylthioadenosine; Hcy, homocysteine; GCL, glutamate cysteine ligase. See also Figure S2.
The principal component analysis (PCA) shown in Figure 2A reveals good clustering of samples within the same group. The effects of the combined treatment dominated the separation in component 1, while effects of CTX alone were responsible for much of components 2 and 3 separation. Overall, the variances emerging early between CTX-treated and -untreated tumors tended to abate at the later time. In contrast, the PCA position of CTX + CD4 AT-treated tumors markedly deviated from that of untreated tumors at the early time point, and the divergences became even greater at the later time point. A high-level heatmap visualization of the metabolomics data (Figure 2B), in which samples are arranged by group and compounds are arranged by major biochemical class (super pathway), shows that the treatments, and especially the combined treatment, elicited major changes in metabolism. Large effects on all metabolite classes were found, but changes in lipids and amino acids metabolites were especially apparent.
The present dataset comprises a total of 601 compounds of known identity (named biochemicals). ANOVA contrasts were used to identify biochemicals that differed significantly between experimental groups, aiming to reveal the magnitude of metabolic changes in tumor, related to a temporal context, caused by CTX or CD4+ T cells. A summary of the numbers of biochemicals that achieved statistical significance (p ≤ 0.05) is shown in Figure 2C. Of note, in most cases the effects of CTX alone on day 10 were very similar to day 5. That is, similar numbers of compounds were affected by CTX at the two time points (CTX effect). In contrast, the number of affected compounds approximately doubled between day 5 and day 10 under the combined treatment condition (combo effect and CD4 effect).
Detailed examination of the metabolomics dataset revealed that CTX + CD4 AT treatment induced profound changes in multiple metabolic pathways related to the biosynthesis of GSH, the most prominent endogenous antioxidant. These pathways, including the methionine cycle, the folate cycle, polyamine synthesis, trans-sulfuration, and glutathione synthesis, are extensively interlinked through metabolite intermediates (Figure 2D). The methionine cycle and the folate cycle, which are coupled through portioning of a carbon unit, constitute a bicyclic metabolic pathway termed one-carbon metabolism. Notably, the major metabolites related to one-carbon metabolism, including methionine, S-adenosylmethionine, S-adenosylhomocysteine, 5-methylthioadenosine, betaine, deoxythymidine monophosphate, and 5-methyltetrahydrofolate, were significantly downregulated after CTX + CD4 AT treatment (Figure 2E). Homocysteine (Hcy) and cystathionine, metabolites in the trans-sulfuration pathway that gives rise to cysteine, were also downregulated in tumor after CTX + CD4 AT (Figure 2F). In terms of GSH synthesis, CTX alone led to an increase in both reduced and oxidized glutathione (GSH and glutathione disulfide [GSSG], respectively) compared with no treatment, whereas CTX + CD4 AT resulted in strong depletion of these compounds (Figure 2G). In addition to GSH reduction, CTX + CD4 AT led to marked decrease of the GSH/GSSG ratio (Figure 2H), indicative of impairment in antioxidant capacity. Furthermore, CTX + CD4 AT treatment reduced polyamine biosynthesis in tumors, especially putrescine and spermidine (Figure 2I).
We also examined how the different treatment conditions affected the major metabolic pathways involved in bioenergetics (Figure S2A). Many of the glycolytic intermediates, including fructose-6-phosphate, 3-phosphoglycerate, and phosphoenolpyruvate, were significantly reduced on day 10 in tumors from CTX + CD4 AT-treated mice (Figure S2B), suggesting a repressed glycolysis. Interestingly, the glucose level was increased on day 10 in the combination treatment group, in line with the idea that impaired glycolysis leads to reduced glucose consumption. Moreover, the lactate levels were not different among the three groups and remained basically unchanged over time, suggesting inactivity of anaerobic glycolysis. In contrast, glutaminolysis and citric acid metabolism did not appear to be negatively affected by CTX + CD4 AT treatment because most of the detected metabolites seemed to either remain unchanged or increase after treatment (Figure S2C). Since tumor glycolysis was inhibited after CTX + CD4 AT treatment, acetyl-CoA needed to fuel the tricarboxylic acid (TCA) cycle may come from fatty acid oxidation. Indeed, marked increase of free fatty acids and long-chain acyl-cholines was detected in tumor after the combination treatment (data not shown), suggesting an increased β-oxidation. Figure S2D shows that the electron carriers nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD) both suffered significant decline, and the ADP levels were much lower in day 10 tumor samples from CTX + CD4 AT-treated mice, suggesting a breakdown of oxidative phosphorylation.
In sum, our metabolomics data revealed that CTX + CD4 AT treatment led to reprogramming of tumor metabolism, characterized by widespread defects in metabolic pathways controlling redox balance, bioenergetics, and biosynthesis.
CTX + CD4 AT Leads to Increased ROS Accumulation in Tumor Cells
To determine whether GSH deficiency in tumor was associated with increased oxidative stress, we resected tumors from mice under different treatment conditions and measured the ROS levels in tumor cells. Figure 3A shows that CD4 AT alone did not alter ROS levels in tumor cells. CTX alone appeared to result in slight increase of ROS in tumor cells, though the differences did not reach statistical significance; in contrast, CTX + CD4 AT led to markedly increased ROS accumulation in tumor cells. It is noteworthy that intensified ROS accumulation was restricted to tumor cells since CD45+ non-tumor cells, mostly leukocytes, exhibited relatively low levels of ROS, even though the leukocytes (including donor T cells) from CTX + CD4 AT-treated mice had somewhat increased ROS. The presence of excessive ROS was further corroborated by readily detectable 8-hydroxy-2′-deoxyguanosine (8-OHDG), a major product of DNA oxidation, in tumors resected from CTX + CD4 AT-treated mice (Figure 3B). Our data indicate that chemotherapy and polyfunctional CD4+ effector cells act in concert to exacerbate oxidative stress in tumor cells.
Figure 3. CTX + CD4 AT Leads to Heightened Oxidative Stress in Tumor Cells.

(A) Detection of ROS in tumor cells. Tumor-bearing mice were untreated (No Tx) or subjected to the indicated treatment. Seven days after T cell transfer, tumors were resected and processed into single-cell suspensions. Cells were stained with CD45.2 to distinguish CD45.2− tumor cells and CD45.2+ non-tumor cells (mostly leukocytes). Intracellular ROS levels were evaluated by CM-H2DCFDA staining. Representative flow cytometry data are shown as overlay of ROS histograms under different treatment conditions. The mean fluorescence intensities (MFI) of CM-H2DCFDA in tumor cells and leukocytes are summarized in bar graph on right.
(B) Resected tumors were subjected to 8-OHDG IF staining.
(C) Single-cell suspensions from resected tumors were flow sorted for CD45.2− tumor cells. Sorted tumor cells were subjected to quantitative real-time PCR in triplicate to evaluate the mRNA levels of the indicated genes. The target gene transcripts are normalized to β-actin, and fold changes relative to the no-treatment sample are shown in bar graphs.
* p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant. Error bars indicate SEM.
Tumor cells are known to respond to oxidative stress by activating the endogenous antioxidant mechanisms (Gorrini et al., 2013; Trachootham et al., 2009). To examine the redox status in tumors, we recovered tumor cells from mice treated under different conditions and examined the expression levels of a panel of genes involved in regulating redox homeostasis. Figure 3C shows that the transcripts of several key antioxidant genes, including Gpx1, Hmxo1, and Sod1, were induced in tumor cells after CTX treatment and were further boosted after CTX + CD4 AT treatment. Transcripts of Gclc and Gclm, subunits of glutamate cysteine ligase (GCL), the rate-limiting enzyme regulating GSH synthesis, were comparable in tumor cells recovered from mice treated with CTX alone or CTX + CD4 AT. The gene expression patterns indicate an attempt by tumor cells to cope with increased oxidative stress after CTX + CD4 AT treatment.
Adoptive T Cell Therapy Intensifies Tumor Oxidative Stress in Orthotopic Tumor Models
One caveat of the above experiments was that subcutaneously implanted CT26HA tumors, growing at a non-physiological location, may not truthfully resemble colon cancer response to therapy in situ. We extended our study to orthotopic tumor models to examine whether ACT causes augmented oxidative stress in cancer cells grown in the relevant anatomical locations. To establish an orthotopic colon cancer model, CT26HA cells were engineered to express luciferase and rat CD2 to allow visualization of tumors in live mice and identification of tumor cells, respectively. Following the experimental schedule depicted in Figure 4A, CT26HA.luci.rCD2 cells were inoculated to mouse cecum by surgery. Two weeks after tumor implantation, presence of tumor in the colon was confirmed by bioluminescence imaging (BLI). Mice were then treated with CTX only or CTX + CD4 AT. Four days after T cell transfer, the donor T cells were readily detectable in tumor (Figure 4B) and were capable of producing TNF-α upon stimulation (Figure 4C). CTX + CD4 AT treatment led to heightened ROS accumulation in cancer cells compared with CTX only, whereas ROS in non-tumor cells (mostly leukocytes) remained at low levels (Figure 4D). Of note, similar results (i.e., tumor infiltration of TNF-α-producing donor T cells, enhanced ROS accumulation, and reduced GSH in tumor cells) were observed in the 4T1HA breast cancer model (Figure S3).
Figure 4. Adoptive T Cell Therapy Intensifies Oxidative Stress in Tumor Cells in Orthotopic Tumor Models.

(A) The schema depicts the experimental procedures in an orthotopic colon cancer model. CT26HA.luci.rCD2 cells were surgically inoculated to mouse cecum. Fourteen days after tumor inoculation, tumor presence in colon was confirmed by BLI. Tumor-bearing mice were treated with CTX only or CTX + CD4 AT.
(B) Detection of donor T cells in tumor. Four days after T cell transfer, tumor masses were resected and processed into single-cell suspensions. The presence of the donor T cells (Thy1.1+) in tumor was evaluated by fluorescence-activated cell sorting (FACS). Numbers in dot plots indicate percentage of donor T cells.
(C) Tumor-infiltrating donor T cells are capable of TNF-α production. T cells in tumor samples were stimulated with PMA and ionomycin for 4 hr before TNF-α intracellular cytokine staining (ICS). Percentage of TNF-α+ donor CD4+ T cells is given in histogram.
(D) ROS levels in tumor cells and non-tumor cells. Tumor-derived single-cell suspension was stained for CD45.2 and rat CD2. ROS levels in tumor cells (CD45.2−rCD2+) and leukocytes (CD45.2+rCD2−) were evaluated by CM-H2DCFDA staining.
(E) The schema depicts the experimental procedures of CD19CAR T cell therapy in A20 B cell lymphoma model. Systemic B cell lymphoma was established by injecting A20.luci.rCD2 cells into mice via tail vein. Seventeen days after tumor injection, tumor burden was measured by BLI (designated as day 0). Mice were randomized into four groups to receive the indicated treatments. By day 3, mice were imaged again before being sacrificed to recover spleens and tumor tissues from the livers.
(F and G) (F) Diagram of the retroviral vector encoding the 1D3-28Z-1.3 CAR. Thy1.1 was inserted after the internal ribosome entry site. Expression of Thy1.1 can be used to evaluate T cell transduction efficiency and as a marker for CAR T cells. Purified mouse CD3+ T cells were stimulated with αCD3/αCD28 mAb-conjugated Dynal beads overnight and then subjected to viral transduction. On day 4 after T cell stimulation, virus-transduced T cells were harvested for FACS analysis and adoptive transfer. Mouse anti-rat-Fab Ab was used to detect the 1D3scFv. TNF-α and IFNγ expression profile of CD19CAR T cells was evaluated by ICS after a 4-hr stimulation with PMA and ionomycin (G).
(H) Frequencies of CD19CAR T cells in spleen and tumor. Spleen- and tumor-derived single-cell suspensions were stained for CD45.1 and Thy1.1 to detect CD19CAR T cells. Results are shown as mean ± SEM of three mice per group.
(I) ROS levels in tumor cells. Tumor-derived single-cell suspensions were stained for CD19 and rat CD2. ROS levels in tumor cells (CD19+rCD2+) were evaluated by CM-H2DCFDA staining. Results are summarized in bar graphs and shown as mean ± SEM of three mice per group.
**p < 0.01, ***p < 0.001. See also Figure S3.
Despite extensive efforts, ACT for colon cancer or breast cancer has yet to become a reality in the clinic, raising the question whether our findings from the above studies are clinically relevant. In recent years, ACT using T cells engineered to express CAR, especially CD19-targeted CAR T cells, has become a viable treatment option for patients with certain types of B cell malignancies (Barrett et al., 2015; Porter et al., 2011; Rosenberg and Restifo, 2015; Sadelain, 2015). Using a retroviral vector carrying a murine CD19-specific CAR design (Kochenderfer et al., 2010), we established a CD19CAR T cell therapy model using A20 B cell lymphoma in immunocompetent mice (Kuczma et al., 2017). As shown in Figure 4E, A20.luci.rCD2 cells were injected to mice via tail vein to establish systemic B cell lymphoma. Seventeen days after tumor inoculation, the presence of lymphoma in multiple organs, primarily liver, was confirmed by BLI (Figure 4E, day 0). Mice were randomized into four groups to receive no treatment, CD19CAR, CTX, or CTX + CD19CAR. More than 70% of T cells expressed CD19CAR after viral transduction (Figure 4F), and the T cells were proficient producers of TNF-α and IFNγ (Figure 4G). By day 3, mice treated with CTX + CD19CAR had reduced tumor signals, while mice in other groups had either increased or unchanged tumor burdens (Figure 4E, day 3). CTX + CD19CAR treatment led to markedly increased presence of tumor-infiltrating CD19CAR T cells (Figure 4H). Importantly, tumor cells recovered from CTX + CD19CAR-treated mice exhibited significantly increased levels of ROS (Figure 4I). Taken together, our results from these orthotopic tumor models suggest that our finding that induction of tumor oxidative stress contributes to treatment efficacy is clinically relevant and has implications for adoptive immunotherapy in general.
TNF-α Synergizes with Chemotherapy to Enhance ROS Production and Tumor Cell Death
We showed that CTX + CD4 AT treatment induced an inflammatory milieu in tumor (Figure 1C). Some of the cytokines, including TNF-α, IFNγ, and IL-1β, are known to induce ROS in cells (Yang et al., 2007). We sought to determine whether these inflammatory cytokines can induce ROS in tumor cells by culturing cells in the presence of recombinant cytokine, either alone or in combination with chemotherapy. CTX is an alkylating agent prodrug that has to be metabolized in the liver to generate the active compounds, excluding its direct use in cell culture. Therefore, we chose to use mafosfamide (maf), a CTX analogue, and melphalan (mel), a mustard alkylating agent with similar mechanism of action as CTX (Dray and Mokyr, 1989; Lu et al., 2015), in cell culture experiments. We used maf or mel at doses (25 μg/mL maf and 50 μg/mL mel) that only cause modest toxicity to tumor cells to mimic the modest and transient antitumor effect exhibited by CTX in vivo. Maf or TNF-α, when used alone, modestly induced ROS, whereas their combination led to significant elevation of ROS levels in CT26 tumor cells (Figure 5A, left panel). A similar pattern of ROS induction was observed when mel was used as chemotherapy (Figure 5A, right panel). In addition, combining TNF-α with mel also resulted in increased ROS levels in 4T1 breast cancer cells, MC38 colon cancer cells, and A20 lymphoma cells (Figure S4A), suggesting that induction of ROS by the combination of alkylating agents and TNF-α is not restricted to a particular cell line. Furthermore, combining chemotherapy and IFNγ or IL-1β failed to induce ROS in tumor cells (Figure S4B), suggesting that TNF-α is a major driver of increased ROS accumulation in chemotherapy-treated tumor cells. Similar to the in vivo situation (Figure 3C), cultured tumor cells responded to oxidative stress by activating the endogenous antioxidant systems, reflected by upregulation of Cat, Gpx1, Gsr1, Hxmo1, Nqo1, Nrf2, Gclc, and Gclm in mel-treated or mel + TNF-α-treated cells (Figure S4C).
Figure 5. TNF-α Synergizes with Chemotherapy to Enhance ROS Production and Tumor Cell Death.

(A) Detection of ROS in cultured CT26 cells. CT26 cells were subjected to the indicated culturing conditions for 18 hr. Mafosfamide (maf) or melphalan (mel) was used as chemotherapy. Cells were stained with CM-H2DCFDA and DAPI to evaluate ROS levels and cell viability, respectively. Gating on live cells (DAPI−), histograms show overlay of CM-H2DCFDA curves under different culturing conditions. The MFIs of CM-H2DCFDA are summarized in bar graphs and shown as mean ± SEM.
(B) TNF-α synergizes with chemotherapy to induce tumor cell death. Percentage of dead cells under each culturing condition was evaluated by DAPI staining. Results are summarized in bar graph and shown as mean ± SEM.
(C) Measurement of GSH and GSH/GSSG ratio in tumor cells. CT26 cells were treated under the indicated conditions for 7 hr. Intracellular GSH and GSSG contents were measured and the GSH/GSSG ratio was calculated. Results are summarized in bar graphs and shown as mean ± SEM.
(D) Supplementation of exogenous GSH diminishes cell death induced by the combination of melphalan and TNF-α. CT26 cells were treated by the combination of mel and TNF-α, in the absence or presence of cell-permeable GSHree or mitochondria-selective antioxidant mitoTEMPO. Cell viability was evaluated by DAPI.
(E) Loss of TNFR1 leads to reduced tumor cells death after mel + TNF-α treatment. TNFR1 knockout CT26HA cells (CT26HA.ΔTNFR1) and the parental CT26HA cells were subjected to the indicated culturing conditions. After overnight incubation, cell viability was evaluated by DAPI stain.
(F–H) (F) Preparation of CM from polyfunctional CD4+ T cells. The schema depicts the experimental procedures. Polyfunctional CD4+ T cells and control CD4+ T cells were prepared following our established protocol. The polyfunctional status of T cells was confirmed by ICS. The expression profiles of TNF-α and IFNγ are shown by dot plots. Conditioned medium from polyfunctional CD4+ T cells (pCD4 CM) or control CD4+ T cells (cCD4CM) were used to culture CT26 cells in the absence or presence or mel. After overnight culture, cells were harvested and examined for intracellular ROS levels by CM-H2DCFDA staining (G) and cell viability by DAPI (H).
(I) Addition of GSHree or TNF-α-neutralizing mAb diminishes cell death induced by the combination of mel and pCD4CM. Data shown are representative of three independent experiments with similar results.
*p < 0.05, **p < 0.01, ***p < 0.001; n.s., not significant. Error bars indicate SEM of triplicate data. See also Figures S4 and S5.
We noticed a correlation between elevated intracellular ROS levels and increased cell death. Figure 5B shows that the combination of chemotherapy (either maf or mel) and TNF-α resulted in significantly enhanced tumor cell death compared with chemotherapy alone, while TNF-α by itself was not toxic. The synergy of chemotherapy and TNF-α in killing tumor cells was reflected by pronounced increases in dead tumor cells (DAPI+ Annexin V+), as well as cells undergoing early apoptosis (DAPI− Annexin V+) (Figure S4D). Because the combination of mel + TNF-α consistently yielded more significant changes in cell viability than maf + TNF-α, we mainly used mel as chemotherapy in subsequent in vitro cell culture experiments for the benefit of clear readout.
Our metabolomics data indicated a GSH deficiency in tumor after CTX + CD4 treatment (Figures 2G and 2H). To examine whether this was the case in the in vitro culture system, we measured the intracellular concentration of GSH and the GSH/GSSG ratio in tumor cells under different treatment conditions. Figure 5C shows that TNF-α had trivial influence on intracellular GSH level or GSH/GSSG ratio, while mel alone modestly reduced GSH and GSH/GSSG ratio; in contrast, the combination of mel and TNF-α resulted in markedly reduced GSH and GSH/GSSG ratio, indicative of heightened oxidative stress. Similar results (i.e., reduction of GSH and decrease of GSH/GSSG ratio) were also observed in 4T1 cells after mel + TNF-α treatment (Figure S4E). The deficiency of GSH in mel + TNF-α-treated tumor cells prompted us to test if supplementation of GSH can attenuate oxidative stress-induced cell death. To this end, CT26 cells were treated with mel + TNF-α in the presence or absence of glutathione reduced ethyl ester (GSHree), a cell-permeable derivative of GSH. Figure 5D shows that GSHree significantly reduced cell death caused by mel + TNF-α, suggesting that GSH deficiency drives excessive oxidative stress, which triggers tumor cell apoptosis. In contrast, mitoTEMPO, a mitochondria-targeted antioxidant, failed to reduce mel + TNF-α-induced cell death, suggesting that ROS are not produced in mitochondria. Harris et al. (2015) recently reported that, when GSH synthesis is reduced, unused glutamate may promote import of cystine via the amino acid transporter system Xc− and activate the thioredoxin (TXN) antioxidant pathway to detoxify ROS in cancer cells. To examine whether this alternative antioxidant pathway was involved in our system, CT26 cells were treated with mel + TNF-α in the presence of sulfasalazine, which reduces cystine uptake by blocking the xCT subunit of system Xc−, or auranofin, which reduces TXN regeneration. Addition of these inhibitors did not enhance mel + TNF-α-induced cell death (Figure S4F), arguing against the involvement of the TXN antioxidant pathway.
It should be noted that after transferring into CTX-conditioned tumor-bearing mice, it took 3–4 days for the donor CD4+ T cells to acquire effector function; i.e., being able to concomitantly produce multiple inflammatory cytokines including TNF-α (Ding et al., 2010, 2012). Since CTX is rapidly converted to the active form and has a half-life of only about 20 min in mice (Carmel and Brown, 1977), it is unlikely that CTX and donor CD4+ T cell-derived TNF-α would act upon tumor cells at the same time. To mimic this scenario, we tested in vitro whether sequential treatment of chemotherapy and TNF-α can still lead to increased tumor cell death. Figure S4G shows that TNF-α exacerbated the death of tumor cells that had been previously exposed to mel (mel/wash/TNF-α versus mel/wash/medium) but had no effect on previously untreated cells (medium/wash/TNF-α). Moreover, only tandem treatment of mel and TNF-α led to reduced level of intracellular GSH in a fraction of tumor cells (Figure S4H). The results suggest that TNF-α-driven ROS induction, which correlates with GSH reduction, occurs only in tumor cells that have been previously exposed to and damaged by chemotherapy.
To mechanistically define the role of TNF-α in ROS induction in chemotherapy-experienced tumor cells, we created TNFR1-deficient cells (CT26HA.ΔTNFR1) using CRSPR/Cas9 technology (Figures S5A–S5E). Figure 5E shows that unresponsiveness to TNF-α drastically reduced tumor cells death induced by mel + TNF-α. In addition, the reduced sensitivity of CT26HA.ΔTNFR1 cells to mel + TNF-α correlated with lower intracellular ROS accumulation (Figure S5F), moderate GSH reduction (Figure S5G), and restored GSH/GSSG ratio (Figure S5H). Collectively, our data provide definitive evidence that TNF-α synergizes with chemotherapy to drive heightened oxidative stress and tumor cell death.
The preceding experiments used recombinant TNF-α in cell culture, omitting the involvement of other cytokines. It was unclear whether polyfunctional CD4+ T cells really relied on TNF-α to promote ROS induction in tumor cells following chemotherapy. We previously reported that antigenic stimulation of CD4+ T cells in the presence of recombinant IL-7 gave rise to polyfunctional CD4+ T cells proficient in TNF-α and IFNγ production (Ding et al., 2016). We prepared polyfunctional CD4+ T cells (pCD4), which were proficient in TNF-α and IFNγ production, and activated control CD4+ T cells (cCD4), which were poor producers of these cytokines (Figure 5F). Conditioned medium (CM) collected from either polyfunctional CD4+ T cells (pCD4CM) or control CD4+ T cells (cCD4CM) was used to culture CT26 tumor cells in the presence or absence of mel. Increased level of ROS was detected in cells cultured in mel + pCD4CM compared with mel + cCD4CM (Figure 5G). Consistent with the increased oxidative stress, mel + pCD4CM exhibited significantly higher toxicity to CT26 cells compared with mel alone, or pCD4CM alone, as well as mel + cCD4CM (Figure 5H). Importantly, the enhanced cell death induced by mel + pCD4CM can be mitigated by GSHree supplementation or TNF-α neutralization (Figure 5I). The results indicate that, among the inflammatory cytokines produced by polyfunctional CD4+ T cells, TNF-α is the key molecule that synergizes with chemotherapy to promote ROS generation and tumor cell death.
p22phox Plays an Important Role in ROS Generation in Tumor Cells Treated with Chemotherapy and TNF-α
NADPH oxidases (NOX enzymes) and the mitochondrial electron transport chain (mETC) are two major sources of cellular ROS production (Chio and Tuveson, 2017). We evaluated the involvement of these two mechanisms in mel + TNF-α-mediated ROS production. Figure 6A shows that mel + TNF-α-treated tumor cells exhibited robust ROS induction as measured by 2′,7′-dichlorofluorescin diacetate (DCFDA), exceeding the ROS levels in mel-treated cells. However, superoxide productions in mitochondria, measured by mitochondria-selective dye mitoSOX, were marginal and equivalent in mel-treated and mel + TNF-α-treated cells, arguing against the notion that mitochondria are the major source of ROS production. To determine whether ROS production was NOX dependent, we created Cyba-deficient cells (CT26HA.ΔCyba) using CRISPR/Cas9 (Figure S6). Cyba encodes for p22phox, which is a subunit of multiple NOX enzymes (NOX1–4) (Bedard and Krause, 2007). We found that CT26HA.ΔCyba cells were more resistant to cell death (Figure 6B), and had lower ROS levels, less GSH reduction, and elevated GSH/GSSG ratio, compared with the parent cells after mel + TNF-α treatment (Figures 6C–6E). The data implicate NOX as the major source of ROS production in tumor after mel + TNF-α treatment.
Figure 6. p22phox Plays an Important Role in ROS Generation in mel + TNF-α-Treated Tumor Cells.

(A) Mitochondria are not the major source of ROS production in mel + TNF-α-treated tumor cells. CT26HA cells were treated as indicated. Cells were evaluated for ROS and superoxide levels by CM-H2DCFDA and mitoSOX staining, respectively.
(B) p22phox-deficient tumor cells are less sensitive to the toxicity of mel + TNF-α. CT26HA.ΔCyba and the parental CT26HA cells were subjected to the indicated culturing conditions. After overnight incubation, cell viability was evaluated by DAPI staining. Error bars indicate SEM.
(C) CT26HA.ΔCyba cells exhibit reduced ROS levels after mel + TNF-α treatment. CT26HA.Δ Cyba and CT26HA cells were treated with mel + TNF-α overnight before evaluating ROS levels by CM-H2DCFDA.
(D and E) (D) Treatment-induced GSH reduction is alleviated in CT26HA.ΔCyba cells. CT26HA.ΔCyba and CT26HA cells were either untreated or treated with mel + TNF-α for 7 hr before being harvested for GSH and GSSG measurements. Percentage GSH reduction, calculated using the formula (GSH in untreated cells – GSH in treated cells)/GSH in untreated cells, is summarized in bar graph and shown as mean ± SEM. Changes in GSH/GSSG ratio are shown in (E).
**p < 0.01, ***p < 0.001. See also Figure S6.
TNF-α-Induced Oxidative Stress Contributes to the Efficacy of CTX + CD4 AT in Mice
We next sought to determine the biological relevance of TNF-α-induced oxidative stress to the curative effect of CTX + CD4 AT observed in our animal model. We reasoned that if TNF-α is critical for tumor rejection, then neutralization of TNF-α would diminish the efficacy of CTX + CD4 treatment. To test this, tumor-bearing mice were given TNF-α-neutralizing monoclonal antibody (mAb) or PBS after CTX + CD4 AT treatment (Figure 7A schema). Strikingly, after TNF-α neutralization, all mice had tumor relapse as opposed to tumor rejection (Figure 7A). Moreover, TNF-α neutralization markedly reduced the presence of 8-OHDG in tumors (Figure 7B), suggesting that TNF-α signaling is a major causal factor of the increased oxidative stress in tumor. Since TNF-α can act on both tumor cells and stromal cells, we sought to determine whether tumor response to TNF-α is sufficient for tumor rejection. For this purpose, mice were inoculated with CT26HA.ΔTNFR1 cells and treated with CTX + CD4 AT. Figure 7C shows that loss of TNF-α responsiveness rendered tumor cells resistant to CTX + CD4 AT treatment. Taken together, the results indicate that TNF-α is the driver of elevated oxidative stress in tumor, which leads to tumor destruction after adoptive immunotherapy. Since our in vitro study indicated the involvement of NOX in ROS generation in tumor cells after mel + TNF-α treatment (Figure 6), we next examined if loss of p22phox altered tumor response to treatment in vivo. Figure 7D shows that CTX + CD4 AT treatment resulted in complete tumor regression in five out of five mice bearing CT26HA tumors but only achieved this in three out of eight mice bearing CT26HA.ΔCyba tumors. Our results indicate that the TNF-α-NOX axis underlies the increased oxidative stress in tumors following CTX + CD4 AT treatment, and disruption of this axis diminishes the treatment efficacy.
Figure 7. Induction of Oxidative Stress in Tumor Contributes to the Efficacy of CTX + CD4 AT in Mice.

(A and B) (A) The schema outlines the timeline of experimental procedures. Tumor-bearing mice were all treated with CTX followed by CD4 AT. Some of these mice received TNF-α-neutralizing mAb injection via intraperitoneal (i.p.) injection, 200 μg per injection. Tumor growth curves of each mouse are shown. Some mice were sacrificed 7 days after T cell transfer. Resected tumors were subjected to 8-OHDG IF staining (B); scale bar, 10 μm.
(C) Loss of TNF-α responsiveness renders tumor cells resistant to CTX + CD4 AT treatment. Mice with established CT26HA or CT26HA.ΔTNFR1 tumors were treated with CTX followed by CD4 AT. Tumor growth curves of each mouse are shown.
(D) CTX + CD4 AT treatment is less effective in treating p22phox-deficient tumors. Mice with established CT26HA or CT26HA.ΔCyba tumors were treated with CTX + CD4 AT. Tumor growth over time was monitored.
(E) Administration of antioxidant NAC impairs the curative effect of CTX + CD4 AT. The schema depicts the experimental design. Tumor-bearing mice were all untreated with CTX + CD4 AT. Some of these mice were fed with drinking water containing 10 mg/mL NAC. Tumor growth curves of each mouse are shown.
(F) Inhibition of GSH synthesis by BSO following CTX mediates improved antitumor effect. The schema depicts the experimental design. Tumor-bearing mice were subjected to the indicated treatment conditions. BSO (20 mM) was provided in drinking water. Tumor growth curves of each mouse are shown. **p < 0.01, ***p < 0.001.
To further assess the significance of oxidative stress in tumor rejection, we examined whether antagonizing oxidative stress by antioxidants would diminish the efficacy of CTX + CD4 AT treatment. To test this, mice with established CT26HA tumors were treated with CTX + CD4 AT. These mice were randomized into the control group (no further treatment) and the experimental group, in which mice were given drinking water containing N-acetylcysteine (NAC), a GSH precursor (Figure 7E schema). Figure 7E shows that mice fed with NAC still had significant tumor masses by day 30, while mice in the control group were cured around day 15, supporting the notion that induction of oxidative stress is essential for tumor elimination after CTX + CD4 AT. Based on these data, we speculated that pro-oxidant drugs, by increasing oxidative stress in tumor cells, may simulate the antitumor effect of polyfunctional CD4+ effector cells. To test this, we replaced CD4 AT with administration of buthionine sulfoximine (BSO), a synthetic amino acid that induces oxidative stress by inhibiting GSH biosynthesis. Figure 7F shows that the combination of BSO and CTX exhibited significantly improved tumor growth control compared with CTX alone or BSO alone, suggesting a synergy between the two drugs. In sum, our data provide clear evidence that ROS accumulated in tumors after adoptive immunotherapy are not merely metabolic byproducts but actively contribute to the treatment efficacy.
DISCUSSION
Intracellular redox homeostasis is regulated by a balance between ROS generation and elimination (Chio and Tuveson, 2017; Marengo et al., 2016). Abnormal ROS accumulation could be due to increased ROS production and/or reduced presence of antioxidants. Mitochondrial respiration and membrane-bound NADPH oxidases are two major cellular sources of ROS (Trachootham et al., 2009). Our in vivo and in vitro data are mutually reinforcing and support the notion that CD4+ T cell-derived TNF-α synergizes with chemotherapy to boost the production of ROS in a p22phox-dependent mechanism. p22phox is an essential component of multiple NOX complexes (NOX1–4), therefore one or more NOX may participate in ROS generation. In fact, we found that both Nox1 and Nox2 gene expressions were quickly upregulated in tumor cells 2 hr after receiving mel or mel + TNF-α treatment but returned to the baseline after 9 hr (Figure S7). During this time frame, mel or mel + TNF-α treatment led to progressive elevation of Nrf2 transcription. Interestingly, increases in transcription of several Nrf2-regulated genes, including Nqo1, Hmox1, and Catalase (Cat), were only evident at later time points after treatment. Our data indicate that NOX-driven ROS production triggers anti-oxidative responses in tumor cells. We attempted to identify the individual NOX accounting for ROS production. However, knocking down individual NOX with small interfering RNA failed to reduce cell death and ROS levels in CT26 cells after mel + TNF-α treatment (data not shown). The results suggest that multiple NOX enzymes may be involved in ROS generation in a mutually compensatory fashion, although the detailed mechanism awaits further investigation. It is worth noting that, besides tumor cell-intrinsic ROS production, phagocytic cell-produced ROS may diffuse into tumor cells and cause ROS accumulation. Iida et al. (2013) reported that in EL4 tumor-bearing mice, ROS produced by infiltrating myeloid cells, CD11b+Gr1hi neutrophils, and F4/80+Gr1int macrophages in particular were important for tumor rejection by oxaliplatin treatment. We showed that depletion of myeloid cells or macrophages did not have any effect on the curative effect of CTX + CD4 AT (Figure S1C), suggesting that tumor rejection observed in our model is mainly dependent on tumor cell-intrinsic ROS accumulation.
As the most abundant and important endogenous antioxidant, GSH is essential for tumor cells to counteract oxidative stress induced by genetic, environmental, or therapeutic causes (Gorrini et al., 2013). Depletion of GSH with various inhibitors appears to be a promising cancer treatment strategy (Cramer et al., 2017; Harris et al., 2015; Raj et al., 2011; Trachootham et al., 2006). We now provide evidence that GSH depletion can be achieved by T cell-based immunotherapy, offering new ways to modulate tumor redox homeostasis toward therapeutic benefit. Unlike most small compound inhibitors, T cell-mediated GSH depletion does not seem to impair the function of the rate-limiting GSH-synthesizing enzyme GCL because the RNA levels of GCL subunits Gclc and Gclm were comparable in tumor cells treated with CTX + CD4 AT or CTX alone. However, after the combination treatment, there were deficits in several intermediate metabolites involved in GSH synthesis, including homocysteine (Hcy), cystathionine, and the sulfur storage compounds hypotaurine and taurine (Figure 2F; data not shown), suggesting a lack of available sulfur precursors to feed the pathway. In addition, low levels of glycine, which emanates from the energy pathways, may have also exacerbated the deficiency in GSH synthesis. The ability of CD4+ effector cells to simultaneously affect multiple metabolic pathways suggests that cellular therapy-mediated tumor metabolic reprogramming is comprehensive and thus may overcome tumor resistance by blocking potential compensatory mechanisms.
TNF-α was once regarded as a promising anticancer agent because of its prominent tumor necrotizing effect (Balkwill, 2009). Extensive in vitro studies have demonstrated that TNF-α can induce apoptosis or necrosis in mouse and human tumor cell lines as well as many other cell types through ROS induction (Blaser et al., 2016). Studies using animal tumor models showed that administration of recombinant TNF-α or TNF-α-inducing endotoxins can induce hemorrhagic tumor necrosis and varying degrees of regression in mice with well-vascularized sarcomas (Havell et al., 1988; North and Havell, 1988). However, systemic TNF-α administration was associated with severe toxicity that limited its clinical application. Currently, TNF-α-based cancer therapy is restricted to the isolated limb perfusion setting in which high doses of TNF-α are delivered locoregionally, in combination with melphalan, to treat patients with advanced melanoma or sarcoma (Verhoef et al., 2007). Paradoxically, the in vivo antitumor effects of TNF-α appear to lie in its antivascular effects rather than its direct cytotoxic effect on tumor cells observed in vitro (Balkwill, 2009; van Horssen et al., 2006). Our study presents clear evidence that tumor cell-intrinsic TNF-α signaling, along with chemotherapy, drives increased ROS production and is essential for the curative outcome of an effective adoptive T cell therapy.
Highlighting the importance of TNF-α in direct tumor killing via ROS, our data nonetheless do not exclude the possibility that TNF-α also participates in tumor vasculature destruction. Indeed, the occurrence of hemorrhagic tumor necrosis, decrease of tumor blood perfusion, and reduction of tumor blood vessels in our tumor model are typical indications of vasculature remodeling. North and Havell (1988) demonstrated that a TNF-α-mediated antivascular effect was necessary for induction of tumor necrosis but was not sufficient to ensure continuous tumor regression (Havell et al., 1988), suggesting the involvement of additional vascular damaging factors. In this regard, IFNγ is known to possess potent antivascular effects (Kammertoens et al., 2017; Qin and Blankenstein, 2000). Moreover, some recent studies have revealed novel mechanisms by which T cell-derived IFNγ remodels the TME to promote antitumor immunity (Ayers et al., 2017; Sharma et al., 2018; Wang et al., 2016). Wang et al. (2016) reported IFNγ derived from tumor-infiltrating CD8+ T cells can act upon stromal fibroblasts to reduce GSH in the TME, thereby abrogating tumor resistance to chemotherapy. Sharma et al. (2018) showed that T cell-derived IFNγ acts upon immature myeloid cells to promote their differentiation into CD103+ immunogenic antigen-presenting cells (APCs) through a p53-dependent mechanism. Here we showed that the host response to IFNγ was required for the curative effect of CTX + CD4 AT treatment (Figure S1B), in line with the notion that IFNγ may contribute to tumor rejection by acting on the TME, including disrupting tumor vasculature, reducing stroma-derived GSH, and promoting immunogenic APCs. It should be noted that effector CD4+ T cells, CD8+ T cells, and CAR T cells are all able to concomitantly produce TNF-α and IFNγ. Indeed, we showed that CD19CAR T cell therapy resulted in intensified oxidative stress in lymphoma cells (Figure 4I), suggesting that our findings may be applicable to adoptive immunotherapy in general. Based on these data, we propose that a curative outcome of adoptive T cell therapy may depend on the ability of T cells to mediate antitumor effects through both tumor cell-intrinsic mechanisms, such as programmed cell death mediated by the TNF-α-NOX-ROS axis, and tumor cell-extrinsic mechanisms, such as TME remodeling by IFNγ.
In summary, our study provides clear evidence that polyfunctional CD4+ effector cells arising after a CD4+ T cell-based adoptive immunotherapy can fundamentally reprogram tumor metabolism, resulting in collapse of the major antioxidant defense system and excessive ROS accumulation in tumor cells. We identify TNF-α as the key T cell-derived factor that synergizes with chemotherapy to drive heightened ROS production in tumor cells through an NOX-dependent mechanism. These findings shed light on how immunotherapy shapes tumor metabolism, and they support the notion that the ability of T cells to tilt tumor redox balance toward oxidative destruction is integral to the efficacy of adoptive immunotherapy.
Limitations of Study
Our mouse model of adoptive CD4+ T cell therapy provides a robust system to investigate the dynamic changes in tumor metabolism during an effective immunotherapy. One unique feature of this model system is that CT26 tumor cells do not express major histocompatibility complex class II molecules and thus cannot be directly killed by CD4+ T cells via the cytolytic mechanism. Therefore, the impact of T cells on tumor metabolism is likely attributable to CD4+ T cell-derived cytokines, which may regulate the expression and activity of a multitude of enzymes that control the various metabolic processes. However, this might be a simplified scenario because T cells, including CD4+, CD8+, and CAR T cells, may directly engage tumor cells and exert effector functions through both cytokines and cytolytic granules. Therefore, the mechanisms by which T cells modulate tumor metabolism may be diverse, depending on the type of T cells and the mode of action of T cells. We also need to point out that some technical challenges, including the limited availability of human tumor tissues during the course of therapy and the lack of suitable markers to distinguish cancer cells from nonmalignant cells, have restricted our work to preclinical models. Therefore, additional investigation is needed to determine whether adoptive cellular therapies in cancer patients also lead to increased oxidative stress in cancer cells. One extended question is whether induction of tumor oxidative stress occurs in other forms of immunotherapy, including immune checkpoint blockade therapy and cancer vaccines. Along this line, one important issue to be addressed is whether intratumoral ROS levels correlate with the efficacy of cancer immunotherapies. In this regard, our study raises the possibility that the use of pro-oxidants may augment the efficacy of immunotherapies that would otherwise be less effective. If this hypothesis is proved true, it may open up new avenues for cancer treatment given the rapid progress of immunotherapies and the large number of clinically applicable pro-oxidants developed in recent years.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse CD4 (Clone: GK1.5) | BioLegend | Cat#:100406; RRID: AB_312691 |
| PerCP anti-mouse CD90.1 (Thy-1.1) (Clone: OX-7) | BioLegend | Cat#: 202512; RRID: AB_1595487 |
| APC anti-mouse INFγ(Clone: XMG1.2) | eBiosciences | Cat#:17-7311-82; RRID: AB_469504 |
| PE Rat anti-mouse TNFα (Clone: MP6-XT22) | BD Biosciences | Cat# 561063; RRID: AB_10563769 |
| APC anti-mouse CD45.2 (Clone: 104) | BioLegend | Cat# 109814; RRID: AB_389211 |
| APC Annexin V | BioLegend | Cat# 640941; RRID: AB_2616657 |
| PE anti-mouse CD120a (Clone: 55R-286) | BioLegend | Cat# 113004; RRID: AB_313533 |
| Mouse monoclonal anti-TNFα (Clone: XT3.11) | BioXcell | Cat# BE0058; RRID: AB_1107764 |
| Mouse monoclonal anti-Gr1 (Clone: RB6-8C5) | BioXcell | Cat# BE0075; RRID: AB_10312146 |
| Rat monoclonal anti-mouse CD31(clone:390) | BD Biosciences | Cat# 558736; RRID: AB_397095 |
| Donkey polyclonal anti-rat Cy3 | Jackson ImmunoResearch | Code# 712-166-153; RRID: AB_2340669 |
| Rabbit anti-mouse cleaved caspase 3 (5A1E) | Cell Signaling Technology | Cat#: 9664T |
| Alexa Fluor 488 Goat anti-rabbit IgG (H+L) | Life Technologies | Cat# A-11008; RRID: AB_143165 |
| Rabbit 8-OHDG polyclonal antibody | Bios Antibodies | Cat# bs-1278R; RRID: AB_10856120 |
| Anti-Cytochrome b245 light chain antibody (polyclonal) | Abcam | Cat# ab75941; RRID: AB_1924907 |
| FITC AffinityPure F(ab′)2 fragment mouse anti-rat IgG (H+L) | Jackson ImmunoResearch | Cat# 212-096-168; RRID: AB_2339204 |
| Bacterial and Virus Strains | ||
| DH5a Competent cell | Thermo Fisher | Cat#: 18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cyclophosphamide | Sigma Aldrich | Cat#:6055-19-2 |
| Mafosfamide Sodium Salt | Santa Cruz Biotech | Cat#: 84211-05-2 |
| Melphalan | Sigma Aldrich | Cat#: 48-82-3 |
| Clodronate Liposomes | Liposoma B.V | N/A |
| CM-H2DCFDA | Thermo Fisher | Cat#: C6827 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Thermo Fisher | Cat#: M36008 |
| Glutathione reduced ethyl ester (GSHree) | Sigma Aldrich | Cat#:92614-59-0 |
| Buthionine Sulfoximine (BSO) | Cayman | Cat#: 83730-53-4 |
| N-Acetyl-L-cysteine | Sigma Aldrich | Cat#: 616-91-1 |
| mitoTEMPO | Sigma Aldrich | Cat#:334850-99-5 |
| Recombinant mouse TNFα | PeproTech | Cat#: 315-01A |
| Recombinant mouse IFNγ | PeproTech | Cat#: 315-05 |
| Recombinant mouse IL1β | PeproTech | Cat#: 211-11B |
| Recombinant mouse IL7 | BioLegend | Cat#: 577804 |
| Leukocyte activation cocktail, with BD GolgiPlug | BD Pharmingen | Cat#: 550583 |
| Recombinant Human Fibronectin Fragment | Takara | Cat. #T100A |
| Evans blue dye | Sigma Aldrich | Cat#: 314-13-6 |
| Formamide | Sigma Aldrich | Cat#:75-12-7 |
| Collagenase type IV | Worthington | Cat#: LS004188 |
| DNase | Roche | N/A |
| ACK lysis buffer | Quality Biological | Cat#: 118-156-101 |
| 0.25% Trypsin | MP Biomedicals | Cat#: 1689649 |
| 10X Annexin V binding buffer | BD Pharmingen | Cat#; 51-66121E |
| Trizol Reagent | Thermo Fisher | Cat#: 15596018 |
| ProLong diamond antifade mountant | Thermo Fisher | Cat#: P36962 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | Cat#: 11668019 |
| Luciferase | ||
| Critical Commercial Assays | ||
| Intracellular GSH Assay Kit | Abcam | ab112132 |
| GSH/GSSG-Glo Assay | Promega | Cat#: V6611 |
| Superscript III first strand synthesis system | Thermo Fisher | Cat#: 18080051 |
| SYBR green mix | Bio-RAD | Cat#: 170-8882 |
| GeneArt Platinum Cas9 Nuclease | Thermo Fisher | Cat#: B25641 |
| GeneArt Precision gRNA Synthesis Kit | Thermo Fisher | Cat#: A29377 |
| Lipofectamine CRISPRMAX Cas9 Transfection Reagent | Thermo Fisher | Cat#: CMAX00015 |
| Gel Extraction Kit | QIAGEN | Cat#: 28706 |
| pGEM-T Easy Vector Systems | Promega | Cat#: A1360 |
| OneTaq DNA Polymerase PCR reagents | NEB | Cat#: M0480L |
| EasySep Mouse T Cell Isolation Kit | STEMCELL Technologies | Cat#: 19851 |
| DynabeadsTM mouse T-activator CD3/CD28 | Invitrogen | Cat#: 11452D |
| Experimental Models: Cell Lines | ||
| CT26 | ATCC | CRL-2638 |
| CT26HA | (Ding et al., 2012) | N/A |
| CT26HA.luci.rCD2 | This paper | N/A |
| A20 | ATCC | TIB-208 |
| A20.luci.rCD2 | This paper | N/A |
| MC38 | (Muroyama et al., 2017) | N/A |
| 4T1 | ATCC | CRL-2539 |
| 4T1HA | (Ding et al., 2012) | N/A |
| CT26HA.ΔTNFR1 | This paper | N/A |
| CT26HA. ΔCyba | This paper | N/A |
| 293FT | ATCC | PTA-5077 |
| Experimental Models: Organisms/Strains | ||
| HA-TCR Tg Mice | (Kirberg et al., 1994) | N/A |
| BALB/c Mice | Charles River Laboratories | 555 |
| Oligonucleotides | ||
| Please see Table S1 for qRT-PCR primer sequences | This paper | N/A |
| Please see Table S2 for sgRNA synthsis oligos and screening primer sequences for CRISPR/Cas9 | This paper | N/A |
| Recombinant DNA | ||
| 1D3scFv.CD28.CD3 | (Kochenderfer et al., 2010) | N/A |
| Mig.luciferase.rCD2 | This paper | N/A |
| pCL-Eco | Addgene | Cat#: 12371 |
| Software and Algorithms | ||
| Prism 4.0 | GraphPad Software | N/A |
| FlowJo software | Treestar | N/A |
| PIMSoft | Perimed | N/A |
| GeneArt CRISPR gRNA Design Tool | Thermo Fisher | N/A |
| Other | ||
| RPMI 1640 medium | HyClone | SH30027.02 |
| DMEM medium | HyClone | SH30243.01 |
| Opti-MEM Medium | Gibco | 31985-062 |
| Fetal Bovine Serum | HyClone | SH30396.03 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Gang Zhou (gzhou@augusta.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture Conditions
Murine colorectal cancer cell line CT26 (ATCC), mammary carcinoma 4T1 (ATCC) and B-cell lymphoma cell line A20 (ATCC) were cultured in RPMI 1640 (HyClone Laboratories) supplemented with 10% fetal bovine serum albumin (FBS), 1% penicillin/streptomycin (HyClone Laboratories), 1% non-essential amino acids and 1% glutamine (Corning) at 37°C in a 5% CO2 incubator. CT26 and 4T1 cells expressing model tumor antigen Influenza hemagglutinin (CT26HA and 4T1HA) were previously described (Ding et al., 2012). CT26HA.luci.rCD2 and A20.luci.rCD2 cells were generated by transducing tumor cells with retrovirus carrying luciferase and rat CD2. To harvest adherent tumor cells, cells were detached using 0.25% Trypsin with 1mM EDTA (MP Biomedicals) and resuspended in PBS or complete medium for mouse inoculations or in vitro culture, respectively.
Mice
BALB/c mice of 6–8 weeks old were purchased from the Charles River Laboratories. TCR-Tg (6.5) mice on a BALB/c (Thy1.1) background expressing an αβTCR specific for amino acids 110–120 from influenza HA presented by MHC class II molecule IEd were described previously (Ding et al., 2010). All mice were housed under specific pathogen-free conditions by Laboratory Animal Services of the Augusta University (AU). All animal experiments and procedures were performed in accordance with the institutional protocol and were approved by the Institutional Animal Care and Use Committee of Augusta University.
METHOD DETAILS
Flow Cytometry Analysis
For analyzing cytokine production by CD4+ T cells, tumor samples were digested and gently dissociated into single-cell suspension. Red blood cells were removed by ACK lysing buffer. After washing with PBS, cells were seeded in 12 well plate and stimulated with PMA and ionomycin for 4 hours. Following stimulation, cells were harvested and stained for surface markers followed by intracellular cytokine staining (ICS). To measure tumor cell viability after culture, cells at ~70% confluence were treated overnight under the specified conditions. Cells were harvested for Annexin V staining following manufacturer’s instruction. Following washing with PBS, cells were resuspended in 400ul 1x binding buffer and labeled with DAPI (0.5ug/ml). DAPI+ cells were counted as dead cells, Annexin V+DAPI− cells were considered apoptotic cells. The levels of reduced GSH in tumor cells were performed using the Intracellular GSH Assay Kit (Abcam) following the manufacturer’s instruction. DAPI was added to each sample before FACS analysis. Data were acquired using a LSRII and analyzed with FlowJo software (Treestar).
Intracellular ROS Measurement
ROS detection was performed using the cell permeant CM-H2DCFDA (Thermo Fisher Scientific). To measure ROS levels in ex vivo tumor samples, resected tumor tissues were digested by a solution containing 500u/ml collagenase type IV (Worthington) and 40 U/ml Dnase (Roche) for 45 mins at 37°C. The homogenate was filtered using a sterile cell strainer and the red blood cells were lysed using ACK lysis buffer. For ROS staining, 1×106 cells were collected in a 96 well plate and washed with PBS once. Cells were stained with 1uM CM-H2DCFDA for 30 minutes at 37°C. Cells were washed with PBS and stained for CD45.2 and DAPI before FACS analysis. ROS levels in live tumor cells were evaluated by gating on CD45.2−DAPI− cells. For detection of intracellular ROS in cultured tumor cells, cells were detached using 0.25% Trypsin with 1mM EDTA and stained for ROS using CM-H2DCFDA following the aforementioned procedure.
In Vitro Cell Culture
50,000 tumor cells in 500 μl volume were seeded to each well in a 24-well plate. Cells at 70–80% confluency were treated overnight under the specified conditions. The final concentrations of reagents used in culture were: 25 μg/ml mafosfamide (maf), 50 μg/ml melphalan (mel), 25 ng/ml recombinant mouse TNFα, 100 ng/ml recombinant mouse IFNγ, 100 ng/ml recombinant mouse IL1b. Antioxidants GSHree and mitoTEMPO were used at 6 mM and 100 μM, respectively. To prepare conditional medium from T cells, polyfunctional CD4+ T cells were generated as previously described (Ding et al., 2016). Briefly, splenocytes from HA-specific TCR-Tg mice (6.5) were stimulated with 2 μg/ml HA peptide in the presence or absence of rmIL7. 7 days later, CD4+ T cells stimulated in the presence of rmIL17 became polyfunctional effector cells (pCD4). CD4+ T cells stimulated without rmIL7 were used as control cells (cCD4). CD4+ T cells were enriched by density gradient centrifugation using Ficoll-Paque. CD4+ T cells were restimulated with peptide-pulsed BALB/c splenocytes for 2 days. Supernatants were collected and used as T cell-conditioned medium for cell culture. At the end of cell culture, cells were detached using 0.25% Trypsin with 1mM EDTA, washed with PBS, and stained with CM-H2DCFDA and DAPI to evaluate ROS level and cell viability, respectively.
Tumor Challenge and In Vivo Treatments
For subcutaneous tumor implantation, 1–2 × 106 tumor cells were inoculated to the right flank of mice. The size of tumors was monitored by caliper measurement of the tumor area every other day, and expressed as the product of two perpendicular diameters in square millimeters. When tumors reached the desired size (120–160mm2 for subcutaneous CT26HA), mice were randomly assigned to different groups to receive the specified treatments. For 4T1HA inoculation, 0.5 × 106 tumor cells were injected into the second mammary fat pad. To establish orthotopic colon cancer, mice were anesthetized and a small abdominal incision was made over the cecum. 2×104 CT26HA.luci.rCD2 cells in 20 ml saline were injected into the cecal wall from the serosal side. Wound was sealed with sterile stainless wound clip using the Autoclip Physicians Kit (Becton Dickson Primary Care Diagnostics). For systemic B-cell lymphoma model, 1×106 CT26HA.luci.rCD2 cells were injected to mice via tail vein. For adoptive T-cell transfer, spleens and lymph nodes from HA-TCR Tg mice were harvested to enrich for CD4+ T cells by MACS (Miltenyi Biotec). A total of 2.5~3 × 106 CD4+ TCR+ T cells were injected intravenously into each recipient. CTX was dissolved in PBS and i.p. injected to mice at the dose of 150 mg/kg. Mice were considered as cured when mice remained tumor-free for at least 30 days after complete tumor regression. N-Acetyl-L-cysteine (NAC) (Sigma) was administered to mice orally in drinking water at the dose of 10 mg/ml. Mice were given NAC-containing water 5 days prior to treatment with CTX+CD4 AT and continued post treatment for 21 days. The NAC-containing water was changed every other day with freshly prepared NAC. 20mM buthionine sulfoximine (BSO) (Cayman) was given to mice in drinking water for 21 days, starting the day CTX was injected to mice.
H&E Immunohistochemistry
Tumor tissue samples were kept in 10% formalin for 24 hours and transferred to 75% ethanol for long term storage. The tissue was dehydrated through a series of graded ethanol baths to displace the water and then embedded into wax blocks. Tissues were then sectioned (4μm thickness) using a rotary microtome (Leica RM 2135) and stained for hematoxylin and Eosin (H&E) following the standard procedure.
Immunofluorescence Analysis
For immunofluorescent (IF) staining, tumor tissue samples were flash frozen in dry ice. The frozen tissues were embedded in OCT in a cryomold and sectioned (7μm thickness) using a microtome cryostat (Leica). Slides were let dry for 15 mins and fixed in 4% paraformaldehyde (CAS: 50-00-0, Sigma) for 10 mins followed by washing with PBS to remove the OCT. For CD31 staining, sections were immersed in blocking buffer (PBS containing 2% bovine serum albumin (BSA)) for 30 minutes. Slides were then incubated with the rat anti-mouse CD31 (BD Biosciences) 1/50-diluted in blocking buffer. After 60 minutes incubation, slides were rinsed three times in PBS and incubated with the secondary antibody, donkey anti-rat Cy3 (Jackson ImmunoResearch), for 50 minutes. Following washing with PBS, sections were covered in ProLong Gold antifade mountant with DAPI (Molecular probes). For cleaved Caspase-3 (cCasp3) staining, sections were permeabilized for 15 minutes using 0.2% Triton X in PBS. Slides were washed with PBS twice, followed by overnight incubation with cleaved caspase-3 rabbit monoclonal antibody (Cell signaling technology) 1/100-diluted in blocking buffer. After washing twice with PBS, slides were incubated with Alexa Fluor 488 conjugated goat anti-rabbit secondary antibody (Thermo Fisher) 1/500-diluted in blocking buffer. Following washing with PBS, sections were covered with ProLong Gold antifade mountant with DAPI. Images were taken using Zeiss 780 upright confocal microscope and analyzed by ImageJ software. For 8-OHDG immunostaining, tumor tissues were formalin fixed and paraffin embedded (FFPE) per the standard protocol. The tissue sections were deparaffinized in xylene and rehydrated in a series of alcohol-water mixtures. Antigen retrieval was performed by placing slides in 10mM sodium citrate (PH 6.0) including Tween-20 and steamed for 30mins. The sections were then blocked in PBS containing 3% BSA for 60 minutes, and incubated with rabbit 8-OHdG polyclonal antibody (Bioss Antibodies) diluted in PBS containing Tween 20 and 3% BSA. After rinsing in PBST 3 times, sections were incubated with the secondary antibody Alexa Fluor555 donkey anti rabbit IgG (Thermo Fisher) in PBST containing 3% BSA for 60 minutes at room temperature. Slides were then rinsed in PBST and mounted with ProLong Gold antifade with DAPI. All images were taken using Zeiss 780 upright confocal microscope and analyzed by ImageJ software.
Measurement of Intracellular GSH Content and GSH/GSSG Ratio
Tumor cells were seeded in a flat-bottom 96-well plate at the density of 10,000 per well one day before the assay. On the day of assay, cells were subjected to the indicated treatments for 7 hours. The GSH/GSSG-Glo Assay Kit (Promega) was used to determine the contents of reduced GSH and its oxidized form GSSG in tumor cells following the manufacturer’s instruction.
RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
Tumor tissue samples from mice under different treatment conditions were resected and a small portion of the tissue was used for total RNA extraction. Tumor samples were homogenized using a motorized homogenizer followed by RNA extraction using TRIzol Reagent (Thermo Fisher Scientific). RNA samples were subject to on-column DNAse treatment to remove contaminating DNA. 1μg of RNA was used to generate cDNA using SuperScript III First-Strand Synthesis System (Thermo Fisher). qRT-PCR was performed using SYBR Green Mix (Bio-Rad), and cDNA amplification was performed on a BioRad iCycler equipped with an iCycler iQ Detection System. To verify that a single product was amplified, a melting curve was generated at the end of each run. β-actin was used to normalize target gene RNA expression levels. All primers were purchased from Integrated DNA Technologies. The sequences of the PCR primers are shown in Table S1.
Metabolomics Analysis
Tumor masses were resected on day 5 and day 10 after the specified treatment. Necrotic tissues, if present, were carefully removed. Resected tumor samples were flash frozen and sent to Metabolon for global metabolic profiling. Five replicate samples from five mice for each condition were analyzed as described below.
Sample Accessioning
Following receipt, samples were inventoried and immediately stored at −80°C. Each sample received was accessioned into the Metabolon LIMS system and was assigned by the LIMS a unique identifier that was associated with the original source identifier only. This identifier was used to track all sample handling, tasks, results, etc. All portions of any sample were automatically assigned their own unique identifiers by the LIMS when a new task was created; the relationship of these samples was also tracked. All samples were maintained at −80°C until processed.
Sample Preparation
Samples were prepared using the automated MicroLab STAR system from Hamilton Company. Several recovery standards were added prior to the first step in the extraction process for QC purposes. A series of organic and aqueous extractions were conducted to remove the protein fraction and recover chemically diverse metabolites. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. The sample extracts were stored overnight under nitrogen before preparation for analysis.
Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS)
All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried then reconstituted in solvents compatible to each of the four methods. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions, chromatographically optimized for more hydrophilic compounds. The second aliquot was also analyzed using acidic positive ion conditions, however it was chromatographically optimized for more hydrophobic compounds. The third aliquot was analyzed using basic negative ion optimized conditions using a separate dedicated C18 column. The fourth aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1×150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM Ammonium Formate, pH 10.8. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion.
Data Extraction, Compound Identification and Bioinformatics
Raw data was extracted, peak-identified and QC processed using Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. More than 3300 commercially available purified standard compounds have been acquired and registered into LIMS for analysis on all platforms for determination of their analytical characteristics. The informatics system consisted of four major components, the Laboratory Information Management System (LIMS), the data extraction and peak-identification software, data processing tools for QC and compound identification, and a collection of information interpretation and visualization tools for use by data analysts. The hardware and software foundations for these informatics components were the LAN backbone, and a database server running Oracle 10.2.0.1 Enterprise Edition.
Statistical Calculation
Selected metabolites were presented as line plot graphics. For pair-wise comparisons Welch’s t-test was used. Analysis of variance (ANOVA) was used for comparison of more than two conditions. A total of 601 compounds were analyzed and ANOVA contrasts were used to identify metabolites that differ significantly between various experimental groups.
Generation of CT26HA.ΔTNFR1 and CT26HA.ΔCyba Cell Lines by CRSIPR/Cas 9
DNA oligonucleotides used for gRNAs were designed using the GeneArt CRISPR gRNA Design Tool. The TNFR1 gene promoter and first exon sequences (NC_000072.6) and Cyba gene 2&3 exon region sequences (NC_000074.6) were put in gRNA design system. The DNA oligonucleotides for sgRNAs were synthesized by Integrated DNA Technologies (http://www.idtdna.com/). sgRNAs were synthesized and purified using GeneArt Precision gRNA Synthesis kit (A29377) following manufacturer’s protocol. The concentration of gRNAs were determined by NanoDrop (Thermo Fisher). One day before transfection, CT26HA cells were seeded in a 24 well plate at the density of 1×105 cells in 500μl DMEM medium (containing FBS but without P/S). The plated cells were transfected with TNFR1 or Cyba sgRNAs together with Cas9 Nuclease protein using Lipofectamine CRISPRMAX Cas9 Transfection Reagent. 48 to 72 hours post transfection, cells were harvested for limiting dilution in 96-well plates to grow single cell clones. After culture for 10–14 days, cell clones were picked for expansion. DNAs were extracted from cell clones for PCR screening to identify the ones with targeted gene editing. The PCR reaction condition is: 95° C 10 min, 36 cycles of 95°C 15″, 58°C 30″, 72°C 45″ and 72°C for 10 min. The primer sequences for PCR amplification of each gene are shown in Table S2. PCR products were analyzed by agarose gel electrophoresis, and DNA fragments with the expected size for each gene were recovered using Gel Extraction Kit (QIAGEN). The recovered DNA fragments were cloned into T-easy vector and transformed into DH5a Competent cells. Plasmids were sent to MCLAB (http://www.mclab.com/) for sequencing to confirm the deletion of the targeted sequences.
Virus Preparation and T Cell Transduction
Detailed protocol for T cell viral transduction was previously described (Kuczma et al., 2017). Briefly, T cells were purified from spleens of BALB/c mice on CD45.1 background using the EasySep Mouse T Cell Isolation Kit (STEMCELL Technologies). T cells were cultured in the presence of hIL2 (30U/ml; NCI) and stimulated with CD3/CD28 activation Dynal beads (Invitrogen) according to manufacturer’s instruction. Activated T cells were transduced twice in two consecutive days with retroviral supernatant using retronectin. One day after the last viral transduction, Dynal beads were removed and cells were washed with PBS before being used for adoptive transfer. The transduction efficiency was evaluated by staining T cells with FITC-labeled mouse anti-rat Fab antibodies or isotype control. 3 × 106 transduced T cells per mouse were used for adoptive transfer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were shown as mean ± SEM unless otherwise indicated. Data were analyzed using Prism 4.0 (GraphPad Software). The statistical significance of the results was determined using unpaired two-tailed Student’s t test. P values less than 0.05 were considered statistically significant.
Supplementary Material
Highlights.
Adoptive CD4+ T cell therapy in mice induces extensive metabolic changes in tumor
Immunotherapy leads to glutathione deficiency and oxidative DNA damage in tumor
TNF-α and chemotherapy synergize to intensify ROS levels in tumor via NADPH oxidase
Reducing tumor oxidative stress diminishes the efficacy of adoptive T cell therapy
Acknowledgments
We thank the Georgia Cancer Center FACS and Imaging Core Facility for cell sorting and imaging analysis. We appreciate the advice from Drs. T. Johnson, Y. He, and M. Shimoda. We thank Dr. H. Xu for assistance in statistical analysis. We are grateful for the technical support provided by Drs. P. Rodriguez, J. Trillo-Tinoco, M. Khan, and S. Vyas. This work was supported by NIH grants R01CA158202 and 1R01CA215523, and the American Cancer Society Research Scholar Grant (RSG-12-169-01-LIB) to G.Z., and R01 HL11879 to B.R.B.
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cmet.2018.05.012.
AUTHOR CONTRIBUTIONS
T.H. and Z.-C.D. performed research, analyzed results, and wrote the paper; W.P., T.L., C.L., T.C., C.X., and H.S. performed research; K.L., Z.H., and N.M. provided critical reagents; Y.H., B.R.B., and D.H.M. edited the paper; and G.Z. designed and performed research, analyzed results, and wrote the paper.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- Barrett DM, Grupp SA, June CH. Chimeric antigen receptor-and TCR-modified T cells enter main street and wall street. J Immunol. 2015;195:755–761. doi: 10.4049/jimmunol.1500751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017;35:7–14. doi: 10.1016/j.cytogfr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Carmel RJ, Brown JM. The effect of cyclophosphamide and other drugs on the incidence of pulmonary metastases in mice. Cancer Res. 1977;37:145–151. [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. 2016;17:364–368. doi: 10.1038/ni.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio IIC, Tuveson DA. ROS in cancer: the burning question. Trends Mol Med. 2017;23:411–429. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, Haraldsen G, Bogen B. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–383. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23:120–127. doi: 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM. Filling the tank: keeping antitumor T cells metabolically fit for the long haul. Cancer Immunol Res. 2016;4:1001–1006. doi: 10.1158/2326-6066.CIR-16-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZC, Blazar BR, Mellor AL, Munn DH, Zhou G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood. 2010;115:2397–2406. doi: 10.1182/blood-2009-11-253336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–2239. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZC, Liu C, Cao Y, Habtetsion T, Kuczma M, Pi W, Kong H, Cacan E, Greer SF, Cui Y, et al. IL-7 signaling imparts polyfunctionality and stemness potential to CD4(+) T cells. Oncoimmunology. 2016;5:e1171445. doi: 10.1080/2162402X.2016.1171445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray S, Mokyr MB. Cyclophosphamide and melphalan as immunopotentiating agents in cancer therapy. Med Oncol Tumor Pharmacother. 1989;6:77–85. doi: 10.1007/BF02985227. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, Corthay A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2:240. doi: 10.1038/ncomms1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med. 1988;167:1067–1085. doi: 10.1084/jem.167.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. 2016;5:29. doi: 10.1186/s40169-016-0110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, Ivanov A, Szymborska A, Patone G, Kunz S, et al. Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature. 2017;545:98–102. doi: 10.1038/nature22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Kaye SB. New antimetabolites in cancer chemotherapy and their clinical impact. Br J Cancer. 1998;78(Suppl 3):1–7. doi: 10.1038/bjc.1998.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirberg J, Baron A, Jakob S, Rolink A, Karjalainen K, von Boehmer H. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J Exp Med. 1994;180:25–34. doi: 10.1084/jem.180.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017;26:94–109. doi: 10.1016/j.cmet.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol. 2017;8:270. doi: 10.3389/fimmu.2017.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczma MP, Ding ZC, Li T, Habtetsion T, Chen T, Hao Z, Bryan L, Singh N, Kochenderfer JN, Zhou G. The impact of antibiotic usage on the efficacy of chemoimmunotherapy is contingent on the source of tumor-reactive T cells. Oncotarget. 2017;8:111931–111942. doi: 10.18632/oncotarget.22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Ding Z, Cao Y, Liu C, Habtetsion T, Yu M, Lemos H, Salman H, Xu H, Mellor AL, Zhou G. Alkylating agent melphalan augments the efficacy of adoptive immunotherapy using tumor-specific CD4+ T cells. J Immunol. 2015;194:2011–2021. doi: 10.4049/jimmunol.1401894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo B, Nitti M, Furfaro AL, Colla R, Ciucis CD, Marinari UM, Pronzato MA, Traverso N, Domenicotti C. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid Med Cell Longev. 2016;2016:6235641. doi: 10.1155/2016/6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31. doi: 10.1038/nrclinonc.2016.60. [DOI] [PubMed] [Google Scholar]
- Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, Carrera-Haro MA, Ghasemzadeh A, Marciscano AE, Velarde E, et al. Stereotactic radiotherapy increases functionally suppressive regulatory T cells in the tumor microenvironment. Cancer Immunol Res. 2017;5:992–1004. doi: 10.1158/2326-6066.CIR-17-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ, Havell EA. The antitumor function of tumor necrosis factor (TNF) II. Analysis of the role of endogenous TNF in endotoxin-induced hemorrhagic necrosis and regression of an established sarcoma J Exp Med. 1988;167:1086–1099. doi: 10.1084/jem.167.3.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36:71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Kalos M, Zheng Z, Levine B, June C. Chimeric antigen receptor therapy for B-cell malignancies. J Cancer. 2011;2:331–332. doi: 10.7150/jca.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12:677–686. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest. 2015;125:3392–3400. doi: 10.1172/JCI80010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:701–703. doi: 10.1016/j.immuni.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen-presenting cells in tumors. Immunity. 2018;48:91–106e6. doi: 10.1016/j.immuni.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YJ, Corona AL, Gemta LF, Vincent BG, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2 doi: 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol. 2017;46:14–22. doi: 10.1016/j.coi.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- Verhoef C, de Wilt JH, Grunhagen DJ, van Geel AN, ten Hagen TL, Eggermont AM. Isolated limb perfusion with melphalan and TNF-alpha in the treatment of extremity sarcoma. Curr Treat Options Oncol. 2007;8:417–427. doi: 10.1007/s11864-007-0044-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH, June CH. Engineering T cells for cancer: our synthetic future. Immunol Rev. 2014;257:7–13. doi: 10.1111/imr.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Kryczek I, Dostal L, Lin H, Tan L, Zhao L, Lu F, Wei S, Maj T, Peng D, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–1105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85:462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.